
Abstract
Spinal cord injury (SCI) remains a major clinical challenge because of the limited regenerative capacity of the adult central nervous system (CNS). Unlike peripheral neurons, CNS neurons often fail to reengage the intrinsic growth programs necessary for long-distance axonal repair after injury. The RE1-Silencing Transcription Factor (REST), also known as Neuron-Restrictive Silencer Factor (NRSF), is a context-dependent transcriptional regulator that promotes neuronal survival in aging and select neurodegenerative settings while inhibiting axon regeneration following injury. To synthesize current understanding, this systematic literature review examined peer-reviewed studies of REST in CNS neurons. Articles from in vivo and in vitro models of CNS injury and neurodegenerative conditions were analyzed for molecular mechanisms, gene regulation, and functional outcomes. The reviewed evidence indicates that REST suppresses regeneration-associated transcription factor networks after CNS injury, limiting axon growth, whereas reduction of REST in injured neurons reactivates pro-regenerative gene programs, enhances axonal elongation, and improves regenerative outcomes in preclinical models. At the same time, broader literature shows that indiscriminate REST inhibition may be harmful in contexts where REST supports autophagy, proteostasis, mitochondrial function, or stress resistance. Collectively, these findings support a more precise view of REST as a context-dependent regulator rather than a uniform therapeutic target. Temporally and spatially targeted REST modulation, therefore, warrants further preclinical investigation as a strategy to enhance CNS repair without disrupting protective functions in other neural contexts.
Keywords: REST/NRSF, CNS injury, Axon regeneration, Neuroprotection, Epigenetic regulation
Introduction
Spinal cord injury (SCI) is recognized as one of the most severe neurological maladies, often leading to lasting motor and sensory deficits due to the limited regenerative capacity of the adult central nervous system (CNS)1,2. Unlike peripheral nerves, which can regenerate axons following injury, CNS neurons typically do not activate the necessary genetic programs for long-distance axon regeneration1,2. Understanding the factors that limit CNS repair is therefore critical for developing strategies to improve recovery after SCI. Historically, research on regeneration failure has emphasized extrinsic barriers present in the injury environment, such as glial scarring and inhibitory molecules that hinder neuronal growth1,2. However, more recent work demonstrated that failed repair also arises from intrinsic limitations within injured neurons, as mature CNS neurons do not readily return to the growth-competent transcriptional state seen in development or after successful peripheral regeneration1,3.
This shift toward intrinsic growth control was shaped by classical regeneration studies showing that neuronal state is functionally important. In adult dorsal root ganglion neurons in mouse models, preconditioning injury enabled central axons to grow into and beyond spinal lesions, demonstrating that growth competence can be reawakened in the adult nervous system4. In this context, the “conditioning injury” paradigm showed that prior peripheral injury enhances the intrinsic growth state of adult neurons and increases their regenerative capacity, rather than leaving regeneration solely to the external injury environment5.
Following these findings, subsequent studies demonstrated that conditioning injury-induced regeneration requires STAT3 activation, that SMAD1 signaling controls axon growth in adult mouse dorsal root ganglion neurons, and that hyperactivated STAT3 further promotes CNS axon repair in vivo6,7,8. STAT3 (Signal Transducer and Activator of Transcription 3) functions as a cytokine-responsive transcription factor that, once activated, translocates to the nucleus and regulates transcriptional programs associated with axon growth and regeneration6,8. Similarly, BMP/SMAD1 (Mothers against decapentaplegic homolog 1) signaling operates through bone morphogenetic protein activation of SMAD1, which regulates transcriptional programs that support axon growth and intrinsic growth capacity7. Together, these pathways convert extracellular injury signals into coordinated transcriptional responses that enable axon growth capacity6,7,8.
Extending these principles to other CNS systems, similar mechanisms are observed in the optic and corticospinal systems in mice, where SOCS3 deletion in adult retinal ganglion cells promotes optic nerve regeneration, PTEN deletion enhances corticospinal axon growth, and combined PTEN/SOCS3 deletion produces robust and sustained axon growth9,10,11. SOCS3 (Suppressor of Cytokine Signaling 3) normally inhibits STAT3 signaling, and its deletion enhances STAT3 signaling, promoting transcriptional programs associated with axon growth9.
In parallel, PTEN (Phosphatase and Tensin Homolog) negatively regulates the PI3K–AKT–mTOR pathway responsible for regulating cap-dependent protein translation through rapamycin, and its deletion in mouse models activates mTOR signaling to enhance intrinsic growth potential and axon elongation10,11. Beyond these pathways, additional pro-regenerative manipulations such as Lin28a upregulation, SOX11 overexpression, and Elk-1 activation likewise demonstrate that neuronal maturity and cell type strongly shape the outcome of transcription factor–based interventions12,13,14.
Among these intrinsic regulators, the RE1-Silencing Transcription Factor (REST), also referred to as Neuron-Restrictive Silencer Factor (NRSF), has been identified as a key molecular player with context-dependent roles. REST/NRSF was initially identified as a DNA-binding silencer that restricts neuronal gene expression in non-neuronal cells by binding RE1/NRSE motifs15,16. At the molecular level, REST is a zinc-finger transcription factor that binds RE1/NRSF DNA motifs near neuronal genes15,16. It recruits corepressors and chromatin modifiers, regulatory proteins that suppress gene expression by altering chromatin structure and reducing DNA accessibility, to silence extensive gene networks through epigenetic mechanisms15,16,17,18,19,20.
