
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers, characterized by a poor prognosis, aggressive progression, and resistance to conventional therapies. Although immunotherapy has successfully treated other cancers, its performance in PDAC is limited due to PDAC tumors having dense stroma and very immunosuppressive microenvironments. Oncolytic virotherapy is a promising field of immunotherapy that uses viruses to directly target and lyse tumor cells and stimulates antitumor immunity. Among several viral candidates for treating PDAC, reovirus has shown much promise because of its safety profile and natural oncolytic properties. However, reovirus has lagged behind other oncolytic viruses (OVs), such as adenoviruses and herpes simplex viruses, in terms of genetic engineering due to reovirus being a segmented double-stranded RNA virus. This paper provides a review of the developments in oncolytic virotherapy and findings from preclinical and clinical studies of OVs, with a focus on reovirus. Furthermore, this review will examine therapeutic transgenes that OVs have been equipped with and propose that reovirus could be modified with the cytokine IL-21 to aid reovirus’s antitumor capabilities. Although the proposed reovirus design is a hypothetical design, it is backed by previous research with other OVs, and available evidence suggests it could reasonably improve reovirus’s performance. Future advancements in reovirus engineering could allow for other transgenes to be expressed, and combination therapy of OVs and other therapies, such as immune checkpoint inhibitors, could be the key to effectively treating PDAC. Oncolytic reoviruses with rational modifications could be important components of future treatments for PDAC.
Keywords: pancreatic cancer, pancreatic ductal adenocarcinoma, oncolytic virus, reovirus, immunotherapy, interleukin-21
Introduction
Current treatments for pancreatic ductal adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer, making up more than 85% of all pancreatic cancer cases1. PDAC is characterized by a very poor prognosis, with a 5-year survival rate of around 9.2% in the US1. These rates, which make PDAC one of the most lethal cancers, are due to both the difficulty in detecting early-stage disease (with the majority of cases being distant stage diseases at diagnosis) and the aggressive nature of the cancer1. Though surgery and chemotherapy have been shown to be effective for early-stage disease, approximately 80% of PDAC cases are unresectable at the time of diagnosis1. The standard therapy for metastatic PDAC has been chemotherapy, specifically gemcitabine-based therapies since 19971,2. While gemcitabine does provide symptomatic benefit for patients, two phase II studies observed partial response (PR) in only 11% of PDAC patients and 6.3% of advanced and metastatic PDAC patients respectively2. Though new chemotherapeutic regimens like FOLFIRINOX (leucovorin (folinic acid), 5-fluorouracil, irinotecan, and oxaliplatin) have become available for patients with PDAC, many patients still receive gemcitabine for treatment because of the increased toxicity of these new combinations3.
Due to the lack of effective treatment options for PDAC, many new therapies, especially various immunotherapies, are being tested. Therapies such as immune checkpoint inhibitors (ICIs) have proven to be effective in treating non-small-cell lung cancer (NSCLC), melanoma, and other solid tumors4. However, ICIs have failed to produce significant results for PDAC in clinical trials. For instance, ipilimumab (Anti-CTLA-4) monotherapy in a phase II trial did not elicit an objective response in any of the patients with PDAC based on Response Evaluation Criteria in Solid Tumors (RECIST)5. Similarly, a phase I trial of BMS-936559 (Anti-PD-L1), another ICI, in patients with various solid tumors showed partial and complete responses in patients with NSCLC, melanoma, renal-cell cancer, and ovarian cancer but reported no objective responses among the 14 enrolled pancreatic cancer patients6. Combination therapy of ipilimumab and gemcitabine in a phase Ib trial resulted in an objective response rate of 14%, higher than that of ipilimumab alone but comparable to the response rate with gemcitabine7. Although ICIs could be effective in a combination therapy for PDAC, ICIs have yet to yield promising results in clinical trials.
Barriers to treatment in pancreatic adenocarcinoma
The inefficacy of treatments for PDAC can partially be attributed to the genetic mutations within cancer cells in PDAC. Some cancers have specific mutations in important genes that respond well to targeted therapies: for instance, some lung carcinomas have EGFR mutations that can be targeted8. Mutations in PDAC meanwhile include oncogenic KRAS mutations and deactivations of multiple tumor suppressor genes such as TP538. Different combinations of these mutations influence resistance to therapies; in addition, genetic analyses of PDAC have shown the intratumoral genetic heterogeneity of PDAC cells, which makes the cancer more likely to develop resistance to treatments8. PDAC is also a cancer with low mutational burden, meaning tumor cells are less likely to express neoantigens that immune cells can identify and distinguish from normal cells1.
Another factor contributing to the difficulty of treating PDAC is its complicated and unusual tumor microenvironment (TME). Many of the cells inside tumors are often cancer-associated fibroblasts (CAFs), which are recruited by cancer cells and surround the tumor; CAFs produce much of the dense extracellular matrix that PDAC is marked by histologically9. The most prominent effect of desmoplasia in PDAC tumors is that the perfusion of drugs, including gemcitabine, is limited in tumors10. The anti-angiogenic effects of the tumor stroma in PDAC further reduce the efficacy of intravenously delivered drugs10. Another important group of cells in tumors are immunosuppressive cells. Regulatory T cells (Treg), M2 macrophages, and myeloid-derived suppressor cells (MDSCs) in the stroma (alongside CAFs) limit the activity of CD8+ T cells in PDAC, maintaining a highly immunosuppressive TME11. Dense stroma and immunosuppressive cells in PDAC tumors limit the efficacy of conventional therapies such as chemotherapies, thus warranting the use of novel therapies that could overcome the problems associated with PDAC.
This review will present an overview of one promising type of immunotherapy that is currently being tested for PDAC along with other cancers: oncolytic viruses (OVs). OVs can directly kill cancer cells, mount antitumor immune responses, and even be modified in numerous ways to increase their efficacy against cancer. The diversity of OVs, in terms of both viral backbones and the spectrum of genetic modifications implemented, shows the potential of the OV platform to create highly specific and potent therapies for challenging cancers to treat such as PDAC. In addition to a review of current OV designs and trials, this review will propose new possible modifications for one promising OV, reovirus, that could potentially improve reovirus’s performance against PDAC.
Methods
A structured literature search was conducted in PubMed and Google Scholar for studies published in English up to July 2025, using the keywords “pancreatic ductal adenocarcinoma”, “oncolytic virus”, and either “adenovirus”, “vaccinia”, “herpes simplex virus”, or “reovirus”. The search included both preclinical and clinical studies investigating OVs for PDAC. Furthermore, reference lists of relevant reviews and trials were screened manually to identify additional studies. Inclusion criteria included studies reporting mechanistic, translational, or clinical data on genetically engineered oncolytic adenoviruses, vaccinia viruses, herpes simplex viruses, or reoviruses targeting PDAC. Exclusion criteria included non-peer-reviewed reports, conference abstracts without full text, and studies unrelated to PDAC or OV therapy. The study selection followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) framework, and a simplified diagram showing the search and screening process is available below (Figure 1).

Evidence assessment
Each included publication was appraised according to study type and design quality. Preclinical studies (in vitro and in vivo) were evaluated for reproducibility and mechanistic relevance, whereas clinical studies were classified by phase and sample size. The overall strength of evidence was summarized by a three-level scale (high, moderate, and exploratory) as presented in Table 1. This grading serves to aid the interpretation of translational maturity for each OV platform for PDAC.
| OV platform | Study type | Key findings | Level of evidence1 |
| Representative studies (References) | |||
| Reovirus (Pelareorep) | Phase I–II human clinical trials (with preclinical support in text) | Safe and immunogenic; limited single-agent efficacy; synergy with chemotherapy or PD-1 blockade | Moderate–High |
| 12,13,14 | |||
| Adenovirus (VCN-01, LOAd703, etc.) | Phase I clinical and translational PDAC studies | Stromal modulation, enhanced penetration, early-phase signals | Moderate–High |
| 15,16,17,18 ,19,20,21 | |||
| Vaccinia virus (CF33, oVV-Smac, etc.) | Preclinical/translational PDAC (plus immunotherapy synergy) | Strong oncolysis and immune modulation; feasibility of genetic arming | Exploratory–Moderate |
| 22,23,24,25 | |||
| HSV (T-VEC, oHSV-CD40L) | Preclinical and early clinical PDAC trials | Potent immune activation; limited PDAC-specific clinical data to date | Exploratory–Moderate |
| 26,27 | |||
Patient-selection framework
For translational and early-phase clinical application, patients would be stratified according to tumor entry-receptor context, specifically sialic-acid expression profiles determined by immunohistochemistry or RNA sequencing8. Baseline anti-reovirus neutralizing-antibody (NAb) titres would be measured to determine dosing frequency and assess the feasibility of systemic delivery11. In cases where intravenous delivery may be insufficient (due to dense stroma or limited vascular access), endoscopic ultrasound (EUS)-guided intratumoral injection would be chosen as the delivery route9,11. Outcome analyses would create groups by receptor status and NAb level to evaluate responsiveness to the therapy and to correlate with pharmacodynamic goals.”
Oncolytic virotherapy: Its history and mechanisms
OVs are novel immunotherapies utilizing, as the name suggests, live, replicating viruses that selectively target, infect and kill cancer cells; OVs can be further altered genetically to be safer and stimulate antitumor immune responses.
The concept of OVs traces back to the 1890s, when it was observed that a patient with leukemia saw improvement after contracting what was likely a flu28. From the 1940s to the 1970s, some viruses like the West Nile virus were found to have natural oncolytic properties, with patients sometimes seeing regressions in their tumors after the so-called virotherapy28. Reovirus is one naturally occurring OV that has been shown to be very safe, meaning even unmodified reoviruses are good OV candidates12,28.
With advances in genetic engineering in the 1990s, new, more effective OVs—including ones derived from human viruses, but with fewer adverse effects—have been created. One example is ONYX-015, an oncolytic adenovirus that was the first genetically engineered OV to be tested in clinical trials. The deletion of the viral gene E1B, which interferes with p53 (cellular apoptotic gene) activation, was removed in ONYX-015. This made the virus selectively replicate in cancer cells (with dysfunctional p53) but not in normal cells with functioning p5328. Modified human viruses like adenoviruses and herpes simplex viruses are used commonly to create OVs today and generally have good safety profiles29.
