
Abstract
T-cell-based immunotherapies have revolutionized cancer treatment, yet their clinical efficacy in solid tumors remains limited. A major barrier is the tumor microenvironment (TME), which imposes nutrient deprivation, hypoxia, and oxidative stress, leading to mitochondrial dysfunction and ultimately T-cell exhaustion. Conventional strategies, such as AKT inhibition or single co-stimulatory antibody treatments, are reactive, applied only after T cells encounter metabolic stress, and fail to address this fundamental challenge. Here, we present a metabolic priming strategy designed to enhance T-cell metabolic and functional fitness prior to tumor encounter. By activating PGC1α with Bezafibrate to enhance mitochondrial biogenesis and applying 4-1BB co-stimulation to amplify activation signals, we trained T cells into “super soldier” with superior metabolic capacity and effector function. Dual-treated T cells demonstrated significant improvements in mitochondrial activity, activation marker expression, and cytokine secretion compared to single-treated or untreated cells. Importantly, these primed T cells exhibited enhanced tumor cell killing and infiltration in both 2D colorectal cancer models and patient-derived 3D tumor organoids. Moreover, this approach offers a cost-effective alternative to expensive personalized CAR-T therapies or single co-stimulatory antibody treatments. By leveraging FDA-approved drugs and well-established co-stimulation techniques, it can be readily incorporated into existing T-cell isolation and expansion protocols without complex genetic modifications. Our findings establish a new paradigm for fundamentally enhancing T-cell anti-tumor function and provide a practical, scalable strategy to improve immunotherapy efficacy in solid tumors.
Introduction
T-cell-based immunotherapies have emerged as a transformative approach in modern cancer treatment1,2; however, their clinical efficacy in solid tumors remains limited3,4. A primary contributing factor is the metabolic stress imposed by the tumor microenvironment (TME)5,6—characterized by nutrient deprivation, hypoxia, and oxidative stress7,8. This harsh environment compromises the mitochondrial function of infiltrating T cells9,10, ultimately leading to their functional impairment, a state known as ‘exhaustion’10,11. This T-cell dysfunction is currently recognized as a critical mechanism underlying immunotherapy failure12,13, underscoring the urgent need for foundational strategies to overcome this challenge.
Previous studies have primarily focused on modulating signaling pathways, such as AKT inhibition14, or activating single co-stimulatory pathways post-T-cell exposure to the TME15. However, these approaches represent reactive strategies, applied after T cells have already encountered metabolic stress, thus failing to provide a fundamental solution. In contrast, this study proposes an innovative paradigm: ‘Metabolic Priming’. This strategy aims to proactively enhance T-cell anti-tumor capacity and persistence by optimizing mitochondrial metabolism and function prior to their direct engagement with the tumor.
Specifically, this research employs a double-target strategy to achieve two synergistic goals. 4-1BB co-stimulation has been previously shown to enhance mitochondrial function and biogenesis in T cells through established signaling pathways. Building on these findings, we incorporate 4-1BB signaling as a complementary activation signal within a broader metabolic priming strategy. So, firstly, we utilized Bezafibrate, a pharmacological activator of PGC1α16,17, to promote mitochondrial biogenesis and fundamentally enhance the metabolic capacity of T cells18,19. This step is akin to “boosting the physical endurance” of T cells. Secondly, 4-1BB co-stimulation was applied to intensify T-cell activation signals20,21, thereby equipping these metabolically enhanced T cells with “combat skills” and preparing them for attack. This integrated approach, combining mitochondrial reinforcement with co-stimulatory activation through Bezafibrate and 4-1BB co-stimulation for Mitochondrial Quality Control, represents a novel application not previously explored in the literature. Our approach fundamentally trains T cells into “super soldier”, capable of recognizing and eliminating tumors while maintaining sustained anti-tumor function.
Therefore, the central hypothesis of this study is that metabolic priming, achieved through the combined application of Bezafibrate and 4-1BB co-stimulation, can enhance the metabolic preparedness and antitumor functionality of T cells before exposure to tumor-associated stress. This strategy transcends simple T-cell activation, offering a potential solution to prevent a major cause of immunotherapy failure by fundamentally improving mitochondrial function, and consequently, enhancing the efficiency of T-cell therapy in solid tumors.
Materials and Methods
Human Peripheral T Cell Isolation and Characterization
Peripheral Blood Mononuclear Cell (PBMC) Isolation
Peripheral blood was collected from healthy donors under informed consent (CGT Global). PBMCs were isolated using Ficoll-Paque density gradient centrifugation. Briefly, blood was diluted with PBS, layered over Ficoll, and centrifuged at 400 × g for 30 minutes at room temperature. The mononuclear cell layer was carefully collected and washed twice with PBS.
T Cell Purification
CD3-positive T cells were purified from PBMCs using fluorescence-activated cell sorting (FACS). Cells were stained with anti-CD3 antibodies and sorted to ensure >95% purity.
T Cell Culture
T cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin. Cells were maintained at 37°C in a humidified incubator with 5% CO₂ and cultured at a density of 1 × 10^6 cells/mL in T25 flasks unless otherwise indicated.
Morphological and Viability Assessment
Isolated T cells were examined under a phase-contrast microscope to confirm characteristic round morphology. Following treatment, T-cell viability was assessed by trypan blue exclusion prior to all functional assays. No significant differences in viability were observed between experimental groups.
Metabolic Priming of T Cells
Treatment Groups
Purified T cells were subjected to metabolic and co-stimulatory priming to generate a “super soldier” phenotype. Four experimental groups were prepared: control (untreated), Bezafibrate only, 4-1BB agonist only, and combination (Bezafibrate + 4-1BB agonist).
Bezafibrate Treatment
Bezafibrate was dissolved in DMSO, and all treatment groups received the same final concentration of vehicle. Control T cells were treated with the corresponding DMSO vehicle alone. T cells were treated with Bezafibrate (PGC1α activator) at a concentration of 50 µM for 48 hours to stimulate mitochondrial biogenesis.
