
Abstract
A mechanistic understanding of antioxidant activity requires detailed evaluation of electronic structure and radical stabilization effects. In this study, a comparative density functional theory (DFT) investigation of three structurally distinct antioxidants: quercetin, curcumin, and ascorbic acid, was conducted at the B3LYP/6-31G(d) level to elucidate intrinsic hydrogen-donation and radical stabilization mechanisms. Fully optimized geometries were confirmed as true minima through harmonic vibrational frequency analysis, and zero-point energy corrections were included in all thermodynamic calculations. Antioxidant behavior was evaluated using bond dissociation enthalpies (BDE), ionization potentials (IP), Natural Bond Orbital (NBO) second-order perturbation stabilization energies, frontier molecular orbital localization, and molecular electrostatic potential (MEP) mapping. Results indicate that antioxidant efficiency cannot be attributed solely to HOMO-LUMO gap magnitude but instead arises from a balance between hydrogen-donation accessibility and post-abstraction radical stabilization. Ascorbic acid exhibits the lowest BDE, reflecting facile hydrogen transfer, whereas quercetin demonstrates superior radical stabilization through extensive π-conjugation and strong lone pair→π* donor-acceptor interactions. Curcumin displays intermediate behavior consistent with partial conjugative stabilization. Integrated analysis across multiple descriptors establishes the intrinsic stabilization trend as quercetin > ascorbic acid > curcumin. These findings provide an electronic framework for understanding structure-activity relationships and guiding the rational design of improved antioxidant systems.
Keywords: Density Functional Theory (DFT), B3LYP, HOMO-LUMO gap, Antioxidants, Curcumin, Quercetin, Ascorbic Acid
Introduction
Oxidative stress, arising from an imbalance between reactive oxygen species (ROS) and endogenous antioxidant defenses, plays a central role in aging and the progression of numerous chronic diseases, including cancer, neurodegenerative disorders, diabetes, and cardiovascular conditions1,2. Antioxidants mitigate oxidative damage by neutralizing radical species through electron or hydrogen atom transfer processes, thereby interrupting radical chain reactions and preserving cellular integrity. Understanding the structural and electronic factors that govern antioxidant efficiency is therefore essential for both mechanistic insight and rational molecular design.
Density Functional Theory (DFT) has become a widely used tool for investigating molecular-level antioxidant reactivity. Many computational studies have relied on frontier molecular orbital descriptors, particularly HOMO and LUMO energies and their energy gap, as indicators of electron-donating ability and chemical reactivity. While these descriptors provide useful qualitative information, antioxidant activity is inherently mechanism-dependent and may proceed via hydrogen atom transfer (HAT), single electron transfer (SET), sequential proton loss electron transfer (SPLET), metal chelation, or mixed pathways depending on the environment and assay conditions3,4,5,6,7. Consequently, reliance on orbital energy gaps alone does not fully capture the thermodynamic and electronic determinants governing radical stabilization. Although numerous DFT investigations have examined individual natural antioxidants, a systematic comparative analysis integrating multiple mechanistically relevant descriptors across structurally distinct antioxidant classes remains limited8,9,10,11,12. In particular, the quantitative relationship between bond dissociation enthalpies (BDEs), ionization potentials (IPs), donor-acceptor interactions derived from Natural Bond Orbital (NBO) analysis, and spatial charge distribution (MEP) has not been consistently explored within a unified framework4,13. Establishing such connections is essential for advancing from descriptor-based ranking toward mechanistically grounded structure-activity relationships.
To address this gap, the present study performs an integrated electronic-structure comparison of three structurally distinct natural antioxidants: quercetin (a flavonoid polyphenol), curcumin (a diarylheptanoid), and ascorbic acid (an enediol lactone). These molecules were selected intentionally to represent three different antioxidant scaffolds with differing degrees of π-conjugation, intramolecular hydrogen bonding, and charge delocalization capacity. Rather than merely confirming known experimental activity trends, this work seeks to identify the electronic origin of radical stabilization by linking hydrogen atom transfer thermodynamics to intramolecular donor-acceptor coupling and charge delocalization strength2,11.