OVs generally work through four mechanisms (Figure 2). The first is replication in and spread throughout a tumor site (usually leading to direct lysis of infected cancer cells i.e. oncolysis)30. As mentioned before with ONYX-015, OVs can be modified to selectively replicate in cancer cells, further enhancing their specificity (a crucial safety feature). The second mechanism is the infection of cells supporting the tumor (tumor-associated stroma cells), such as endothelial cells and, in the case of PDAC, cancer-associated fibroblasts9,11. In the third mechanism, the deaths of cancer cells can trigger an antitumor immune response and introduce more tumor-infiltrating lymphocytes—in other words, turning an immunologically ‘cold’ TME into a ‘hot’ TME with more immune activity30. Sometimes, the antitumor immune response caused by oncolytic virotherapy can achieve significant regressions in uninjected tumors with similar tumor-associated antigens, which is known as the ‘abscopal’ effect—an effect especially important in metastatic cancers28. A phase III trial of T-VEC (talimogene laherparepvec, an oncolytic herpes simplex virus 1 for late-stage melanoma) observed the abscopal effect, with 34% of uninjected non-visceral lesions shrank by 50% or more30. Finally, OVs can be modified with transgenes for additional therapeutic effect. For instance, the aforementioned oncolytic herpes simplex virus, T-VEC, was equipped with granulocyte-macrophage colony-stimulating factor (GM-CSF), which has the effect of boosting the antitumor immune response through the recruitment of dendritic cells (DCs)30. OVs can be armed to express a range of cytokines, chemokines, and other genes to become more effective.
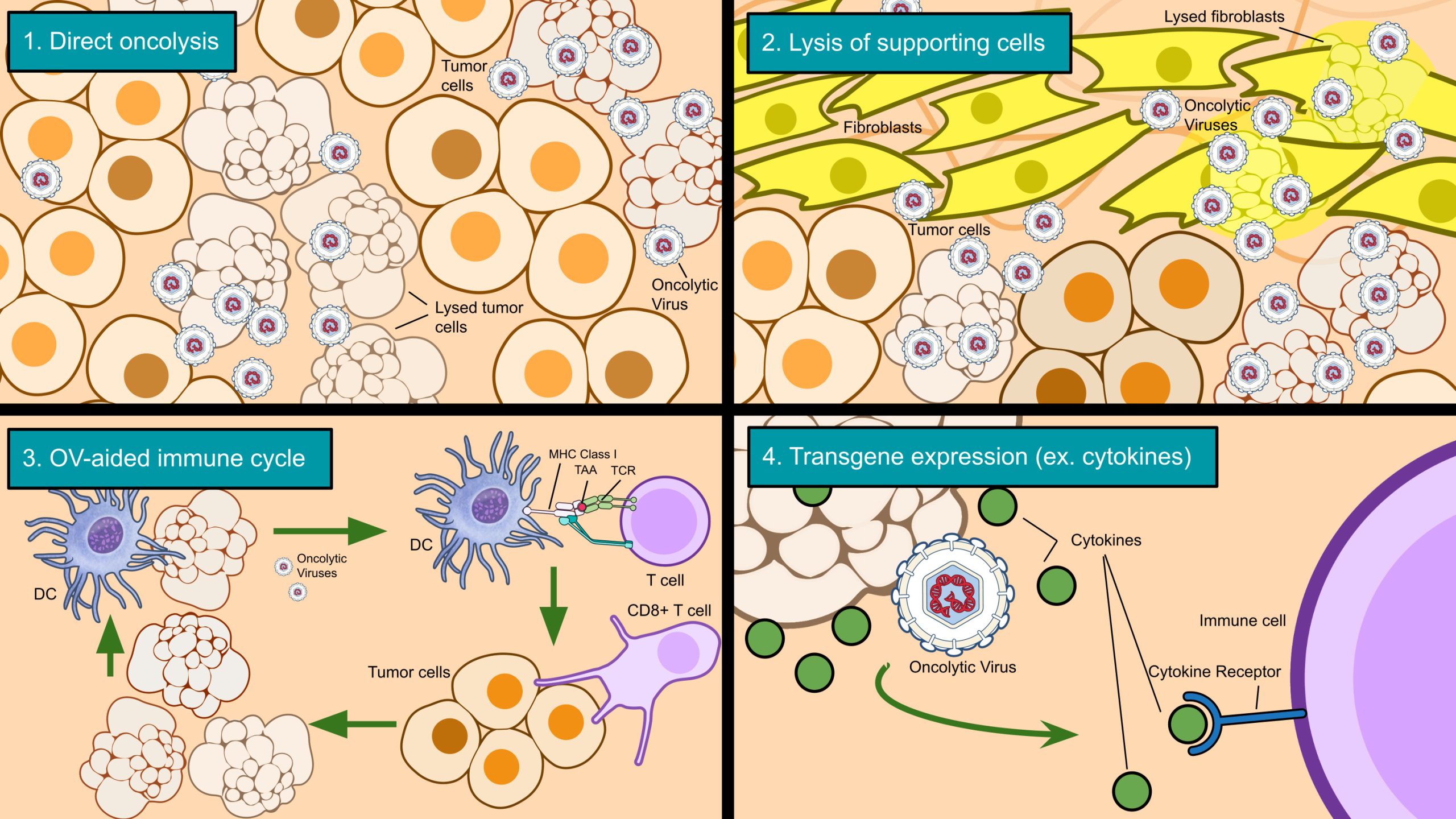
Approved oncolytic viruses
Currently, the only OV approved by the FDA is T-VEC (talimogene laherparepvec) for recurrent, unresectable melanoma. Globally, there are a few more available OVs, including G47 (teserpaturev) as an adjuvant therapy for residual or recurrent glioblastoma in Japan30. Both viruses are derived from herpes simplex virus 1. This section will discuss the modifications of these two viruses and how the modifications have improved the viruses’ performance in their respective cancers.
(teserpaturev) as an adjuvant therapy for residual or recurrent glioblastoma in Japan30. Both viruses are derived from herpes simplex virus 1. This section will discuss the modifications of these two viruses and how the modifications have improved the viruses’ performance in their respective cancers.
T-VEC and G47 share two same genetic alterations, specifically in the genes gamma 1(34.5) and  47. The deletion of ICP34.5 increases the virus’s specificity, and the deletion of ICP47 allows more MHC class I expression on infected cells, enabling a stronger antitumor immune response30. The same genes are also deleted in G47;
47. The deletion of ICP34.5 increases the virus’s specificity, and the deletion of ICP47 allows more MHC class I expression on infected cells, enabling a stronger antitumor immune response30. The same genes are also deleted in G47;  34.5 has further been shown to be a factor for neurovirulence (infection of normal and nondividing cells in the nervous system) so its removal in G47, an OV for glioblastoma, is an important safety feature28. In addition to these two modifications, T-VEC is also armed with a therapeutic gene: GM-CSF, which stimulates the maturation of progenitor cells into DCs30. Preclinical trials showed that GM-CSF expression indeed improved the virus’s performance30.
34.5 has further been shown to be a factor for neurovirulence (infection of normal and nondividing cells in the nervous system) so its removal in G47, an OV for glioblastoma, is an important safety feature28. In addition to these two modifications, T-VEC is also armed with a therapeutic gene: GM-CSF, which stimulates the maturation of progenitor cells into DCs30. Preclinical trials showed that GM-CSF expression indeed improved the virus’s performance30.
These modifications have undoubtedly increased the efficacy of T-VEC and G47 for melanoma and glioblastoma. The median overall survival (OS) for melanoma patients treated with T-VEC in a phase III trial was 23.3 months, while the control group receiving GM-CSF had an OS of 18.9 months30. Meanwhile, the median OS for glioblastoma patients treated with G47 in a phase II trial was reported to be 20.2 months, compared to an OS of 5.0 months for various chemotherapies30.
One important aspect of OV engineering is safety: few patients in clinical trials of both viruses experienced grade  3 adverse events30. Another very important aspect is an enhanced antitumor immune response: biopsies found increased counts of tumor-infiltrating CD8+ T cells in phase II trials for both viruses30. Few studies have engineered reoviruses to improve antitumor immune responses, however, so this evidence from T-VEC and G47 (along with other modified OVs) suggests that oncolytic reoviruses could be improved genetically for better effect on PDAC.
3 adverse events30. Another very important aspect is an enhanced antitumor immune response: biopsies found increased counts of tumor-infiltrating CD8+ T cells in phase II trials for both viruses30. Few studies have engineered reoviruses to improve antitumor immune responses, however, so this evidence from T-VEC and G47 (along with other modified OVs) suggests that oncolytic reoviruses could be improved genetically for better effect on PDAC.
OVs currently being tested for the treatment of pancreatic ductal adenocarcinoma
Currently, no OVs have been approved for PDAC; nonetheless, several promising viruses with various viral backbones and modifications are currently in preclinical and clinical trials. This section of the review will review five OVs being tested for PDAC and their modifications—including therapeutic transgenes that have been inserted to improve performance in PDAC tumors.
Oncolytic adenoviruses, herpes simplex viruses, and poxviruses for PDAC
Commonly used OV backbones include adenoviruses (oAds), herpes simplex viruses (oHSVs), and poxviruses. Unlike reoviruses, natural strains of these three viruses do cause illnesses, so they must be attenuated and retargeted to cancer cells. Although reoviruses may have an advantage in terms of safety, the three currently used viruses are easier to manipulate genetically. The three viruses all have linear, double-stranded DNA genomes, but reoviruses have segmented, double-stranded RNA (dsRNA) genomes26. Here, this review will examine different transgenes that oAds, oHSVs, and oncolytic poxviruses have been equipped with.
Typically, the first modifications to oAds, oHSVs, and oncolytic poxviruses are for decreasing virulence and increasing tumor specificity. This can be done through natural attenuation, as was the case with Canerpaturev (HF10), a spontaneously occurring HSV-1 mutant28. A chimeric poxvirus, CF33, was also created from co-infecting cells with several poxviruses and chosen for its enhanced oncolytic abilities over its parental viruses25s. Many OVs, like T-VEC, are instead genetically modified in order to selectively or conditionally replicate, usually via the deletion or modification of key genes causing virulence—for example, the gamma 1(34.5) gene in oHSVs, or the E1A and E1B genes in oAds28. These changes can be referred to as “detargeting” and “retargeting” OVs.