4-1BB Co-stimulation
For 4-1BB stimulation, T cells were cultured with immobilized anti-4-1BB agonist antibody (Acro Biosystem; 41B-H5256) at 1 µg/mL for 48 hours as part of the priming procedure. The antibody was immobilized using a plate-bound format, and no additional anti-CD3 stimulation or secondary cross-linking antibody was included during this step. This configuration was selected to provide a defined 4-1BB-associated priming signal prior to tumor co-culture.
After the 48-hour priming period, T cells were collected and washed 2–3 times with PBS to remove residual Bezafibrate and 4-1BB agonist before co-culture with HCT116, SW480 cells or patient-derived organoids.
Following treatment, T cell morphology was examined using microscopy to ensure integrity was maintained after interventions.
qRT-PCR analysis
Total RNA was isolated from control or primed T cells after 48 h of treatment using an QIAGEN RNeasy extraction kit according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from equal amounts of RNA using a reverse transcription kit. Quantitative real-time PCR was performed using SYBR Green master mix on a real-time PCR system. Relative gene expression was calculated using the 2-ΔΔCt method and normalized to an internal housekeeping gene (GAPDH). PGC-1α(PPARGC1A) Forward : AGCCTCTTTGCCCAGATCTT, Reverse : GGCAATCCGTCTTCATCCAC; GAPDH Forward : CCAAGGAGTAAGACCCCTGG, Reverse : TGGTTGAGCACAGGGTACTT
ATP viability assay
To assess metabolic activity/viability, intracellular ATP levels were measured in control or primed T cells after 48 h of treatment using a luminescence-based ATP assay kit (Promega) according to the manufacturer’s instructions. Briefly, equal numbers of cells were plated in replicate wells, incubated with ATP detection reagent, and luminescence was measured using a microplate reader. ATP levels were normalized to the control group and presented as relative viability.
Western Blot Analysis
Protein Extraction and Immunoblotting
T cells from each treatment group were lysed using RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentration was determined using a BCA assay. Equal amounts of protein were separated via SDS-PAGE and transferred to PVDF membranes.
Target Proteins
Membranes were probed with antibodies against PGC1α (cell signaling technology;#2178) and AKT/p-AKT (cell signaling technology;#9272, #9271). HRP-conjugated secondary antibodies were used for detection, and chemiluminescent signals were captured using an imaging system. Quantification of PGC1α and p-AKT protein levels from three independent experiments.
All primary antibodies used in this study were validated by the manufacturers for the indicated applications, and were used according to the manufacturers’ recommended protocols.
Mitochondrial Activity Assays
MitoTracker Staining
To evaluate mitochondrial mass and membrane potential, T cells were incubated with MitoTracker Red CMXRos dye at 100 nM for 30 minutes at 37°C. Cells were washed and analyzed either by flow cytometry or fluorescence microscopy.
Analysis
Fluorescence intensity was quantified to compare mitochondrial activity among control, single-treated, and dual-treated groups.
T Cell Activation Marker Analysis
Flow Cytometry
Activation markers CD25 and CD69 were assessed by staining T cells with fluorescently labeled antibodies. Stained cells were analyzed using a flow cytometer, and the percentage of marker-positive cells was recorded.
Cytokine Secretion Assays
ELISA
T cell effector function was evaluated by measuring cytokine secretion. Culture supernatants were collected from each group and analyzed using ELISA kits for IFN-γ, TNF-α, and IL-2, following the manufacturer’s instructions.
2D Tumor Cell Co-culture Assays
Co-culture with HCT116 Cells
To assess cytotoxic activity, baseline or primed T cells were co-cultured with HCT116 colorectal cancer cells at a ratio of 10:1 (T cell:tumor cell) for 24–48 hours. Control conditions included tumor cells cultured alone and T cells cultured alone to define baseline viability and cytotoxicity.
Co-culture with SW480 Cells
To assess cytotoxic activity, baseline or primed T cells were co-cultured with SW480 colorectal cancer cells at a ratio of 10:1 (T cell:tumor cell) for 24–48 hours. Control conditions included tumor cells cultured alone and T cells cultured alone to define baseline viability and cytotoxicity.
Colony Forming Assay
After co-culture, tumor cell viability was evaluated by a colony forming assay. Cells were fixed, stained, and colony numbers were counted.
LDH Release Assay
Cytotoxicity was further quantified by measuring lactate dehydrogenase (LDH) release from lysed tumor cells using a commercial LDH assay kit.
3D Tumor Organoid Assays
Patient-derived Organoid Culture
Colorectal cancer organoids were established from one patient tumor samples (Korea Cell Line Bank) and maintained in Matrigel-based 3D culture.
Co-culture with Primed T Cells
Primed or baseline T cells were added to organoid cultures and incubated for 72 hours. Organoid growth was monitored by measuring size and number under a microscope.
Immunohistochemistry
Organoids were fixed, embedded, and sectioned for staining with Granzyme B antibody to assess T cell infiltration and cytotoxic activity.
Statistical analysis
All quantitative data are presented as mean ± SD unless otherwise indicated. Statistical analyses were performed using GraphPad Prism. Comparisons between multiple groups were conducted using one-way ANOVA followed by Tukey’s post hoc test. A p-value < 0.05 was considered statistically significant.