Calculations were performed at the B3LYP/6-31G(d) level of theory to evaluate optimized geometries, vibrational stability, frontier orbital characteristics, bond dissociation enthalpies for key O-H sites, ionization potentials, molecular electrostatic potentials, and second-order perturbation stabilization energies from NBO analysis. By correlating thermodynamic hydrogen donation propensity with quantitative measures of electronic delocalization, this study aims to clarify the mechanistic factors that differentiate antioxidant scaffolds at the quantum chemical level. Through this integrated approach, the work provides a mechanistically oriented framework for understanding antioxidant efficiency and establishes electronic structure criteria that may guide the rational design of next-generation radical-scavenging molecules.
Computational Methodology
All quantum chemical calculations were performed using the Gaussian 09 program suite (Revision D.01). Initial molecular structures of ascorbic acid, quercetin, and curcumin were constructed based on experimentally reported geometries from the literature and subsequently pre-optimized using molecular mechanics before DFT refinement. Given the conformational flexibility of quercetin (multiple hydroxyl rotamers) and curcumin (flexible linker and methoxy rotations), a systematic conformational search was performed before DFT optimization. Rotational scans were conducted around phenolic O-H bonds, methoxy substituents in curcumin, central linker dihedral angles in curcumin, and relevant hydroxyl dihedrals in quercetin. For each molecule, 8-15 distinct conformers were generated and optimized at the B3LYP/6-31G(d) level. The conformer with the lowest electronic energy, including zero-point energy (ZPE) correction, was selected for all reported electronic, thermodynamic, NBO, and MEP analyses. All reported HOMO-LUMO energies correspond to these lowest-energy conformers. Curcumin was found to preferentially adopt the enol tautomer in the gas phase, consistent with prior theoretical studies. All reported results correspond to this tautomeric form.
All geometry optimizations were carried out using DFT with the B3LYP hybrid exchange-correlation functional in conjunction with the 6-31G(d) basis set3,14,15. This level of theory was selected because B3LYP has been extensively benchmarked for organic radical systems, provides reliable trends in bond dissociation enthalpies and ionization potentials, and offers a practical balance between computational cost and accuracy for molecules of this size15. Literature benchmarks report typical orbital energy uncertainties of approximately ±0.2-0.3 eV at this level. Although range-separated functionals such as ωB97X-D and larger basis sets may improve quantitative accuracy, B3LYP/6-31G(d) is sufficient for qualitative comparative analysis across structurally related organic systems. All geometries were fully optimized without symmetry constraints. An ultrafine integration grid (99 radial shells and 590 angular points) was employed. The self-consistent field (SCF) convergence criterion was set to 10⁻⁸ Hartree. Geometry optimizations were performed using Gaussian’s default tight convergence criteria for maximum and root-mean-square (RMS) forces. Harmonic vibrational frequency calculations were performed at the same level of theory to confirm the absence of imaginary frequencies, verify that all optimized structures are true minima on the potential energy surface, obtain zero-point energy corrections, and extract the thermochemical corrections required for enthalpy calculations. Neutral molecules were treated as closed-shell singlets (charge = 0, multiplicity = 1). Hydrogen-abstracted radicals were computed using unrestricted DFT (UB3LYP) with doublet multiplicity (charge = 0, multiplicity = 2). Radical cations, used for ionization potential calculations, were also treated as doublets (charge = +1, multiplicity = 2). Spin contamination was monitored in all open-shell calculations and found to be negligible, with ⟨S²⟩ values approximately 0.75 for doublet species.
Bond Dissociation Enthalpy (BDE)
O–H bond dissociation enthalpies were calculated using the adiabatic enthalpy difference:
(1) 
All species, including neutral molecules and radicals, were fully optimized and verified as true minima prior to enthalpy extraction. Zero-point energy corrections were included in all reported BDE values
Ionization Potential (IP)
Adiabatic ionization potentials were calculated as:
(2) 
Both neutral and cationic species were fully optimized and confirmed via frequency analysis before thermochemical data extraction.