Collectively, these studies establish intrinsic growth control as a central determinant of CNS repair and set the stage for examining whether or not REST functions as a transcriptional repressor of these programs following CNS injury21. This regulatory role becomes particularly evident in the adult CNS. In the aging brain and in the cases of certain neurodegenerative diseases, REST can act protectively by repressing genes associated with oxidative stress, apoptosis, toxic protein accumulation, and loss of cellular homeostasis22,23,24,25,26,27.
Conversely, emerging evidence suggests that REST’s repressive activity may become maladaptive after CNS injury. In adult mouse models of corticospinal tract injury and optic nerve crush, REST is upregulated and suppresses a core regeneration-associated transcription factor network required for axon repair21. This network includes transcription factors such as Jun, STAT3, SOX11, SMAD1, and ATF3, which are essential for activating regeneration-associated gene programs and are repressed following injury21. The central issue, then, is not whether REST is simply beneficial or harmful, but how the same transcriptional regulator shifts function across injury, aging, ischemia, and neurodegeneration. This paper synthesizes current evidence on REST’s molecular mechanisms, its context-dependent effects in neural tissue, and the implications of targeting REST for therapeutic intervention after CNS injury.
Methods
Literature Search Strategy
A systematic literature review with narrative synthesis was conducted in accordance with the PRISMA 2020 reporting guidelines to examine the role of REST/NRSF in neuroprotection and axonal regeneration within the adult central nervous system. Searches were performed in PubMed, Scopus, and Web of Science between June and August 2025.
The primary database search covered studies published between 2000 and 2025. This range was selected to prioritize studies using modern molecular, transcriptomic, epigenetic, and systems-level approaches. Because REST/NRSF was first identified in 1995, seminal earlier studies were not excluded outright. Foundational papers published between 1995 and 1999 were identified through backward citation screening of eligible articles and were retained when necessary to establish REST discovery, RE1 binding, and corepressor recruitment. The primary focus of this literature review is the therapeutic implications of REST mechanisms and their manipulation, rather than the initial establishment of the pathways they utilize, and so the search range was kept to 2000-2025, barring these exceptions.
Search terms combined REST-related terminology with terms related to CNS injury, neurodegeneration, and epigenetic regulation. The core Boolean search structure was as follows:
(“REST” OR “NRSF” OR “RE1-silencing transcription factor” OR “neuron-restrictive silencer factor”) AND (“spinal cord injury” OR “optic nerve injury” OR “CNS injury” OR “axon regeneration” OR “neurite outgrowth”) AND (“neuroprotection” OR “neurodegeneration” OR “aging” OR “ischemia” OR “stroke”) AND (“epigenetic regulation” OR “transcriptional repression” OR “chromatin remodeling” OR “regeneration-associated genes”)
Search syntax was adapted as needed for each database. Reference lists of eligible papers were also screened manually to identify additional relevant studies. Manual reference screening was conducted by reviewing the reference lists of eligible articles and relevant reviews to identify additional studies not captured through the database search.
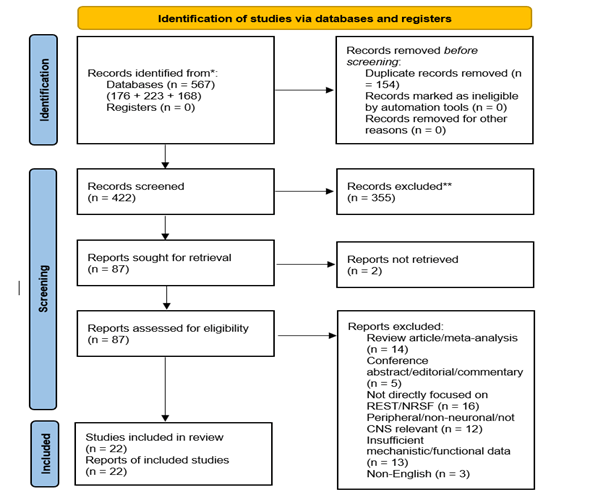
The database search identified 176 records from PubMed, 223 from Scopus, and 168 from Web of Science, with 9 additional records identified through manual reference screening, for a total of 576 records. After removal of 154 duplicates, 422 records remained for title and abstract screening. The full selection process, including reasons for exclusion, is summarized in the PRISMA flow diagram (Figure 1). In the final manuscript, 22 direct primary REST/NRSF studies met the criteria for inclusion in the systematicqualitative synthesis.
Eligibility Criteria
Studies were included if they were peer-reviewed primary research articles that directly examined REST/NRSF in central nervous system neurons or CNS-relevant neuronal models and investigated molecular mechanisms, transcriptional regulation, epigenetic control, neuroprotection, neurodegeneration, or axon regeneration using in vivo or in vitro systems relevant to spinal cord injury, optic nerve injury, aging, ischemia, or neurodegenerative disease.
Studies were also included if they were foundational primary mechanistic investigations necessary to establish REST/NRSF discovery, RE1 binding, corepressor recruitment, chromatin-based repression, or context-dependent transcriptional regulation relevant to neuronal gene control.This allowed inclusion of both contemporary disease-focused studies and foundational mechanistic studies required to interpret REST/NRSF function across contexts.Only studies published in English were considered.