Results
Isolation and Characterization of Human Peripheral T Cells
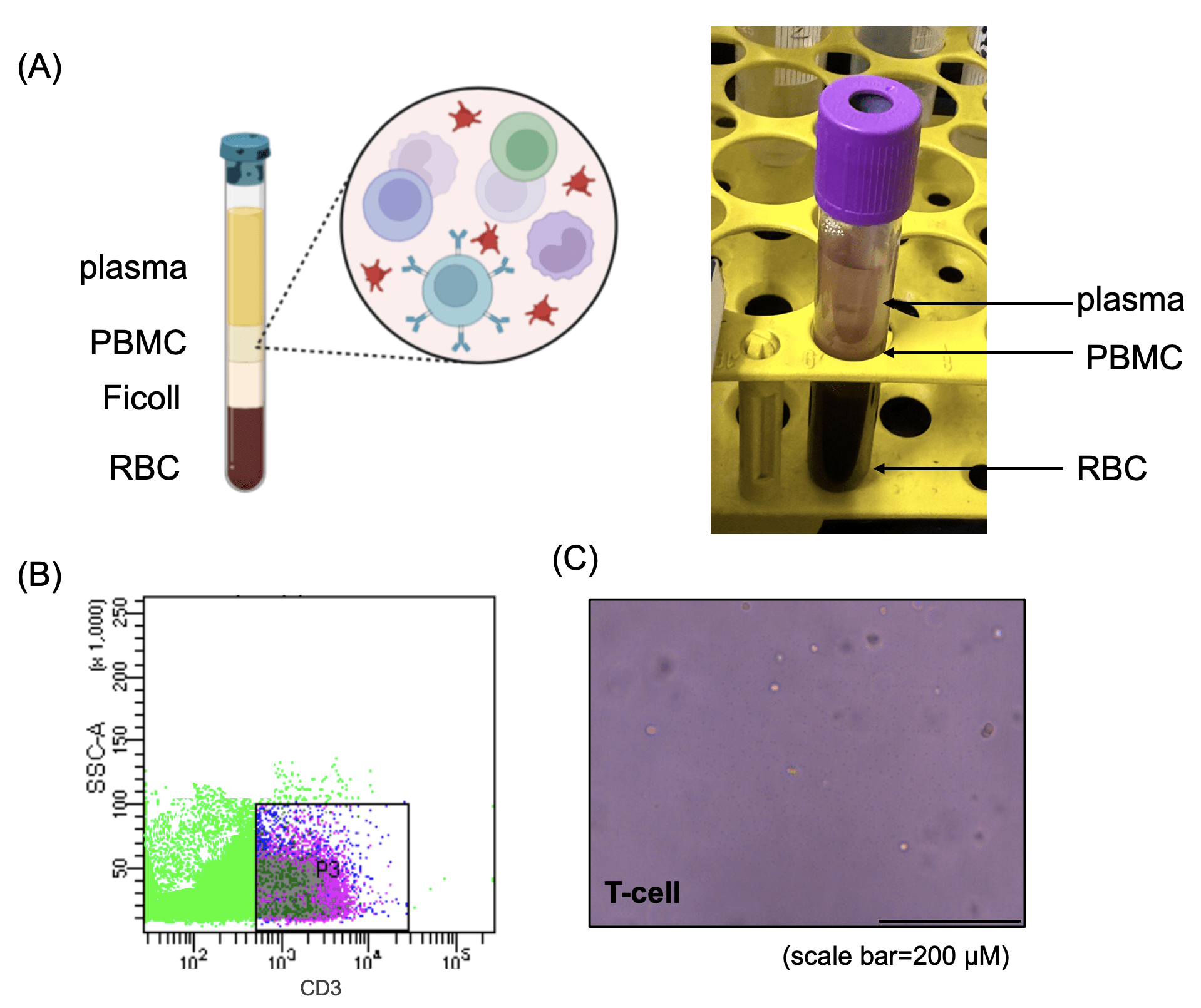
(B) Flow cytometry analysis of isolated T cells, stained with CD3 antibodies, showing distinct T-cell populations.
(C) Representative image of purified T cells, confirming their characteristic round morphology under phase-contrast microscopy. Scale bar = 200 μm.
To obtain primary T cells for subsequent experiments, peripheral blood was collected from healthy donors and PBMCs were isolated using Ficoll density gradient centrifugation (Fig. 1a). T cells were then purified via FACS by selecting CD3-positive populations (Fig. 1b). Morphological assessment confirmed that the isolated T cells displayed characteristic round shapes and were maintained in a clean culture environment, indicating high purity and viability (Fig. 1c).
Metabolic Priming of T Cells to Generate “Super Soldier” Phenotype
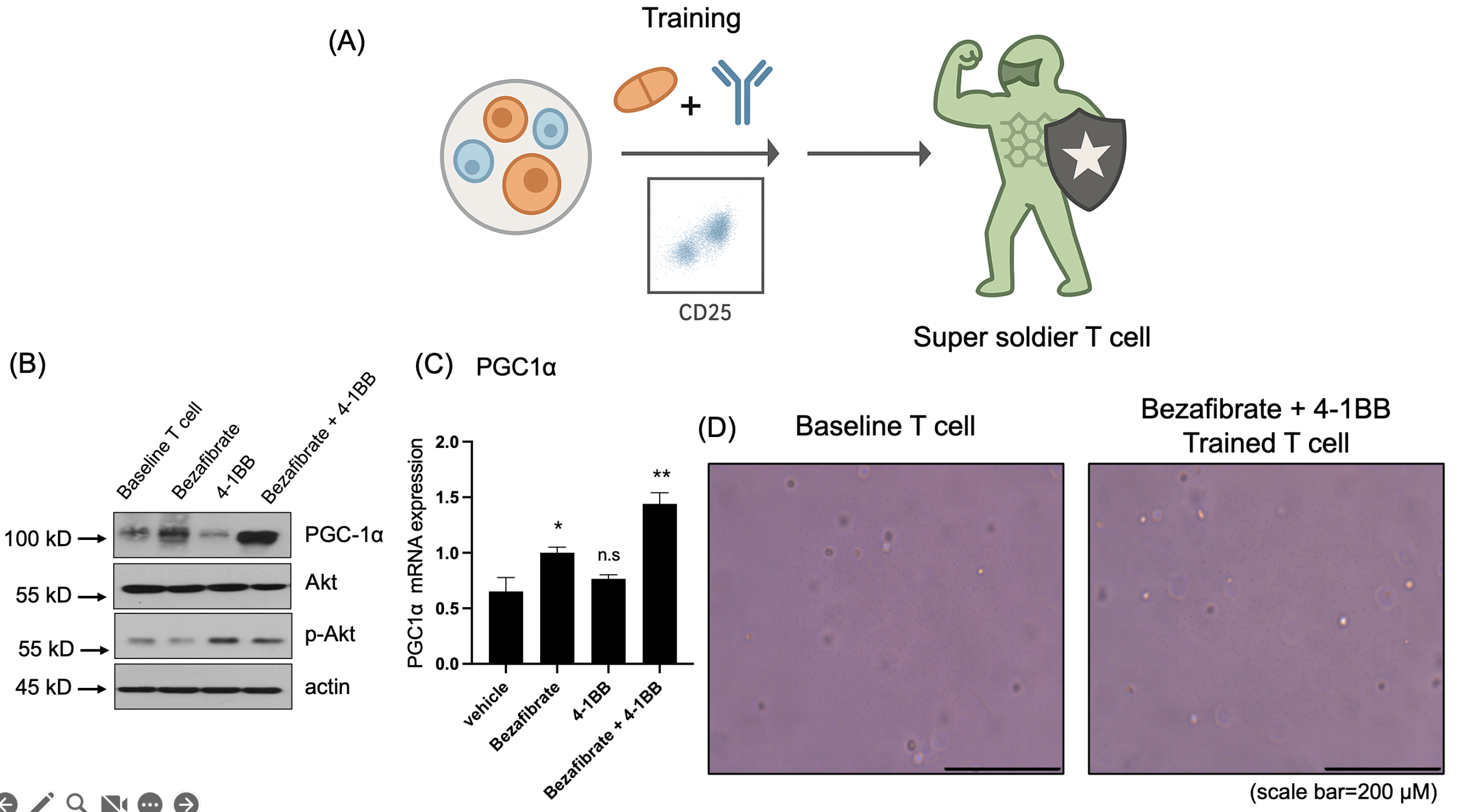
(B) Western blot analysis showing PGC1α upregulation in the Bezafibrate-treated group and increased phosphorylation of AKT (p-AKT) in the 4-1BB-treated group.