The first genetically modified OVs, such as HSV-1 dlsptk (oHSV) and ONYX-015 (oAd), only had detargeting and retargeting changes to them28; since then, newer generation OVs have been encoded with therapeutic transgenes to enhance their efficacy (Table 2). oAds, oHSVs, and oncolytic poxviruses have so far been modified to express a range of transgenes, including but not limited to suicide genes (ex. HSV thymidine kinase, adenovirus death protein), cytokines (ex. Interleukins (ILs), interferons (IFNs)), tumor suppressor genes (ex. ST13), and co-stimulatory ligands (ex. CD40L). Several of these viruses have been tested in preclinical models, and a few have been tested in clinical trials.
| Cytokines | Role | Virus(es) |
| References | ||
| Granulocyte-macrophage colony stimulating factor (GM-CSF) | Stimulates maturation of progenitor cells into dendritic cells | Talimogene laherparepvec (T-VEC) (HSV) OrienX010 (HSVGM-CSF) (HSV) |
| 26,31 | ||
| Interleukin-2 (IL-2) | Stimulates antigen-specific T cell growth and proliferation | OAd-TNFa-IL2 (Ad) |
| 32 | ||
| IL-10 | Inhibits antiviral immune response; stimulates antitumor immune response | VVTK-IL-10 (VACV) |
| 33 | ||
| IL-12 | Activates and primes both innate and adaptive immune systems | Ad5-yCD/mutTKSR39rep-hIL12 (Ad) |
| 34 | ||
| IL-21 | Activates T cells, inhibits Treg growth | VVL-21 (VACV) |
| 22 | ||
| Interferon ꞵ (IFN-ꞵ) | Inhibits tumor growth, activates immune system | SJ-815 (VACV) |
| 35 | ||
| Tumor Necrosis Factor ɑ (TNF-ɑ) | Induces apoptosis and stimulates immune system | OAd-TNFa-IL2 (Ad) |
| 32 | ||
| Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) | Causes apoptosis in tumor cells | CD55-ST13-TRAIL |
| 18 | ||
| Co-stimulatory ligands | Role | Virus(es) |
| References | ||
| CD40L (CD154) | Stimulates antigen presentation and T cell activation | oHSV-CD40L (HSV) LOAd703 (Ad) |
| 16,27 | ||
| 4-1BBL (CD137L) | Stimulates T cell proliferation, cytokine production | LOAd703 (Ad) |
| 16 | ||
| Immune checkpoint inhibitors (co-inhibitory ligands) | Role | Virus(es) |
| References | ||
| Anti-PD-L1 | PD-1/PD-L1 interaction suppresses T cells | CF33-hNIS-AntiPDL1 (orthopoxvirus) |
| 25 | ||
| Miscellaneous | Role | Virus(es) |
| References | ||
| Sodium iodide symporter (NIS) | Reporter gene that, with radioactive iodine isotope intake, allows for screening and enhances cytotoxicity of virus | CF33-hNIS-AntiPDL1 (orthopoxvirus) |
| 25 | ||
| Yeast cytosine deaminase/HSV thymidine kinase | Enhances chemotherapy and radiotherapy; suicide gene | Ad5-yCD/mutTKSR39rep (Ad) |
| 34 | ||
| Adenovirus Death Protein (ADP) | Enhances cytotoxicity of virus; causes cell lysis | Ad5-yCD/mutTKSR39rep-ADP (Ad) |
| 34 | ||
| Second mitochondrial-derived activator of caspase (Smac) | Blocks apoptosis inhibition | oVV-Smac (VACV) |
| 23 | ||
| Hyaluronidase | Degrades ECM (hyaluronan) | VCN-01 (Ad) |
| 20 | ||
| Relaxin | Degrades ECM (collagen) | YDC002 (Ad) oAd/RLX-PCDP (Ad) |
| 15,17 | ||
| ST13 | Tumor suppressor gene | CD55-ST13-TRAIL (Ad) |
| 18 | ||
| Ad = adenovirus; HSV = herpes simplex virus; VACV = vaccinia virus; Bolded = used in a clinical trial | ||
One common group of transgenes expressed by OVs is cytokines, which serve to stimulate antitumor immune responses. A diverse array of cytokines has been tested in PDAC OVs, including GM-CSF, ILs, IFNs, cytokines from the tumor necrosis factor superfamily, and chemokines. Of these, multiple ILs (IL-2, IL-10, IL-12, IL-21) have been expressed by OVs. One of these viruses, an adenovirus expressing IL-12, has been tested in a phase I trial with metastatic PDAC patients34. The study reported a median survival of 18.1 months among patients receiving the highest dose and elevated levels of IL-12, IFN, and CXCL10 (cytokines important for NK cell and CD8+ T cell activation) in patients’ blood serum. Importantly, the study hypothesized that IL-12 expressed by infected tumor cells, in addition to its role as a cytokine, acted as a neoantigen that immune cells could identify, creating an immunologically hot TME34. Thus, based on the outcomes of this clinical trial, the usage of cytokines in OVs could enhance immune responses against PDAC not only as a signalling molecule but also as a way for immune cells to distinguish cancer cells.
Another group of transgenes used in OVs is co-stimulatory ligands. The oncolytic adenovirus LOAd703 is armed with two co-stimulatory ligands, CD40L and 4-1BBL, that attach onto the receptors CD40 and 4-1BB respectively16. CD40 and 4-1BB are expressed by some cells in the tumor stroma (specifically, stellate cells and immune cells like MDSCs and anti-inflammatory type II macrophages). The stimulation of these two receptors activates multiple pathways, like the MAPK pathway, involved in cellular processes and immune responses16. Gene therapy with CD40L was previously shown to increase key cytokine (IL-12 and IFN) levels and immune infiltration into tumors, meaning OV expression of CD40L could also achieve similar immune activation27. The study indeed observed that various cell types in the PDAC tumor (stellate cells, DCs, and tumor cells) expressed important chemokines for immune responses in vitro.
Other transgenes include ICIs such as an anti-PD-L1 single-chain antibody fragment in a poxvirus or suicide genes to improve viral cytotoxicity25,34. Two interesting transgenes that have been used are the genes encoding for relaxin, which suppresses collagen production and stimulates collagenase, and the genes for PH20, a soluble human hyaluronidase. Both were encoded in their respective viruses (the oAds YDC002 and VCN-01) in order to combat a problem especially important in PDAC: the desmoplastic ECM, which can prevent drug perfusion15,20. In vitro testing of YDC002 and gemcitabine in tumor spheroid models showed decreased levels of collagen, fibronectin, and elastin expression, and the study observed synergistic effect between YDC002 and gemcitabine—the degradation of the ECM by YDC002 made gemcitabine more potent against tumor cells15.
Recently, oncolytic adenoviruses have demonstrated encouraging activity in PDAC and clinical advances. LOAd703 (LOKON001 Arm 1; NCT02705196) combined with gemcitabine/nab-paclitaxel achieved an objective response rate (ORR) of 44% (8/18; 95% CI 25–66) and a disease control rate (DCR) of 94%16. VCN-01, a hyaluronidase-expressing oncolytic adenovirus designed to degrade stromal components and enhance viral spread, produced approximately 50% ORR in combination cohorts with gemcitabine/nab-paclitaxel in its phase I trial20. Momentum for oncolytic adenoviruses for PDAC has continued as VCN-01 received U.S. FDA Fast Track designation in 2024, LOAd703 in 2025, and the VIRAGE phase 2b trial reported positive top-line results in 202516,20.
Oncolytic reoviruses for PDAC: Pelareorep
Currently, only one reovirus has been used in clinical trials for PDAC: Pelareorep, also known as Reolysin®12. Although Pelareorep is an unmodified reovirus, it has shown very promising results in clinical trials for PDAC and other cancers; in fact, Pelareorep currently is designated as an FDA orphan drug for glioma, ovarian, pancreatic, gastric, and other cancers36. In this section, this review will examine reovirus mechanisms and the results of pre-clinical and clinical trials of Pelareorep for patients with PDAC.
Mammalian reovirus infects a large proportion of humans, with approximately 50% of children aged 5-6 being seropositive for antibodies against reovirus37. Most cases of infection are asymptomatic or result in mild respiratory or gastrointestinal discomfort (with the name ‘reovirus’ coming from ‘respiratory enteric orphan virus’), although reovirus infection can cause severe disease in immunocompromised individuals and children and has been linked to celiac disease (an autoimmune disease)36,38.
Out of the four strains of mammalian orthoreovirus, mammalian orthoreovirus Type 3 Dearing (simply referred to as reovirus) has been used the most for oncolytic reoviruses (including Pelareorep)36. The virus’s dsRNA genome has 10 segments classified by their molecular weights (3 L segments, 3 M segments, and four S segments), with the overall genome length being around 23.5k base pairs (bp)36. The S1 segment is of interest, as it encodes for the attachment protein σ136. Differences in S1 distinguish serotypes of mammalian orthoreovirus (Types 1, 2 and 3) and influence virus tropism36. S1 has been manipulated genetically to broaden cell tropism (infecting cancer cells unable to be infected by wild-type reovirus) and even express transgenes39,40.
Reovirus (mammalian orthoreovirus Type 3 Dearing) possesses natural oncolytic properties, replicating in and lysing cancer cells with activated Ras41,42. Specifically, it has been shown that Ras activation results in the inhibition of dsRNA-activated protein kinase R (PKR)42. In normal cells, PKR is activated by viral dsRNA in cells and leads to an antiviral response (through shutting down viral protein synthesis); however, in cancer cells with Ras mutations, this antiviral response cannot be activated due to PKR inhibition, meaning reovirus is conditionally replicative42. Although this explanation covers why Ras-mutated cells can be sensitive to reovirus infection and subsequent death, other studies have shown that (i) some Ras-mutated cells are resistant to reovirus-mediated death and (ii) cytopathic effect (lysis due to reovirus infection) may not be connected to PKR activation nor Ras expression43. The exact mechanisms by which reovirus oncolysis preferentially affects Ras mutant cells likely include more factors than just PKR inhibition and are still being studied36.
Because 92% of PDAC tumors express mutations in the KRas oncogene, PDAC is potentially a good target for oncolytic reoviruses8. An in vitro study of Pelareorep, a wild-type mammalian orthoreovirus Type 3 Dearing, indeed showed that reovirus replication was higher in KRas-transfected pancreatic epithelial cell lines than in KRas-negative cell lines41. Additionally, endoplasmic reticulum (ER) stress-related gene expression was significantly higher in the KRAS-transfected cells, suggesting that apoptosis can occur because of ER stress that reovirus induces41. These results confirmed Pelareorep’s ability to target pancreatic cancer cells, and further clinical trials have explored Pelareorep’s efficacy for treating PDAC.