NBO analysis was performed using NBO version 3.1 as implemented in Gaussian 0916,17. Second-order perturbation stabilization energies E(2) were extracted to evaluate donor-acceptor interactions. Only interactions with E(2) ≥ 5 kcal/mol were considered significant. Particular attention was given to lone pair (LP) donor to π* acceptor interactions associated with hydrogen-donating sites. Default NBO population thresholds were employed. All NBO analyses were conducted on optimized lowest-energy conformers. MEP surfaces were generated from optimized neutral geometries at the B3LYP/6-31G(d) level. Electrostatic potentials were mapped onto electron density surfaces defined at 0.002 a.u. Visualizations were produced using GaussView 5.0, and consistent color scale ranges were maintained across all molecules to enable direct comparison. All calculations were performed in the gas phase. Solvent effects, which may significantly influence antioxidant mechanisms such as sequential proton loss electron transfer (SPLET), were not included in the present study and are discussed explicitly as a limitation.
Results and Discussions
The molecular structures of quercetin, curcumin, and ascorbic acid were fully optimized, and harmonic vibrational frequency analyses were performed at the B3LYP/6-31G(d) level of theory to confirm that all stationary points correspond to true minima on the potential energy surface. The discussion integrates key geometric characteristics with derived thermodynamic and electronic properties, with particular emphasis on ZPE corrections and radical stabilization effects. Given the reported uncertainty of approximately ±0.2-0.3 eV for orbital energies at this level of theory, the calculated HOMO energy differences are interpreted in terms of broader electronic tendencies rather than strict quantitative distinctions. Accordingly, mechanistic conclusions are drawn from the collective analysis of multiple descriptors, including BDE, IP, NBO stabilization energies, MEP, and orbital localization patterns.
Structural Optimization and ZPE
Geometry optimization revealed distinct structural frameworks that directly influence radical stabilization capacity. Quercetin adopts a predominantly planar configuration across its A, B, and C rings (Figure 1a), enabling extensive π-conjugation throughout the polyphenolic scaffold. An intramolecular hydrogen bond between the 5-hydroxyl group and the carbonyl oxygen forms a stable six-membered pseudo-ring, enhancing structural rigidity and promoting efficient charge redistribution following hydrogen abstraction. Curcumin optimizes in its enol tautomeric form (Figure 1b), consistent with previous theoretical studies. The conjugated heptadienone linker supports partial delocalization of electron density, although slight flexibility along the central chain reduces uniformity of conjugation relative to quercetin.
| Molecule | Key Structural Features | Planarity | Intramolecular H-Bonds | ZPE-Corrected Energy (Hartree) |
| Quercetin | Planar A, B, C rings, extended π-conjugation | High | 5-OH → C=O (pseudo-ring) | -1083 |
| Curcumin | Near-planar enol tautomer, conjugated heptadienone linker | Moderate | Enol bridge H-bond | -1262 |
| Ascorbic acid | Puckered lactone ring, 3D H-bonding network | Low | Local H-bonding in enediol | -684 |
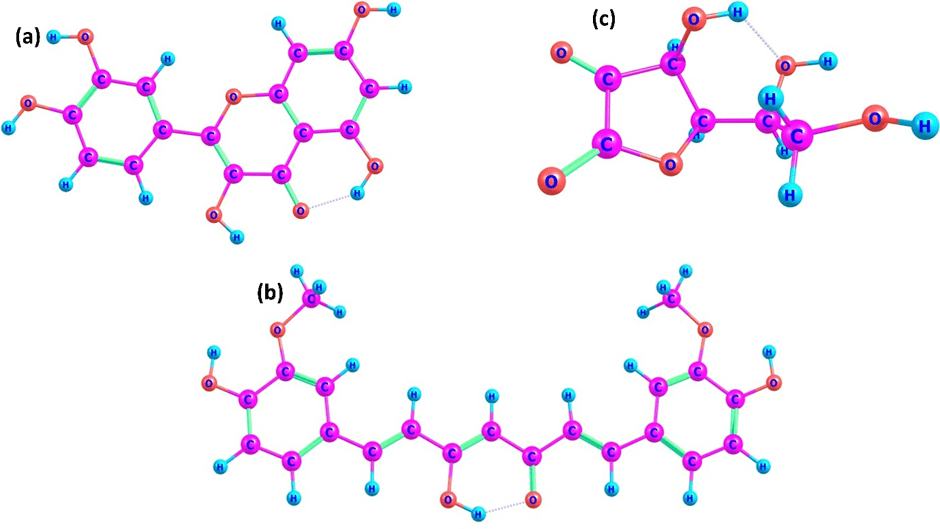
In contrast, ascorbic acid (Figure 1c) exhibits a puckered lactone ring with a three-dimensional hydrogen-bonding network. While the enediol moiety serves as the primary redox-active site, the absence of extended aromatic conjugation limits long-range delocalization of unpaired electron density after hydrogen donation. Frequency analyses confirmed that all geometries correspond to true minima. ZPE corrections were included for all neutral, radical, and cationic species. Although absolute ZPE-corrected energies scale with molecular size, relative ZPE differences between neutral and radical species provide meaningful insight into accessible hydrogen abstraction pathways (Table 1). The rigid, planar, and conjugated scaffold of quercetin therefore provides a structural advantage for post-abstraction radical stabilization.