Studies were excluded from PRISMA if they were review articles, meta-analyses, editorials, conference abstracts, or other non-primary literature; if they focused exclusively on the peripheral nervous system without CNS relevance; if they primarily examined non-neuronal cell types without relevance to REST/NRSF biology; or if they did not directly investigate REST/NRSF function or regulation.
To preserve conceptual continuity in the manuscript, a small number of review articles and broad background papers were retained in the reference list for framing only. In addition, mechanistically relevant primary studies on axon regeneration pathways that did not directly manipulate or measure REST/NRSF were retained for contextual interpretation of regeneration-associated transcription factor networks, but these studies were not counted in the systematic evidence set.
Study Selection and Duplicate Removal
Records were imported into Zotero, de-duplicated using its built-in duplicate detection tools, and cross-checked manually by comparing title, author list, year, journal, and DOI to ensure accuracy. Screening was conducted in two stages, consisting of title and abstract screening for topic relevance, followed by full-text eligibility assessment using prespecified inclusion and exclusion criteria.
Of the 422 records screened by title and abstract, 335 were excluded because they did not meet the predefined eligibility criteria. Specifically, studies were removed at this stage if they were clearly non-primary literature, focused on peripheral rather than central nervous system models, examined non-neuronal cell types without relevance to REST/NRSF biology, or did not directly investigate REST/NRSF function or regulation. In many cases, exclusion was based on insufficient topical alignment identified at the title and abstract level, including studies centered on unrelated transcription factors, non-neural tissues, or general epigenetic mechanisms without relevance to the topic.
87 full-text reports were sought for retrieval, of which 2 could not be retrieved, leaving 85 full-text articles assessed for eligibility. Of these, 63 were excluded due to being review articles or meta-analyses (n = 14), conference abstracts, editorials, or commentaries (n = 5), not directly focused on REST/NRSF (n = 16), peripheral-only or not CNS-relevant (n = 12), insufficient mechanistic or functional data (n = 13), or non-English publications (n = 3). A total of 22 primary studies were included in the final qualitative synthesis.
Because this was a single-author review, formal inter-reviewer agreement was not applicable. To improve consistency and reduce selection bias, all potentially eligible studies were re-evaluated in a second screening pass using the same prespecified inclusion and exclusion criteria. Study inclusion was based on direct REST/NRSF relevance, CNS neuronal context, and availability of extractable mechanistic or functional data.
Data Extraction
For each study included in the systematic synthesis, information was extracted on authors and year of publication, experimental model and species, CNS context, REST/NRSF measurement or manipulation strategy, molecular cofactors and interacting regulators, downstream genes and transcriptional pathways, transcriptomic and epigenetic findings, and functional outcomes including neuronal survival, axonal growth, and regenerative capacity. The primary outcomes of interest were REST/NRSF-associated effects on neuronal survival/neuroprotection and axonal growth or regenerative capacity. Secondary outcomes included downstream gene-expression changes, transcription factor network effects, chromatin or epigenetic remodeling, and other mechanistic findings relevant to REST/NRSF function. No common quantitative effect measure was prespecified because the review synthesized heterogeneous mechanistic and preclinical studies narratively rather than through pooled statistical comparison. Possible sources of heterogeneity were explored narratively by comparing findings across biological context, model organism, injury or disease paradigm, neuronal subtype, and REST/NRSF manipulation strategy. Because this was a single-author review, data extraction was performed by one reviewer. No study investigators were contacted for additional information or data clarification.
Data Synthesis and Analysis
A narrative qualitative synthesis was used because the included studies were heterogeneous in model system, injury or disease context, REST manipulation strategy, and outcome measures, making meta-analysis inappropriate. The 22 direct primary REST/NRSF studies were grouped into four predefined themes: molecular mechanisms of REST-mediated transcriptional repression, neuroprotective roles of REST in aging and neurodegeneration, inhibitory effects of REST on axon regeneration after CNS injury, and therapeutic implications of context-dependent REST modulation. This structure was chosen to reflect the biological argument of the manuscript and the dual role of REST as both a chromatin-associated transcriptional regulator and a context-dependent determinant of neuronal survival and regenerative capacity. Primary regeneration studies involving STAT3, SMAD1, SOX11, PTEN/SOCS3, Lin28a, and Elk-1 were cited narratively to interpret regeneration-associated transcription factor networks, but were not included in the core systematic evidence set.
Quality Assessment
Because the included evidence base was methodologically heterogeneous and included in vitro mechanistic studies, animal models, mixed-design studies, and human tissue analyses rather than a uniform set of randomized intervention studies, a single standardized risk-of-bias tool was not applied across all included studies. Although animal-study tools are available, they likewise would not capture the full range of study designs included in this review, particularly foundational in vitro studies, mixed human-animal designs, and genomic or mechanistic analyses. Instead, methodological limitations were assessed qualitatively for each included study using a structured set of review-specific criteria: clarity of study design, appropriateness of the model system to the research question, directness of REST/NRSF measurement or manipulation, adequacy of controls or comparators, alignment between mechanistic findings and functional outcomes, and transparency of outcome reporting. These judgments were used to inform narrative interpretation of the evidence rather than to exclude studies after eligibility assessment.
