
Abstract
This study evaluates the performance and interpretability of featurizer model combinations for predicting aqueous solubility in drug discovery. Using the AquaSolDB-clean dataset (about 10,000 molecules), the Light Gradient Boosting Machine (LightGBM), Random Forest, and Extreme Gradient Boosting Machine (XGBoost) models were assessed with Linear Regression as a baseline model, in conjunction with Extended-Connectivity Fingerprints (ECFP), Chem Bidirectional Encoder Representations from Transformers (ChemBERTa 2.0), and Graph Convolutional Networks (GCN). Among all combinations, LightGBM and ChemBERTa achieved the best performance with a root mean squared error (RMSE) of 1.181 (95% CI: 1.138–1.224). Paired t-tests against all other model-featurizer combinations confirmed that this combination performed significantly better than all others except XGBoost + ChemBERTa and Linear Regression + ChemBERTa, for which the differences were not statistically significant (p = 0.12 and p = 0.17, respectively). The study also investigates molecular substructures driving solubility predictions using Shapley Additive Explanations (SHAP), finding specific chemical structures contributing to accurate predictions. Confidence intervals are reported for each combination, statistical significance testing is performed using paired t-tests, and a critical analysis of featurizer contributions is provided, offering interpretability through feature attribution methods.
Introduction
Aqueous solubility is a critical physicochemical property that plays a key role in many scientific and industrial applications, particularly in drug discovery, chemical engineering, and environmental science1. In pharmaceutical development, solubility directly affects drug absorption and bioavailability. Poor solubility is one of the leading causes of failure in drug candidates, making accurate prediction of solubility an important step in early-stage screening.
Traditional methods for measuring solubility rely on experimental techniques, which are often timeconsuming and expensive2. As a result, there has been growing interest in developing computational approaches that can efficiently predict solubility from molecular structure. Machine learning models have become a popular solution due to their ability to learn complex relationships between chemical features and their corresponding solubility values.
Recent work has explored a wide range of modeling approaches, including basic linear regression models, ensemble learning methods, deep neural networks, and graph-based models. Common representations employed by these studies include molecular descriptors, fingerprints, SMILES-based embeddings, and graph structures, each capturing different aspects of chemical information. However, existing studies often focus on a focused or single set of models or representations, making it difficult to determine which approach among the many that exist is most efficient. In addition, many high-performing methods rely on complex feature engineering techniques or domain-specific knowledge, which can limit their practical applicability. There is a need for a comparative study of the three main featurization types: graph-based, SMILES-based, and fingerprint-based, across different model structures including gradient boosting, decision trees, and graphbased deep learning. This study aims to address these challenges by evaluating multiple machine learning models for solubility prediction using a consistent dataset and evaluation strategy. Different molecular representations, including sequence-based embeddings, graph-based features, and fingerprint-based features, are explored to assess their impact on performance. In addition, model performance is evaluated using cross-validation and statistical analysis to ensure reliability. The following question is aimed to be answered through the study: How do different molecular featurizers, from graph-based to language-based, affect the performance and interpretability of a machine learning model in predicting solubility?
Related Work
Solubility prediction has been widely studied using both traditional machine learning and deep learning approaches, with a strong focus on identifying effective model architectures and molecular representations. Early approaches relied on linear and statistical models3. More recent work has explored ensemble methods, graph-based learning, transformer-based architectures, and common regression models such as LightGBM and Random Forest4,5.
Several studies have demonstrated the effectiveness of ensemble and tree-based models for solubility prediction. LightGBM has been shown to outperform multiple baseline models including partial least squares, ridge regression, k-nearest neighbors, decision trees, extra trees, random forests, and support vector machines, while also revealing relationships between structural features and solubility6. Other work comparing random forests, LightGBM, Least Absolute Shrinkage and Selection Operator (LASSO), and partial least squares found that LASSO achieved the best predictive performance, while random forests offered a strong balance between model complexity and predictive ability7. Ensemble QSPR models combining random forest and XGBoost have also been shown to improve accuracy compared to individual models8. Additional studies using random forest, XGBoost, LightGBM, and CatBoost further highlight the importance of ensemble methods, while also exploring interpretability in these models9. These works establish ensemble learning as a strong baseline for solubility prediction.
Deep learning approaches have also been widely explored to capture more complex relationships. Transformer based models such as SolTranNet have demonstrated improved performance over traditional linear models by learning contextual representations from SMILES strings, although larger models were not always beneficial for prediction performance10. Other work using convolutional neural networks, deep neural networks, and generalized regression neural networks found that GRNN achieved the best performance when modeling solubility under varying temperature and pressure conditions11. These studies show that deep learning models can effectively learn nonlinear relationships from molecular data.