(C) qRT-PCR analysis of PGC1α mRNA expression in treated T cells. Data are presented as mean ± SD. n = 3 independent biological replicates. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test. *p < 0.05, **p < 0.01, n.s=non-significant
(D) Representative images of T cells before and after treatment. Scale bar = 200 μm.
To evaluate whether T cells could be metabolically primed into a “super soldier” state, isolated T cells were treated with Bezafibrate (PGC1α activator) and/or 4-1BB agonist (Fig. 2a). Western blot analysis of the resulting treatment groups (control, Bezafibrate only, 4-1BB only, and combination) revealed Bezafibrate treatment increased PGC1α expression, consistent with activation of mitochondrial biogenesis-associated programs. Concurrently, 4-1BB stimulation increased AKT phosphorylation, consistent with engagement of activation-related signaling in T cells (Fig. 2b). In addition, qRT-PCR analysis demonstrated that PGC1α mRNA expression was also increased in Bezafibrate-treated T cells (Fig. 2c). Morphological inspection before and after treatment indicated that the metabolic and co-stimulatory interventions did not compromise T cell integrity (Fig. 2d). Collectively, these data confirmed that the dual-target priming strategy effectively modulated T cell metabolic and activation pathways.
Metabolic and Functional Characterization of Primed T Cells
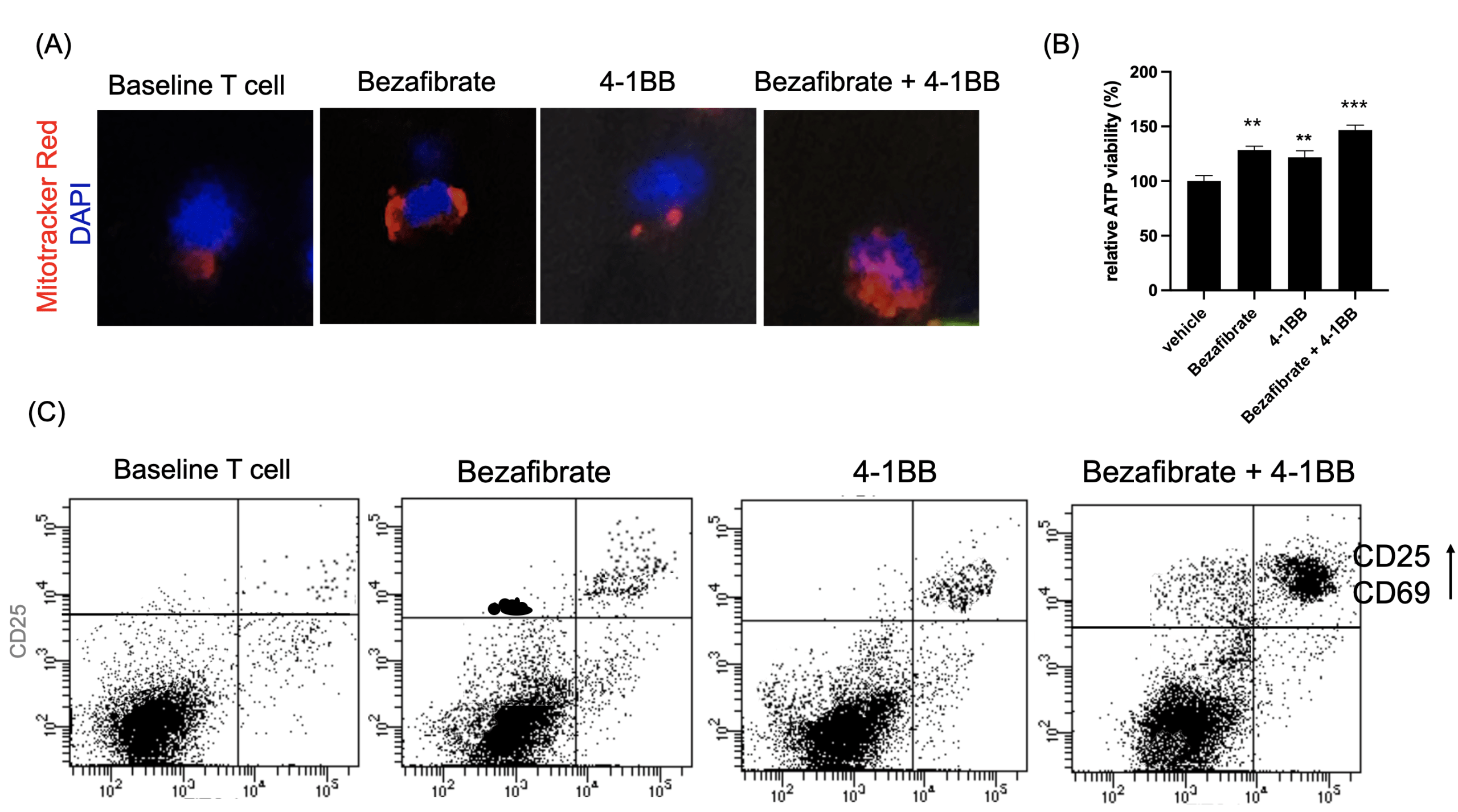
(B) Relative ATP levels measured after priming.