Reporting bias assessment
No formal assessment of reporting bias due to missing results was performed. Because this review synthesized a heterogeneous preclinical literature narratively rather than through meta-analysis, standard quantitative methods for assessing publication bias or selective outcome reporting were not applicable. Instead, reporting bias was noted as a general limitation of the evidence base, particularly where studies emphasized positive mechanistic findings, provided limited discussion of null or inconsistent results, or reported limited methodological detail for key outcomes.
Certainty Assessment
No formal certainty-of-evidence framework, such as GRADE (Grading of Recommendations Assessment, Development, and Evaluation), was applied. Given the mechanistic and heterogeneous nature of the included preclinical studies, certainty was considered narratively in light of consistency, biological plausibility, model relevance, and convergence of molecular and functional findings.
PRISMA Reporting and Evidence Set Composition
Study identification, screening, exclusion, and inclusion were documented in a PRISMA 2020 flow diagram, and a completed PRISMA 2020 checklist was prepared as supplementary material. 32 peer-reviewed primary research studies were cited overall, including 22 REST/NRSF studies in the systematic synthesis and 10 additional primary studies used for contextual interpretation. The remaining 6 sources were background or review articles, with 1 source being the PRISMA reporting guideline. This distinction is important because PRISMA 2020 emphasizes transparent reporting of study identification, selection, and synthesis; accordingly, the review distinguishes the systematic evidence set from contextual and background citations. A structured summary of all studies included in the systematic qualitative synthesis is provided in Table 1 to allow evaluation of study design, model systems, REST/NRSF manipulation methods, key findings, and brief methodological limitations relevant to narrative interpretation.
| Author(s) | Year | Study Type | Animal or Cell Model used | REST/NRSF manipulation or assessment | Key findings(s) | Key methodological limitations / bias concerns |
| Chong et al | 1995 | In vitro + developmental expression analysis | Non-neuronal and neuronal cell systems; developing mouse embryos | REST cDNA cloning; recombinant REST expression; dominant-negative REST; expression mapping | Identified REST as a DNA-binding silencer that represses neuronal gene expression outside neurons | Foundational in vitro and developmental study; limited direct relevance to adult CNS injury or disease outcomes |
| Schoenherr and Anderson et al | 1995 | In vitro + tissue expression analysis | Cell-based DNA-binding and reporter systems; embryonic/non-neuronal tissues | NRSF cDNA isolation; NRSE-binding and expression analyses | Defined NRSF as a coordinate repressor of multiple neuron-specific genes | Cell-based and tissue-expression study; limited direct functional relevance to adult CNS pathology or regeneration |
| Andrés et al | 1999 | In vitro | Human protein/cell systems expressing REST and CoREST | CoREST identification; interaction mapping; repression assays | Established CoREST as a functional REST corepressor required for neural gene repression | In vitro mechanistic design; limited in vivo validation and limited functional disease-context evidence |
| Grimes et al | 2000 | In vitro + developmental expression analysis | Mammalian cell systems; mouse embryo expression comparison | REST–mSin3A interaction mapping; repression interference assays | Showed mSin3A is a functional component of the REST-CoREST repressor complex | Mechanistic cell-system study; limited direct evidence for organism-level neuronal outcomes |
| Bruce et al | 2004 | Computational + in vitro genomic analysis | Human, mouse, and Fugu genomes; human U373 cells | Genome-wide RE1/NRSE mapping; REST occupancy assays | Showed that REST target-site occupancy is selective and depends on context and REST levels | Genomic and occupancy-focused study; limited direct functional validation in injury or disease models |
| Roopra et al | 2004 | In vitro | Non-neuronal cell lines | G9a recruitment/interference studies | Demonstrated that REST recruits G9a to generate localized H3K9 methylation and silence neuronal genes | In vitro non-neuronal cell-line study; limited translational relevance to CNS neurons in vivo |
| Ballas et al | 2005 | In vitro / developmental | Mouse pluripotent-to-neural stem/progenitor-to-neuron differentiation systems | REST/corepressor chromatin occupancy across differentiation | Showed REST repression is dynamic and developmentally regulated rather than permanently fixed | Developmental differentiation model; limited direct applicability to adult injury or neurodegenerative settings |
| Mulligan et al | 2008 | In vitro | Human cell culture systems, including immortalized primary human cells | CDYL identification; RNAi knockdown; complex mapping | Identified CDYL as a bridge between REST and histone methyltransferases, extending REST’s chromatin-repressive machinery | Cell-culture mechanistic study; limited in vivo confirmation and limited functional outcome data |
| Lu et al | 2014 | Mixed: human tissue + in vivo + in vitro | Aging human cortical/hippocampal neurons; conditional mouse brain REST deletion model; C. elegans SPR-4 model; primary human cortical neurons | Endogenous REST profiling; REST knockdown/overexpression; conditional deletion | Showed REST is induced in normal aging, lost in AD, and protective against oxidative and Aβ-related stress | Mixed-model design strengthens breadth but limits direct comparability across systems and endpoints |