Frontier Molecular Orbitals to Support Electronic Insight
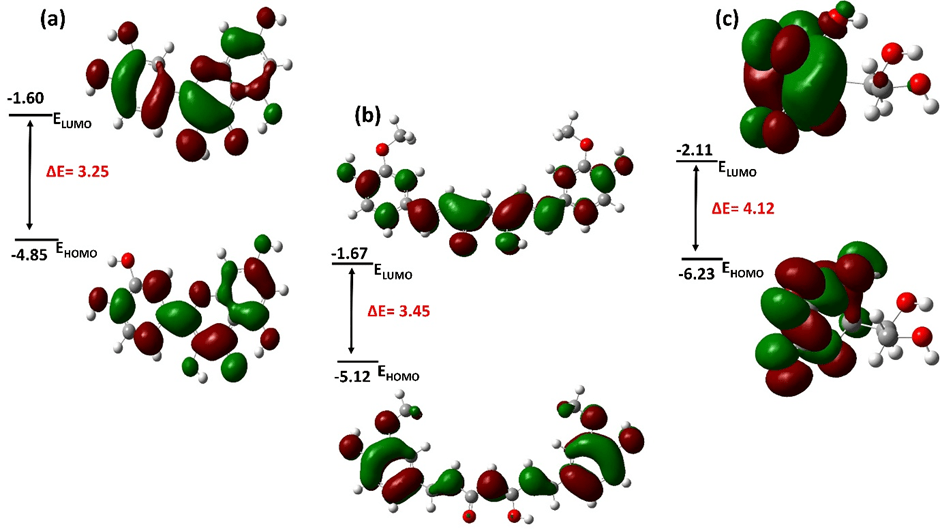
Frontier molecular orbitals were analyzed to evaluate charge redistribution capacity rather than as standalone predictors of antioxidant activity. Antioxidant mechanisms may proceed via HAT, SET, or combined pathways depending on environmental conditions. Quercetin exhibits the highest HOMO energy and the smallest HOMO-LUMO gap, suggesting enhanced electronic softness and greater polarizability (Figure 2, Table 2). However, given the methodological uncertainty at this level of theory, the 0.27 eV HOMO difference between quercetin and curcumin lies near the expected error range. Therefore, these values are interpreted qualitatively. Visualization of the HOMO reveals strong localization over the catechol moiety in quercetin, identifying this region as the most probable oxidation site. In curcumin, the HOMO extends along the conjugated linker and phenolic rings, though with less concentrated density at individual hydroxyl sites. In ascorbic acid, the HOMO remains largely confined to the enediol fragment, indicating limited delocalization potential. Thus, the critical distinction is not the orbital gap magnitude itself, but the degree of HOMO delocalization at hydrogen-donating sites, which directly influences radical stabilization following HAT.
