
Abstract
The goal of this study was to identify the effectiveness of ML210 on two types of sarcomas: alveolar (ASPS-KY) and rhabdomyosarcoma (SJCRH30). We conducted this study with the intention of analyzing a potential protein inhibitor for these forms of cancer. ML210 is a prodrug known to inhibit the enzyme glutathione peroxidase 4 (GPX4), whose function in preventing ferroptosis is relied upon by cancer cells for survival. When present, this inhibitor undergoes various transformations to acquire the necessary structure for GPX4 inhibition. ASPS-KY and SJCRH30 cell lines were grown in the laboratory and treated with ML210, and the CellTiter-Glo and Crystal Violet assays were performed to analyze cell viability. Results from the CTG assays showed a decrease in cell viability corresponding to increasing ML210 concentrations (0.003–10 μM), notably at marked reductions beginning at 0.3 μM. Calculated IC50 values were approximately 0.1137 μM for SJCRH30 cells and 0.295 μM for ASPS-KY cells. Visual representations from the Crystal Violet displayed a decreasing gradient with intensified ML210 concentrations (0.1–0.5 μM) in both cell lines. Because of scheduling difficulties, a Western Blot analysis was not conducted to confirm decreasing GPX4 levels with increasing ML210 concentrations. Though it is known that ML210 inhibits GPX4 activity, we recommend future researchers to conduct a Western Blot and ferroptosis rescue to confirm that GPX4 is inhibited specifically in ASPS-KY and SJCRH30 cells and such reduced viability is due to ferroptosis.
Keywords: ML210, sarcoma, alveolar sarcoma, rhabdomyosarcoma, prodrug, glutathione peroxidase 4, ferroptosis, inhibition, CellTiter-Glo, Crystal Violet
Introduction
Sarcoma is a rare form of cancer that makes up about 1% of all adult cancers and affects both adults and children1. It originates in the bones and soft tissue such as muscle, fat, tendons, cartilage, ligaments, blood vessels, and nerves. Sarcomas develop due to genetic alterations in DNA, allowing the cancerous cells to grow in an uncontrollable manner2. Other risk factors contributing to its formation include chemical exposure, radiation, and lymphedema. Tests such as the Computed Tomography (CT) scan, ultrasound, Magnetic Resonance Imaging (MRI), and Positron Emission Tomography (PET) scan can be performed to properly diagnose and identify sarcomas3.
Alveolar soft-part sarcoma (ASPS) is known as one of the rarest forms of sarcomas, constituting approximately 0.5% to 1% of all soft tissue sarcomas. It is found primarily in teenagers and young adults under the age of 404. This form of cancer can be found in areas including the neck, head, and arms. Unlike other sarcomas, ASPS tumors tend to grow at a slower rate and can be difficult to identify in its early stages since it typically begins as a painless lump. By the time the disease is diagnosed, it will have most likely metastasized to other parts of the body. Treatment for ASPS includes surgery, radiation therapy, and targeted therapy, depending on the location of the tumor5.
On the other hand, rhabdomyosarcoma (RMS) is a fast-growing and aggressive form of cancer that typically arises in skeletal muscles, mainly affecting children and teenagers. Individuals under the age of 20 account for about 350 cases each year in the U.S6. However, rhabdomyosarcoma can be present in adults as well, representing less than 1% of all solid tumor malignancies7. Treatment for this cancer often involves surgery, chemotherapy, and radiation therapy6.
The rarity and complexity of sarcomas make it an extremely difficult disease to diagnose and treat, leaving many questions unanswered. Unfortunately, only about 48.5% of patients diagnosed with rare cancers like sarcoma live beyond 5 years8. This underscores the desperate need for improvements in treatments for rare cancers such as RMS and ASPS.