| Perera et al | 2015 | In vitro / molecular mechanistic | Mouse retina and neuronal molecular systems | TET3–REST interaction assays; overexpression/co-expression studies | Showed REST can recruit TET3 and support context-specific hydroxymethylation and gene activation | Mechanistic neuronal-system study; limited direct functional injury or disease outcome assessment |
| Zuccato et al | 2003 | Mixed: cells + mouse + human tissue | Huntington’s disease cellular systems; mouse models; human HD brain | Comparison of wild-type vs mutant huntingtin effects on REST localization and target-gene repression | Showed wild-type huntingtin restrains REST/NRSF from inappropriate nuclear repression | Mixed Huntington’s disease models provide disease relevance, but broader generalizability beyond HD is limited |
| Zuccato et al | 2007 | Mixed: cells + in vivo+ human tissue | Hdh knock-in cells; R6/2 and knock-in mouse models; postmortem human HD cortex | REST occupancy mapping; attenuation of REST binding | Showed widespread abnormal REST occupancy in HD and restoration of BDNF with reduced REST binding | Mixed-model and postmortem design; causal interpretation in human tissue remains limited |
| Calderone et al | 2003 | In vivo + mechanistic | Adult rat global ischemia model; vulnerable hippocampal neurons | Endogenous REST induction after ischemia; promoter/gene-expression analyses | Showed global ischemia induces REST and represses GluR2 in neurons destined to die | Single acute ischemia model; limited generalizability beyond this injury context |
| Formisano et al | 2007 | In vivo + molecular | Post-ischemic hippocampal neurons | Endogenous ischemia-induced REST-associated epigenetic remodeling | Showed ischemic injury promotes epigenetic reprogramming of neuronal gene expression linked to REST activity | Molecular injury study with limited direct functional outcome assessment |
| Noh et al | 2012 | In vivo + mechanistic intervention | Clinically relevant global ischemia model; post-ischemic hippocampus | Lentiviral REST RNAi; dominant-negative REST; chromatin/remodeling assays | Demonstrated that REST-dependent epigenetic remodeling is causally linked to ischemia-induced neuronal death | Strong mechanistic intervention design, but findings are still limited to ischemia-specific preclinical models |
| Morris-Blanco et al | 2019 | In vivo | Adult rat focal cerebral ischemia model (MCAO) | REST knockdown with intracerebral siRNA | Showed that REST inhibition reduces cortical ischemic damage and improves outcome after stroke | Animal stroke model only; direct translational relevance to humans remains limited |
| Chen et al | 2017 | In vitro | Mouse striatal-derived STHdh Q111/Q111 Huntington’s disease cells | ASO-induced exon-3 skipping; siRNA-mediated REST reduction | Reduced nuclear REST and restored neuronal gene expression in an HD cell model | In vitro Huntington’s disease cell model; limited in vivo validation |
| Cheng et al | 2022 | Mixed: systems analysis + in vivo | Adult mouse corticospinal tract spinal cord injury model; adult mouse optic nerve crush model | REST network inference; conditional REST deletion; inhibitory manipulation in injured neurons | Identified REST as an intrinsic suppressor of CNS regeneration and showed enhanced regenerative outcomes after REST reduction | Mechanistically strong preclinical study, but conclusions rely on a limited set of adult mouse injury models |
| Aron et al | 2023 | Mixed: human tissue + in vivo | Cognitively intact aging human cortex; J20 and 3xTg Alzheimer’s disease mouse models | REST deletion and viral overexpression; early pathology profiling | Defined REST as a neurodegeneration checkpoint that limits early AD pathology | Mixed human and mouse AD-related systems; disease-stage and cross-model comparability may vary |
| Rocchi et al | 2021 | In vitro | Primary mouse neurons | REST deficiency in neurons; rescue of autophagic flux | Showed REST loss impairs autophagy, increases oxidative stress, and promotes neuronal senescence | Primary-neuron in vitro model; limited organism-level functional validation |
| Ryan et al | 2021 | Mixed: in vivo+ human cell model + in vitro | α-synuclein-overexpressing BAC-transgenic mice; human iPSC-derived dopaminergic neurons | Endogenous REST comparison; CRISPR REST knockout; REST overexpression | Showed REST protects dopaminergic neurons from mitochondrial and α-synuclein-related pathology | Mixed-model design improves triangulation, but cross-system heterogeneity limits direct comparability |
| Pajarillo et al | 2024 | In vivo | Dopaminergic REST conditional knockout mice with manganese exposure; AAV-based REST restoration in substantia nigra | REST-cKO; neuronal AAV REST rescue | Showed dopaminergic REST is protective against manganese-induced neurotoxicity | Context-specific neurotoxicity model; broader generalizability to other neurodegenerative contexts remains limited |
Several included studies used mixed designs combining human tissue, animal models, and cell systems. Accordingly, Table 1 lists all major systems used when necessary rather than assigning each study to a single model category.
Results
Molecular Mechanisms of REST/NRSF
REST/NRSF functions as a central regulator of transcriptional repression, but its activity is not uniformly repressive across all contexts. Instead, REST exhibits selective, context-dependent regulation of target genes, with differential binding and transcriptional effects depending on cell type, developmental stage, and physiological conditions15,16,28,29.