Graph-based learning methods have gained attention due to their ability to directly model molecular structure. Studies using graph convolutional networks and graph attention mechanisms have shown strong performance, especially on high-quality datasets, although these models were sensitive to noise and errors12. In contrast, descriptor-based approaches have been shown to be more stable, with large-scale analysis identifying hundreds of molecular descriptors that significantly contribute to solubility prediction12. Additional work comparing molecular descriptors and Morgan fingerprints with random forest models showed that descriptor-based features outperform fingerprint-based representations, while also providing interpretability through SHAP analysis13. More recent work has also explored combining multiple representations, including electrostatic potential maps, molecular graphs, and tabular descriptors, where tabular features combined with XGBoost achieved the best performance14. These findings highlight the importance of feature representation in solubility prediction.
Beyond purely data-driven approaches, hybrid methods that incorporate thermodynamic modeling have also been explored. COSMO-SAC and COSMO-RS models have been combined with machine learning techniques to improve predictive accuracy. For example, Gaussian process regression has been used to correct deviations in COSMO-SAC predictions, significantly improving alignment with experimental data15. Similarly, neural networks combined with COSMO-RS achieved strong predictive performance while requiring less data16. Other work has proposed integrating machine learning predictions with thermodynamic properties such as activity coefficients, fusion enthalpy, and melting point to improve solubility estimation17. These approaches show that incorporating physical chemistry knowledge can improve prediction quality.
Additional studies have explored specialized feature engineering and modeling techniques. Regression methods combining LASSO and support vector regression achieved improved accuracy for high-temperature solubility prediction18. Molecular dynamics simulations have also been used to extract physicochemical features, which were then used in models such as random forest, extra trees, XGBoost, and gradient boosting, with gradient boosting achieving the best performance19. Other work using COSMO-RS with descriptors derived from quantum chemical methods applied models such as association rule mining, decision trees, and neural networks, showing that specific descriptor types have varying levels of influence on solubility prediction20. While these approaches improve performance, they often require complex feature extraction processes.
Despite the large body of work, several important gaps remain. First, many studies focus on a limited set of model types, rather than performing a direct comparison across fundamentally different model structures under the same experimental setup. Second, there is no clear consensus on the most effective molecular representation, with different studies favoring SMILES-based embeddings, graph-based features, molecular descriptors, or fingerprints. Third, many high-performing approaches rely on complex feature engineering techniques such as molecular dynamics simulations or thermodynamic modeling, which limits their practical applicability. Finally, interpretability is not consistently applied across different modeling approaches, making it difficult to fully understand model behavior.
To address these limitations, three models of varying structures, paired with molecular fingerprints like ECFP, ChemBERTa 2.0, and GCN have been tested under the same conditions on the same dataset to find optimal model-featurizer pairings21,22,23.
Methods
Dataset and Featurizers
AquaSolDB-clean or AqSolDBc is a curated version of the widely-used AquaSolDB dataset24. By removing repeating data points and values below the limit of detection or quantification, the probability of error is reduced. The clean dataset contains approximately ten thousand values and includes many molecular features, including SMILES strings. Duplicates and extreme outliers were removed. The used feature from the dataset includes the SMILES strings and the predictions were compared to the experimentally measured logS values. Potential limitations include its limited size and chemical diversity relative to other datasets (e.g., ESOL, ZINC).
Featurizers- In this study, a diverse set of molecular featurizers were employed to capture complex chemical information. ECFP converts molecular structures into binary vectors, encoding substructural features important for solubility prediction. This fingerprint-based approach is commonly used due to its ability to represent the detailed connectivity patterns of atoms. ChemBERTa 2.0, a language based featurizer, applies transformer models to interpret molecular structures formatted as SMILES strings. It is an experienced featurizer, benefiting from extensive pre-training on vast chemical datasets, enabling it to record complex patterns within molecular sequences. Lastly, the GCN represents molecules as graphs, where the nodes and edges correspond to atoms and bonds. This graph-based method provides a detailed understanding of the influence of structural configurations on chemical behavior. These featurizers were chosen to span symbolic, language-based, and structural paradigms. Future work could expand this to include DeepChem’s featurizers such as Mol2Vec, MACCS, and others.
Models
To evaluate the influence of model architecture on solubility prediction, four different machine learning models were implemented. These included tree-based ensemble methods like Random Forest, LightGBM, and XGBoost, and a baseline Linear Regression model representing a traditional QSAR-style approach. These models were chosen to reflect a range of capacities for capturing non-linear relationships and molecular complexity. However, each model type comes with its limitations. For example, linear regression may underfit complex patterns, while tree-based models can overfit small datasets or become biased toward dominant substructures.
