
Abstract
Diffuse Midline Glioma, or DMG, is an incurable high-grade pediatric brain tumor with a prognosis of less than one year. While the most defining mutation is H3K27M, mutations within the ACVR1, JAK1, and JAK2 genes are also associated with DMG. After years of extensive research, Jazz Pharmaceutical’s recent development of the ONC201 drug for those with a histone mutation marks a significant breakthrough by improving survival by approximately 10 months compared to the initial prognosis. However, there is still much room for improvement in advancing DMG treatment. Past research has noted overactivation of the bone morphogenetic protein (BMP) and JAK/STAT pathways as a common characteristic of DMG. As a result, inhibition of these pathways should be explored as a potential treatment method. Momelotinib, an ACVR1/JAK1/JAK2 inhibitor FDA-approved for the treatment of myelofibrosis, is a viable candidate. While previous research has examined other pathways to treat DMG, the use of momelotinib has not been previously examined in this context. PyRx, PyMol, and Biova Discovery Studio Visualizer were used to test the binding of momelotinib to relevant protein structures obtained from Protein Data Bank. Momelotinib was able to bind to both WT ACVR1, WT JAK1, and mutant ACVR1 with high binding affinities. This indicates momelotinib’s potential to bind to these proteins. However, further laboratory and clinical testing is necessary to truly confirm momelotinib’s potential to be used in treatment of DMG. While previous papers have confirmed momelotinib’s ability to bind to WT ACVR1, binding to mutant ACVR1 has not been previously explored.
Keywords: Diffuse Midline Glioma, Pediatric Brain Tumor, Momelotinib, Drug Repurposing, ACVR1, JAK/STAT, BMP
Introduction
Diffuse Midline Glioma, or DMG, is a high-grade pediatric brain tumor commonly found in children between the ages of 3 to 10 years. They are found within the midline structures, including the thalamus, brainstem, and spinal cord. Unlike many other pediatric brain cancers, DMG tumors exhibit a diffuse growth pattern, invading nearby cells rather than forming a mass. Additionally, they are mainly composed of astrocyte glial cells1. It is characterized by the histone H3K27M mutation, with a substitution of lysine to a methionine that occurs in approximately 70% of DMG cases2. This mutation, which occurs in the area of the histone H3 that is responsible for regulating gene expression, has been found to lead to an overall loss in gene repression and promote active gene transcription1.
The diffusive nature and the sensitive location makes the cancer exceptionally difficult to treat, setting it apart from other cancers. While chemotherapy and radiation therapy are primary treatment options, tumors recur in nearly 100% of patients after treatment, meaning that DMG is often considered to be an incurable disease. Despite DMG only manifesting in 1-2 per 100,000 children annually in the U.S, its median survival of less than a year makes it the leading cause of brain tumor-related deaths in children3. In contrast, oligodendroglioma, a more common type of brain cancer, accounts for 2-5% of all brain tumors but has a 5-year survival rate of approximately 50%4. Furthermore, non-small cell lung cancer, the second most common cancer type, but yet the leading cause of cancer death, has a 5-year survival rate of 26.4%5. Through these statistics, it can be seen that the survival rate for DMG is substantially lower compared to these other cancers. Diffuse Intrinsic Pontine Glioma (DIPG), a subtype of DMG located in the pons, has an even poorer prognosis.
While this histone mutation is the most common, previous research has identified various other mutated genes and dysregulated pathways that seem to commonly appear in DMG. Past research has pinpointed DMG as being heavily influenced by the BMP pathway as well as the JAK/STAT pathway, which play crucial roles in the skeletal and immune system, respectively. Specifically, gain of function mutations in ACVR1, a driver of the BMP pathway, were found in 25% of those diagnosed with DMG2,6. Similarly, overactivation of STAT3 was identified in many instances7. Increased activation of the JAK/STAT pathway has also been indicated to play a role in influencing the prognosis of patients with gliomas, as STAT3 overactivation is associated with a shorter prognosis7,8.
Despite the many years spent researching DMG, extensive work in both clinical and laboratory settings has failed to yield significant clinical advances. Surgery is often not considered an option due to the tumors’ diffusive nature and delicate location. Chemotherapy and radiation therapy remain the standard treatment options; however, tumors recur after treatment in almost 100% of patients3.
Recent research has made progress in improving treatment options for DMG. One prominent example is Jazz Pharmaceuticals’ recent development of the ONC201 drug for those with the histone mutation. The mechanism of ONC201 is unique. It targets the mitochondrial serine protease ClpP, thus decreasing mitochondrial membrane potential, increasing mitochondrial generation of reactive oxygen species, and inducing mitochondrial morphologic aberrations. Additionally, the drug triggers apoptosis and the integrated cell response9. Altogether, these factors contribute to ONC201’s efficacy in DMG treatment. Clinical trials have shown that this new drug appears to extend overall survival to approximately 21 months, compared to historical controls of just 12 months10. While this is nearly double the previous prognosis, there is still clear room for improvement. Due to the wide number of pathways that have been found to be dysregulated in DMG and the poor prognosis of this cancer, it is essential to further examine those pathways to look for potential treatment options.
The goal of this research is to highlight possible avenues for further research by using in silico modeling to test the binding affinities of momelotinib, which based on prior research has high potential to be used in treatment of DMG. This research is intended to be hypothesis-generating. As this study is entirely in silico, preliminary findings must be validated with further in vitro and in vivo testing. PyRx, PyMol, and Biova Discovery Studio visualizer were used to calculate the binding affinities of momelotinib to ACVR1 and JAK proteins. This is the first known study of momelotinib binding to mutant ACVR1. Due to its in silico nature, the scope of this study is severely limited. Cell lines do not always follow the behaviors predicted by in silico models, and modeling softwares do not always account for external factors. Additionally, the study was limited by the protein structures that were readily available online, meaning that not all proteins were able to be tested. Despite this, this study highlights avenues for further in vivo and in vitro research that hopefully will improve the treatment of DMG.