At the molecular level, early in vitro and cell-based studies established the foundation of this mechanism by identifying REST as a DNA-binding silencer that recognizes RE1/NRSE motifs near neuronal genes and restricts their expression outside the neuronal lineage15,16. This discovery established that REST acts through sequence-specific binding to RE1/NRSE motifs, rather than indiscriminate genome-wide repression, enabling selective regulation of neuronal gene expression15,16,29. Subsequent in vitro and mammalian cell system work clarified how REST enforces this repression at the chromatin level. Rather than acting independently, REST functions as a scaffold that recruits corepressor complexes, including CoREST and mSin3A, which connect REST to chromatin-modifying machinery17,18. This shifted the view of REST from a simple DNA-binding repressor to a coordinator of multi-protein complexes capable of altering chromatin accessibility and transcriptional activity17,18,29.
Further mechanistic details emerged from in vitro and molecular studies, which identified additional cofactors that reinforce REST-mediated repression. Recruitment of histone-modifying enzymes such as G9a and CDYL links REST to histone methylation, particularly the deposition of repressive marks such as H3K9 methylation, which is associated with reduced transcriptional accessibility at neuronal genes19,20. In addition, in pathological contexts such as ischemic injury, REST-associated complexes have also been shown to include HDAC1/2 and MeCP2 at target promoters, demonstrating that the composition of REST-mediated repression is not fixed but adapts to the cellular environment30. Together, these findings establish that REST regulates gene expression through coordinated chromatin remodeling rather than a single repressive mechanism.
Consistent with this, the developmental literature further shows that REST-mediated repression is dynamic. During neurogenesis, as examined in mouse pluripotent-to-neuronal stem/progenitor-to-neuron differentiation systems, REST and its corepressors maintain neuronal genes in a repressed but poised state in progenitor cells and then dissociate as differentiation proceeds, allowing neuronal gene expression to be activated at the appropriate stage31. This indicates that REST plays a role in regulating the timing of gene expression during development, rather than irreversibly silencing target genes.
At a broader scale, genome-wide analyses in human and mouse genomic systems provide additional insight into why REST can produce different biological outcomes. REST target-site occupancy varies with cellular REST levels, and because REST binds many loci across the genome, its effects extend beyond individual genes to broader transcriptional programs that shape neuronal identity and cellular state28,29. Importantly, REST is not limited to repression in all contexts. It has also been shown to recruit TET3 in neurons, enabling context-specific hydroxy methylation and downstream gene activation, further supporting its role as a flexible regulator of gene expression rather than a purely repressive factor32.
Taken together, these in vitro, genomic, and developmental model studies support a more precise view of REST as a context-dependent chromatin-associated regulator. Its function depends on the combination of cofactors present, the chromatin environment, and the physiological state of the cell. This flexibility is essential for interpreting its role in disease and injury because the same regulatory system that maintains neuronal identity or supports stress responses in one context can suppress regeneration-associated transcriptional programs in another29,32,21. This synthesis was informed primarily by in vitro, developmental, and genomic studies, which provided detailed mechanistic insight but were limited in direct functional relevance to adult CNS injury or disease outcomes.
REST in Neuroprotection
The same context dependence that allows REST to inhibit regeneration after injury also helps explain why it can be protective in other settings. In the aging brain, REST is a part of a broader stress-response program that supports neuronal stability over time. Nuclear REST levels increase during normal aging in human cortical and hippocampal neurons, and higher REST is associated with cognitive preservation and longevity22. This relationship is supported across multiple model systems: REST protects primary human cortical neurons, its conditional deletion in the mouse brain leads to age-related neurodegeneration, and a related REST-like protein in Caenorhabditis elegans promotes stress resistance22. Together, these findings suggest that REST contributes to neuronal resilience in a conserved and biologically meaningful way rather than acting as a passive transcriptional repressor22.
At a cellular level, loss of REST in primary mouse neurons during physiological aging impairs autophagy, increases oxidative stress, and promotes cellular senescence, indicating that REST supports core homeostatic processes that maintain neuronal function25. Consistent with this, broader analyses, primarily based on review-level synthesis of preclinical and human data, have framed REST as a stress-responsive regulator that can limit pathways associated with apoptosis, excitotoxicity, and oxidative damage, although these conclusions are drawn from review-level synthesis rather than single experimental systems23,33.
Related review studies drawing on animal models and human observational data in stress and depression contexts similarly describe REST as a factor that supports neuronal homeostasis under physiological stress conditions, reinforcing its role in maintaining cellular balance beyond injury-specific models34.
This protective function is especially well characterized in Alzheimer’s disease. REST has been described as a “neurodegeneration checkpoint,” meaning a protective response that is activated early in disease progression to limit downstream damage24. In both cognitively intact aging humans and Alzheimer’s disease mouse models, REST increases at the onset of amyloid-beta deposition and tau accumulation24. In this context, REST suppresses pathways that contribute to pathology, including gamma-secretase activity, tau kinase signaling, and pro-apoptotic gene expression, thereby reducing protein aggregation and neuronal death. When REST is removed, these protective effects are lost, leading to accelerated amyloid accumulation, tau pathology, neurodegeneration, and cognitive decline, whereas increasing REST expression suppresses disease progression24. Together with aging data, these findings support a model in which REST actively maintains neuronal integrity in neurodegenerative conditions rather than functioning as a nonspecific transcriptional repressor22,24.