(C) Flow cytometry analysis of activation markers CD25 and CD69.
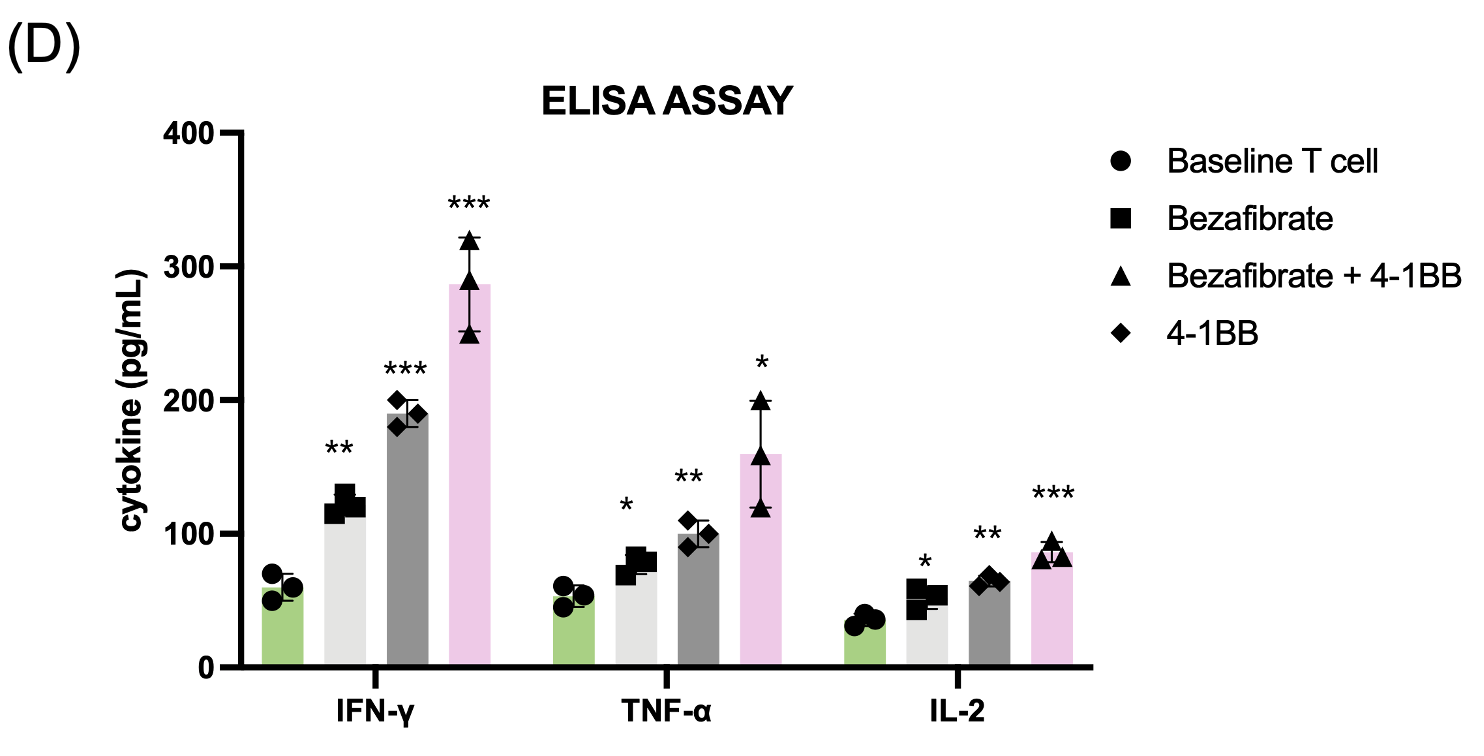
(D) Cytokine secretion analysis via ELISA, demonstrating significantly higher levels of IFN-γ, TNF-α, and IL-2 in dual-treated T cells.
In these figures, data are presented as mean ± SD. n = 3 independent biological replicates. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test. *p < 0.05, **p < 0.01, ***p < 0.001.