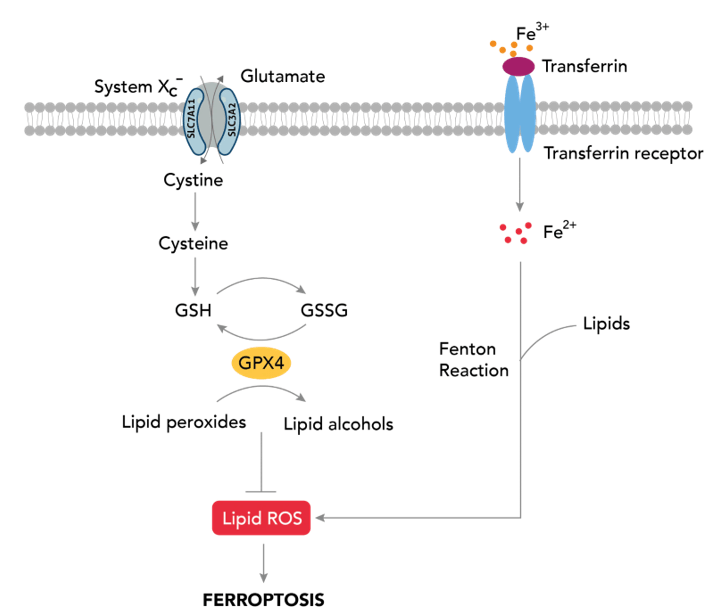
Ferroptosis is an iron-dependent form of regulated cell death driven by lipid peroxidation10. Glutathione peroxidase 4 (GPX4) is a selenoprotein that belongs to the glutathione peroxidase family and plays an important role in regulating and maintaining homeostasis. Cancerous cells often overexpress and rely on GPX4 for the prevention of ferroptosis11. GPX4 has been identified as a key regulator that protects cells from ferroptosis by reducing lipid peroxides to non-toxic alcohols12,13.
In the ferroptosis pathway, ferric iron (Fe³⁺) bounded to transferrin is imported into cells through the transferrin receptor (TfR). Inside the endosome, it is reduced to ferrous iron (Fe²⁺) before being released into the cytosol. Through the Fenton Reaction, Fe2+ reacts with hydrogen peroxide (H₂O₂) to form reactive oxygen species (ROS). These ROS then interact with polyunsaturated fatty acids (PUFAs) to form lipid peroxides, a process known as lipid peroxidation. The accumulation of lipid peroxides poses a threat to cancer cells, as it ultimately leads to ferroptosis14,15. This build-up, however, can be mitigated by GPX4. Firstly, its active site selenol (SeH) is oxidized to selenenic acid (SeOH) by lipid peroxides. This step is not depicted in the schematic.. This oxidation results in the reduction of such toxic lipid peroxides to non-toxic alcohols. To regenerate the active selenol of GPX4, two equivalents of glutathione (GSH) are required. The first equivalent reacts with selenic acid to form GPX4-Se-SG, a selenium-glutathione intermediate. The second GSH equivalent then reduces this intermediate back to the active selenol form, releasing glutathione disulfide (GSSG) as a byproduct and allowing GPX4 to repeat the process. By detoxifying lipid peroxides, GPX4 suppresses ferroptosis, thereby enabling continued proliferation of cancer cells16.
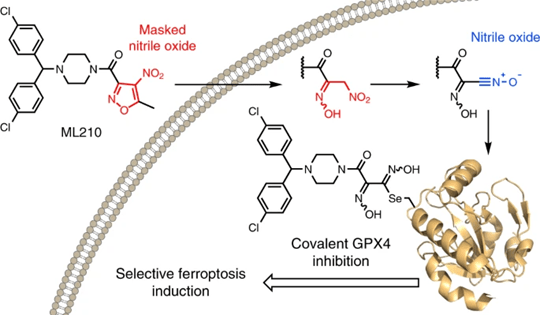
The structure of GPX4 features a highly specific active site, making it difficult to target with traditional small-molecule binding. ML210, however, is a prodrug that is capable of undergoing various transformations necessary to covalently bind to and inhibit GPX4. This prodrug possesses a masked nitroisoxazole group that exhibits structural features that are vulnerable to chemical changes. Since it is not reactive in its initial state, a cellular transformation is required to become an active electrophile that inhibits its target protein17.
Once present in the cell, ML210 is first transformed into an intermediate metabolite known as JKE-1674. This conversion occurs when the nitroisoxazole ring of ML210 is replaced with an α-nitroketoxime through hydrolysis. Still, similar to ML210, JKE-1674 does not contain the appropriate structure for GPX4 inhibition and requires a second transformation17.
JKE-1674 is further metabolized into active nitrile oxide electrophile JKE-1777 through a process of dehydration. This protein-reactive electrophile is capable of covalently binding to GPX417. As a result, GPX4 inhibition leads to an accumulation of lipid peroxides, thereby triggering ferroptosis.
