
Audrey Kwan1,7, Aishwarya Yuvaraj2,7, Shloka Raghavan3,7, Ansh Rai4,7, Tyler Shern4,7, Krithikaa Premnath5,7, Ria Kolala4,7, Ishani Ashok4,7, Edward Njoo7
(1) Dublin High School, Dublin, CA; (2) American High School, Fremont, CA; (3) Amador Valley High School, Pleasanton, CA; (4) Mission San Jose High School, Fremont, CA; (5) Dougherty Valley High School, San Ramon, CA; (6) Amador Valley High School, Pleasanton, CA; (7) Center for Advanced Study, Aspiring Scholars Directed Research Program, Fremont, CA
Abstract
Model organisms are often used in biological testing because they have been widely studied and are easy to maintain and breed in a laboratory. However, the validity of screening clinical candidate compounds in model organisms depends on the degree to which interactions between the small molecule and its biochemical target in a non-human system simulates that of the human homolog. In practice, sequence similarity is often used to predict the similarity between a biochemical system in humans and its homologous counterpart in model organisms, excluding structural information about the physical interactions between a small molecule and its biochemical target. One such target is the kinesin Eg5, which plays a critical role in bipolar microtubule assembly during division of human cells. Inhibition of Eg5 by the small molecule monastrol has been investigated for potential antiproliferative applications in cancer therapy, but it is unclear how studies performed in non-mammalian model organisms might translate in a human system. Here, we identify trends in nucleotide and amino acid sequence similarity between the human gene KIF11 and its encoded protein, the kinesin Eg5, in comparison to homologous counterparts in ten model organisms. Moreover, we perform homology modeling of the allosteric binding pocket to identify a structural basis for predicted changes in ligand binding affinity. Our results indicate that, for both nucleotides and amino acids, there is a weak correlation between sequence similarity and binding affinity within model organisms. Thus, sequence similarities may not be the most accurate means of representing how well a model organism will model the human system. Identifying specific structural changes within target proteins and analyzing how those changes affect binding of the ligand would more accurately indicate how characteristic a model organism’s system is of human models.
Introduction
Model organisms which are often used to study and understand biological processes, prior to clinical trials in humans, are widely used in development of new medicines or therapies.13 They are selected based on their genetic similarity to humans and the ease in manipulating them for experimental reasons.12 Model organisms can be single-celled organisms like bacteria (Escherichia coli), or multicellular organisms, such as house mice (Mus musculus) and zebrafish (Danio rerio).
In 2020, it is estimated that there will be 1.8 million new cases of cancer and over 600,000 cancer-related deaths in the United States.11 With the high mortality rate of the disease, there has been a strong emphasis on creating new or improving known anti-cancer treatments. Such research has led to the creation of many small molecule therapeutics in cancer treatment, such as doxorubicin, cabozantinib, and lapatinib.8
Monastrol, a small molecule dihydropyrimidine (DHPM), was discovered by the Mitchison group in 2000 and has been evaluated as a potential antiproliferative agent with clinical potential.5 The molecule has been shown to allosterically inhibit the mitotic kinesin Eg5, a motor protein that ensures microtubule motility for successful cell division.7 The inhibition of this protein with monastrol results in mitotic arrest and ultimately cell death.5 The crystal structure of monastrol bound to the human kinesin Eg5 has also been reported on Protein Data Bank (PDB:1X88) and it was found that the ligand forms hydrogen bonding interactions with glutamic acid (GLU) 116 and GLU118 along with engaging in various hydrophobic interactions in its allosteric binding pocket.6 There have already been many research studies on kinesin-like proteins in various organisms, which include research performed by Sakowicz and co-workers on the antitumor aspects of CK0106023, a small molecule kinesin inhibitor, on mouse models,10 and research performed by Exterier and coworkers on the obstruction of tumor development via inhibition of KIF11, the kinesin family member 11 gene, with dimethylenastron (DMN) in zebrafish and chick models,1 The model organisms included in this research are house mice (Mus musculus), zebrafish (Danio rerio), thale cress (Arabidopsis thaliana), fruit flies (Drosophila melanogaster), fission yeast (Saccharomyces cerevisiae), Brewer’s yeast (Schizosaccharomyces pombe), roundworms (Caenorhabditis elegans), blood parasites (Leishmania donovani), slime mold (Dictyostelium discoideum), and soybeans (Glycine max). Previous studies have shown that the loop 5 and alpha 3 domains of kinesin-like proteins are poorly conserved in invertebrates, and thus the protein is inactive in the binding of monastrol in invertebrate models.6
Homology modeling is the structural prediction of a protein without a reported crystal structure based on known structures of similar, homologous proteins.2 This provides structural insight for proteins without having to obtain structures via X-ray crystallography or cryogenic electron microscopy (cryo-EM), both of which are resource and time-intensive.