Bone Morphogenic Proteins in DMG
One pathway commonly dysregulated in DMG is the BMP signaling pathway, which is part of the TGF-Beta superfamily. TGF-Beta is a superfamily of signaling proteins. Initially discovered to play a role in bone formation, it is now seen how they are involved in regulating various physiological processes11. In its pathway, BMPs bind to the cell surface receptors and form a heterotetrameric complex with two dimers of type I and type II serine/threonine kinase receptors. ACVR1 is a type 1 BMP receptor, and thus plays a significant role in BMP signaling. Upon formation of the complex, the type II receptor transphosphorylates the type I receptor, activating it and thus leading to phosphorylation of the SMADs. R-SMADs then form a complex with SMAD4 to function as a transcription factor in the nucleus, regulating gene expression12.
Normally, ACVR1 activation is heavily regulated and depends on ligand binding. However, mutations within the glycine-serine (GS) domain or kinase domain of the type 1 ACVR1 receptor can allow this protein to phosphorylate without the ligand, leading to continuous and uncontrolled pathway activation and increased expression in the SMAD transcriptional targets, thus leading to abnormal cell proliferation. Mutations within the ACVR1 gene have been found to be especially prominent in patients with DIPG, with various studies noting them to be present in approximately 20-30% of cases2,13. Through genome sequencing, Fontebasso et al. were able to identify specific recurrent missense substitution mutations within ACVR1, including R206H within the GS domain, and R258G, G328E, G328V, and G356D within the kinase domain14.
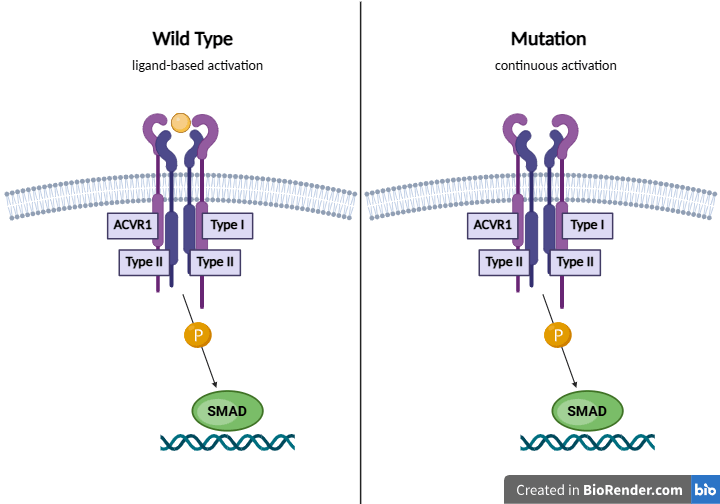
While overactivation of the ACVR1 pathway contributes to tumor progression, ACVR1 mutations cannot cause tumor formation on their own. Past in vivo studies have noted that continued ACVR1 activation promotes the mesenchymal profile of cells by increasing STAT3 signaling, and this effect is exacerbated by the H3K27M mutation15. Thus, while the H3K27M mutation is the driving mutation of the cancer, ACVR1 worsens its effects.
Due to the increased activation of the pathway in mutated ACVR1, a small molecule ACVR1 inhibitor that is able to penetrate the blood brain barrier is ideal for inhibiting BMP pathway overactivation6. Because ACVR1 mutations and BMP pathway dysregulation are relatively common occurrences in DMG, it is essential for this pathway to be further investigated to look for potential treatment options. Various molecules, such as momelotinib, Apigenin, and Diosmetin, work to target the BMP pathway16. Therefore, such drugs could be repurposed for DMG treatment. Throughout this paper, momelotinib’s potential to be repurposed in this manner will be explored.
Momelotinib and the BMP and JAK/STAT Pathway
Mutations in ACVR1 have been noted to be present in up to 21-30% of DMG patients2. Because these mutations have been thought to have an oncogenic role, an ACVR1 inhibitor should be explored for DMG treatment potential2. Momelotinib is a small molecule JAK1/JAK2/ACVR1 inhibitor that is FDA-approved for myelofibrosis and is a dual-inhibitor of both the JAK/STAT pathway and the BMP pathway. Both myelofibrosis and DMG share similar characteristics in regards to their pathways, as both are often characterized by BMP and JAK/STAT pathway overactivation17. An assay conducted by Asshof et al. noted the IC50 of JAK1, JAK2, and ACVR1 to be 26.9 nM, 1.4 nM, and 3.4 nM, respectively18. While these data cannot be directly compared against each other, they showcase the overall potency of the drug. Similarly to DMG, BMPs have been found to be overexpressed in myelofibrosis19. Because momelotinib inhibits the action of ACVR1, the main driver of the BMP pathway, ACVR1 inhibition reduces BMP pathway activation, as seen in myelofibrosis to reduce hepcidin production18. Thus, momelotinib could be explored in DMG to inhibit ACVR1 and BMP pathway overactivation. In addition to being an ACVR1 inhibitor, momelotinib also serves as a JAK1/JAK2 inhibitor. The JAK family, which consists of JAK1, JAK2, JAK3, and Tky2, play a large role in the JAK/STAT pathway. In this pathway, activation of JAK by a ligand triggers STAT phosphorylation. Once phosphorylated, dimerized STATs enter the nucleus to activate/repress transcription of target genes20.
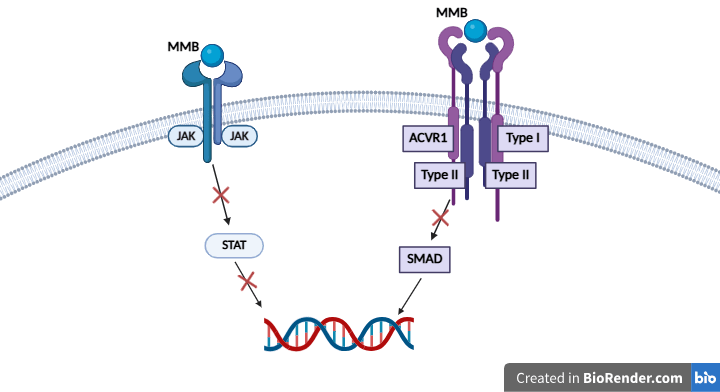
In an in vitro study conducted by Zhang et al., researchers screened a library of drugs using patient-derived DMG cell lines7. When the researchers tested various JAK2/STAT3 inhibitors, they found that many of them displayed a high efficacy, and thus STAT3 was identified as a significant target in DMG. Additionally, the researchers noted the prevalence of STAT3 mutations in DMG patients, finding that in a dataset of 211 various pediatric gliomas, including DMG, overexpression of STAT3 was linked to shorter survival, further emphasizing the role of STAT3 in DMG7. In order to inhibit STAT3 overactivity in DMG patients with STAT3 mutations, a JAK inhibitor, upstream from STAT, is ideal.