Although ML210 is effective in mitigating the abundance of cancer cells, we wanted to assess its impact specifically on alveolar sarcoma and rhabdomyosarcoma cells. In order to test our hypothesis that ML210 presents a plausible treatment for RMS and ASPS, we performed the CellTiter-Glo and Crystal Violet assays to analyze cell viability.
Methods
This was an experimental study that used ASPS-KY cells and SJCRH30 cells in various tests. Conditions were ran in quadruplets, with three technical replicates per experiment and three biological repeats on separate days.
Cell Lines
ASPS-KY cells were obtained from the RIKEN BioResource Research Center (BRC). SJCRH30 cells were obtained from the American Type Culture Collection. Both cell lines were grown in RPMI 1640 Medium, which was obtained from Fisher Scientific and supplemented with 10% fetal bovine serum (FBS). The ASPS-KY cells were derived from the lungs of a 27-year-old Japanese female with alveolar soft part sarcoma. The SJCRH30 cells were taken from the bone marrow of a 17-year-old Caucasian male with rhabdomyosarcoma. Splitting cells involved aspiration of old media, rinsing with phosphate-buffered Saline (PBS), adding enough trypsin to coat the plate, separating the cells in 2 equal amounts into two plates, and adding new media. Cells were incubated in a 37°C and 5% CO2 incubator.
Drug
ML210 was obtained from MedChemExpress. It was dissolved in DMSO to make a 10 mM stock solution. For all experiments, vehicle control wells received an equivalent concentration of DMSO. To make our drug concentrations for our CTG assays, we created a serial dilution starting with 10 mM of ML210 stock. We made 8 microcentrifuge tubes with the following concentrations: 0.003 μM, 0.01 μM, 0.03 μM, 0.1 μM, 0.3 μM, 1 μM, 3 μM, 10 μM. The drug was diluted using RPMI 1640 media, and each tube had a total volume of 900 μL.
To make our drug concentrations for the Crystal Violet assays, we used 0.5 μM as our initial concentration, which was made from 10 mM of stock ML210. The total volume, including media and drug, of the 500 nM tube was 20 mL. ML210 was then diluted using RPMI 1640 media to final concentrations of 0.1 μM, 0.2 μM, 0.3 μM, 0.4 μM, and 0.5 μM, with each tube having a final volume of 8 mL.
CellTiter-Glo Assay
For CellTiter-Glo (CTG) (Promega) experiments, cells were seeded in quadruplicate in 96-well black plates at a concentration of 2*10^3 cells per well in 180 μL of growth medium. 24 hours after seeding, cells were treated with increasing concentrations of 20 μL ML210 (0.003–10 μM) for 72 hours and maintained at 37°C and 5% CO2. Cell viability was then read on a Centro LB960 microplate luminometer (Berthold Technologies) according to the manufacturer’s instructions, with the exception that 25 uL of CTG reagent was used per well. For analysis, the untreated wells were averaged and that value represented 100% viability. Raw luminescence values for each individual well were then divided by the untreated average and multiplied bv 100 to obtain individual viability percentages for each well. Background subtraction was not performed. Untreated wells (cells with no ML210) were averaged to define the control baseline.
Crystal Violet Assay
We first plated 50,000 ASPS-KY cells and 50,000 SJCRH30 cells into two separate 6-well plates. The cells were then incubated in a 37°C and 5% CO2 incubator for 24 hours. Next, we added ML210 at different concentrations to each well in the following order, respectively: 0 μM, 0.1 μM, 0.2 μM, 0.3 μM, 0.4 μM, and 0.5 μM. The 0 μM was defined as our control well. Every 3 days, the growth medium containing the indicated concentrations of drug was replenished to ensure sufficient nutrients. Once the cells in the No Rx control well approached full confluence as determined by microscope, the cells were fixed with 0.5 mL glutaraldehyde into each well. The plates were then left at room temperature for 10 minutes. Next, we aspirated the glutaraldehyde-media mix and performed two rinses with dH2O, aspirating in between each wash. After rinsing the wells, we added 0.1% crystal violet (Sigma-Aldrich), enough to fully cover each well completely, and let the cells incubate at room temperature for 30 minutes. Then, we aspirated off the crystal violet and conducted another dH2O wash. Finally, we allowed the cells to dry inverted overnight before analyzing. In our experiment, we utilized the crystal violet assay as a visual analysis of cell viability. The gradient formed from the dye represented the abundance of cellular material. Glutaraldehyde fixation was performed according to an established laboratory protocol; however, the stock concentration and preparation details were not recorded. Crystal Violet staining was used for qualitative visualization only and was not quantified.