| Compound | HOMO (eV) | LUMO (eV) | ΔHOMO-LUMO (eV) |
| Ascorbic acid | -6.23 | -2.11 | 4.12 |
| Quercetin | -4.85 | -1.60 | 3.25 |
| Curcumin | -5.12 | -1.67 | 3.45 |
Mechanistic Implications for Antioxidant Activity
| Molecule | Key O-H Site | BDE (kcal/mol) | IP (eV) | H-Donation Ability | Electron Donation Ability |
| Quercetin | 4’-OH (B ring) | 76 | 7.48 | Moderate-High | High |
| Curcumin | Phenol O-H | 82 | 8.16 | Low | Low |
| Ascorbic acid | O1–H | 72 | 7.6 | High | Moderate |
BDE provides a direct measure of hydrogen atom transfer feasibility (Table 3). Lower BDE values indicate more favorable hydrogen abstraction3. Ascorbic acid exhibits the lowest BDE, reflecting highly accessible hydrogen donation at the enediol site. Quercetin displays slightly higher BDE values but benefits from significantly enhanced post-abstraction radical stabilization. Curcumin shows the highest BDE among the three. This distinction highlights an important mechanistic nuance: antioxidant efficiency depends not only on hydrogen accessibility, but also on stabilization of the resulting radical intermediate2,11.
While ascorbic acid donates hydrogen readily, its radical is more localized. Quercetin, despite a slightly higher BDE, stabilizes the resulting radical more effectively through extensive π-delocalization and intramolecular hydrogen bonding5,4. IP values indicate that quercetin most readily undergoes electron removal, followed by ascorbic acid and curcumin. However, in the gas phase, the HAT mechanism is expected to dominate over SET. Thus, IP is interpreted as a secondary descriptor of electronic softness rather than as the primary determinant of antioxidant capacity.
Molecular Electrostatic Potential Analysis
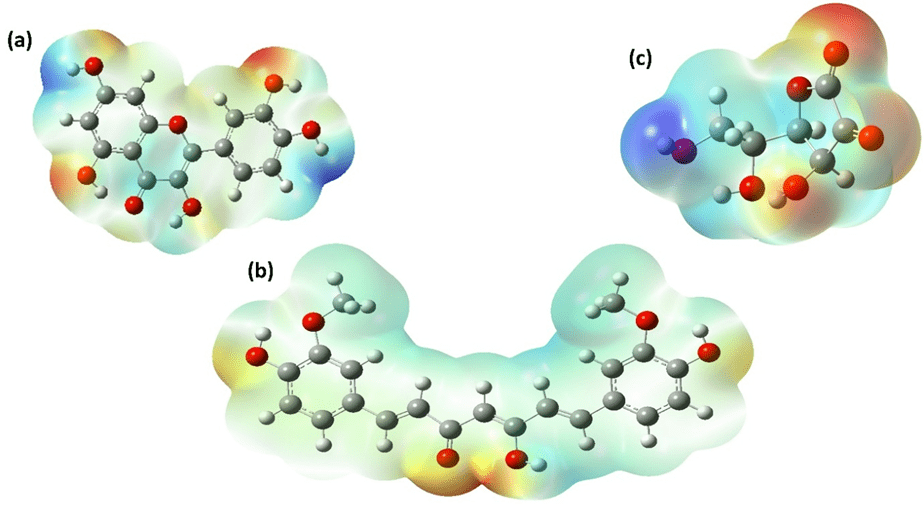
MEP surfaces (Figure 3) reveal the reactive landscape of each molecule. Regions of strongly negative electrostatic potential are concentrated around hydroxyl oxygen atoms. Quercetin displays extensive negative potential across its polyphenolic scaffold, particularly in the catechol region, consistent with strong hydrogen-donating and electron redistribution capability. Curcumin shows moderately negative regions near phenolic and enolic oxygens, while ascorbic acid exhibits more localized negative potential confined to the enediol moiety. These spatial differences align with BDE trends and HOMO localization patterns, supporting the mechanistic interpretation derived from thermodynamic analysis.