This research aims at providing structural insight into which model organisms are the most suitable for testing the efficiency of small molecule drug candidates that target the human kinesin Eg5, such as monastrol. This is pursued by conducting a sequence analysis of the DNA and protein sequences of the kinesin-like proteins in the model organisms, and subsequent homology modeling of the binding pocket with point substitutions of residues in or near the reported binding site. The homology modeling used in this research aims to provide structural insight into potential differences in the interactions between monastrol and human kinesin Eg5. The method also aims to show how alterations of residues that shape the binding site affect ligand binding. We hypothesize that model organisms whose kinesin protein and nucleotide sequences most closely align with the human kinesin Eg5 sequence will generate similar results in the binding affinity of monastrol. The results of this study provide structural insight into this idea.
Methods
Nucleotide similarity
The order of the four different nucleotides in DNA is unique in each organism. In order to determine the similarity of kinesin-like proteins in model organisms compared to that in humans, a nucleotide comparison was conducted. The comparison measured the similarity of the DNA that encoded the kinesin-like protein in humans to each of the model organisms. The nucleotide sequence of the kinesin family member 11 (KIF11) protein in Homo sapiens (Humans) was acquired through the NCBI database.4 The nucleotide sequences for the kinesin-like proteins of each of the ten model organisms were then found on NCBI and run through nucleotide BLAST (write full form of BLAST here, then no need to write it in Results section). The kinesin-like proteins for each model organism are as follows: kinesin-like protein at 61F (Klp61F) in Drosophila melanogaster (fruit fly), P-Loop containing nucleoside triphosphate hydrolases superfamily protein (AT2G28620) in Arabidopsis thaliana (thale cress), kinesin family member 11 (KIF11) in Mus musculus (house mouse), Kip1p (KIP1) in Saccharomyces cerevisiae (brewer’s yeast), kinesin family member 11 (KIF11) in Danio rerio (zebrafish), kinesin-like protein (bmk-1) in Caenorhabditis elegans (roundworm), kinesin-like protein Cut7 (cut7) in Schizosaccharomyces pombe (fission yeast), OSM3-like kinesin, putative (LDBPK_170890) in Leishmania donovani (blood parasite), kinesin family member 13 (KIF13) in Dictyostelium discoideum (slime mold), and kinesin-like protein KIN-5C (LOC100800246) in Glycine max (soybean). Snapgene Viewer was used to visualize each nucleotide sequence. The European Bioinformatics Institute’s (EBI) EMBOSS needle and Pairwise Sequence Alignment were used to compare the nucleotide sequences of the kinesin-like proteins in each model organism to the human KIF11 nucleotide sequence.
Protein Similarity
The amino acids that make up the monastrol binding pocket in each kinesin-like protein are likely to vary between species. A protein similarity comparison was conducted to analyze how different the binding pocket proteins were between each model organism compared to that of humans. The protein sequence of the human kinesin Eg5 protein was obtained through the Protein Data Bank (PDB:1X88). Protein BLAST was used to find the respective protein sequences of each kinesin-like protein in each model organism. The protein sequences were compared using the European Bioinformatics Institute’s (EBI) EMBOSS needle and Pairwise Sequence Alignment.
Significant Proteins in Binding Pocket
The changes of thirteen significant amino acids in the monastrol binding pocket were identified. The crystal structure of the human kinesin Eg5 protein (PDB:1X88) was obtained through the PDB. The crystal structure was visualized through UCSF Chimera, where thirteen amino acids that defined the binding pocket were identified. The significant amino acids of each model organisms’ kinesin-like protein were characterized based on their differences in charge, hydrophobicity, and hydrophilicity.