Inhibition of these overactivated pathways is expected to have several downstream effects. In the BMP pathway, inhibiting ACVR1 has been found to reduce SMAD phosphorylation21. Thus, it should be expected to reduce cell proliferation. Additionally, inhibition of JAK should reduce the overexpression of STAT, the downstream target of JAK. Since STAT was previously identified as a major driver of poor outcomes, JAK inhibition should mitigate these outcomes.
Overall, due to its function as an inhibitor for ACVR1, JAK1, and JAK2, momelotinib has potential to be implemented for treatment of DMG. Testing the drug’s properties in silico is advantageous to inform in vitro and in vivo experimentation.
Methods
Protein Data Bank was used to locate the crystal structures of the proteins. Specifically, structure ID numbers 3H9R (WT ACVR1), 9L04 (mutant ACVR1), 5KHW (WT JAK1), and 1UBQ (Ubiquitin) were used in the various simulations. Pubchem was utilized to download the chemical structure of momelotinib, as well as the compounds Itacnosertib and glucose that were used for the controls. Next, Biova Discovery Studio Visualizer was used to remove the water molecules and any co-crystallized ligands that were used to stabilize the protein during the crystallization process. FKBP12 was removed from 3H9R.
Then, the compound and protein were loaded into the PyRx Vina Wizard software. OpenBabbel was used to minimize the energy of the ligand, convert the protein into .pdbqt format, and simulate a docking. The exhaustiveness setting on Vina Wizard was 8. The grid box size was maximized, as blind docking was used. Protonation states and tautomers were assigned during the conversion of the ligands to .pdbqt format in preparation for docking. No residues were flexible. The PyRx docking yielded 9 poses for each protein-ligand pair, modes 0-8. The top ranked poses are shown in the results section above.
After docking was conducted, Biova Discovery Studio Visualizer was used to find the coordinates of where the co-crystallized ligand originally bonded to the receptor, as well as the binding coordinates of the new predicted pose. The 2 sets of coordinates were compared to ensure that they fell within a few angstroms of each other, validating that the workflow was not producing nonspecific binding results.
While the PyRx software gave an overview of the compound’s position relative to the protein, PyMol was used to better visualize how the ligand fit within the protein, as well as how it interacted with the surrounding atoms of the protein. The general ranges and trends of the binding affinities were evaluated to measure the probability of binding to the drug. No ethical considerations were necessary, as the study was conducted entirely in silico.
Results
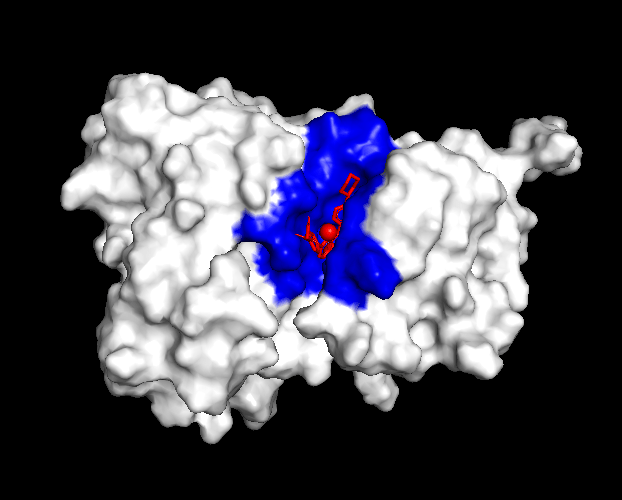
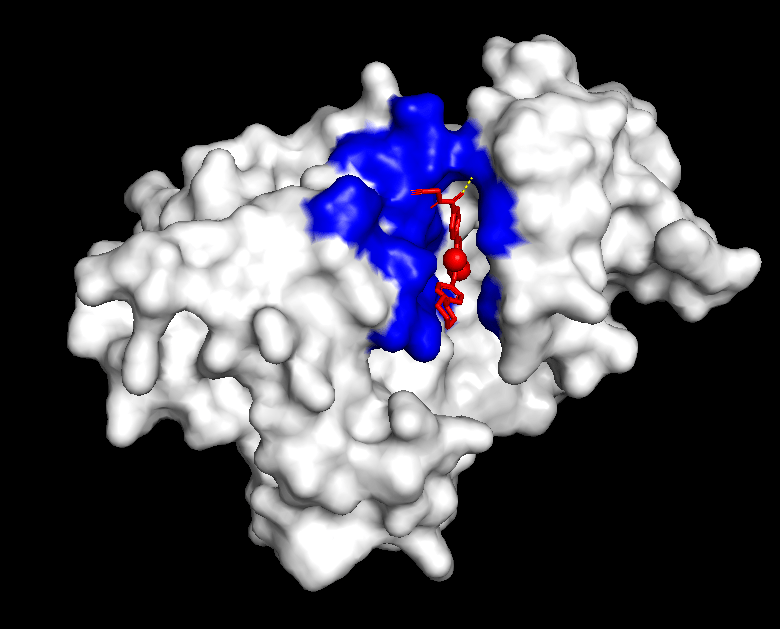
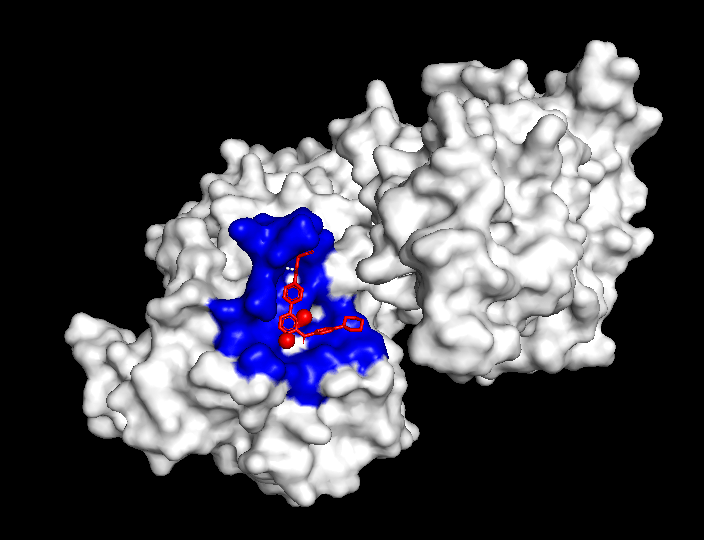
The compound’s properties and interaction with other proteins were tested through various softwares such as PyRx, PyMol, and more. Through this in silico testing, the goal was to evaluate the potential of momelotinib to bind to the ACVR1 and JAK1 proteins, as well as analyze how mutations could impact these binding affinities. In order to achieve this, the testing consisted of binding simulations through PyRx, as well as visual representations through PyMol. momelotinib is predicted to bind to both WT (Figure 1) and R206H mutant (Figure 2) ACVR1, as shown by their high binding affinities (Table 1 and 2).