Natural Bond Orbital Analysis
NBO analysis was conducted to quantify donor-acceptor interactions via second-order perturbation energies E(2) (Table 4). Larger E(2) values indicate stronger intramolecular charge transfer and improved radical stabilization. Quercetin exhibits the highest stabilization energies (E(2) ≈ 31 kcal/mol), primarily arising from LP(O)→π* interactions within the aromatic framework. These strong interactions enable effective delocalization of unpaired electron density across the conjugated scaffold, providing a mechanistic basis for enhanced radical stabilization. Curcumin shows intermediate stabilization energies (E(2) ≈ 18 kcal/mol), dominated by interactions involving the carbonyl oxygen and conjugated linker. Although these interactions support partial delocalization, the stabilization is less extensive than in quercetin. Ascorbic acid demonstrates comparatively weaker donor–acceptor interactions (E(2) ≈ 10 kcal/mol), largely confined to the enediol region. The absence of extended aromatic conjugation limits long-range charge redistribution. The magnitude of NBO stabilization energies follows the trend: Quercetin > Curcumin > Ascorbic acid. Importantly, this trend mirrors the degree of π-conjugation and HOMO delocalization, reinforcing the central role of electronic delocalization in stabilizing radical intermediates.
| Molecule | Donor → Acceptor Interaction | Donor Occupancy (e) | Acceptor Occupancy (e) | E(2) (kcal/mol) |
| Quercetin | LP(Ocatechol) → π*(C=Caromatic) | 1.98 | 0.32 | 31.2 |
| LP(Ocarbonyl) → π*(C=Cring) | 1.97 | 0.28 | 27.5 | |
| π(C=C) → π*(C=O) | 1.89 | 0.41 | 19.4 | |
| Curcumin | LP(Ocarbonyl) → π*(C=Clinker) | 1.98 | 0.35 | 18.6 |
| LP(Ophenol) → π*(C=Caromatic) | 1.97 | 0.30 | 15.3 | |
| π(C=C) → π*(C=Odiketone) | 1.88 | 0.38 | 13.7 | |
| Ascorbic Acid | LP(Oenediol) → π*(C=C) | 1.98 | 0.29 | 10.4 |
| LP(Olactone) → σ*(C–O) | 1.96 | 0.07 | 7.9 |
Integrated Mechanistic Framework
Across multiple electronic descriptors such as geometry, ZPE differences, HOMO localization, BDE, IP, MEP distribution, and NBO stabilization energies, a coherent mechanistic picture emerges. Ascorbic acid exhibits the most accessible hydrogen donation (lowest BDE), but radical stabilization remains localized. Curcumin shows moderate hydrogen donation and moderate delocalization. Quercetin combines strong hydrogen-donating capability with extensive radical stabilization through conjugation and donor-acceptor interactions. When both hydrogen accessibility and radical stabilization are considered collectively, the overall intrinsic antioxidant potential follows: Quercetin > Ascorbic acid > Curcumin. This ranking reflects a balance between the thermodynamic feasibility of hydrogen abstraction and the electronic capacity to delocalize and stabilize the resulting radical species. The results align qualitatively with experimental observations and highlight that extended π-conjugation, intramolecular hydrogen bonding, and strong donor-acceptor coupling act synergistically to enhance antioxidant efficiency. The present electronic-structure analysis therefore provides a rational framework for understanding structure-activity relationships and guiding the design of next-generation antioxidant systems.
Limitations of the Study
While the present work provides a mechanistically grounded comparative electronic-structure analysis of three representative antioxidant scaffolds, several limitations must be acknowledged to properly contextualize the findings. First, all calculations were performed in the gas phase at the B3LYP/6-31G(d) level of theory18. Antioxidant activity in biological and experimental systems typically occurs in solution, where solvent polarity, hydrogen bonding, dielectric effects, and pH significantly influence reaction energetics7,19,18. In nonpolar environments, the HAT mechanism often dominates, whereas in SPLET mechanism may become thermodynamically favourable7,13.
Solvation can substantially reduce ionization potentials, modify bond dissociation enthalpies, and alter relative reaction free energies. Because no implicit solvent model (e.g., PCM or SMD) or explicit solvent representation was included, the reported thermodynamic parameters reflect intrinsic gas-phase behavior rather than solution-phase free energies18. Future investigations incorporating continuum solvation models and Gibbs free energy (ΔG) calculations will be essential to evaluate mechanistic shifts under biologically relevant conditions.