Molecular Docking
The crystal structures of each of the kinesin-like proteins for the model organisms used in this study were not reported on the PDB. As a result, each amino acid was manually altered through homology modeling. This was performed using UCSF Chimera to adjust the human kinesin Eg5 protein to resemble the structure of the kinesin-like proteins from each model organism. Only the residues lining the binding pocket were altered in UCSF Chimera. For certain kinesin-like proteins, some of the thirteen significant amino acids were not listed on BLAST. Unlisted amino acids were changed into glycine on UCSF Chimera to remove the side chain while preserving the general secondary structure. The grid box center of the binding pocket was obtained through UCSF Chimera as well, and the x, y, z coordinates of the center are 46.146, 25.739, 111.901 respectively. The x, y, z coordinates of the grid box size are 60, 42, 58 respectively. These docking parameters were validated and optimized by re-docking monastrol to the crystal structure of Eg5, which returned a docked binding pose with a high degree of similarity with that which was reported in the original crystal structure. SwissDock, a web-based docking server, was used to dock monastrol to each of the kinesin-like proteins in the different model organisms to find the binding affinities, binding poses, and RMSD values of each of the model organisms.3 The RMSD value was calculated using UCSF Chimera by comparing the ligand binding poses of each of the model organisms with the ligand binding pose of the crystal structure of monastrol bound to the human kinesin Eg5 (PDB:1X88). This was used to determine the degree of similarity in the binding poses of monastrol bound to each homology model compared to that of wild type human Eg5.
Results
Nucleotide Similarity
An initial comparison was conducted on Basic Local Alignment Search Tool (BLAST) to determine the degree of nucleotide similarity in the sequence that codes for kinesin Eg5 in humans, with homologous genes in model organisms. The KIF11 gene sequence was obtained from the NCBI database.4 The similarity of the nucleotide sequences is shown in Figure 1. The mouse model had the highest nucleotide similarity, whereas the other model organisms had a similarity between 30-50%.
Protein Similarity
A comparison of protein sequence similarity was also conducted between the model organisms’ kinesin-like proteins and the human kinesin Eg5 protein. The human kinesin Eg5 protein sequence was found on the Protein Data Bank (PDB:1X88) and the kinesin-like protein sequences of the model organisms were found through Protein BLAST. The comparison was done by running the protein sequences through the European Bioinformatics Institute’s (EBI) EMBOSS needle and Pairwise Sequence Alignment. The percent similarity of the amino acid sequences is shown in Figure 1. The mouse model had the highest protein similarity and the remaining models had a similarity between 20-60%.

Figure 1a: Nucleotide sequence similarity in kinesin-like proteins in model organisms in comparison with human kinesin Eg5

Figure 1b: Protein sequence similarity in kinesin-like proteins in model organisms in comparison with human kinesin Eg5
Significant proteins in the binding pocket
A depiction of the human kinesin Eg5 bound to monastrol is shown in Figure 2, along with the chemical structure of the ligand. Thirteen amino acids in the human kinesin Eg5 protein were selected based on their significance in the topography of the allosteric binding pocket. The amino acid name and position are indicated in Table 1. It was determined that the house mice protein sequence had the least variation in significant amino acids compared to the sequences of the other model organisms, with the entirety of the residues lining the binding pocket fully conserved. The zebrafish protein sequence had the second least variation, while the blood parasite sequence had the most variation in significant amino acids.

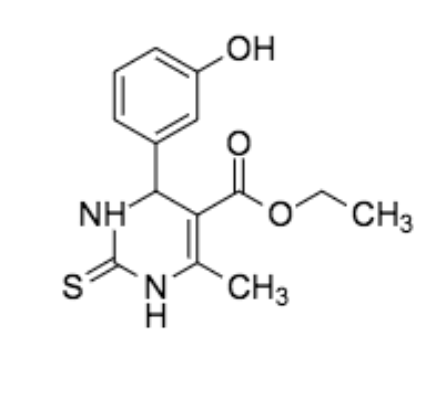
Figure 2: Crystal structure of human kinesin Eg5 bound to Mg-ADP and monastrol obtained from Protein Data Bank (PDB: 1X88); Chemical Structure of monastrol

Molecular Docking
In order to assess the impact of structural changes in the binding pocket of kinesin-like proteins across the ten selected model organisms on ligand binding affinity, monastrol was docked to the homology models of the kinesin-like proteins of each model organism. Select overlays of the docked ligand are depicted in Figure 5.
Figure 3 depicts a comparison of the RMSD values for the lowest <#DELTA>G value (highest binding affinity) of each binding pose. Figure 4 illustrates the binding affinity (<#DELTA> G) of the binding pose for the lowest RMSD value.