Following metabolic priming, T cell functional status was assessed. Mitochondrial activity measured by MitoTracker staining was highest in the dual-treated group, indicating enhanced mitochondrial mass and membrane potential (Fig. 3a). To obtain a more direct functional readout of mitochondrial metabolism, we additionally measured intracellular ATP levels and found that ATP production was significantly increased in Bezafibrate-treated T cells, with the highest levels observed in the dual-treated group (Fig. 3b) Flow cytometry analysis of activation markers CD25 and CD69 demonstrated significant upregulation in dual-treated T cells compared to single-treated or control groups (Fig. 3c). Functional assessment via cytokine ELISA revealed that dual-treated T cells secreted markedly higher levels of IFN-γ, TNF-α, and IL-2 (Fig. 3d), indicating robust effector function. Cytokine concentrations were quantified and are reported as pg/mL. This indicates that the trained T cells are prepared to function as “super soldier,” capable of eliciting a stronger immune response than T cells in the single-treatment groups
Antitumor Efficacy in 2D Tumor Cell Models
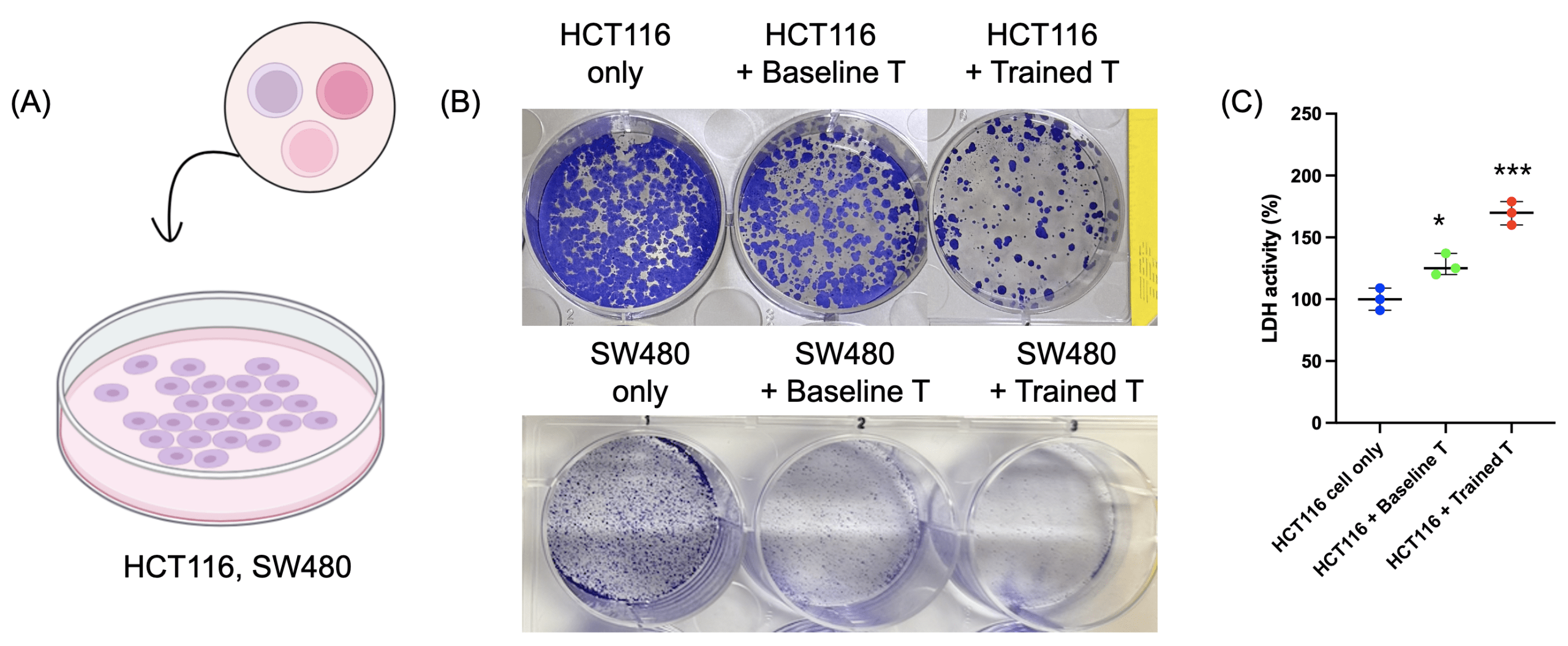
(B) Colony-forming assay results showing HCT116 cells (upper panel) and SW480 cells (bottom panel) viability in the dual-treated T cell group compared to baseline T cells or tumor cells alone.
(C) LDH release assay indicating cytotoxicity of T cells.
Data are presented as mean ± SD. n = 3 independent biological replicates. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparison test. *p < 0.05, ***p < 0.001.
To determine whether the enhanced functional potential of “super soldier” translated to antitumor activity, we co-cultured HCT116 and SW480 colorectal cancer cells with baseline or primed T cells (Fig. 4a). Colony-forming assay results showing reduced tumor cell viability in the dual-treated T cell group compared to baseline T cells or tumor cells alone (Fig. 4b). LDH release assays further confirmed increased tumor cell lysis in the dual-treated T cell condition (Fig. 4c). These results indicate that metabolic priming enhances T cell-mediated cytotoxicity against tumor cells in a 2D model system.
Antitumor Efficacy and Infiltration in 3D Tumor Organoid Models
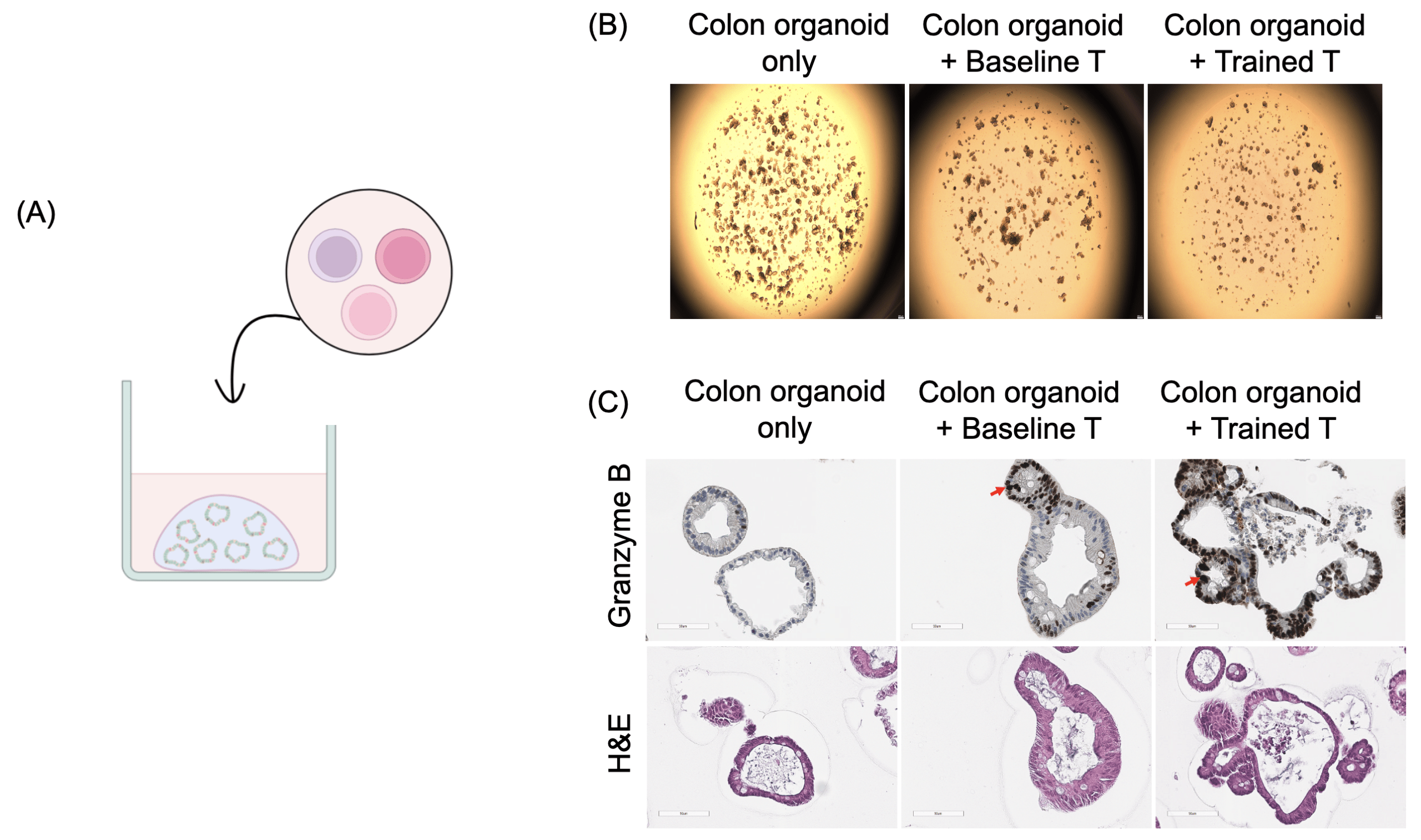
(B) Growth suppression of organoids observed in the dual-treated T cell group compared to baseline T cells or organoids cultured alone.
(C) Immunohistochemical staining for Granzyme B showing infiltration of primed T cells into organoid structures, with active cytotoxicity against tumor cells observed in the dual-treatment group.
To more closely recapitulate the tumor microenvironment, we tested “super soldier” in a single patient-derived 3D colorectal cancer organoids (Fig. 5a). Dual-treated T cells significantly suppressed organoid growth compared to baseline T cells or organoids cultured alone (Fig. 5b). Immunohistochemical staining for Granzyme B demonstrated successful infiltration of primed T cells into organoid structures, accompanied by active cytotoxic activity against tumor cells (Fig. 5c). These findings confirm that metabolically primed T cells maintain superior antitumor efficacy in physiologically relevant 3D tumor models and support the potential clinical applicability of this dual-target priming strategy.
Discussion
T-cell-based immunotherapies have revolutionized cancer treatment, yet their clinical efficacy, particularly in solid tumors, remains constrained by the metabolic barriers of the tumor microenvironment (TME). Exposure to this harsh environment leads to impaired mitochondrial function and ultimately T-cell exhaustion11,22, representing a critical challenge in current immunotherapy. Previous research has largely focused on reactive strategies, such as modulating single signaling pathways like AKT inhibition14,23, or activating co-stimulatory pathways after T cells have encountered metabolic stress24,25. To overcome these limitations, our study introduces an innovative strategy: ‘Metabolic Priming’, aiming to proactively optimize and enhance T-cell metabolic capacity before their direct confrontation with the tumor.