| Binding Affinity | Mode | RMSD lower bound | RMSD upper bound |
| -9.4 | 0 | 0.0 | 0.0 |
| -8.9 | 1 | 1.757 | 2.989 |
| -8.8 | 2 | 2.915 | 5.478 |
| -8.7 | 3 | 2.514 | 4.421 |
| -8.2 | 4 | 2.411 | 8.212 |
| -8.2 | 5 | 3.052 | 4.54 |
| -7.9 | 6 | 1.818 | 8.06 |
| -7.5 | 7 | 3.896 | 7.5 |
| -7.3 | 8 | 15.218 | 18.151 |
| Binding Affinity | Mode | RMSD lower bound | RMSD upper bound |
| -7.9 | 0 | 0.0 | 0.0 |
| -7.1 | 1 | 19.976 | 24.158 |
| -6.8 | 2 | 15.499 | 18.493 |
| -6.8 | 3 | 15.167 | 18.533 |
| -6.7 | 4 | 16.61 | 19.735 |
| -6.7 | 5 | 30.451 | 32.632 |
| -6.6 | 6 | 17.882 | 21.978 |
| -6.6 | 7 | 15.218 | 18.852 |
| -6.5 | 8 | 16.05 | 18.722 |
Consistent with previous research, the simulation predicts momelotinib to bind to WT JAK1 (Figure 3), as showcased by the high binding affinities (Table 3). The binding affinities of JAK1 are similar to those of the mutant ACVR1 protein (Figure 2, Table 2), indicating a high probability of binding for both proteins.
| Binding Affinity | Mode | RMSD lower bound | RMSD upper bound |
| -7.2 | 0 | 0.0 | 0.0 |
| -7.1 | 1 | 29.496 | 32.665 |
| -6.9 | 2 | 44.853 | 50.624 |
| -6.7 | 3 | 35.555 | 38.796 |
| -6.6 | 4 | 5.942 | 9.671 |
| -6.5 | 5 | 29.49 | 34.97 |
| -6.4 | 6 | 26.273 | 28.97 |
| -6.2 | 7 | 26.638 | 28.867 |
| -6.2 | 8 | 28.66 | 31.71 |
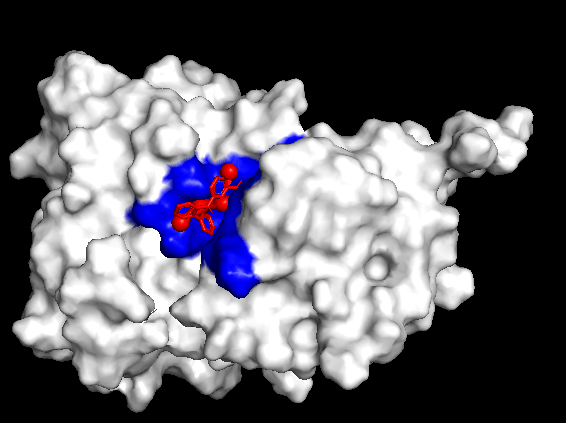
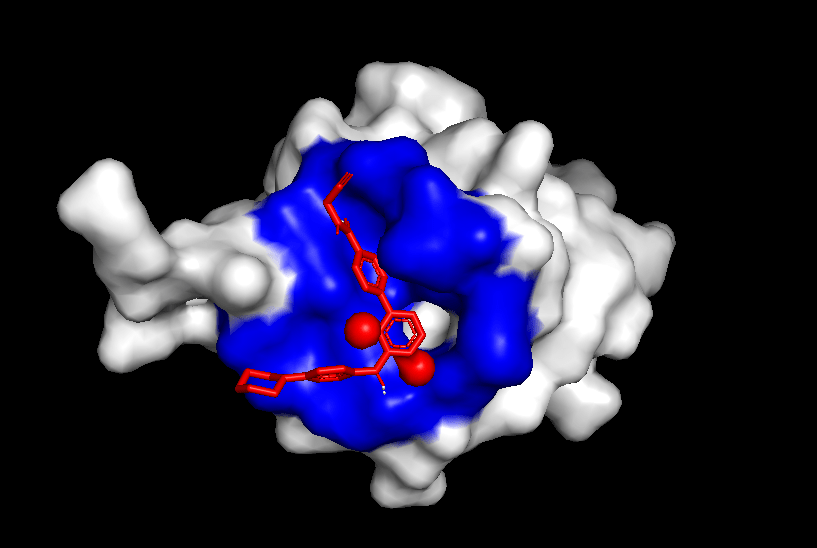
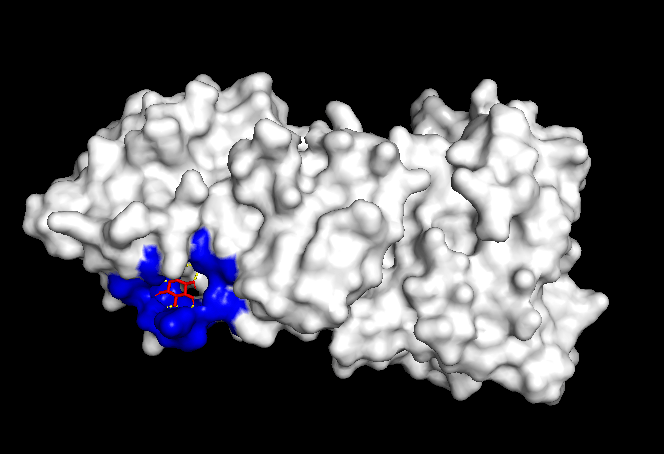
Finally, to test the reliability of the simulations, both positive and negative controls were tested. When Itacnosertib, a known ACVR1 inhibitor (Figure 4) was bound to momelotinib, the binding affinities were relatively high (Table 4). When momelotinib was bound to Ubiquitin (Figure 5), which is not known to have any interaction, the binding affinities were relatively low (Table 5). Similarly, when glucose was bound to momelotinib (Figure 6), which is not expected to have any reaction, the binding affinities were also relatively low (Table 6).