Second, the mechanistic scope of the present study primarily emphasizes the HAT pathway, with ionization potentials included to probe single electron transfer (SET) tendencies4,13. Proton affinities (PA) and electron transfer enthalpies (ETE), which are necessary to fully characterize the SPLET mechanism, were not computed. Because antioxidant behavior is strongly assay-dependent and environment-dependent, a comprehensive mechanistic evaluation would require PA calculations, ETE determination, and complete reaction free energy profiles in solvent. Accordingly, the mechanistic conclusions drawn here are specific to intrinsic hydrogen-donation thermodynamics and radical stabilization effects under isolated conditions.
Third, only three antioxidant molecules were examined. These were intentionally selected to represent structurally distinct antioxidant classes: quercetin as a polyphenolic flavonoid, curcumin as a conjugated diarylheptanoid, and ascorbic acid as an enediol lactone. Nevertheless, the limited dataset restricts broad statistical generalization. The present work should therefore be interpreted as a comparative mechanistic proof-of-concept study rather than a universal predictive model for all antioxidant systems. Expansion of the dataset to include additional phenolic acids, flavanols, and synthetic antioxidants would strengthen structure-activity generalization. Fourth, although B3LYP/6-31G(d) is widely used and provides reliable qualitative trends for organic radical systems, it has known limitations14. The functional may slightly underestimate ionization potentials, underestimate long-range charge-transfer effects, and lack explicit dispersion corrections. More modern functionals such as ωB97X-D or M06-2X, combined with larger basis sets, could improve quantitative accuracy. However, because this study focuses on relative trends across structurally related systems, B3LYP/6-31G(d) remains appropriate for comparative analysis. Finally, all calculations were performed using optimized static geometries at 0 K. Dynamic effects, conformational averaging at finite temperature, and explicit intermolecular interactions were not considered. Radical stability and reactivity may be influenced by entropic contributions and dynamic solvent interactions, which fall beyond the scope of the present study.
Despite these limitations, the consistent trends observed across bond dissociation enthalpies, ionization potentials, NBO stabilization energies, HOMO localization patterns, and molecular electrostatic potential mapping provide a coherent qualitative framework for understanding intrinsic radical stabilization capacity across distinct antioxidant scaffolds. Future work incorporating solvent models, free energy calculations, and expanded molecular datasets will enable the development of a more comprehensive and predictive antioxidant design framework.
Conclusions
This study presents a comparative electronic-structure analysis of three structurally distinct antioxidants: quercetin, curcumin, and ascorbic acid, using density functional theory at the B3LYP/6-31G(d) level. By integrating structural optimization, ZPE corrections, BDE, IP, NBO analysis, frontier molecular orbital localization, and MEP mapping, a coherent mechanistic framework for intrinsic antioxidant behavior has been established. The results demonstrate that antioxidant efficiency cannot be attributed to a single descriptor such as the HOMO-LUMO gap. Instead, it emerges from a balance between hydrogen-donation accessibility and the ability to stabilize the resulting radical species. Ascorbic acid exhibits the lowest O-H bond dissociation enthalpy, indicating highly accessible hydrogen donation. However, its radical stabilization is largely localized within the enediol moiety due to limited extended conjugation. Curcumin displays moderate hydrogen-donation ability and partial delocalization along its conjugated linker. In contrast, quercetin combines favorable hydrogen abstraction energetics with extensive π-delocalization and strong donor–acceptor interactions, as evidenced by the highest NBO second-order stabilization energies and pronounced HOMO localization at catechol sites.
The collective analysis across multiple electronic descriptors consistently indicates that radical stabilization capacity follows the trend: Quercetin > Ascorbic acid > Curcumin. This ordering reflects the synergistic influence of extended π-conjugation, intramolecular hydrogen bonding, and efficient intramolecular charge redistribution. Importantly, the agreement among independent thermodynamic and electronic parameters strengthens the reliability of the mechanistic interpretation. Although the calculations were performed in the gas phase and therefore represent intrinsic molecular behavior, the findings provide valuable insight into the structural features that govern antioxidant performance. Extended conjugation and strong lone pair to π* donor-acceptor interactions emerge as key determinants of radical stabilization. These insights offer a rational electronic basis for antioxidant design and highlight the utility of computational chemistry in guiding the development and screening of next-generation antioxidant systems.