The core of this research is the integration of a double-target strategy—combining Bezafibrate to support mitochondrial biogenesis-associated programs with 4-1BB co-stimulation to enhance activation-associated signaling—to train T cells into “super soldier.” Our experimental results demonstrated that Bezafibrate-mediated PGC1α activation effectively stimulated mitochondrial biogenesis within T cells, while 4-1BB co-stimulation enhanced the AKT signaling pathway, thereby promoting cell survival and activation. Notably, the dual-treated group exhibited the most significant increases in mitochondrial function, activation marker expression, and cytokine secretion. Furthermore, these “super soldier” displayed superior anti-tumor efficacy and tumor infiltration capabilities in both 2D and 3D tumor models. These findings strongly suggest that proactive metabolic priming fundamentally enhances T-cell anti-tumor capacity and persistence, capable of eliciting a more potent immune response compared to single-treatment strategies.
Moreover, the approach proposed in this study offers a relatively cost-effective advantage compared to high-cost personalized CAR-T therapies or single co-stimulatory antibody strategies. Bezafibrate is an FDA-approved drug already in clinical use for hyperlipidemia, and 4-1BB co-stimulation is a well-established method for T-cell activation. Consequently, this strategy allows for the maximization of T-cell therapeutic efficacy by simply adding drug treatment and co-stimulation steps to existing T-cell isolation and expansion protocols, circumventing the need for expensive or complex genetic manipulation technologies. This offers a more realistic and economical immunotherapy approach for solid tumor patients, based on validated “super soldier”, thus holding significant implications for broadening access to advanced immunotherapies.
However, several limitations of the present study should be acknowledged. First, healthy donor–derived bulk CD3⁺ T cells were evaluated in co-culture with allogeneic tumor cell lines and patient-derived organoids, raising the possibility that part of the observed cytotoxicity reflects alloreactive responses rather than strictly tumor-antigen–specific killing. Nevertheless, because identical donor–tumor mismatch conditions were maintained across all experimental groups, the relative differences among control, single-treated, and metabolically primed T cells are more likely to represent treatment-associated changes in metabolic fitness and effector function than differences in antigen recognition. Second, although metabolic priming increased PGC1α expression, ATP production, AKT phosphorylation, and MitoTracker Red CMXRos signal, these findings do not comprehensively establish mitochondrial biogenesis or fully delineate the downstream signaling pathways induced by Bezafibrate and 4-1BB stimulation. Further studies evaluating PGC1α nuclear localization, canonical mitochondrial target genes, mtDNA copy number, oxygen consumption rate, and broader signaling pathways such as ERK, mTOR, and NF-κB would provide a more complete mechanistic framework. Third, the tumor co-culture experiments were conducted under standard culture conditions rather than under experimentally defined tumor microenvironment–mimetic metabolic stress, such as hypoxia, low nutrient availability, elevated lactate, or acidic pH. Accordingly, our data support the conclusion that metabolic priming enhances T-cell metabolic and functional fitness before tumor encounter, but do not yet directly prove a selective advantage under defined metabolic stress conditions. Future studies using antigen-matched or autologous systems in combination with tumor microenvironment–like stress models will be important to strengthen both the mechanistic depth and translational relevance of this approach. In addition, the present study did not include broader phenotypic profiling of effector quality and persistence, such as intracellular Perforin, proliferation, apoptosis, exhaustion markers, or memory-associated markers. Therefore, while our findings support enhanced acute functional fitness, they do not yet fully define the durability or differentiation status of the primed T-cell phenotype.