An important suggestion that preclinical trials in vivo made was using reovirus in combination therapies. Pelareorep plus bortezomib (an approved proteasome inhibitor that causes apoptosis) in xenograft models of pancreatic cancer resulted in significant reductions in tumor burden compared to monotherapies of the two42. Hence, although early reovirus clinical trials (for various cancers) used reovirus monotherapies, many clinical trials including ones for PDAC) have been testing combination therapies36,41.
| Virus / Regimen | Trial ID / Reference number | N (evaluable) | ORR (95% CI) | DCR / CBR | Median PFS (95% CI) | Median OS (95% CI) |
| Pelareorep + Gemcitabine (Phase II, single-arm) | NCT00998322;12 | 34 (all treated) | 2.9% (1/34); CI NR | 58% CBR at 12 wks | 3.4 mo (2.1–4.4) | 10.2 mo (4.8–17.4) |
| Pelareorep + Pembrolizumab + Chemo (Phase Ib) | NCT02620423;13 | 11 enrolled; 10 efficacy-evaluable | 10% (1/10); CI NR | 30% DCR (PR 1 + SD 2/10) | NR | NR |
| Pelareorep + Pembrolizumab (Phase II; terminated for low accrual) | NCT03723915;13 | 12 | 8.3% (1/12); CI NR | 41.7% (PR 1 + SD 4 /12) | NR | NR |
| Pelareorep + Carboplatin/Paclitaxel vs Chemo (Randomized Phase II) | NCT01280058;14 | Arm A 36; Arm B 37 | PR 19% overall (95% CI 11–30) | DCR 56% (Arm A) vs 59% (Arm B) | 4.9 mo (3.0–6.3) vs 5.2 mo (2.3–6.2) | 7.3 mo (4.8–11.2) vs 8.8 mo (6.6–11.8) |
| Pelareorep + Modified FOLFIRINOX ± Atezolizumab (Phase I/II; ongoing) | EudraCT 2020-003996-15;13 | Safety run-in completed | NR (ongoing) | NR | NR | NR |
| DCR = disease control rate; FOLFIRINOX = chemotherapy regimen of leucovorin (folinic acid), 5-fluorouracil, irinotecan, and oxaliplatin; NR = no response; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; SD = stable disease | ||||||
Among the four completed reovirus clinical trials, one of the most promising trials has been a phase II trial of Pelareorep and gemcitabine12. Out of 34 chemotherapy-naïve advanced/metastatic PDAC patients, 91% presented with metastases12. The median OS was 10.2 months, and the 1-year and 2-year survival rates were 45% and 24%12. For comparison, the median OS with the chemotherapeutic regimen FOLFIRINOX is 11.1 months with 1-year and 2-year survival rates of 48.4% and approximately 10%3.
One notable detail in the Pelareorep study was toxicity. Grade 3 hematological toxicities experienced by patients were anemia (26%), neutropenia (27%), and thrombocytopenia (6%)12. Meanwhile, a phase II-III trial of FOLFIRINOX reported 7.8%, 45.7%, and 9.1% of patients in the FOLFIRINOX group experiencing grade 3 anemia, neutropenia, and thrombocytopenia respectively3. In the Pelareorep study, the most common grade 3 nonhematological adverse event was fatigue (9%), with no grade 4 nonhematological adverse events. In the FOLFIRINOX study, the most common grade 3 nonhematological toxicities included fatigue (23.6%), vomiting (14.5%), diarrhea (12.7%), and sensory neuropathy (9.0%).
Although FOLFIRINOX has been shown to have higher survival rates compared to gemcitabine (and gemcitabine plus nab-paclitaxel), its associated toxicity means that today, the regimen is typically reserved only for patients  76 years old with good performance health status3. In a phase II trial, Pelareorep combined with gemcitabine achieved a median OS of 10.2 months with acceptable toxicity12. Although this outcome falls within the survival range reported for standard chemotherapies such as FOLFIRINOX and gemcitabine/nab-paclitaxel, because these observations come from different patient populations, it is difficult to gauge how Pelareorep compares with other treatments. Overall, the results of the Pelareorep study indicate that reovirus therapies are feasible and generally well tolerated, warranting further evaluation in controlled studies. In addition, when compared to gemcitabine-based therapies, the Pelareorep regimen had a smaller dose of gemcitabine (800 mg/m2 in gemcitabine plus Pelareorep vs. 1000 mg/m2 in gemcitabine plus nab-paclitaxel) and consequently less toxicity12.
76 years old with good performance health status3. In a phase II trial, Pelareorep combined with gemcitabine achieved a median OS of 10.2 months with acceptable toxicity12. Although this outcome falls within the survival range reported for standard chemotherapies such as FOLFIRINOX and gemcitabine/nab-paclitaxel, because these observations come from different patient populations, it is difficult to gauge how Pelareorep compares with other treatments. Overall, the results of the Pelareorep study indicate that reovirus therapies are feasible and generally well tolerated, warranting further evaluation in controlled studies. In addition, when compared to gemcitabine-based therapies, the Pelareorep regimen had a smaller dose of gemcitabine (800 mg/m2 in gemcitabine plus Pelareorep vs. 1000 mg/m2 in gemcitabine plus nab-paclitaxel) and consequently less toxicity12.
Across clinical trials, Pelareorep has demonstrated consistent results of disease stabilization with occasional partial responses and a favorable safety profile12,14,36. Although these outcomes indicate that unmodified reovirus monotherapy achieves limited tumor regression, they also highlight its ability to alter the TME and synergize with chemotherapy or immune checkpoint inhibitors13,14. The modest efficacy likely reflects barriers to systemic delivery in PDAC, including desmoplastic stroma, neutralizing antibodies, and an immunologically “cold” TME with poor T-cell infiltration11.
Among OV platforms, reovirus offers several advantages that make it an attractive candidate for future therapeutic engineering. Its segmented double-stranded RNA genome provides high replication fidelity and safety with minimal risk of genomic integration, while its inherent tumor selectivity through Ras-pathway activation allows preferential replication in more than 90% of PDAC tumors harboring oncogenic KRAS mutations42. In contrast to DNA viruses like adenovirus or vaccinia, which require complex transgene regulation, or HSV, which relies on endoscopic injection, reovirus is able to be administered systemically and has a favorable toxicity profile even in the presence of pre-existing immunity22,26,30,36. These biological and translational characteristics make reovirus a favorable platform for future genetic modifications. The next section will describe one such hypothetical reovirus that is armed with a therapeutic transgene.
Hypothetical design of an IL-21–expressing reovirus for pancreatic ductal adenocarcinoma
This section proposes a hypothetical reovirus design expressing the cytokine IL-21 that could improve reovirus’s efficacy against PDAC. The selection of IL-21 as the transgene is based on its balanced immunostimulatory profile and suitability for the reovirus genome. Compared with IL-12 and GM-CSF, IL-21 promotes strong CD8⁺ T-cell and NK-cell activation while limiting regulatory T-cell expansion and maintaining a lower risk of cytokine-related toxicity22,44. IL-15 also enhances cytotoxic lymphocytes but primarily expands memory subsets with less direct effector activation in the PDAC context. Furthermore, the compact size of the IL-21 coding sequence (~0.5 kb) allows stable incorporation into the reovirus genome, whose segmented RNA structure restricts large inserts40. Collectively, these properties make IL-21 an attractive payload to amplify local antitumor immunity without compromising viral fitness or safety of the reovirus backbone.
Although IL-21 has been linked to tumor invasion and poorer prognosis in some PDAC contexts, these effects have not been linked to IL-21 expressed by OVs22. In the proposed reovirus design, IL-21 expression is confined to infected tumor cells, restricting diffusion and systemic activity and stimulating localized antitumor immune activation rather than a sustained and potentially dangerous response.
To reduce the risk of immunopathology caused by IL-21, the design incorporates multiple safety layers. IL-21 expression is limited to tumor-infected cells through the intrinsic tumor-selective replication of reovirus29,44. EUS-guided intratumoral injections, conservative doses, and defined treatment schedules can be used to maintain localized exposure. In preclinical PDAC models, serum IL-21 levels, pancreatic histopathology, and cytokine profiles can be prospectively monitored to detect excessive inflammation or fibrosis11. These measures aim to preserve IL-21–mediated cytotoxic lymphocyte recruitment while preventing any chance of pancreatic injury.
Although the immunostimulatory activity of IL-21 was first demonstrated in an oncolytic vaccinia model, the results cannot be directly extrapolated to reovirus, as the two platforms differ substantially in genome structure, innate immune sensing, and replication mechanisms22,45,46. Reovirus replicates exclusively in the cytoplasm and tolerates only small genetic inserts due to its segmented double-stranded RNA genome, whereas vaccinia possesses a large DNA genome with more capacity for transgene expression22,41. Consequently, cytokine incorporation into reovirus requires precise balancing of genome length and segment stability38.
Our proposed configuration confines IL-21 (~0.5 kb) to the S1 segment and links it via a self-cleaving 2A peptide to preserve viral packaging and replication kinetics40. This hypothetical design seeks to achieve effective cytokine expression while maintaining the safety and tumor selectivity intrinsic to the reovirus backbone. Ultimately, the design’s efficacy and the utility of the IL-21 insert would have to be experimentally validated. This review will examine how reovirus has been genetically engineered so far for oncolytic virotherapy as well as research on key cytokines before discussing how existing research provides rationale for an IL-21–expressing reovirus.
Genetic engineering of reovirus
As mentioned before, reovirus has a segmented dsRNA genome, which has historically made it less amenable for genetic engineering compared to other viruses. Nevertheless, a plasmid-based reverse genetics system for reovirus has been developed, tested and used since 200745,46. The system uses 10 separate plasmids, each having a bacteriophage T7 RNA polymerase promoter, hepatitis delta virus ribozyme sequences, and the complementary DNA (cDNA) of one of the reovirus genome segments (with the 10 plasmids matching the 10 segments). Recombinant viruses derived from co-transfection of murine cells with the plasmids have been shown to match natural reovirus isolates46.
This plasmid-based system was used to create a GM-CSF-expressing reovirus in one study40. The researchers in the study had previously tested a reovirus expressing the gene E4orf4, an adenovirus gene that increases viral replication and can cause tumor-selective cell death40. However, E4orf4 expression by itself did not seem to improve oncolysis, so an immunomodulator, the cytokine GM-CSF, was chosen next for expression on reovirus.