Experimental Setup
The input features for the model included only the SMILES strings representing molecular structure, and no variables that were direct indicators of solubility were utilized to prevent data leakage. To evaluate generalization to novel chemical scaffolds, a scaffold splitting strategy with an 80/10/10 split for training, validation, and testing was adopted. This ratio is widely accepted as a strong balance between training capacity and realistic generalization. Scaffold splitting ensures that molecules with similar core structures are grouped together, making the test set more challenging and reflective of real-world generalization scenarios compared to random splits. To enhance statistical reliability, 5-fold cross-validation was applied across all model–featurizer combinations. For each fold, RMSE was computed, reporting both the mean and standard deviation to quantify model performance and variance. To determine whether differences between models were statistically significant, RMSE distributions were simulated and pairwise t-tests were conducted against the best performing model. Finally, for interpretability, a SHAP analysis was performed on the featurizers to highlight feature contributions.
Feature Attribution
Feature attribution was performed using multiple complementary techniques tailored to the molecular representation. For ECFP-based models, global importance was derived from gain-based rankings averaged across cross-validation folds. Fingerprint bits were then mapped back to chemical substructures using RDKit, and atom-level heatmaps were generated to visualize molecular fragments25. For ChemBERTa embeddings, SHAP was applied to the XGBoost classifier trained over pooled embeddings. This produced global feature rankings across embedding dimensions. To provide chemical interpretability, token-level attributions were projected back onto atoms, and color-coded heatmaps were generated to highlight contributions that increased or decreased solubility predictions. For the GCN models, important atoms were identified using a combination of SHAP analysis and per-atom occlusion, where each atom was temporarily masked to observe its effect on the prediction. The resulting importance scores were visualized as red and blue highlights on each molecule, illustrating the functional groups that most strongly influenced the model’s decision.
Results
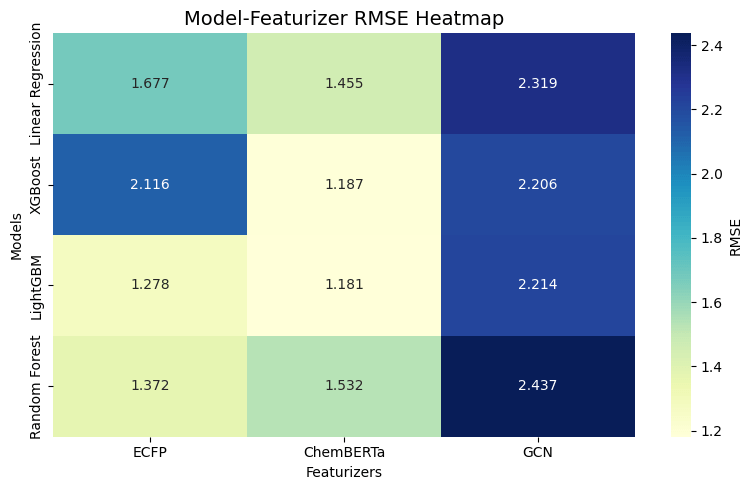
Performance Comparison
The experiments revealed substantial variation in predictive performance across model–featurizer combinations (Fig. 2). The best-performing configurations were LightGBM combined with ChemBERTa and XGBoost combined with ChemBERTa, achieving mean RMSE values of 1.181 (95% CI: 1.138–1.224) and 1.187 (95% CI: 1.147–1.227), respectively. The overlapping confidence intervals indicate that the difference between these two models is not statistically significant.
The statistical analysis further confirms ChemBERTa’s effectiveness as a featurizer. Pairwise t-tests using LightGBM + ChemBERTa as the reference model show that every non-ChemBERTa combination, including GCN-based models, ECFP-based models, and linear baselines, performed significantly worse (p < 0.01 for all comparisons). Only the two ChemBERTa combinations, XGBoost + ChemBERTa (p = 0.12468) and Linear Regression + ChemBERTa (p = 0.167114), failed to show a statistically significant difference.
Among traditional fingerprint approaches, ECFP showed competitive performance, particularly when paired with LightGBM (RMSE 1.278, 95% CI: 1.230–1.326) and Random Forest (RMSE 1.372, 95% CI: 1.312–1.432). Linear Regression models generally lagged behind tree-based methods, even when paired with ChemBERTa. In contrast, models using the GCN featurizer consistently underperformed, with RMSE values ranging from 2.206 (95% CI: 2.153–2.259) to 2.437 (95% CI: 2.418–2.456).
Although Graph Convolutional Networks (GCNs) are theoretically well suited for molecular property prediction, the specific GCN implementation used in this study underperformed due to several architectural and data-related limitations. The model employed only atomic number as a single node feature, lacking important chemical information such as valence, aromaticity, hybridization, and formal charge. Additionally, bond features were not encoded, preventing the model from distinguishing between different bond types. These limitations restrict the ability of the GCN to learn meaningful structural representations.