In the human model, the ligand forms a hydrogen bond between the nitrogen on its thiourea functional group and GLU116, and between the oxygen located on the aryl group and GLU118. In the fruit fly model, arginine (ARG) 119 changes to GLU119, causing the ligand to change its binding pose in a way that allows the sulfur on the thiourea of the ligand to create a hydrogen bond with the residue. GLU119 is further away from the center of the binding pocket than GLU116 and GLU118, causing the binding affinity of the fruit fly model to be lower than the one for the human model. The ligand also lacks the hydrogen bonds seen in the human model with GLU116 and GLU118 because the residues have changed to become valine (VAL) 116 and asparagine (ASN) 118. The loss of these hydrogen bonds and the hydrogen bonding interaction with GLU119 creates a low binding affinity for the fruit fly model, which is consistent in both its lowest <#DELTA>G value and the <#DELTA>G value associated with its lowest RMSD value. However, the high RMSD values depicted in both Figure 3 and Figure 4 indicate that the ligand is shifted in its orientation in the binding pocket of the protein. This is consistent with prior experimental reports that monastrol is inactive against Drosophila melanogaster due to poor conservation in the binding pocket.7
The zebrafish model had a lowest <#DELTA>G value that was slightly lower than the human model’s lowest <#DELTA>G value. At its lowest <#DELTA>G value, the ligand loses its hydrogen bonding interaction with GLU116 and GLU118, but at the <#DELTA>G value for its lowest RMSD, the ligand maintains a hydrogen bonding interaction with GLU116. The variations between the different binding poses for the zebrafish model is minimal and is consistent with previously reported observations that monastrol is active in preventing successful mitosis in Danio rerio cells.6 In both Figure 3 and Figure 4, the house mouse model had a <#DELTA>G value of -7.89 kcal/mol, which matched the <#DELTA>G value of the human model. The house mouse model had two hydrogen bonds between the ligand and the amino acids within the binding pocket, both of which are also apparent in the human model. Consequently, the maintenance of the hydrogen bonds portrayed in the human model aids in maintaining similar binding affinities.
As shown in Figure 3, the slime mold model has a lowest <#DELTA>G value of -8.50 kcal/mol. However, the <#DELTA>G value of the model is approximately -7.88 kcal/mol at the lowest RMSD value, as shown in Figure 4. In the binding poses of the ligand for both <#DELTA>G values, the ligand maintains a hydrogen bonding interaction between the nitrogen on its thiourea functional group and serine (SER) 116. However, in the binding pose at the lowest binding affinity, the ligand creates an additional hydrogen bonding interaction between the 3-hydroxy group and lysine (LYS) 118. The binding pose with the <#DELTA>G value that corresponded with the lowest RMSD is likely to be more predictive of the actual binding pose of the ligand, meaning that the actual binding pose is likely to not include a hydrogen bonding interaction with LYS118. For the thale cress model, a different condition occurs. The ligand forms no hydrogen bonding interactions at any of its binding poses, which suggest that other positional factors are affecting the ligand’s low binding affinity. While this may cause the ligand to adopt a different pose with comparable or improved predicted binding affinity, as binding the poses differ at RMSD values of 2.754 and 2.502, it is unclear whether changes in the binding pose perturbs the ligand’s ability to act as an allosteric inhibitor.
For all the model organisms, regions relatively deeper in the binding pocket had more hydrophobic interactions while the remaining portions had neutral regions. Overall, the best binders displayed more hydrophobic interactions in their binding pockets while the less successful binders had more neutral and hydrophilic regions located deep inside their binding regions. Thus, a correlation can be made between hydrophobic interactions and lower <#DELTA>G values for the lowest RMSD value.

Figure 3: Percent deviation in lowest <#DELTA>G value (kcal/mol) and RMSD in altered kinesin-like proteins of each model organism in comparison with human kinesin Eg5. Provides a measurement for the lowest binding affinity of the ligand in each model organism, as well as a scale of how close the binding affinity is to the human model.

Figure 4: Percent deviation in <#DELTA>G value (kcal/mol) and lowest RMSD in altered kinesin-like proteins of each model organism in comparison with human kinesin Eg5. Provides a measurement for the binding affinity of the ligand in each model organism at which it is closest to the binding affinity is to the human model.

Figure 5: Monastrol bound to Kinesin Eg5 (protein code 1X88) (gray) overlaid with the altered kinesin-like proteins (blue) of model organisms in the following order: house mouse (a), fruit fly (b), thale cress (c), zebrafish (d), slime mold (e)
There is a weak correlation between sequence similarity and both percent change in <#DELTA>G values and RMSD values. Generally, as sequence similarities increase, the percent change in <#DELTA>G values and RMSD values decrease. However, the correlation appears to be weak and inconsistent among the model organisms. Thus, sequence similarities may not be the most accurate means of representing how well an interaction within a model organism will model the human system. Instead, identifying specific structural changes within model organism proteins and how those changes affect binding of the ligand would more accurately indicate how characteristic a model organism’s system is of human models.