| Binding Affinity | Mode | RMSD lower bound | RMSD upper bound |
| -9.5 | 0 | 0.0 | 0.0 |
| -9.5 | 1 | 3.365 | 7.431 |
| -9.5 | 2 | 3.696 | 7.69 |
| -9.5 | 3 | 2.137 | 5.313 |
| -9.1 | 4 | 3.195 | 9.047 |
| -9.0 | 5 | 1.927 | 3.436 |
| -8.8 | 6 | 2.938 | 6.79 |
| -8.5 | 7 | 3.548 | 6.968 |
| -8.5 | 8 | 2.416 | 9.128 |
| Binding Affinity | Mode | RMSD lower bound | RMSD upper bound |
| -6.4 | 0 | 0.0 | 0.0 |
| -6.2 | 1 | 1.719 | 2.241 |
| -5.9 | 2 | 17.602 | 21.486 |
| -5.8 | 3 | 20.938 | 23.243 |
| -5.8 | 4 | 19.858 | 23.542 |
| -5.6 | 5 | 17.917 | 19.284 |
| -5.5 | 6 | 20.565 | 24.317 |
| -5.5 | 7 | 22.455 | 25.692 |
| -5.4 | 8 | 18.959 | 22.033 |
| Binding Affinity | Mode | RMSD lower bound | RMSD upper bound |
| -5.5 | 0 | 0.0 | 0.0 |
| -5.4 | 1 | 1.183 | 3.186 |
| -5.4 | 2 | 18.535 | 20.642 |
| -5.3 | 3 | 1.289 | 2.781 |
| -5.3 | 4 | 34.922 | 36.575 |
| -5.2 | 5 | 16.383 | 19.238 |
| -5.2 | 6 | 18.826 | 20.565 |
| -5.2 | 7 | 1.38 | 3.698 |
| -5.1 | 8 | 18.189 | 19.354 |
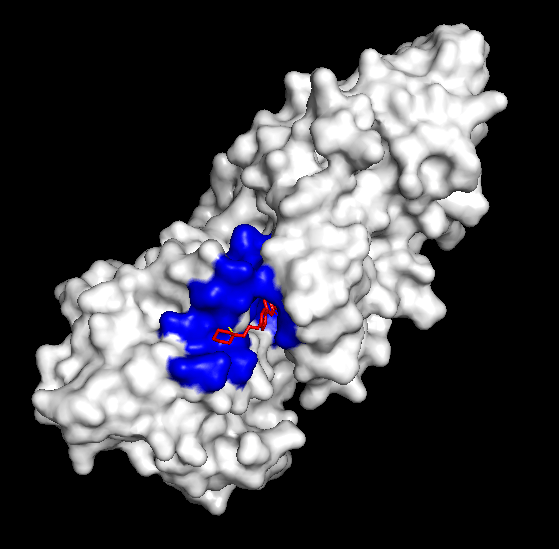
| Binding Affinity | Mode | RMSD lower bound | RMSD upper bound |
| -9.7 | 0 | 0.0 | 0.0 |
| -9.6 | 1 | 1.319 | 2.312 |
| -9.5 | 2 | 7.139 | 11.784 |
| -9.3 | 3 | 6.052 | 11.316 |
| -9.1 | 4 | 5.692 | 12.803 |
| -9.0 | 5 | 7.076 | 10.765 |
| -9.0 | 6 | 7.01 | 12.817 |
| -8.9 | 7 | 5.634 | 11.56 |
| -8.9 | 8 | 5.414 | 11.2 |
Discussion
Overall, the results of the simulation are consistent with what previous research has shown. However, it is important to note that due to the limited research that has been previously conducted in DMG, most evidence for momelotinib’s mechanism of action is derived from non-DMG disease models, specifically from myelofibrosis. While inferences can be drawn from these other disease models, the exact impacts in DMG remain unclear.
From the general patterns seen in the data, it can be seen that momelotinib binds relatively well to both the WT ACVR1 and WT JAK1 protein, with the binding affinities of the preferred poses being -9.3 and -7.2, respectively. Generally, a binding affinity of less than -6.0 kcal/mol represents an active drug, while a score of greater than -6.0 kcal/mol represents an inactive drug22. Previous research has already identified momelotinib to be an inhibitor of the ACVR1 and JAK proteins, consistent with the in silico results. The controls reaffirm the results of the simulation, as the binding affinities of the positive controls were relatively high (-9.9 for Itacnosertib bound to ACVR1), while the affinities of the negative controls were comparatively low (-6.4 for momelotinib bound to Ubiquitin, and -5.5 for glucose bound to ACVR1). Additionally, dorsomorphin, the co-crystallized ligand in 3H9R, was removed and redocked to ACVR1, the original receptor. The docking had relatively high binding affinities, thus validating the methods. However, when this predicted binding pose of dorsomorphin was compared against the experimental pose, the RMSD = 6.968. This indicates that the docking failed to reproduce the experimental pose, highlighting a significant limitation of the workflow.
T-tests were conducted on the binding affinities of the controls against WT ACVR1, mutant ACVR1, and WT JAK1. When comparing WT ACVR1, mutant ACVR1, and WT JAK1 individually against negative controls, p was less than 0.05, indicating a significant difference when they were compared against the negative controls. P was also less than 0.05 when the positive controls and WT ACVR1, mutant ACVR1, and WT JAK1, indicating a statistically significant difference between the groups. While this means that there was a significant difference between the positive controls and WT ACVR1, mutant ACVR1, and WT JAK1, all compounds exhibited strong binding scores, indicating potential binding despite differences in scores.