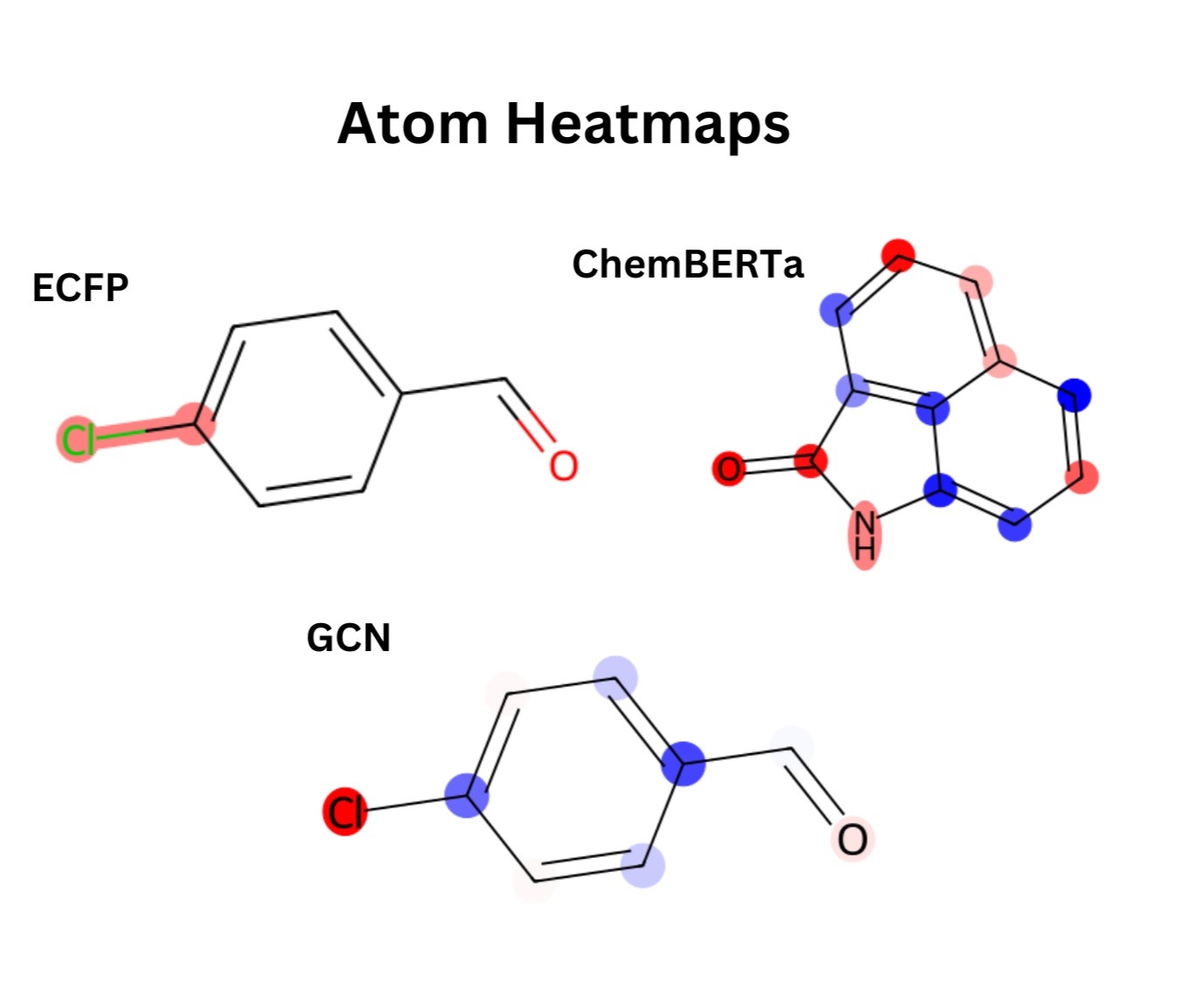
Important Substructures
For the ECFP models, averaging feature importance across folds shows that only a few bits matter most, with bit 561 being the top signal. From Figure 3, it can be seen that when this bit is mapped back to chemical space, it corresponds strongly to an aryl chloride. This suggests that halogenated aromatic systems strongly drive the model’s predictions.