Statistical Analysis
For CTG data, statistical comparisons between treated wells and untreated control groups were performed using an unpaired two-tailed t-test assuming equal (sample size=10 for control; sample size=4 for treated groups) to show statistical significance between the means of the control group and treated groups. For each cell line, three representative concentrations were selected for statistical analysis: (1) a concentration when viability reduction happened (ns), (2) a concentration at the first concentration of significance, and (3) a concentration resulting in near-complete loss of viability. A p-value less than 0.05 was considered statistically significant.
Results
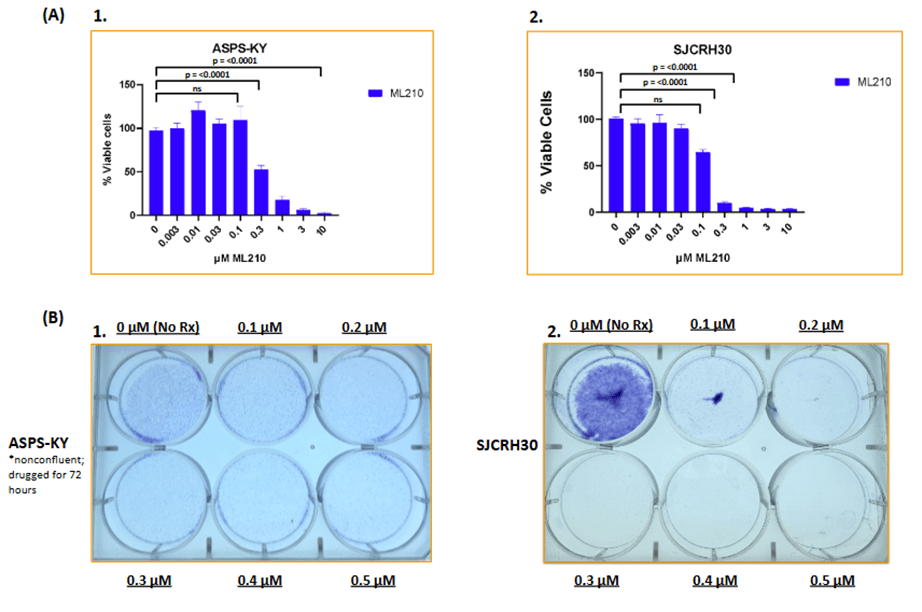
ML210 demonstrates noticeable effects in both ASPS-KY and SJCRH30 cell lines. Based on the CTG assay (A1), ASPS-KY cells exhibited a reduction in cell concentration from 0.1 μM to 0.3 μM, indicating a susceptibility to reduced cell viability. The visual gradient from the Crystal Violet assay (B1) indicates the effect of ML210 in ASPS-KY cells by portraying reduced cell concentration at high intensities of ML210 such as 0.3 μM, 0.4 μM, and 0.5 μM when compared to 0 μM. Similarly, CTG data of SJCRH30 cells (A2) showed a relatively steady decrease in cell viability from 0 μM to 0.1 μM, followed by a dramatic reduction at 0.3 μM. Moreover, results from the Crystal Violet (B2) assay demonstrated the effect of ML210 in SJCRH30 cells by revealing a weak presence of a purple gradient from 0.3 μM to 0.5 μM when compared with the No Rx well. Using CTG data, the IC50 defined as the concentration of ML210 required to reduce cell viability by 50%, was calculated to be approximately 0.1137 μM for SJCRH30 cells and approximately 0.295 μM for ASPS-KY cells.
For SJCRH30 cells, statistical analysis was performed at 0.1 μM, 0.3 μM, and 1 μM. Such concentrations resulted in significant reduction in cell viability compared to untreated controls, as p < 0.05 for all concentrations. Similarly, in ASPS-KY cells, statistical analysis was performed at 0.1 μM, 0.3 μM, and 10 μM and saw statistically significant viability reduction in each concentration. Concentrations from 0.003 μM to 0.03 μM saw no statistically significant difference.