One significant aspect of these simulations are the results of the R206H mutant ACVR1. Previous research notes that momelotinib is able to bind to WT ACVR1 – however, no research exists to indicate the binding potential to mutant ACVR1, specifically R206H mutation that is frequently found in DMG cases. The results of the simulation show that the binding affinities of momelotinib to the mutant ACVR1 were relatively high (-7.9), and similar to the affinities of the WT ACVR1 (-9.3).
Additionally, the RMSD values given by AutoDock Vina compare how similar the positions of different predicted poses are. However, it is important to note that high RMSD values produced by AutoDock Vina above 30 can indicate unstable binding. Because RMSD indicates changes in the conformations of the different ligand poses, high values indicate poses that vary greatly and are thus unstable. High RMSD values were seen in the binding of momelotinib to WT JAK as well as R206H mutant ACVR1. Thus, despite the relatively high binding affinities, the high RMSD values should be taken into consideration.
Another important detail to note is the somewhat high binding affinity of momelotinib to ubiquitin, which was meant as a negative control. While the binding affinities were still lower than those of momelotinib and the positive control, they most likely indicate nonspecific docking or score inflation.
Testing in this manner does not come without limitations. For example, not all aspects of a drug can be tested with computer simulations, and simulations can only be as good as the models of which they are based. Additionally, differences in cell characteristics from person to person cannot be measured through a computer simulation, so simulation results are not completely accurate. Despite this, computer simulations provide a good starting point for drug discovery. Thus, while definite conclusions cannot be drawn, momelotinib’s potential for treatment of DMG should further be explored.
Future Research and Next Steps
In vitro and in vivo testing should be performed in the laboratory and clinical settings to truly understand the potential of this drug to target DMG cancers.
Primarily, a viability assay should be performed to test for DMG cancer cell death when treated with momelotinib. To perform a viability assay, cell lines must be acquired. This can be accomplished either through commercial suppliers, or by establishing a new cell line using patient cells after obtaining patient consent. Because momelotinib is an ACVR1 inhibitor, it is essential to ensure that the cell lines are validated for ACVR1 mutations – otherwise, the results will not truly be representative as the pathway would not be overactivated in WT cells. In their study evaluating the role of the STAT3 mutation in DMG, Zhang et al. (2022) used CellTiter-Blue Cell Viability Assay7. However, assays such as CellTiter-Blue and MTT that rely on cell metabolism should be avoided, as cell metabolism has been found to be altered in DMG cancer cells23. On the other hand, SRB assays, which rely on protein concentration rather than metabolism to measure cell death, are frequently used in cancer drug screening, making SRB a strong method to ensure accurate results24. In the analysis of the assay, it is essential for a variety of key data points, such as the IC50, various time points, and phenotype to be noted.
However, implementing momelotinib for DMG treatment poses a variety of challenges. Primarily, the largest challenge is ensuring that the drug can cross the blood brain barrier. The FDA Center for Drug Evaluation and Research noted that momelotinib, despite being classified as a small molecule, transferred across the blood brain barrier at a relatively low rate, with a blood-to-plasma ratio for this drug being estimated at 0.215 at 60 minutes25. This rate indicates that momelotinib penetrates the blood brain barrier at a relatively low rate, posing a significant challenge for efficacy. However, because momelotinib has never been tested for use in the brain, there is a lack of research in this area. Thus, further research must be conducted to explore momelotinib’s ability to penetrate the blood brain barrier.
The low rate of penetration may be due to the presence of efflux transporters that target momelotinib and remove it from the cell. In vitro studies have identified that momelotinib acts as a substrate for various efflux drug transporters, such as P-glycoprotein and Breast Cancer Resistance Protein26. One possible solution is to use efflux transporter inhibitors, which would involve pharmacologically inhibiting the efflux transporters such as P-glycoprotein and Breast Cancer Resistance Proteins. Various inhibitors for these drugs currently exist. By using inhibitors such as these, it would address the root cause of the issue and allow for more effective transport of momelotinib across the blood-brain barrier. However, the side effects of these inhibitors should be considered and further researched. For example, drug interactions could have a toxic impact on the body, or toxins could build up in the cell due to the compromised efflux transporters. Therefore, these inhibitors should go through extensive testing in order to isolate which inhibitors are best suited for this usage.
Another potential challenge is gaining FDA approval for this specific drug usage. Although momelotinib has already been FDA-approved for the treatment of myelofibrosis, gaining FDA approval for this new indication may be a time-consuming process, particularly due to the clinical trials required. However, drug repurposing is more productive and economically cheaper, as noted by Ashburn and Thor27. Although, as stated in a literature review conducted by Krishnamurthy et al., exactly how much of a difference repurposing makes is unclear, presenting another limitation28.
Additionally, momelotinib has only previously been used in adult populations. However, DMG is primarily a pediatric cancer. The impacts of any drug on the body can greatly differ between pediatric and adult patients. Pharmacokinetic processes, including absorption, distribution, metabolism and excretion, often vary between children and adults29. Thus, the effects of momelotinib in pediatric populations must be examined through vigorous testing before clinical use.
Furthermore, the drug will likely be most effective in patient populations with overactivation in either the BMP or JAK/STAT pathway. This may be caused by an ACVR1 mutation, but it also may be caused by other mutations within the BMP or JAK/STAT pathways. Thus, for this drug to be effective, widespread genetic testing must be implemented for those with the disease to ensure that the drug is only administered to those within the proper patient population, as it may be ineffective otherwise. However, there are a variety of challenges preventing widespread genetic testing to locate the proper patient population. For instance, the extremely sensitive and fragile location of the tumor can make it difficult to determine what mutations are present within the cancerous cells. One potential solution to this is to use liquid biopsies to analyze the DNA for mutations in a minimally invasive manner. An added benefit of these biopsies is that they would enable physicians to monitor how the tumor responds to treatment over time, which is not available with current diagnostic tools30. Of note, the ability of liquid biopsies to be used in a clinical setting continues to be tested.