Future studies using autologous patient T cells, multiple tumor-matched organoids, or T cells expressing defined tumor-specific TCRs will be essential to determine how metabolic priming influences antigen-specific anti-tumor immunity in more physiologically relevant settings.
Furthermore, while only a single concentration and treatment duration of Bezafibrate and 4-1BB agonist were examined in this study, these conditions were selected based on prior reports demonstrating robust metabolic and co-stimulatory effects in human T cells. A systematic dose–response and time-course optimization will be important in future studies to define the minimal effective dose and therapeutic window.
References
- Morrissey, K., Yuraszeck, T., Li, C.-C., Zhang, Y. & Kasichayanula, S. Immunotherapy and Novel Combinations in Oncology: Current Landscape, Challenges, and Opportunities. Clinical and Translational Science 9, 89-104 (2016). [↩]
- Pilipow, K., et al. IL15 and T-cell Stemness in T-cell–Based Cancer Immunotherapy. Cancer Research 75, 5187-5193 (2015). [↩]
- Wagner, J., Wickman, E., DeRenzo, C. & Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Molecular Therapy 28, 2320-2339 (2020). [↩]
- Tang, X., et al. Magnetic–Acoustic Sequentially Actuated CAR T Cell Microrobots for Precision Navigation and In Situ Antitumor Immunoactivation. Advanced Materials 35, 2211509 (2023). [↩]
- Le Bourgeois, T., et al. Targeting T Cell Metabolism for Improvement of Cancer Immunotherapy. Frontiers in Oncology Volume 8 – 2018(2018). [↩]
- Evans, J.V., et al. Improving combination therapies: targeting A2B-adenosine receptor to modulate metabolic tumor microenvironment and immunosuppression. JNCI: Journal of the National Cancer Institute 115, 1404-1419 (2023). [↩]
- Reynolds, T.Y., Rockwell, S. & Glazer, P.M. Genetic instability induced by the tumor microenvironment. Cancer research 56 24, 5754-5757 (1996). [↩]
- Han, Y., Jo, H., Cho, J.H., Dhanasekaran, D.N. & Song, Y.S. Resveratrol as a Tumor-Suppressive Nutraceutical Modulating Tumor Microenvironment and Malignant Behaviors of Cancer. International Journal of Molecular Sciences 20, 925 (2019). [↩]
- Siska, P.J., et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2(2017). [↩]
- Scharping, N.E., et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374-388 (2016). [↩] [↩]
- Yu, Y.-R., et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nature Immunology 21, 1540-1551 (2020). [↩] [↩]
- Jiang, P., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nature Medicine 24, 1550-1558 (2018). [↩]
- Tsai, H.F. & Hsu, P.N. Cancer immunotherapy by targeting immune checkpoints: mechanism of T cell dysfunction in cancer immunity and new therapeutic targets. J Biomed Sci 24, 35 (2017). [↩]
- van der Waart, A.B., et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 124, 3490-3500 (2014). [↩] [↩]
- Sailer, N., et al. T-Cells Expressing a Highly Potent PRAME-Specific T-Cell Receptor in Combination with a Chimeric PD1-41BB Co-Stimulatory Receptor Show a Favorable Preclinical Safety Profile and Strong Anti-Tumor Reactivity. Cancers 14, 1998 (2022). [↩]
- Kamata, S., et al. Different Coactivator Recruitment to Human PPARα/δ/γ Ligand-Binding Domains by Eight PPAR Agonists to Treat Nonalcoholic Fatty Liver Disease. Biomedicines 12, 624 (2024). [↩]
- Augustyniak, J., et al. Bezafibrate Upregulates Mitochondrial Biogenesis and Influence Neural Differentiation of Human-Induced Pluripotent Stem Cells. Molecular Neurobiology 56, 4346-4363 (2019). [↩]
- Tan, S., et al. Platelet factor 4 enhances CD4+ T effector memory cell responses via Akt‐PGC1α‐TFAM signaling‐mediated mitochondrial biogenesis. Journal of Thrombosis and Haemostasis 18, 2685-2700 (2020). [↩]
- Li, W. & Zhang, L. Rewiring Mitochondrial Metabolism for CD8+ T Cell Memory Formation and Effective Cancer Immunotherapy. Frontiers in Immunology Volume 11 – 2020(2020). [↩]
- Campana, D., Schwarz, H. & Imai, C. 4-1BB Chimeric Antigen Receptors. The Cancer Journal 20, 134-140 (2014). [↩]
- Kroon, H.M., et al. 4-1BB Costimulation of Effector T Cells for Adoptive Immunotherapy of Cancer: Involvement of Bcl Gene Family Members. Journal of Immunotherapy 30, 406-416 (2007). [↩]
- Franco, F., Jaccard, A., Romero, P., Yu, Y.-R. & Ho, P.-C. Metabolic and epigenetic regulation of T-cell exhaustion. Nature Metabolism 2, 1001-1012 (2020). [↩]
- He, Y., et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduction and Targeted Therapy 6, 425 (2021). [↩]
- Menk, A.V., et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. Journal of Experimental Medicine 215, 1091-1100 (2018). [↩]
- Chester, C., Sanmamed, M.F., Wang, J. & Melero, I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood 131, 49-57 (2018). [↩]