The attachment protein  1 (which is encoded by the gene segment S1) on reovirus binds to two molecules on the host cell surface, that being sialic acid (with low affinity) and junctional adhesion molecule A (JAM-A) (with higher affinity)39. In particular, the head domain of 1 binds to JAM-A. JAM-A is important for cell entry by wild-type reovirus, meaning tumor cells with low JAM-A expression are less prone to infection47. Previously, spontaneously occurring reovirus mutants that could infect cells regardless of JAM-A expression, named jin-1, jin-2, and jin-3 (JAM-A independent), were created47. The third variant, jin-3, features just a single mutation (G196R, where glycine is replaced by arginine).
1 (which is encoded by the gene segment S1) on reovirus binds to two molecules on the host cell surface, that being sialic acid (with low affinity) and junctional adhesion molecule A (JAM-A) (with higher affinity)39. In particular, the head domain of 1 binds to JAM-A. JAM-A is important for cell entry by wild-type reovirus, meaning tumor cells with low JAM-A expression are less prone to infection47. Previously, spontaneously occurring reovirus mutants that could infect cells regardless of JAM-A expression, named jin-1, jin-2, and jin-3 (JAM-A independent), were created47. The third variant, jin-3, features just a single mutation (G196R, where glycine is replaced by arginine).
Reovirus has a limited coding capacity and cannot carry hundreds of additional nucleotides, meaning the addition of a transgene to reovirus would have to be balanced out by a deletion in a gene38. The jin-3 mutant allows for a deletion in S1: as jin-3 infects cells independently of JAM-A expression and instead only depends on sialic acid expression, the S1 segment encoding for the head domain of 1 (which normally binds JAM-A) becomes unnecessary and can be replaced by a transgene.
Using this strategy, researchers successfully created a GM-CSF-expressing reovirus, replacing the S1 segment encoding for the 1 head domain and part of the 3’ untranslated region (UTR). In place of the deleted segments, researchers placed a hexahistidine-tag, a porcine teschovirus-1 2A (P2A) sequence, and then either human GM-CSF (hsGM-CSF, 432 bp), murine GM-CSF (mmGM-CSF, 423 bp), or a fluorescent protein for control (iLOV, 402 bp)40. The S1 segments of the recombinant viruses rS1-mmGMCSF and rS1-hsGMCSF were 1407 bp and 1416 bp long respectively. For comparison, the S1 segment of wild-type reovirus is 1416 bp long.
Cell lines (human embryonic retinoblast-derived “911” cells) were able to produce mmGM-CSF when infected with rS1-mmGMCSF. Mouse bone marrow leukocytes were exposed to the supernatants of the infected 911 cells to gauge the functionality of the secreted GM-CSF. Higher numbers of mature and immature DCs were created from the leukocytes treated with the rS1-mmGMCSF supernatant compared to the leukocytes treated with the rS1-iLOV supernatant, indicating that the secreted murine GM-CSF is indeed functional40.
Unlike rS1-mmGMCSF, which was stably expressed throughout viral propagation, in vitro testing of rS1-hsGMCSF revealed that (i) genetic deletions occurred in the gene encoding for hsGM-CSF and (ii) these deletion variants actually outperformed the original rS1-hsGMCSF in terms of replication in 911 cells. The researchers hypothesized that either the hsGM-CSF gene or protein had negative influences on viral replication40.
Another key observation in the study was from testing rS1-mmGMCSF in vivo with the KPC3 mouse pancreatic tumor model. Compared to mice treated with rS1-iLOV, mice treated with rS1-mmGMCSF had significantly higher levels of CD86 expression among CD11c+ cells (DCs) in their tumor-draining lymph nodes. Mice treated with rS1-mmGMCSF also had significantly more CD62L– cells (effector memory T cells) among CD4+ (helper T) and CD8+ cells in their spleens. However, within the tumors of the mice, no significant difference was shown for T cell activation markers in the CD11c+ cell population. Furthermore, the study was unable to determine whether treatment with rS1-mmGMCSF improved survival or reduced tumor mass40.
Preclinical trials with rS1-mmGMCSF and rS1-hsGMCSF show that it is possible for reovirus to express a functional transgene. However, it seems that modified reoviruses created from current methods are not entirely reliable and are limited in how many modifications can be made, at least when compared to other OVs (like oAds) that have successfully expressed two or more transgenes that are also larger in size than GM-CSF. As new research fully uncovers the specific roles of, interactions between, and replication signals for each gene segment, better engineered reoviruses could potentially be created38,48.
Choosing the optimal cytokine for expression on reovirus
KPC3 mouse pancreatic tumor models treated with rS1-mmGMCSF suggested that while GM-CSF increased levels of T cell activation markers in the spleen and tumor-draining lymph nodes, there was a negligible increase of T cell activation markers in the pancreatic tumor itself compared to rS1-iLOV. This could potentially be due to the TME of PDAC, which is known to suppress CD8+ T cell activity by using immune cells. In this section, this review will examine different cytokines (including GM-CSF), their speculated roles in pancreatic tumors, and why IL-21 is a good contender for expression on reovirus.
GM-CSF (granulocyte-macrophage colony-stimulating factor) has been used on multiple OVs for PDAC. Out of several OVs expressing a cytokine that have been used in clinical trials for PDAC, two OVs (T-VEC, OrienX010) express GM-CSF. For certain cancers such as melanoma, GM-CSF expressed by OVs serves to primarily promote the development of hematopoietic stem cell progenitors into DCs that can in turn start an effective antitumor immune response30. Yet, for PDAC, GM-CSF may be causing the opposite effect, aiding tumor growth instead of an antitumor response. Researchers examined the generation of Gr-1+ CD11b+ cells (myeloid-derived suppressor cells, MDSCs) in 10 different cell lines49. As expected, c-kit+ splenocytes (progenitor cells isolated from the mice’s spleens) only proliferated into MDSCs in pancreatic cancer cell lines but not in normal cell lines. The study noted that out of 11 proteins analyzed for in the cell lines, tumor-derived GM-CSF was the only protein with high expression in pancreatic cancer cell lines but not in normal cell lines. Indeed, GM-CSF was shown to cause c-kit+ splenocytes to proliferate into MDSCs in vitro, while using an anti-GM-CSF antibody blocked proliferation49. An analysis of PDAC tumor samples from patients further showed that in 19 out of 20 samples, the majority of tumor cells expressed GM-CSF49.
MDSCs block CD8+ T cell activity, so this research suggests that, in the case of PDAC, introducing GM-CSF into the tumor could ultimately block an effective antitumor immune response. This could perhaps explain part of why in vivo testing of rS1-mmGMCSF revealed insignificant levels of T cell activation markers in the tumor. For the reasons mentioned, it was decided not to express GM-CSF in our hypothetical reovirus design.
ILs are a group of cytokines also commonly used in OVs. An IL-12 expressing oAd (Ad5-yCD/mutTKSR39rep-hIL12) has been used in a clinical trial for pancreatic cancer as well34. So far, several ILs have been expressed on oAds, oHSVs and vaccinia viruses, that being IL-2, IL-10, IL-12, and IL-21 (Table 2). In addition, IL-15 has been expressed on an oncolytic vesicular stomatitis virus28.
It was decided not to use IL-2 or IL-10 in the reovirus design. As with GM-CSF, IL-2 likely has a complicated overall effect on PDAC tumors. Although IL-2 has been shown to improve DC tumor infiltration and T cell antitumor activity, it is a potent activator of Treg cells, which are one of the immunosuppressive cell types in the PDAC TME11,33,50. Meanwhile, high levels of serum IL-10 has been linked to a worse prognosis in PDAC patients, and studies have shown that peripheral NK cells in these patients are a different phenotype than normal NK cells, have increased production of IL-10, and are immunosuppressive, aiding tumor growth and evasion51. An IL-10–armed vaccinia virus seems to primarily have inhibited viral clearance by immune cells but not directly affect antitumor immune responses33. As with GM-CSF and many other cytokines, current research suggests that both IL-2 and IL-10 induce multiple effects, sometimes increasing immune activity in PDAC tumors but also utilized by the tumor itself or tumor-associated cells.
IL-12 is known to stimulate NK, CD4+, and CD8+ cells and make these cells produce IFN- and also the chemokine CXCL10, both of which are important for antitumor immune activity34. Indeed, IL-12 is to our knowledge the only interleukin to have been expressed on an OV in a clinical trial for PDAC34. Therapy with an IL-12 expressing oAd indeed resulted in elevated counts of serum NK, CD4+, and CD8+ cells in patients. However, in terms of reovirus engineering, IL-12 may not be a good option to use. When designing Ad5-yCD/mutTKSR39rep-hIL12, researchers used a 1.6k bp coding sequence encoding for the two parts of IL-12 (IL-12A, also called p35, and IL-12B, also called p40)34. The method used by Kemp et al in creating rS1-GMCSF only leaves 522 bp in the reovirus S1 segment that can be replaced by transgenes, however, so IL-12 may be physically unable to be encoded by reovirus40. Newer methods of modifying reovirus, perhaps by deleting additional genes and allowing larger transgenes to be used, could make an IL-12 expressing reovirus possible in the future.
Hence, the selection was narrowed down to IL-15 and IL-21 that could be expressed on the hypothetical reovirus design. Both cytokines are from the IL-2 family of cytokines and have similar gene lengths encoding around 162 amino acids22,28. In comparison, GM-CSF consists of 144 amino acids (and hence its coding sequence is 432 bp as was in rS1-hsGMCSF)40. From these two cytokines, IL-21 was chosen, a cytokine primarily secreted by CD4+ T cells that is closely involved in B cell activation as well as CD8+ T cell activity22. Still, if an IL-21–expressing reovirus was not suitable for some reason or did not perform well, an IL-15–expressing design could be used instead.