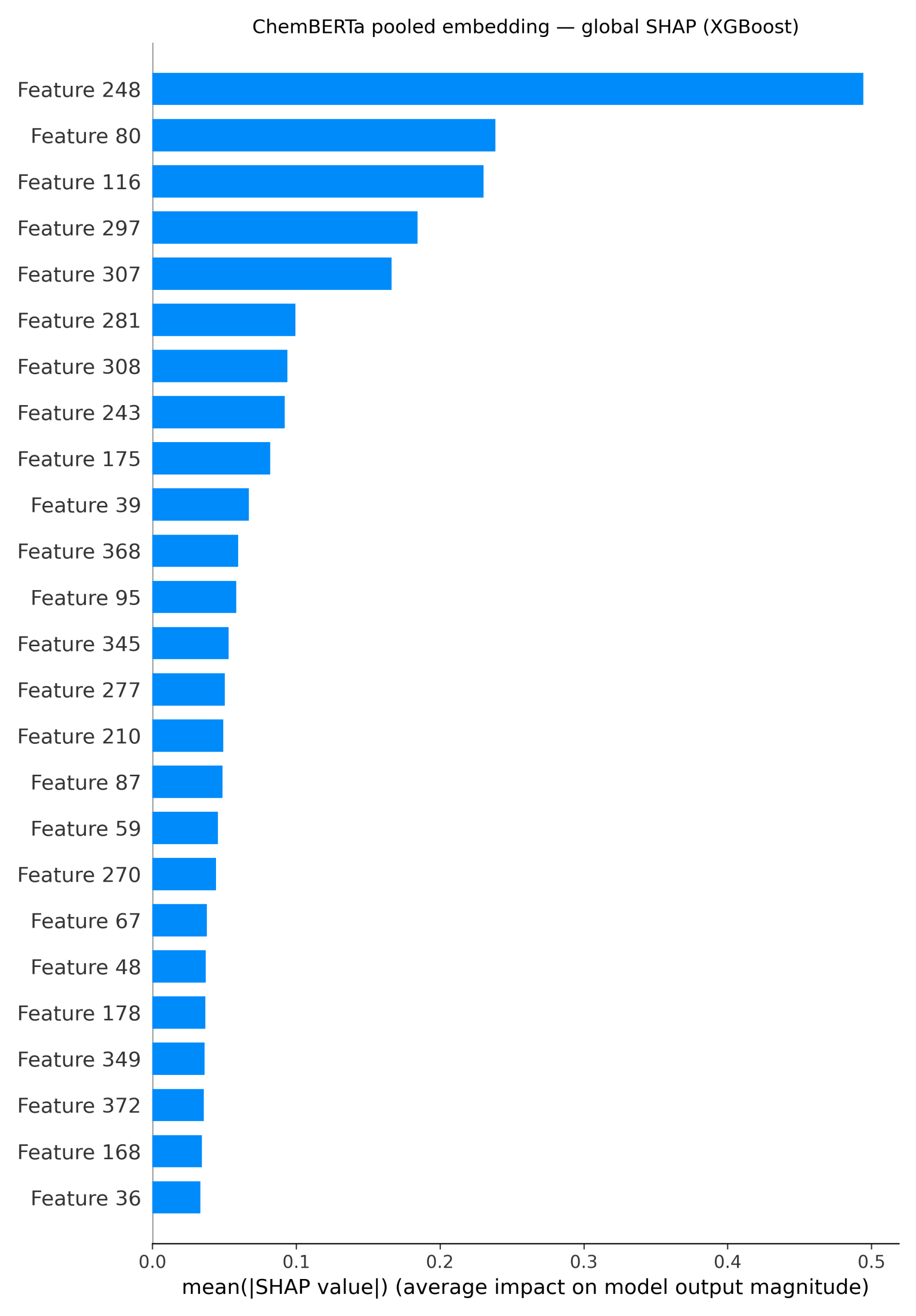
It can be seen in Figure 4, that for the ChemBERTa + XGBoost model, global SHAP values on the embeddings reveal that just a handful of dimensions (such as features 248, 80, and 116) carry most of the signal. To connect these features back to chemistry, the attributions are projected onto the molecule itself. The resulting atom-level heatmap (Figure 3) shows that the carbonyl oxygen and nearby nitrogen atoms are the main drivers, while several aromatic carbons instead reduce the prediction.