Finally, there is a significant lack of both preclinical and clinical evidence supporting the use of momelotinib in DMG treatment. Primarily, conducting docking purely in silico poses significant limitations, as no simulation can replicate true biological activity. Thus, these simulations must be followed with further laboratory and clinical testing. Additionally, there is a lack of DMG specific data on momelotinib, as the compound has never previously been evaluated in the context of brain cancer treatment. Furthermore, while myelofibrosis and DMG share common pathway overactivations, they differ in their locations and mechanisms. Because most research on momelotinib comes from myelofibrosis disease models, this poses a significant issue.
Conclusion
Overall, momelotinib may have potential to be used in the treatment of DMG. The BMP pathway, which consists of type I and type II receptors that phosphorylate SMADs to regulate gene expression, has been found to be mutated in many cases of DMG. Specifically, ACVR1 mutations within this pathway have been found to lead to BMP pathway overactivation. Additionally, the JAK/STAT pathway has also been found to be commonly overactivated in DMG patients. In silico testing also showed momelotinib as having high binding affinities when bound to both WT and mutant ACVR1, as well as JAK1 – however, the high RMSD values could indicate unstable binding. While no definite conclusions can currently be made, momelotinib should be further investigated through laboratory trial and clinical study as a potential treatment for DMG patients with overexpressed BMP or JAK/STAT pathways.
References
- S. Induni, H. Soderholm, K. B. Pointer. Prognostic factors in H3K27M-mutant diffuse midline gliomas. International Journal of Radiation Oncology Biology Physics. Vol. 120, 2024, https://doi.org/10.1016/j.ijrobp.2024.07.1518. [↩] [↩]
- E. Hayden, H. Holliday, R. Lehmann, A. Khan, M. Tsoli, B. S. Rayner, D. S. Ziegler. Therapeutic targets in diffuse midline gliomas—an emerging landscape. Cancers. Vol. 13, pg. 6251, 2021, https://doi.org/10.3390/cancers13246251. [↩] [↩] [↩] [↩] [↩]
- A. Noon, S. Galban. Therapeutic avenues for targeting treatment challenges of diffuse midline gliomas. Neoplasia. Vol. 40, 2023, https://doi.org/10.1016/j.neo.2023.100899. [↩] [↩]
- S. Liu, X. Liu, Y. Xiao, S. Chen, W. Zhuang. Prognostic factors associated with survival in patients with anaplastic oligodendroglioma. PLOS ONE. Vol. 14, 2019, https://doi.org/10.1371/journal.pone.0211513. [↩]
- A. K. Ganti, A. B. Klein, I. Cotarla, B. Seal, E. Chou. Update of incidence, prevalence, survival, and initial treatment in patients with non–small cell lung cancer in the US. JAMA Oncology. Vol. 7, pg. 1824–1832, 2021, https://doi.org/10.1001/jamaoncol.2021.4932. [↩]
- K. R. Taylor, M. Vinci, A. N. Bullock, C. Jones. ACVR1 mutations in DIPG: Lessons learned from FOP. Cancer Research. Vol. 74, pg. 4565–4570, 2014, https://doi.org/10.1158/0008-5472.CAN-14-1298. [↩] [↩]
- L. Zhang, C. L. Nesvick, C. A. Day, J. Choi, V. M. Lu, T. Peterson, E. A. Power, J. B. Anderson, F. H. Hamdan, P. A. Decker, R. Simons, J. P. Welby, R. Siada, J. Ge, T. Kaptzan, S. A. Johnsen, E. H. Hinchcliffe, D. J. Daniels. STAT3 is a biologically relevant therapeutic target in H3K27M-mutant diffuse midline glioma. Neuro-Oncology. Vol. 24, pg. 1700–1711, 2022, https://doi.org/10.1093/neuonc/noac093. [↩] [↩] [↩] [↩] [↩]
- Y. Tu, Y. Zhong, J. Fu, Y. Cao, G. Fu, X. Tian, B. Wang. Activation of JAK/STAT signal pathway predicts poor prognosis of patients with gliomas. Medical Oncology. Vol. 28, pg. 15–23, 2011, https://doi.org/10.1007/s12032-010-9435-1. [↩]
- B. Purow. ONC201 and ONC206: metabolically ClipPing the wings of diffuse midline glioma. Neuro-Oncology. Vol. 24, pg. 1452-1453, 2022, https://doi.org/10.1093/neuonc/noac103. [↩]
- I. Arrillaga-Romany, A. Lassman, S. L. McGovern, S. Mueller, B. Nabors, M. van den Bent, M. A. Vogelbaum, J. E. Allen, A. S. Melemed, R. S. Tarapore, P. Y. Wen, T. Cloughesy. Action: A randomized phase 3 study of ONC201 (dordaviprone) in patients with newly diagnosed H3 K27M-mutant diffuse glioma. Neuro-Oncology, Vol. 26, 2024, https://doi.org/10.1093/neuonc/noae031. [↩]
- J. L. Wrana. Signaling by the TGF-β superfamily. Cold Spring Harbor Perspectives in Biology. Vol. 5, pg. a011197, 2013, https://doi.org/10.1101/cshperspect.a011197. [↩]
- R. N. Wang, J. Green, Z. Wang, Y. Deng, M. Qiao, M. Peabody, Q. Zhang, J. Ye, Z. Yan, S. Denduluri, O. Idowu, M. Li, C. Shen, A. Hu, R. C. Haydon, R. Kang, J. Mok, M. J. Lee, H. L. Luu, L. L. Shi. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes & Diseases. Vol. 1, pg. 87–105, 2014, https://doi.org/10.1016/j.gendis.2014.07.005. [↩]
- K. R. Taylor, A. Mackay, N. Truffaux, Y. Butterfield, O. Morozova, C. Philippe, D. Castel, C. S. Grasso, M. Vinci, D. Carvalho, A. M. Carcaboso, C. de Torres, O. Cruz, J. Mora, N. Entz-Werle, W. J. Ingram, M. Monje, D. Hargrave, A. N. Bullock, J. Grill. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nature Genetics. Vol. 46, pg. 457–461, 2014, https://doi.org/10.1038/ng.2925. [↩]