Discussion
The house mouse and zebrafish homology models had the highest similarity in both protein and nucleotide sequences between its kinesin-like protein, Kif11, and the human homolog, kinesin Eg5. Moreover, this translated to the greatest degree of similarity in the binding pocket topography with the human kinesin Eg5 protein. The house mouse model had the same binding affinity as the human model because both had the same amino acids in the binding pocket. It also had the lowest RMSD value, indicating that its binding pose is very accurate and almost entirely matches up with the binding pose of the crystal structure of the human kinesin Eg5 protein. These results suggest using house mice models or zebrafish models for studying allosteric inhibitors for kinesin as they demonstrate the greatest amount of similarity to how these molecules would behave in the human system. These results are also consistent with prior research that indicates that both house mice and zebrafish cells undergoing mitosis respond to monastrol in manners similar to that of human cells.6 The altered kinesin-like protein of thale cress docked to monastrol had the highest binding affinity. However, the RMSD value was relatively high, suggesting that the virtually docked conformation does not closely resemble the crystal structure of monastrol docked to the human protein.
While sequence similarities of nucleotide base pairs and protein residues may give an approximation of the similarity between a human biochemical system and a homologous system in a model organism, structural changes on the targeted protein-ligand interface provide greater insight into the biophysical basis for drug binding, and by extension, biological activity. This is due to the fact that the identities of the amino acids within the binding pocket may have a greater effect on the binding affinity of the ligand than the sheer number of sequential changes to the protein.
Previous studies have indicated that monastrol is a poor inhibitor of cell viability in invertebrates. However, the results of this research have shown that invertebrate and vertebrate models have similar binding affinities when the ligand is docked into the altered kinesin binding pocket. This is because the homology modeling used in this study only altered the residues in the primary structure of the protein. While the alterations to the primary structure of the protein created minor differences between the binding affinities in each model, the RMSD values were very large. This suggests that the binding affinity of the ligand to the kinesin-like proteins within each model organism may not be solely attributed to substitutions within the residues of the protein.
References
1. Exertier, Prisca, et al. “Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5.” Oncotarget 4.12 (2013): 2302.
2. Gromiha, M. Michael, Raju Nagarajan, and Samuel Selvaraj. “Protein Structural Bioinformatics: An Overview.” (2019): 445-459.
3. Grosdidier, A., Zoete, V., & Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic acids research, 39(Web Server issue), W270–W277.
4. “KIF11 Kinesin Family Member 11 [Homo Sapiens (Human)] – Gene – NCBI.” National Center for Biotechnology Information, U.S. National Library of Medicine, www.ncbi.nlm.nih.gov/gene/3832.
5. Maliga, Zoltan, Tarun M. Kapoor, and Timothy J. Mitchison. “Evidence that monastrol is an allosteric inhibitor of the mitotic kinesin Eg5.” Chemistry & biology 9.9 (2002): 989-996.
6. Maliga, Zoltan, and Timothy J. Mitchison. “Small-molecule and mutational analysis of allosteric Eg5 inhibition by monastrol.” BMC chemical biology 6.1 (2006): 2.
7. Mayer, Thomas U., et al. “Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen.” Science 286.5441 (1999): 971-974.
8. Pathak, Akshat, et al. “Present and Future Prospect of Small Molecule & Related Targeted Therapy Against Human Cancer.” Vivechan international journal of research 9.1 (2018): 36.
9. Sanders, Jeremy. Veusz – A Scientific Plotting Package, Version 3.2.1., 2003, GitHub, 2020, https://veusz.github.io/download/
10. Sakowicz, Roman, et al. “Antitumor activity of a kinesin inhibitor.” Cancer research 64.9 (2004): 3276-3280.
11. Siegel, Rebecca L., Kimberly D. Miller, and Ahmedin Jemal. “Cancer statistics, 2020.” CA: A Cancer Journal for Clinicians 70.1 (2020): 7-30.
12. “Using Research Organisms to Study Health and Disease.” National Institute of General Medical Sciences, U.S. Department of Health and Human Services, https://www.nigms.nih.gov/education/fact-sheets/Pages/using-research-organisms.aspx.
13. “What Are Model Organisms?” Facts, The Public Engagement Team at the Wellcome Genome Campus, 3 Mar. 2017, https://www.yourgenome.org/facts/what-are-model-organisms.



{kind=link}