Generating an IL-21–expressing reovirus
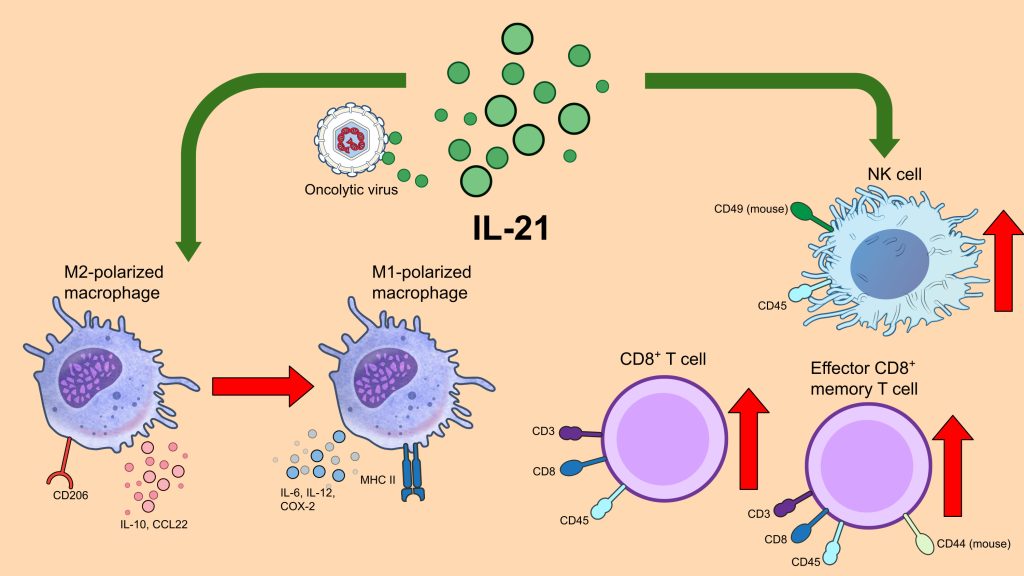
An oncolytic vaccinia virus expressing IL-21, VVL-21, showed promising effects in a preclinical trial22. In particular, the study noted that the immunosuppressive macrophages in pancreatic tumor models were impacted by IL-21. In the PDAC TME, tumor-associated macrophages are M2-like tumor-supporting types, but IL-21 produced by VVL-21 was able to repolarize these macrophages into M1 macrophages, which possess antitumor properties instead22. Tumor-associated (M2-like) macrophages have been speculated to promote antiviral immune responses in other studies, so this repolarization effect induced by IL-21 may further aid oncolytic activity22. This observation with VVL-21 could perhaps be replicated in an IL-21 expressing–reovirus.
An IL-21–expressing reovirus could be generated following a similar reovirus design as rS1-GMCSF that Kemp et al devised. The new S1 segment would consist of (i) the 768 bp that includes the 5’ untranslated region, the open reading frame for the σ1s protein, and the G196R mutation present in jin-3 mutant reovirus; (ii) a hexahistidine tag; (iii) the sequence for thosea asigna virus 2A peptide (T2A); (iv) 486 bp for the coding sequence of IL-21; (v) a stop codon; (vi) and finally, 98 bp of the 3’ untranslated region that is necessary for packaging52. The resulting S1 segment would be approximately 1436 bp long, which is 19 bp longer than the wild-type S1 segment.
Reovirus reverse genetics would be performed using a plasmid-based rescue system with the jin-3 strain, which harbors a G196R mutation in the σ1 protein, enabling JAM-A–independent entry45,46,47. The S1 segment would be modified to accommodate an IL-21 insert, linked via a Thosea asigna virus 2A (T2A) peptide39,48. The resulting recombinant genome would maintain a balanced overall genome length.
To address challenges with engineering feasibility and insert stability, the S1 segment was selected as the head-domain deletion in the JAM-A–independent jin-3 backbone provides sufficient coding space for a cytokine without affecting receptor binding or packaging39,47,48. Incorporation of the short T2A self-cleaving peptide ensures the translation of IL-21 as a separate product and not a part of the viral assembly39,40. To mitigate the chances of instability previously observed with large or human cytokine inserts, the design restricts the transgene to <0.6 kb, preserving genome length parity with wild-type S139,47,48. Basic quality-control steps would include sequence confirmation after serial passages (5), quantification of IL-21 expression by ELISA, and replication kinetics compared with the parental jin-3 virus to verify genetic stability and preserved infectivity29,45,46.
It should be noted that, like other cytokines, IL-21 can be harnessed by cancer cells to aid cancer development in some tumors. A study found that Th17-like cells in PDAC tumors secreted IL-21, with high density of IL-21+ immune cells in tissue samples being correlated to worse prognosis53. In the same study, adding IL-21 to tumor cells aided tumor invasion in vitro. However, other research suggests that IL-21 is still important for antitumor immunity in PDAC, and the overall effect of IL-21 may ultimately be beneficial. In immunocompetent mouse models, VVL-21 indeed induced greater tumor regressions and improved survival compared to the control virus VVLTK-STCN1L (VVL-21 is an IL-21–expressing variant of VVLTK-STCN1L)22. As was done with VVL-21, in vivo testing with an immunocompetent model (with unmodified reovirus as control) would be crucial in assessing the efficacy of the IL-21–expressing reovirus.
Hence, from the analyses of VVL-21 preclinical trials, it seems that IL-21 expression on an OV has several antitumor effects in the context of PDAC tumors (Figure 3). Firstly, it can polarize immunosuppressive M2-like macrophages in the TME into antitumor M1 macrophages. In addition, IL-21 can also significantly increase counts of CD8+ T cells, effector memory CD8+ T cells, and NK cells, all of which are important for antitumor immunity, in the tumor and the blood22.
Finally, a combination therapy of the IL-21–expressing reovirus and an immune checkpoint inhibitor (ICI) could achieve synergistic effect and improve response to treatment. Several studies of OVs, including VVL-21, have pointed out higher expression of PD-L1 by tumor cells following oncolytic virotherapy, likely due to the TME becoming immunologically ‘hot’22,27. Whereas ICI monotherapy may not elicit a significant effect in PDAC due to its ‘cold’ TME, administering ICIs together with OVs can significantly improve immune activity. In fact, clinical trials of Pelareorep for PDAC have been using ICIs like pembrolizumab (anti-PD-1) and atezolizumab (anti-PD-L1) in combination already12,13. It can be hypothesized that the IL-21–expressing reovirus could also be used in combination with ICIs for improved efficacy.
Safety and delivery considerations
Cytokine-armed OVs carry an inherent risk of systemic inflammation, particularly with potent T-cell stimulators such as IL-21. To mitigate these risks, transgene expression is restricted by using the tumor-selective replication of reovirus and placing the IL-21 insert under native viral control elements, favoring intratumoral and not systemic release29,30. Conservative dosing and local delivery help to further limit exposure. Previous experiences with IL-12- and IL-2-expressing OVs demonstrated that excessive systemic cytokine exposure and resulting effects, rather than local immune stimulation, were the likely causes of toxicity44,34,50. These precedents guided our approach toward moderate transgene expression balanced with viral fitness and safety, making sure the virus can create an effective yet controllable immune response.
Effective delivery of oncolytic reovirus to pancreatic tumors remains a major challenge due to the tumor sites’ dense desmoplastic stroma, high interstitial pressure, and deep anatomical location, all of which hinder viral dissemination9,10,11. Moreover, pre-existing neutralizing antibodies against reovirus are common in the adult population and could potentially target circulating virions following intravenous delivery37,38.
To overcome these physical and immunological barriers, several complementary strategies can be used. Repeated intratumoral dosing or EUS-guided injection achieves higher local titers and bypasses stromal resistance while minimizing systemic inactivation19. On the other hand, intravenous administration may require additional measures—such as carrier-cell delivery using mesenchymal stromal or immune cells—to shield virions from neutralization and promote tumor-homing dissemination17. Transient immunomodulation with agents such as low-dose cyclophosphamide can maximize OV effects and prevent antiviral responses36. Collectively, these safety- and delivery-oriented adaptations provide a rational framework for using IL-21-armed reoviruses to achieve an effective yet controlled immune activation in PDAC’s challenging TME.
Limitations of the evidence base
Current evidence supporting OV therapy for PDAC is largely from small, early-phase, and often single-arm studies, some of which combine OVs with existing chemotherapy or immunotherapy44,34,54. As a result, there is a substantial risk of selection bias and cross-trial variability that limits the generalizability of reported outcomes30,44. Moreover, differences in viral backbones, dosing routes, and patient populations complicate direct comparisons between trials. While preclinical data are mechanistically robust, translation to clinical efficacy remains constrained by the heterogeneity of study design and endpoint reporting. Future randomized and biomarker-guided trials will be essential to validate the observed signals and define the true therapeutic value of any novel OV for treating PDAC29,30.
Conclusion
PDAC is one of the most lethal cancers, with patients often being unresponsive to conventional therapies. Immunotherapies have shown efficacy against some cancers and hold promise for cancers like PDAC, which have very immunologically ‘cold’ TMEs. Oncolytic virotherapy is one especially promising immunotherapy for PDAC because it operates through multiple mechanisms, such as direct lysis of tumor cells and activating antitumor immunity, and is diverse in terms of viral backbones and genetic modifications. In the case of PDAC, viroengineering has been used most extensively on oAds, oHSVs, and oncolytic poxviruses to improve their performance. Reovirus is a segmented dsRNA virus with natural tumor selectivity that has shown promise as a therapeutic agent for PDAC but has lagged behind in therapeutic transgene expression.
This review proposed a hypothetical design of a modified reovirus expressing the cytokine IL-21 that, based on current research, could improve reovirus’s efficacy for PDAC. Although the hypothetical design is not an experimentally tested design, rationale was provided on how genetic engineering of reovirus, specifically the expression of IL-21, could improve reovirus’s performance. An IL-21–expressing reovirus in combination with ICIs or other agents, such as chemotherapeutic agents, could achieve synergistic effect for treating PDAC. Future genetically engineered reoviruses could utilize IL-21 or other transgenes for enhanced capabilities, and further research on reovirus engineering could allow for more efficient packaging and expression of transgenes.
Acknowledgements
I greatly appreciate Klaus Okkenhaug from the University of Cambridge for being my mentor, introducing me to the field of immunology, and guiding me through the process of writing this paper. In addition, I am thankful for the team at Lumiere Education.