References
- B. Halliwell and J. M. C. Gutteridge, Oxford University Press, 2015, https://doi.org/10.1093/acprof:oso/9780198717478.001.0001. [↩]
- C. A. Rice-Evans, N. J. Miller, and G. Paganga, Free Radic. Biol. Med., Vol. 20 pg. 933–956, 1996, https://doi.org/10.1016/0891-5849(95)02227-9. [↩] [↩] [↩]
- J. S. Wright, E. R. Johnson, and G. A. DiLabio, J. Am. Chem. Soc., Vol. 123, pg. 1173–1183, 2001, https://doi.org/10.1021/ja002455u. [↩] [↩] [↩]
- M. Leopoldini, T. Marino, N. Russo, and M. Toscano, J. Phys. Chem. A, vol. 108, pg. 4916–4922, 2004, https://doi.org/10.1021/jp037247d. [↩] [↩] [↩] [↩]
- M. Leopoldini, N. Russo, and M. Toscano, Food Chem., vol. 125, pg. 288–306, 2011, https://doi.org/10.1016/j.foodchem.2010.08.012. [↩] [↩]
- M. Cordova-Gomez, A. Galano, and J. R. Alvarez-Idaboy, RSC Adv., vol. 3, pg. 20209, 2013, https://doi.org/10.1039/c3ra42923g. [↩]
- G. Litwinienko and K. U. Ingold, J. Org. Chem., Vol. 68, pg. 3433–3438, 2003, https://doi.org/10.1021/jo026917t. [↩] [↩] [↩]
- N. Nenadis, O. Lazaridou, and M. Z. Tsimidou, J. Agric. Food Chem., Vol. 55, pg. 5452–5460, 2007, https://doi.org/10.1021/jf070473q. [↩]
- Z. Marković et al., Food Chem., Vol. 135, pg. 2070–2077, 2012, https://doi.org/10.1016/j.foodchem.2012.05.119. [↩]
- J.-M. Zhang, Y. Zhang, K.-W. Xu, and V. Ji, Solid State Commun., Vol. 139, pg. 87–91, 2006, https://doi.org/10.1016/j.ssc.2006.05.026. [↩]
- G. Mazzone, M. Toscano, and N. Russo, J. Agric. Food Chem., Vol. 61, pg. 9650–9657, 2013, https://doi.org/10.1021/jf403262k. [↩] [↩] [↩]
- N. Nenadis and M. Z. Tsimidou, Food Research International, Vol. 48, pg. 538–543, 2012, https://doi.org/10.1016/j.foodres.2012.05.014. [↩]
- A. Galano, RSC Adv., Vol. 6, pg. 22951–22963, 2016, https://doi.org/10.1039/C6RA00549G. [↩] [↩] [↩]
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., Vol. 4, pg. 1849–1868, 2008, https://doi.org/10.1021/ct800246v. [↩] [↩]
- J.-D. Chai and M. Head-Gordon, Physical Chemistry Chemical Physics, Vol. 10, pg. 6615, 2008, https://doi.org/10.1039/b810189b. [↩] [↩]
- F. Weinhold and C. R. Landis, Chem. Educ. Res. Pract., Vol. 2, pg. 91–104, 2001, https://doi.org/10.1039/B1RP90011K. [↩]
- A. E. Reed, L. A. Curtiss, and F. Weinhold, Chem. Rev., Vol. 88, pg. 899–926, 1988, https://doi.org/10.1021/cr00088a005. [↩]
- C. J. Cramer and D. G. Truhlar, Chem. Rev., Vol. 99, pg. 2161–2200, 1999, https://doi.org/10.1021/cr960149m. [↩] [↩] [↩]
- A. Galano and M. Francisco-Marquez, J. Phys. Chem. B, Vol. 113, pp. 16077–16081, 2009, https://doi.org/10.1021/jp907906h. [↩]



{kind=link}