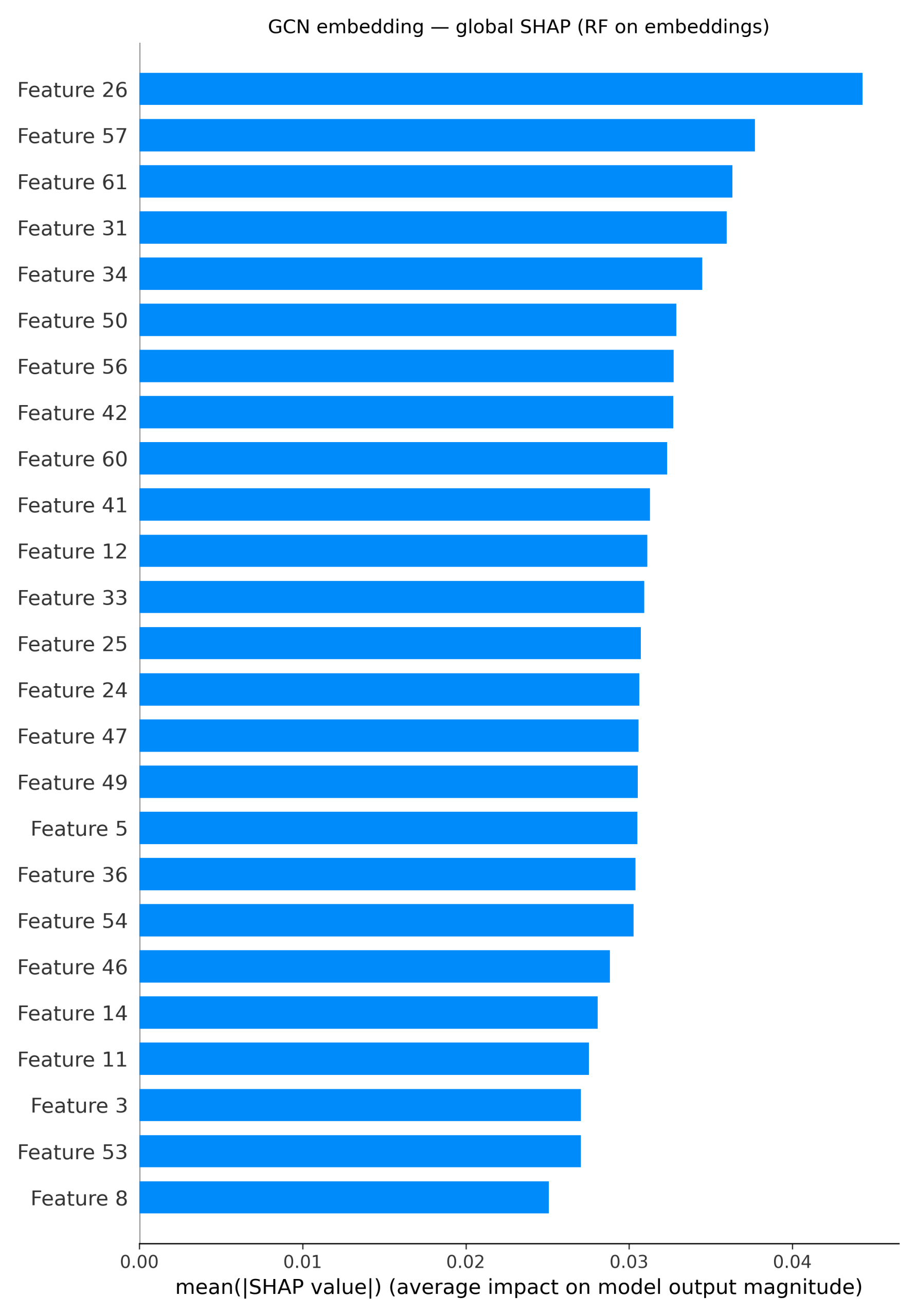
For the GCN pipelines, the global SHAP bar plot (Figure 5) over GCN embeddings shows a distributed profile. Instead of a single dominant feature, numerous embedding dimensions showed moderate importance. These signals were localized through per-atom occlusion on the XGBoost model and generated corresponding atom-level heatmaps. In this example, the chlorine and the oxygen are the most important atoms (shown in red), while some of the ring carbons reduce the prediction (shown in blue). This pattern aligns with the trends observed in the ECFP and ChemBERTa models, with heteroatoms and halogens driving solubility-related signals.
Discussion
The results demonstrate that model performance in solubility prediction is strongly influenced by the choice of molecular representation and model architecture. Across all experiments, ChemBERTa-based molecular representations consistently produced the lowest RMSE values when combined with models such as XGBoost and LightGBM. The narrow confidence intervals observed for these models indicate stable performance across cross validation folds, suggesting that these representations capture meaningful structural information consistently. In contrast, other featurization approaches, particularly graph-based representations, resulted in higher prediction errors despite having stable confidence intervals. When it comes to feature representation, studies combining multiple representations, such as electrostatic potential maps, graph features, and tabular descriptors, have shown that representation choice plays a central role in predictive performance14. The results of this study reinforce the idea that richer representations generally lead to better model outcomes.
These findings are consistent with prior work showing that representations from SMILES strings can effectively encode molecular structure. For example, transformer-based models such as SolTranNet have demonstrated improved performance over traditional linear models by capturing contextual relationships within SMILES sequences10. The strong performance of ChemBERTa in this study supports this idea, indicating that pretrained language models can provide features helpful for downstream prediction tasks. In addition, the effectiveness of gradient boosting methods aligns with previous studies where LightGBM and other ensemble models outperformed a range of baseline approaches6,11,14. These models are able to recognize complex feature interactions while maintaining strong generalization, contributing to their consistent performance.