A different pattern emerges in Huntington’s disease, where dysregulation of REST, rather than its simple presence or absence, contributes to disease pathology. Under normal conditions, wild-type huntingtin protein restricts REST from entering the nucleus. In Huntington’s disease, this control is lost, allowing REST to accumulate in the nucleus and repress neuronal genes inappropriately in mouse Huntington disease cells35. This repression affects genes critical for neuronal survival, including BDNF, a neurotrophic factor that supports neuronal maintenance and function. Genome-wide analyses of postmortem Huntington’s disease brain tissue show altered REST binding at target genes, and reducing REST occupancy restores BDNF expression36. Further cellular studies demonstrate that modifying REST regulation, such as through exon-3 skipping in mutant huntingtin models, can reduce nuclear REST levels and restore neuronal gene expression, suggesting a potential pathway for correcting disease-associated transcriptional changes37.
However, REST is not uniformly protective across all CNS insults, and the ischemia literature highlights this limitation. After global ischemia in adult rat models, REST is induced in vulnerable hippocampal neurons, where it suppresses GluR2 and contributes to transcriptional changes associated with neuronal death38. Subsequent in vivo rodent studies show that REST participates in epigenetic reprogramming of neuronal genes following ischemic injury, including changes in mu opioid receptor expression, indicating that REST-driven chromatin remodeling is part of the injury response39. In ischemic stroke models, REST assembles with chromatin-modifying complexes at target promoters, and reducing REST activity rescues neuronal survival, further demonstrating its role in injury-associated gene repression30. Similarly, in focal cerebral ischemia, REST induction is linked to cortical damage, and its inhibition reduces injury severity and improves functional recovery40. These findings show that REST can shift from a protective regulator in aging to a harmful driver of transcriptional repression in acute injury contexts.
A more protective pattern reappears in dopaminergic systems. Reduced REST levels are associated with mitochondrial dysfunction and alpha-synuclein pathology in both mouse models and patient-derived dopaminergic neurons, whereas increasing REST expression mitigates these effects through pathways linked to mitochondrial regulation26. In manganese-induced neurotoxicity, loss of REST worsens behavioral and cellular outcomes, while restoring REST expression reduces oxidative stress and neuronal damage27. These studies, across mouse models and human-derived neuronal systems, reinforce the idea that REST can support neuronal survival under certain stress conditions, particularly those involving mitochondrial dysfunction and protein aggregation.
Overall, neuroprotection literature supports a more nuanced conclusion. REST contributes to neuronal stability in aging, Alzheimer’s disease, and certain dopaminergic stress models by supporting processes such as autophagy, stress resistance, and mitochondrial function22,24,25,26,27. By contrast, in ischemic injury, REST contributes to harmful transcriptional reprogramming and neuronal loss30,38,39,40. This distinction underscores why REST must be understood as a context-dependent regulator rather than a uniformly neuroprotective factor and sets the stage for its opposing role in axon regeneration after CNS injury23,33. This synthesis draws on heterogeneous evidence spanning human tissue, mouse, rat, and cell-based systems, with interpretive caution warranted because methodological quality was variable and no standardized risk-of-bias tool was applied.
REST as a Suppressor of Axon Regeneration After Spinal Cord Injury
In clear contrast to its neuroprotective roles in aging and certain disease contexts, REST functions as an intrinsic barrier to axonal regeneration in the injured adult central nervous system21,33. Following spinal cord injury, REST is upregulated in mature CNS neurons, particularly within corticospinal pathways, and this increase is associated with suppression of gene programs required for axon regrowth21.
Systems-level analyses in mouse injury models have clarified how this repression occurs. REST has been identified as a transcriptional regulator of pro-regenerative gene networks in both complete spinal cord injury models affecting corticospinal neurons and optic nerve crush models affecting retinal ganglion cells21. Rather than broadly describing regeneration failure, these analyses defined a specific regeneration-associated transcription factor network that is suppressed in the injured CNS. This network includes Jun, STAT3, SOX11, SMAD1, and ATF3, which are known to promote axon growth in peripheral or developmentally permissive contexts21.
The suppression of this network is both temporally structured and mechanistically specific. In corticospinal neurons after spinal cord injury, REST deletion leads to increased expression of Jun and Atf3 by day 3, followed by increased SMAD1, SOX11, and Stat3 by day 7. This pattern indicates that REST acts early to prevent activation of key regenerative programs rather than simply maintaining a repressed state21. In addition, REST foot printing was detected near multiple RAG-TF loci, supporting at least partial direct transcriptional repression of these genes rather than a purely indirect regulatory effect21.
Functional experiments in adult mouse models further demonstrate that REST actively limits regeneration. Conditional deletion of REST in adult mouse corticospinal neurons reactivates pro-regenerative transcriptional programs and increases expression of genes involved in cytoskeletal organization, vesicle trafficking, and growth-associated signaling21. This pattern is consistent with restoration of a broader axon growth state rather than changes in a small number of isolated genes21. These molecular changes translate into measurable anatomical and behavioral outcomes. Injured corticospinal axons show increased regrowth across lesion sites, consistent with improved regenerative outcomes after injury21. Similar effects are observed in optic nerve injury models, where REST suppression in retinal ganglion cells promotes both neuronal survival and axonal regeneration21.