- A. M. Fontebasso, S. Papillon-Cavanagh, J. Schwartzentruber, H. Nikbakht, N. Gerges, P. Fiset, D. Bechet, D. Faury, N. De Jay, L.A. Ramkissoon, A. Corcoran, D. R. Jones, D. Sturm, P. Johann, T. Tomita, S. Goldman, M. G. Nagib, A. Bendel, L. Goumnerova, D.C Bowers. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nature Genetics, Vol. 46, pg. 462–466, 2014, https://doi.org/10.1038/ng.2950. [↩]
- C. M. Hoeman, F. J. Cordero, G. Hu, K. Misuraca, M. M. Romero, H. J. Cardona, J. Nazarian, R. Hashizume, R. McLendon, P. Yu, D. Procissi, S. Gadd, O. J. Becher. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nature Communications. Vol. 10, 2019, https://doi.org/10.1038/s41467-019-08823-9. [↩]
- K. Vrijens, W. Lin, J. Cui, D. Farmer, J. Low, E. Pronier, F.-Y. Zeng, A. A. Shelat, K. Guy, M. R. Taylor, T. Chen, M. F. Roussel. Identification of small molecule activators of BMP signaling. PLOS ONE. Vol. 8, 2013, https://doi.org/10.1371/journal.pone.0059045. [↩]
- A. Bruzzese, E. A. Martino, C. Labanca, F. Mendicino, E. Lucia, V. Olivito, A. Zimbo, V. Fragliasso, A. Neri, F. Morabito, E. Vigna, M. Gentile. Momelotinib in myelofibrosis. Expert Opinion on Pharmacotherapy. Vol. 25, pg. 521–528, 2024, https://doi.org/10.1080/14656566.2024.2343780. [↩]
- M. Asshoff, V. Petzer, M. R. Warr, D. Haschka, P. Tymoszuk, E. Demetz, M. Seifert, W. Posch, M. Nairz, P. Maciejewski, P. Fowles, C. J. Burns, G. Smith, K.-U. Wagner, G. Weiss, J. A. Whitney, I. Theurl. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. Vol. 129, pg. 1823–1830, 2017, https://doi.org/10.1182/blood-2016-09-740092. [↩] [↩]
- O. Bock, J. Höftmann, K. Theophile, K. Hussein, B. Wiese, J. Schlué, H. Kreipe. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. The American Journal of Pathology. Vol. 172, pg. 951–960, 2008, https://doi.org/10.2353/ajpath.2008.071030. [↩]
- J. S. Rawlings, K. M. Rosler, D. A. Harrison. The JAK/STAT signaling pathway. Journal of Cell Science. Vol. 117, pg. 1281–1283, 2004, https://doi.org/10.1242/jcs.00963. [↩]
- S. T. Oh, R. A. Mesa, C. N. Harrison, P. Bose, A. T. Gerds, V. Gupta, B. L. Scott, J. Kiladjian, A. Lucchesi, T. Kong, S. A. Buckley, S. Tyavanagimatt, B. G. Harder, K. Roman-Torres, J. Smith, A. R. Craig, J. Mascarenhas, S. Verstovsek. Pacritinib is a potent ACVR1 inhibitor with significant anemia benefit in patients with myelofibrosis. Blood Advances. Vol. 7, pg. 5835-5842, 2023, https://doi.org/10.1182/bloodadvances.2023010151. [↩]
- A. Nath, A. Kumer, F. Zaben and M. W. Khan. Investigating the binding affinity, molecular dynamics, and ADMET properties of 2,3-dihydrobenzofuran derivatives as an inhibitor of fungi, bacteria, and virus protein. Beni-Suef University Journal of Basic and Applied Sciences. Vol. 10, 2021, https://doi.org/10.1186/s43088-021-00117-8. [↩]
- J. Park, C. Chung. Epigenetic and metabolic changes in diffuse intrinsic pontine glioma. Brain Tumor Research and Treatment. Vol. 11, pg. 86–93, 2023, https://doi.org/10.14791/btrt.2023.0011. [↩]
- M. S. Shakil, Z. Rana, M. Hanif, R. J. Rosengren. Key considerations when using the sulforhodamine B assay for screening novel anticancer agents. Anti-Cancer Drugs. Vol. 33, pg. 6–10, 2022, https://doi.org/10.1097/CAD.0000000000001131. [↩]
- Center for Drug Evaluation and Research. Application number: 216873Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/216873Orig1s000IntegratedR.pdf, 2023. [↩]
- Y. L. Ho, P. Gorycki, G. Ferron-Brady, P. Martin, G. Vlasakakis. Clinical assessment of momelotinib drug–drug interactions via CYP3A metabolism and transporters. Clinical and Translational Science. Vol. 17, 2024, https://doi.org/10.1111/cts.13799. [↩]
- T. T. Ashburn, K. B. Thor. Drug repositioning: Identifying and developing new uses for existing drugs. Nature Reviews Drug Discovery. Vol. 3, pg. 673–683, 2004, https://doi.org/10.1038/nrd1468. [↩]
- N. Krishnamurthy, A. A. Grimshaw, S. A. Axson, S. H. Choe, J. E. Miller. Drug repurposing: A systematic review on root causes, barriers and facilitators. BMC Health Services Research. Vol. 22, 2022, https://doi.org/10.1186/s12913-022-08272-z. [↩]
- E. Fernandez, R. Perez, A. Hernandez, P. Tejada, M. Arteta, J. T. Ramos. Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics. Vol. 3, 2011, https://doi.org/10.3390/pharmaceutics3010053. [↩]
- E. Panditharatna, L. B. Kilburn, M. S. Aboian, M. Kambhampati, H. Gordish-Dressman, S. N. Magge, N. Gupta, J. S. Myseros, E. I. Hwang, C. Kline, J. R. Crawford, K. E. Warren, S. Cha, W. S. Liang, M. E. Berens, R. J. Packer, A. C. Resnick, M. Prados, S. Mueller, J. Nazarian. Clinically relevant and minimally invasive tumor surveillance of pediatric diffuse midline gliomas using patient derived liquid biopsy. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. Vol. 24, pg. 5850–5859, 2018, https://doi.org/10.1158/1078-0432.CCR-18-1345. [↩]