The underperformance of graph-based models in this study contrasts with prior work where graph neural networks achieved strong results for molecular property prediction12. This difference can be explained by the simplicity of the GCN implementation used in this work. The model relied only on atomic number as the node feature and did not include bond features or additional chemical descriptors. As a result, the model lacked important information such as bond type, aromaticity, and hybridization, which are known to be critical for capturing molecular structure. This limitation likely restricted the model’s ability to learn meaningful graph representations, leading to reduced predictive performance. This observation suggests that while graph-based models have strong theoretical advantages, their effectiveness depends heavily on the quality and richness of the input features.
Despite these findings, several limitations should be noted. First, the study was conducted on a single, small dataset, which may limit the generalizability of the results to other chemical domains or datasets with different distributions. Second, the GCN architecture used in this work was relatively simple and did not incorporate advanced features such as edge attributes, attention mechanisms, or deeper network structures. Third, ChemBERTa embeddings were used without fine-tuning, possibly affecting their performance slightly. Finally, while multiple model types were compared, hyperparameter optimization was not exhaustively explored, which could influence performance outcomes.
These limitations also point to directions for future work. Incorporating richer graph features, experimenting with a larger dataset and more advanced graph neural network architectures could improve the performance of graph-based models. In addition, combining multiple representations, such as graph features and learned embeddings, could provide deeper information and improve predictive accuracy.
Overall, this study demonstrates that representation choice is a key factor in solubility prediction. ChemBERTa-based embeddings combined with gradient boosting methods provide a strong balance between accuracy and stability, while simpler graph-based approaches may require more advanced feature engineering to achieve competitive performance. These results provide useful guidance for selecting models and representations in molecular property prediction tasks.
Conclusion
This study shows that molecular representation plays a central role in solubility prediction, with ChemBERTa features combined with gradient-boosting models achieving the best overall performance. The results highlight that selecting an optimal pairing of featurizer and model can significantly improve predictive accuracy. In addition, interpretability analysis confirms that the models capture chemically meaningful patterns, consistent with known factors such as hydrogen bonding and aromaticity. Some limitations should be noted. The study was conducted on a single dataset, which may limit generalizability to other chemical spaces. The performance of graph-based models was also worse than expected, likely due to simplified feature design and architectural choices rather than inherent limitations of graph neural networks. Future work can explore more advanced representations, including improved graph-based models and hybrid approaches that combine multiple feature types. Fine-tuning pretrained models and evaluating performance across more diverse datasets may further improve generalizability and applicability.
References
- Qing, R., Hao, S., Smorodina, E., Jin, D., Zalevsky, A., Zhang, S. Protein design: From the aspect of water solubility and stability. Chemical Reviews, 122(18), 14085–14179, 2022. [↩]
- Dara, S., Dhamercherla, S., Jadav, S. S., Babu, C. H. M., & Ahsan, M. J. Machine learning in drug discovery: a review. Artificial Intelligence Review, 55(3), 1947–1999, 2022. [↩]
- Gozalbes, R., and Pineda-Lucena, A. QSAR-based solubility model for drug-like compounds. Bioorganic & Medicinal Chemistry, 18(19), 7078–7084, 2010. [↩]
- Li, M., Chen, H., Zhang, H., Zeng, M., Chen, B., Guan, L. Prediction of the aqueous solubility of compounds based on light gradient boosting machines with molecular fingerprints and the cuckoo search algorithm. ACS Omega, 7(46), 42027–42035, 2022. [↩]
- Palmer, D. S., O’Boyle, N. M., Glen, R. C., and Mitchell, J. B. O. Random forest models to predict aqueous solubility. Journal of Chemical Information and Modeling, 47(1), 150–158, 2007. [↩]
- Ye, Z., and Ouyang, D. Prediction of small-molecule compound solubility in organic solvents by machine learning algorithms. Journal of Cheminformatics, 13(1), 2021, https://doi.org/10.1186/s13321-021-00575-3. [↩] [↩]