- Evidence levels were graded as High (multicenter human trials with reproducible outcomes), Moderate (well-designed preclinical or early-phase clinical studies), or Exploratory (conceptual or preliminary preclinical findings). ↩︎
References
- A. Adamska, A. Domenichini, M. Falasca. Pancreatic ductal adenocarcinoma: current and evolving therapies. Int J Mol Sci 18, 1338 (2017). https://doi.org/10.3390/ijms18071338. [↩] [↩] [↩] [↩] [↩] [↩]
- H.A. Burris, M.J. Moore, J. Andersen, M.R. Green, M.L. Rothenberg, M.R. Modiano, M.C. Cripps, R.K. Portenoy, A.M. Storniolo, P. Tarassoff, R. Nelson, F.A. Dorr, C.D. Stephens, D.D. Von Hoff. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15, 2403–2413 (1997). https://doi.org/10.1200/JCO.1997.15.6.2403. [↩] [↩]
- T. Conroy, F. Desseigne, M. Ychou, O. Bouché, R. Guimbaud, Y. Bécouarn, A. Adenis, J.-L. Raoul, S. Gourgou-Bourgade, C. De La Fouchardière, J. Bennouna, J.-B. Bachet, F. Khemissa-Akouz, D. Péré-Vergé, C. Delbaldo, E. Assenat, B. Chauffert, P. Michel, C. Montoto-Grillot, M. Ducreux. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364, 1817–1825 (2011). https://doi.org/10.1056/NEJMoa1011923. [↩] [↩] [↩] [↩]
- Q. Huang, Y. Zheng, Z. Gao, L. Yuan, Y. Sun, H. Chen. Comparative efficacy and safety of PD-1/PD-L1 inhibitors for patients with solid tumors: a systematic review and Bayesian network meta-analysis. J Cancer 12, 1133–1143 (2021). https://doi.org/10.7150/jca.49325. [↩]
- R.E. Royal, C. Levy, K. Turner, A. Mathur, M. Hughes, U.S. Kammula, R.M. Sherry, S.L. Topalian, J.C. Yang, I. Lowy, S.A. Rosenberg. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 33, 828–833 (2010). https://doi.org/10.1097/CJI.0b013e3181eec14c. [↩]
- J.R. Brahmer, S.S. Tykodi, L.Q.M. Chow, W.-J. Hwu, S.L. Topalian, P. Hwu, C.G. Drake, L.H. Camacho, J. Kauh, K. Odunsi, H.C. Pitot, O. Hamid, S. Bhatia, R. Martins, K. Eaton, S. Chen, T.M. Salay, S. Alaparthy, J.F. Grosso, A.J. Korman, S.M. Parker, S. Agrawal, S.M. Goldberg, D.M. Pardoll, A. Gupta, J.M. Wigginton. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med 366, 2455–2465 (2012). https://doi.org/10.1056/NEJMoa1200694. [↩]
- S.D. Kamath, A. Kalyan, S. Kircher, H. Nimeiri, A.J. Fought, A. Benson, M. Mulcahy. Ipilimumab and gemcitabine for advanced pancreatic cancer: a phase Ib study. Oncologist 25, e808–e815 (2020). https://doi.org/10.1634/theoncologist.2019-0473. [↩]
- J. Peng, B.-F. Sun, C.-Y. Chen, J.-Y. Zhou, Y.-S. Chen, H. Chen, L. Liu, D. Huang, J. Jiang, G.-S. Cui, Y. Yang, W. Wang, D. Guo, M. Dai, J. Guo, T. Zhang, Q. Liao, Y. Liu, Y.-L. Zhao, D.-L. Han, Y. Zhao, Y.-G. Yang, W. Wu. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29, 725–738 (2019). https://doi.org/10.1038/s41422-019-0195-y. [↩] [↩] [↩] [↩] [↩]
- G.S. Karagiannis, T. Poutahidis, S.E. Erdman, R. Kirsch, R.H. Riddell, E.P. Diamandis. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res 10, 1403–1418 (2012). https://doi.org/10.1158/1541-7786.MCR-12-0307. [↩] [↩] [↩] [↩]
- K.P. Olive, M.A. Jacobetz, C.J. Davidson, A. Gopinathan, D. McIntyre, D. Honess, B. Madhu, M.A. Goldgraben, M.E. Caldwell, D. Allard, K.K. Frese, G. DeNicola, C. Feig, C. Combs, S.P. Winter, H. Ireland-Zecchini, S. Reichelt, W.J. Howat, A. Chang, M. Dhara, L. Wang, F. Rückert, R. Grützmann, C. Pilarsky, K. Izeradjene, S.R. Hingorani, P. Huang, S.E. Davies, W. Plunkett, M. Egorin, R.H. Hruban, N. Whitebread, K. McGovern, J. Adams, C. Iacobuzio-Donahue, J. Griffiths, D.A. Tuveson. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009). https://doi.org/10.1126/science.1171362. [↩] [↩] [↩]
- P. Sarantis, E. Koustas, A. Papadimitropoulou, A.G. Papavassiliou, M.V. Karamouzis. Pancreatic ductal adenocarcinoma: treatment hurdles, tumor microenvironment and immunotherapy. World J Gastrointest Oncol 12, 173–181 (2020). https://doi.org/10.4251/wjgo.v12.i2.173. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- D. Mahalingam, S. Goel, S. Aparo, S. Patel Arora, N. Noronha, H. Tran, R. Chakrabarty, G. Selvaggi, A. Gutierrez, M. Coffey, S. Nawrocki, G. Nuovo, M. Mita. A phase II study of Pelareorep (REOLYSIN®) in combination with gemcitabine for patients with advanced pancreatic adenocarcinoma. Cancers 10, 160 (2018). https://doi.org/10.3390/cancers10060160. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- D. Mahalingam, S. Chen, P. Xie, H. Loghmani, T. Heineman, A. Kalyan, S. Kircher, I.B. Helenowski, X. Mi, V. Maurer, M. Coffey, M. Mulcahy, A. Benson, B. Zhang. Combination of pembrolizumab and Pelareorep promotes anti-tumour immunity in advanced pancreatic adenocarcinoma (PDAC). Br J Cancer 129, 782–790 (2023). https://doi.org/10.1038/s41416-023-02344-5. [↩] [↩] [↩] [↩] [↩] [↩]
- A.M. Noonan, M.R. Farren, S.M. Geyer, Y. Huang, S. Tahiri, D. Ahn, S. Mikhail, K.K. Ciombor, S. Pant, S. Aparo, J. Sexton, J.L. Marshall, T.A. Mace, C.S. Wu, B. El-Rayes, C.D. Timmers, J. Zwiebel, G.B. Lesinski, M.A. Villalona-Calero, T.S. Bekaii-Saab. Randomized phase 2 trial of the oncolytic virus Pelareorep (reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol Ther 24, 1150–1158 (2016). https://doi.org/10.1038/mt.2016.66. [↩] [↩] [↩] [↩]
- K.H. Jung, I.-K. Choi, H.-S. Lee, H.H. Yan, M.K. Son, H.M. Ahn, J. Hong, C.-O. Yun, S.-S. Hong. Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett 396, 155–166 (2017). https://doi.org/10.1016/j.canlet.2017.03.009. [↩] [↩] [↩] [↩]
- B.L. Musher, E.K. Rowinsky, B.G. Smaglo, W. Abidi, M. Othman, K. Patel, S. Jawaid, J. Jing, A. Brisco, A.M. Leen, M. Wu, L.C. Sandin, J. Wenthe, E. Eriksson, G.J. Ullenhag, B. Grilley, J. Leja-Jarblad, S.G. Hilsenbeck, M.K. Brenner, A.S.I. Loskog. LOAd703, an oncolytic virus-based immunostimulatory gene therapy, combined with chemotherapy for unresectable or metastatic pancreatic cancer (LOKON001): results from arm 1 of a non-randomised, single-centre, phase 1/2 study. Lancet Oncol 25, 488-500 (2024). https://doi.org/10.1016/S1470-2045(24)00079-2. [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Y. Na, J.-P. Nam, J. Hong, E. Oh, H.C. Shin, H.S. Kim, S.W. Kim, C.-O. Yun. Systemic administration of human mesenchymal stromal cells infected with polymer-coated oncolytic adenovirus induces efficient pancreatic tumor homing and infiltration. J Control Release 305, 75–88 (2019). https://doi.org/10.1016/j.jconrel.2019.04.040. [↩] [↩] [↩]
- Y. Zhang, M. Ye, F. Huang, S. Wang, H. Wang, X. Mou, Y. Wang. Oncolytic adenovirus expressing ST13 increases antitumor effect of tumor necrosis factor-related apoptosis-inducing ligand against pancreatic ductal adenocarcinoma. Hum Gene Ther 31, 891–903 (2020). https://doi.org/10.1089/hum.2020.024. [↩] [↩] [↩]
- J. Lee, D.W. Shin, H. Park, J. Kim, Y. Youn, J.H. Kim, J. Kim, J.-H. Hwang. Tolerability and safety of EUS-injected adenovirus-mediated double-suicide gene therapy with chemotherapy in locally advanced pancreatic cancer: a phase 1 trial. Gastrointest Endosc 92, 1044-1052.e1 (2020). https://doi.org/10.1016/j.gie.2020.02.012. [↩] [↩]
- R. Garcia-Carbonero, M. Bazan-Peregrino, M. Gil-Martín, R. Álvarez, T. Macarulla, M.C. Riesco-Martinez, H. Verdaguer, C. Guillén-Ponce, M. Farrera-Sal, R. Moreno, A. Mato-Berciano, M.V. Maliandi, S. Torres-Manjon, M. Costa, N. Del Pozo, J. Martínez De Villarreal, F.X. Real, N. Vidal, G. Capella, R. Alemany, E. Blasi, C. Blasco, M. Cascalló, R. Salazar. Phase I, multicenter, open-label study of intravenous VCN-01 oncolytic adenovirus with or without nab-paclitaxel plus gemcitabine in patients with advanced solid tumors. J Immunother Cancer 10, e003255 (2022). https://doi.org/10.1136/jitc-2021-003255. [↩] [↩] [↩] [↩] [↩]
- E. Eriksson, I. Milenova, J. Wenthe, M. Ståhle, J. Leja-Jarblad, G. Ullenhag, A. Dimberg, R. Moreno, R. Alemany, A. Loskog. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin Cancer Res 23, 5846–5857 (2017). https://doi.org/10.1158/1078-0432.CCR-17-0285. [↩]
- G. Marelli, L.S. Chard Dunmall, M. Yuan, C. Di Gioia, J. Miao, Z. Cheng, Z. Zhang, P. Liu, J. Ahmed, R. Gangeswaran, N. Lemoine, Y. Wang. A systemically deliverable vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J Immunother Cancer 9, e001624 (2021). https://doi.org/10.1136/jitc-2020-001624. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- W. Chen, W. Fan, G. Ru, F. Huang, X. Lu, X. Zhang, X. Mou, S. Wang. Gemcitabine combined with an engineered oncolytic vaccinia virus exhibits a synergistic suppressive effect on the tumor growth of pancreatic cancer. Oncol Rep 41, 67-76 (2019). https://doi.org/10.3892/or.2018.6817. [↩] [↩]