Discussion
Our findings suggest that ML210 has an apparent effect on reducing cell viability in ASPS-KY and SJCRH30 cells. CTG data, followed by qualitative Crystal Violet data, of the ASPS-KY cell line support such findings by showing lower quantitative and visual results in cell viability as ML210 concentrations were intensified. This trend was also present in both types of assays for the SJCRH30 cell line. Notably, the significant decrease in cell viability at 0.3 μM observed in the graphs of both CTG assays, followed by p values less than 0.05, suggests that 0.3 μM may represent a critical concentration at which ML210 can most effectively reduce cell proliferation. Collectively, these results support our hypothesis that ML210 successfully diminishes cell viability in both ASPS-KY and SJCRH30 cell lines. To expand on the existing findings of the prodrug’s effects on cancer cells in general, our study shows ML210’s effect specifically in alveolar sarcoma and rhabdomyosarcoma.
Although previous studies have demonstrated ML210’s effectiveness in inhibiting GPX4 activity, we could not confirm specific inhibition in ASPS-KY and SJCRH30 cells due to the absence of a Western Blot analysis. Scheduling restrictions prevented us from performing this analysis to verify GPX4 expression, leaving it unclear whether the observed decreased cell viability was caused by GPX4 inhibition. Additionally, we could not perform ferroptosis rescue, and therefore we cannot conclude that this reduced cell viability was a result of ferroptosis specifically. These findings motivate further mechanistic investigation rather than confirm ferroptosis. In addition, the Crystal Violet of the ASPS-KY cell line was only treated for 24 hours (compared to 7 days for the SJCRH30 cells) due to time constraints. Crystal Violet data therefore limits direct comparison between cell lines. The data at 0.01 μM ML210 in ASPS-KY CTG data presented a percent viability that was unexpected in our experiment. This could have been caused by cell plating or treatment errors. However, though faint, there is still a visible gradient that indicates the occurrence of decreased cell viability of ASPS-KY cells. Regardless, we believe that our CTG and Crystal Violet data still show that ML210 plays a role in inducing cell viability in both alveolar sarcoma and rhabdomyosarcoma cells. This can lay a foundation for future research projects investigating plausible treatments for ASPS and RMS. A Western Blot that shows declining GPX4 expression with increasing ML210 concentrations, along with performing ferroptosis rescue, will further support this study by proving that the specific form of cell death induced by ML210 is ferroptosis. Repeating a Crystal Violet assay for the ASPS-KY cell line for the appropriate duration (until control wells are confluent) will also provide stronger evidence of cell death. Background subtraction could also be performed using wells that contain media with no cells or drug. Additionally, we recommend that further experiments to determine ML210’s effects on non-cancerous cells will be necessary before comparing its effect on cancer cells lines versus normal cells.
Acknowledgements
This research was conducted as part of the VCU CHiSEL Program at the Philips Institute for Oral Health Research. I would like to express my sincere gratitude to everyone in the Faber Lab for their support, guidance, and generosity throughout my time. I would also like to thank Ron for all his help and guidance during experimentation. Many thanks to Dr. Anthony Faber and Dr. Krista Dalton for granting me the extraordinary opportunity to learn and grow in such an inspiring environment. Lastly, I would like to thank my family for their support throughout this process.
References
- Z. Burningham, M. Hashibe, L. Spector, J. D. Schiffman. The Epidemiology of Sarcoma. Clin Sarcoma Res. 2, 14 (2012), doi: 10.1186/2045-3329-2-14. [↩]