- Lovrić, M., Pavlović, K., Žuvela, P., Spataru, A., Lučić, B., Kern, R., and Wong, M. W. Machine learning in prediction of intrinsic aqueous solubility of drug-like compounds: Generalization, complexity, or predictive ability? Journal of Chemometrics, 35, e3349, 2021, https://doi.org/10.1002/cem.3349. [↩]
- Hu, P., Jiao, Z., Zhang, Z., and Wang, Q. Development of solubility prediction models with ensemble learning. Industrial & Engineering Chemistry Research, 60(30), 11627–11635, 2021. [↩]
- Xue, N., Zhang, Y., and Liu, S. Evaluation of Machine Learning Models for Aqueous Solubility Prediction in Drug Discovery. 2024 7th International Conference on Artificial Intelligence and Big Data (ICAIBD), 26–33, 2024, https://doi.org/10.1109/ICAIBD62003.2024.10604556. [↩]
- Francoeur, P. G., and Koes, D. R. SolTranNet–a machine learning tool for fast aqueous solubility prediction. Journal of Chemical Information and Modeling, 61(6), 2530–2536, 2021, https://doi.org/10.1021/acs.jcim.1c00331. [↩] [↩]
- An, F., Sayed, B. T., Parra, R. M., Hamad, M. H., Sivaraman, R., Zanjani Foumani, Z., Rushchitc, A. A., El-Maghawry, E., Alzhrani, R. M., Alshehri, S., et al. Machine learning model for prediction of drug solubility in supercritical solvent: Modeling and Experimental Validation. Journal of Molecular Liquids, 363, 119901, 2022, https://doi.org/10.1016/j.molliq.2022.119901. [↩] [↩]
- Zheng, T., Mitchell, J. B., and Dobson, S. Revisiting the application of machine learning approaches in predicting aqueous solubility. ACS Omega, 9(32), 35209–35222, 2024. [↩] [↩] [↩]
- Tayyebi, A., Alshami, A. S., Rabiei, Z., Yu, X., Ismail, N., Talukder, M. J., and Power, J. Prediction of organic compound aqueous solubility using machine learning: A comparison study of descriptor-based and fingerprints-based models. Journal of Cheminformatics, 15(1), 2023, https://doi.org/10.1186/s13321-023-00752-6. [↩]
- Ghanavati, M. A., Ahmadi, S., and Rohani, S. A machine learning approach for the prediction of aqueous solubility of pharmaceuticals: A comparative model and dataset analysis. Digital Discovery, 3(10), 2085–2104, 2024, https://doi.org/10.1039/d4dd00065j. [↩] [↩] [↩]
- Oliveira, G., Wegner, P. H., de Lima Carvalho, P. V., Voll, F. A., de Paula Scheer, A., de Pelegrini Soares, R., and Farias, F. O. Machine learning-enhanced Cosmo-SAC for accurate solubility predictions. Fluid Phase Equilibria, 600, 114535, 2026, https://doi.org/10.1016/j.fluid.2025.114535. [↩]
- Mac Fhionnlaoich, N., Zeglinski, J., Simon, M., Wood, B., Davin, S., and Glennon, B. A hybrid approach to aqueous solubility prediction using COSMO-RS and Machine Learning. Chemical Engineering Research and Design, 209, 67–71, 2024, https://doi.org/10.1016/j.cherd.2024.07.050. [↩]
- Al Ibrahim, E., Morgan, N., Müller, S., Motati, S., and Green, W. H. Accurately predicting solubility curves via a thermodynamic cycle, machine learning, and Solvent Ensembles. Journal of the American Chemical Society, 147(49), 45057–45069, 2025, https://doi.org/10.1021/jacs.5c13746. [↩]
- Osada, M., Tamura, K., and Shimada, I. Prediction of the solubility of organic compounds in high-temperature water using machine learning. The Journal of Supercritical Fluids, 190, 105733, 2022, https://doi.org/10.1016/j.supflu.2022.105733. [↩]
- Sodaei, Z., Ekrami, S., and Hashemianzadeh, S. M. Machine learning analysis of molecular dynamics properties influencing drug solubility. Scientific Reports, 15(1), 2025, https://doi.org/10.1038/s41598-025-11392-1. [↩]
- Can, E., Jalal, A., Zirhlioglu, I. G., Uzun, A., and Yildirim, R. Predicting water solubility in ionic liquids using machine learning towards design of Hydro-Philic/phobic ionic liquids. Journal of Molecular Liquids, 332, 115848, 2021, https://doi.org/10.1016/j.molliq.2021.115848. [↩]
- Axen, S. D., Huang, X.-P., C´aceres, E. L., Gendelev, L., Roth, B. L., and Keiser, M. J. A simple representation of three-dimensional molecular structure. Journal of Medicinal Chemistry, 60(17), 7393–7409, 2017. [↩]
- Lang, A., et al. Fine-Tuning ChemBERTa-2 for Aqueous Solubility Prediction. ResearchGate, 2023. [↩]
- Deng, C., Liang, L., Xing, G., Hua, Y., Lu, T., Zhang, Y., Chen, Y., and Liu, H. Multi-channel GCN ensembled machine learning model for molecular aqueous solubility prediction on a clean dataset. Molecular Diversity, 27(3), 1023–1035, 2023. [↩]
- Llompart, P., Minoletti, C., Baybekov, S., Horvath, D., Marcou, G., and Varnek, A. Will we ever be able to accurately predict solubility? Scientific Data, 11(1), 303, 2024. [↩]
- Scalfani, V. F., Patel, V. D., and Fernandez, A. M. Visualizing chemical space networks with RDKit and NetworkX. Journal of Cheminformatics, 14(1), 87, 2022. [↩]


 and a Developing (India) Economy")
{kind=link}