- Z. Liu, R. Ravindranathan, P. Kalinski, Z.S. Guo, D.L. Bartlett. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun 8, 14754 (2017). https://doi.org/10.1038/ncomms14754. [↩]
- Y. Woo, Z. Zhang, A. Yang, S. Chaurasiya, A.K. Park, J. Lu, S.-I. Kim, S.G. Warner, D. Von Hoff, Y. Fong. Novel chimeric immuno-oncolytic virus CF33-hNIS-antiPDL1 for the treatment of pancreatic cancer. Journal of the American College of Surgeons 230, 709–717 (2020). https://doi.org/10.1016/j.jamcollsurg.2019.12.027. [↩] [↩] [↩] [↩] [↩]
- K. Runcie, Y. Bracero, A. Samouha, G. Manji, H.E. Remotti, T.A. Gonda, Y. Saenger. Phase I study of intratumoral injection of talimogene laherparepvec for the treatment of advanced pancreatic cancer. Oncologist 30, oyae200 (2025). https://doi.org/10.1093/oncolo/oyae200. [↩] [↩] [↩] [↩]
- R. Wang, J. Chen, W. Wang, Z. Zhao, H. Wang, S. Liu, F. Li, Y. Wan, J. Yin, R. Wang, Y. Li, C. Zhang, H. Zhang, Y. Cao. CD40L-armed oncolytic herpes simplex virus suppresses pancreatic ductal adenocarcinoma by facilitating the tumor microenvironment favorable to cytotoxic T cell response in the syngeneic mouse model. J Immunother Cancer 10, e003809 (2022). https://doi.org/10.1136/jitc-2021-003809. [↩] [↩] [↩] [↩]
- S. Zhang, S.D. Rabkin. The discovery and development of oncolytic viruses: are they the future of cancer immunotherapy? Expert Opin Drug Discov 16, 391–410 (2021). https://doi.org/10.1080/17460441.2021.1850689. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- S. Gujar, J.G. Pol, V. Kumar, M. Lizarralde-Guerrero, P. Konda, G. Kroemer, J.C. Bell. Tutorial: design, production and testing of oncolytic viruses for cancer immunotherapy. Nat Protoc 19, 2540–2570 (2024). https://doi.org/10.1038/s41596-024-00985-1. [↩] [↩] [↩] [↩] [↩]
- S.Z. Shalhout, D.M. Miller, K.S. Emerick, H.L. Kaufman. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol 20, 160–177 (2023). https://doi.org/10.1038/s41571-022-00719-w. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- H. Liu. Preclinical evaluation of herpes simplex virus armed with granulocyte-macrophage colony-stimulating factor in pancreatic carcinoma. World J Gastroenterol 19, 5138 (2013). https://doi.org/10.3748/wjg.v19.i31.5138. [↩]
- K. Watanabe, Y. Luo, T. Da, S. Guedan, M. Ruella, J. Scholler, B. Keith, R.M. Young, B. Engels, S. Sorsa, M. Siurala, R. Havunen, S. Tähtinen, A. Hemminki, C.H. June. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 3, e99573 (2018). https://doi.org/10.1172/jci.insight.99573. [↩] [↩]
- L.S. Chard, N.R. Lemoine, Y. Wang. New role of interleukin-10 in enhancing the antitumor efficacy of oncolytic vaccinia virus for treatment of pancreatic cancer. OncoImmunology 4, e1038689 (2015). https://doi.org/10.1080/2162402X.2015.1038689. [↩] [↩] [↩]
- K.N. Barton, F. Siddiqui, R. Pompa, S.O. Freytag, G. Khan, I. Dobrosotskaya, M. Ajlouni, Y. Zhang, J. Cheng, B. Movsas, D. Kwon. Phase I trial of oncolytic adenovirus-mediated cytotoxic and interleukin-12 gene therapy for the treatment of metastatic pancreatic cancer. Mol Ther Oncolytics 20, 94–104 (2021). https://doi.org/10.1016/j.omto.2020.11.006. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- E. Cho, S.M.B.U. Islam, F. Jiang, J.-E. Park, B. Lee, N.D. Kim, T.-H. Hwang. Characterization of oncolytic vaccinia virus harboring the human IFNB1 and CES2 transgenes. Cancer Res Treat 52, 309–319 (2020). https://doi.org/10.4143/crt.2019.161. [↩]
- L. Müller, R. Berkeley, T. Barr, E. Ilett, F. Errington-Mais. Past, present and future of oncolytic reovirus. Cancers 12, 3219 (2020). https://doi.org/10.3390/cancers12113219. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- J.H. Tai, J.V. Williams, K.M. Edwards, P.F. Wright, D.E. Crowe Jr. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis 191, 1221–1224 (2005). https://doi.org/10.1086/428911. [↩] [↩]
- M.R. Eledge, M.D. Zita, K.W. Boehme. Reovirus: friend and foe. Curr Clin Microbiol Rep 6, 132–138 (2019). https://doi.org/10.1007/s40588-019-00121-8. [↩] [↩] [↩] [↩] [↩]
- T. Kawagishi, Y. Kanai, R. Nouda, I. Fukui, J.A. Nurdin, Y. Matsuura, T. Kobayashi. Generation of genetically RGD σ1-modified oncolytic reovirus that enhances JAM-A-independent infection of tumor cells. J Virol 94, e01703-20 (2020). https://doi.org/10.1128/JVI.01703-20. [↩] [↩] [↩] [↩] [↩] [↩]
- V. Kemp, D.J.M. Van Den Wollenberg, M.G.M. Camps, T. Van Hall, P. Kinderman, N. Pronk-van Montfoort, R.C. Hoeben. Arming oncolytic reovirus with GM-CSF gene to enhance immunity. Cancer Gene Ther 26, 268–281 (2019). https://doi.org/10.1038/s41417-018-0063-9. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- J.S. Carew, C.M. Espitia, W. Zhao, K.R. Kelly, M. Coffey, J.W. Freeman, S.T. Nawrocki. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis 4, e728–e728 (2013). https://doi.org/10.1038/cddis.2013.259. [↩] [↩] [↩] [↩] [↩]
- J.E. Strong, M.C. Coffey, D. Tang, P. Sabinin, P.W.K. Lee. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J 17, 3351–3362 (1998). https://doi.org/10.1093/emboj/17.12.3351. [↩] [↩] [↩] [↩] [↩]
- L. Song, T. Ohnuma, I.H. Gelman, J.F. Holland. Reovirus infection of cancer cells is not due to activated Ras pathway. Cancer Gene Ther 16, 382–382 (2009). https://doi.org/10.1038/cgt.2008.84. [↩]
- M. Nisar, R.Z. Paracha, S. Adil, S.N. Qureshi, H.A. Janjua. An extensive review on preclinical and clinical trials of oncolytic viruses therapy for pancreatic cancer. Front Oncol 12, 875188 (2022). https://doi.org/10.3389/fonc.2022.875188. [↩] [↩] [↩] [↩] [↩]
- T. Kobayashi, A.A.R. Antar, K.W. Boehme, P. Danthi, E.A. Eby, K.M. Guglielmi, G.H. Holm, E.M. Johnson, M.S. Maginnis, S. Naik, W.B. Skelton, J.D. Wetzel, G.J. Wilson, J.D. Chappell, T.S. Dermody. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1, 147–157 (2007). https://doi.org/10.1016/j.chom.2007.03.003. [↩] [↩] [↩] [↩]
- T. Kobayashi, L.S. Ooms, M. Ikizler, J.D. Chappell, T.S. Dermody. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398, 194–200 (2010). https://doi.org/10.1016/j.virol.2009.11.037. [↩] [↩] [↩] [↩] [↩]
- D.J.M. van den Wollenberg, I.J.C. Dautzenberg, S.K. van den Hengel, S.J. Cramer, R.J. de Groot, R.C. Hoeben. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS ONE 7, e48064 (2012). https://doi.org/10.1371/journal.pone.0048064. [↩] [↩] [↩] [↩] [↩]
- M. Pan, A.L. Alvarez-Cabrera, J.S. Kang, L. Wang, C. Fan, Z.H. Zhou. Asymmetric reconstruction of mammalian reovirus reveals interactions among RNA, transcriptional factor µ2 and capsid proteins. Nat Commun 12, 4176 (2021). https://doi.org/10.1038/s41467-021-24455-4. [↩] [↩] [↩] [↩]
- L.J. Bayne, G.L. Beatty, N. Jhala, C.E. Clark, A.D. Rhim, B.Z. Stanger, R.H. Vonderheide. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835 (2012). https://doi.org/10.1016/j.ccr.2012.04.025. [↩] [↩] [↩]
- T. Gong, X. Huang, Z. Wang, Y. Chu, L. Wang, Q. Wang. IL-2 promotes expansion and intratumoral accumulation of tumor infiltrating dendritic cells in pancreatic cancer. Cancer Immunol Immunother 73, 84 (2024). https://doi.org/10.1007/s00262-024-03669-7. [↩] [↩]
- F. Marcon, J. Zuo, H. Pearce, S. Nicol, S. Margielewska-Davies, M. Farhat, B. Mahon, G. Middleton, R. Brown, K.J. Roberts, P. Moss. NK cells in pancreatic cancer demonstrate impaired cytotoxicity and a regulatory IL-10 phenotype. OncoImmunology 9, 1845424 (2020). https://doi.org/10.1080/2162402X.2020.1845424. [↩]
- M.R. Roner, J. Roehr. The 3′ sequences required for incorporation of an engineered ssRNA into the reovirus genome. Virol J 3, 1 (2006). https://doi.org/10.1186/1743-422X-3-1. [↩]
- A. Linnebacher, P. Mayer, N. Marnet, F. Bergmann, E. Herpel, S. Revia, L. Yin, L. Liu, T. Hackert, T. Giese, I. Herr, M.M. Gaida. Interleukin 21 receptor/ligand interaction is linked to disease progression in pancreatic cancer. Cells 8, 1104 (2019). https://doi.org/10.3390/cells8091104. [↩]
- I.P. Taylor, J.A. Lopez. Oncolytic adenoviruses and the treatment of pancreatic cancer: a review of clinical trials. J Cancer Res Clin Oncol 149, 8117–8129 (2023). https://doi.org/10.1007/s00432-023-04735-w. [↩]



{kind=link}