- D. Hanahan, R. A. Weinberg. Hallmarks of Cancer: The Next Generation. Cell. 144, 646 – 674 (2011), doi: 10.1016/j.cell.2011.02.013. [↩]
- A. M. Vibhakar, J. A. Cassels, R. Botchu, W. J. Rennie, A. Shah. Imaging update on soft tissue sarcoma. J Clin Orthop Trauma. 22, 101568 (2021), doi: 10.1016/j.jcot.2021.101568. [↩]
- Y. J. Cho, J. Y. Kim. Alveolar Soft Part Sarcoma: Clinical Presentation, Treatment and Outcome in a Series of 19 Patients. Clin Orthop Surg. 6, 80-86 (2014), doi: 10.4055/cios.2014.6.1.80. [↩]
- A. Singh, S. Gupta, S. Ghosh, M. B. Yuwanati. Alveolar soft part sarcoma. BMJ Case Rep. (2014), doi: 10.1136/bcr-2013-203386. [↩]
- A. Zarrabi, D. Perrin, M. Kavoosi, M. Sommer, S. Sezen, P. Mehrbod, B. Bhushan, F. Machaj, J. Rosik, P. Kawalec, S. Afifi, S. M. Bolandi, P. Koleini, M. Taheri, T. Madrakian, M. J. Łos, B. Lindsey, N. Cakir, A. Zarepour, K. Hushmandi, A. Fallah, B. Koc, A. Khosravi, M. Ahmadi, S. Logue, G. Orive, S. Pecic, J. W. Gordon, S. Ghavami. Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies. Cancers (Basel). 15, 5269 (2023), doi: 10.3390/cancers15215269. [↩] [↩]
- D. Egas-Bejar, W. W. Huh. Rhabdomyosarcoma in adolescent and young adult patients: current perspectives. Adolesc Health Med Ther. 5, 115-125 (2014), doi: 10.2147/AHMT.S44582. [↩]
- C. Drabbe, D. J. Grünhagen, W. J. Van Houdt, P. M. Braam, VLMN. Soomers, JA. Van der Hage, JJ. De Haan, KBMI. Keymeulen, O. Husson, WTA. Van der Graaf. Diagnosed with a Rare Cancer: Experiences of Adult Sarcoma Survivors with the Healthcare System—Results from the SURVSARC Study. Cancers (Basel). 13, 679 (2021), doi: 10.3390/cancers13040679. [↩]
- D. Sakthivel. The role of ferroptosis in human diseases. https://www.ptglab.com/news/blog/the-role-of-ferroptosis-in-human-diseases/, (no date). [↩]
- S. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison 3rd, B. R. Stockwell. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149, 1060-72 (2012), doi: 10.1016/j.cell.2012.03.042. [↩]
- K. Weaver, R. Skouta. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines. 10, 891 (2022), doi: 10.3390/biomedicines10040891. [↩]
- W. S. Yang, R. SriRamaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji, C. B. Clish, L. M. Brown, A. W. Girotti, V. W. Cornish, S. L. Schreiber, B. R. Stockwell. Regulation of ferroptotic cancer cell death by GPX4. Cell. 156, 317-331 (2014), doi: 10.1016/j.cell.2013.12.010. [↩]
- V. S. Viswanathan, M. J. Ryan, H. D. Dhruv, S. Gill, O. M. Eichhoff, B. Seashore-Ludlow, S. D. Kaffenberger, J. K. Eaton, K. Shimada, A. J. Aguirre, S. R. Viswanathan, S. Chattopadhyay, P. Tamayo, W. S. Yang, M. G. Rees, S. Chen, Z. V. Boskovic, S. Javaid, C. Huang, X. Wu, Y. Tseng, E. M. Roider, D. Gao, J. M. Cleary, B. M. Wolpin, J. P. Mesirov, D. A. Haber, J. A. Engelman, J. S. Boehm, J. D. Kotz, C. S. Hon, Y. Chen, W. C. Hahn, M. P. Levesque, J. G. Doench, M. E. Berens, A. F. Shamji, P. A. Clemons, B. R. Stockwell, S. L. Schreiber. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 547, 453-457 (2017), doi: 10.1038/nature23007. [↩]
- A. Arjunan, R. Rajan. Noise and brain. Physiology & Behavior. 227, 113136 (2020), https://doi.org/10.1016/j.physbeh.2020.113136. [↩]
- Hf. Yan, T. Zou, Qz. Tuo, S. Xu, H. Li, A.A. Belaidi, P. Lei. Ferroptosis: mechanisms and links with diseases. Signal Transduction and Targeted Therapy. 6, 49 (2021). [↩]
- J. Pei, X. Pan, G. Wei, Y. Hua. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front Pharmacol. 14, 1147414 (2023), doi: 10.1038/s41392-020-00428-9. [↩]
- J. K. Eaton, L. Furst, R. A. Ruberto, D. Moosmayer, A. Hilpmann, M. J. Ryan, K. Zimmermann, L. L. Cai, M. Niehues, V. Badock, A. Kramm, S. Chen, R. C. Hillig, P. A. Clemons, S. Gradl, C. Montagnon, K. E. Lazarski, S. Christian, B. Bajrami, R. Neuhaus, A. L. Eheim, V. S. Viswanathan, S. L. Schreiber. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 16, 497–506 (2020), doi: 10.1038/s41589-020-0501-5. [↩] [↩] [↩] [↩]



{kind=link}