Network-level analyses in mouse corticospinal and optic nerve injury models reinforce this interpretation. Weighted gene co-expression network analysis identified REST-regulated gene modules linked to regeneration-associated pathways, including TGF-β, GPCR, MAPK, and integrin signaling. This finding indicates that REST represses coordinated gene networks rather than isolated downstream targets21. Together, these results support the conclusion that REST functions as an active transcriptional brake on regeneration-associated programs in the injured CNS. Within the systematic evidence set, the clearest regeneration-specific evidence comes from Cheng et al (2022), which identifies REST as an intrinsic suppressor of CNS regeneration in adult mouse corticospinal and optic nerve injury models21.
Beyond the included REST-specific studies, contextual regeneration literature further shows why REST-dependent effects must be interpreted in relation to other intrinsic growth regulators. Multiple transcription factors and signaling pathways, including STAT3, SMAD1, PTEN/SOCS3, Lin28a, SOX11, and Elk-1, influence regenerative capacity across different experimental systems6,7,8,9,10,11,12,13,14. These studies were cited for contextual interpretation of regeneration-associated transcription factor networks and were not part of the core systematic REST/NRSF evidence set.
For a contextual comparison, STAT3 activation is required for conditioning injury-induced regeneration in mouse dorsal root ganglion neurons, and increasing STAT3 activity further enhances CNS axon regeneration in vivo6,8. Similarly, SMAD1 signaling promotes axon growth in adult mouse sensory neurons, while deletion of PTEN and SOCS3 enhances regeneration in corticospinal and optic nerve systems7,9,10,11.
However, these effects are not uniform across all neuronal types. For example, SOX11 overexpression promotes regeneration in some mouse retinal ganglion cell subtypes but is toxic to others. Elk-1 has been identified as a downstream regulator of PTEN signaling, and is part of regeneration-associated transcriptional networks suppressed by REST13,14,21.
These findings across multiple animal models highlight an important limitation. Transcription factor-based interventions can produce different outcomes depending on neuronal subtype, developmental stage, and the specific endpoint being measured.
Taken together, REST should be understood as part of a broader network of intrinsic growth regulators. Its role as a transcriptional brake emerges specifically in the context of CNS injury, where it suppresses coordinated regeneration-associated gene programs21. However, the effects of REST modulation, as interpreted from the direct REST evidence together with the broader regeneration studies, depend on cellular context and must be interpreted alongside other signaling pathways that shape regenerative capacity13,14,21. This synthesis is driven primarily by a limited but mechanistically strong preclinical evidence base, especially adult mouse corticospinal and optic nerve injury models, though generalizability is constrained by model specificity.
Comparing Neuroprotection and Regeneration
When the neuroprotection and regeneration literature are considered together, REST is best understood as neither a uniformly beneficial nor a uniformly harmful factor, but as a context-dependent regulator (Table 2). Its effects depend on cell type, injury state, disease class, temporal stage, and the gene networks available for repression or activation21,22,23,24,33. In aging and selected neurodegenerative settings, REST supports neuronal stability by repressing stress-associated and pro-death pathways and by helping preserve homeostatic processes22,24,25,26,27.
However, post-injury, it suppresses regeneration-associated transcriptional factors, including Jun, STAT3, SOX11, SMAD1, and ATF3, as well as broader gene modules involved in cytoskeletal remodeling, vesicle trafficking, and growth-associated signaling21.
The basis of this divergence is not that REST uses entirely different molecular machinery in each setting. Rather, the available preclinical and human data suggest that REST deploys a similar chromatin-associated regulatory toolkit across contexts, while the biological outcome depends on which target genes are engaged and when repression occurs. In aging and Alzheimer’s disease, REST induction is associated with repression of pathways linked to oxidative stress, apoptosis, amyloid-related processing, and tau-associated toxicity, thereby favoring neuronal survival22,24. After CNS injury, however, REST induction suppresses the very transcriptional programs required for regenerative re-entry, shifting the balance away from structural repair (Table 2)21.
Importantly, the neuroprotective side of REST is not universal (Table 2). The ischemia literature shows that REST can become maladaptive in acute injury settings, where it contributes to harmful epigenetic reprogramming and neuronal loss28,30,39,40.
This exception strengthens rather than weakens the central argument of this review: REST should not be viewed as a simple on-or-off therapeutic target, because its function changes with injury type, neuronal subtype and disease stage23,33. Viewed together, these findings suggest that CNS neurons, as long-lived postmitotic cells, may prioritize transcriptional stability over structural plasticity, and REST may reflect this trade-off.
Table 2 synthesizes findings across heterogeneous contributing studies, including animal models, in vitro systems, and mixed human tissue designs; accordingly, the patterns shown should be interpreted in light of differences in model system, endpoint selection, and degree of functional validation.
| Physiological or pathological context | Specific model organism(s) or system(s) in which the role was demonstrated | REST activity state | Illustrative REST-linked targets or processes | Dominant biological effect | Representative source(s) |
| Aging brain | Aging human cortical and hippocampal neurons; conditional mouse brain REST deletion model; C. elegans SPR-4 ortholog model; primary mouse neurons | Increased nuclear REST in normal aging | Repression of oxidative stress- and apoptosis-associated pathways; support of stress resistance and homeostasis | Neuroprotection and preservation of neuronal integrity | Lu et al, 2014; Rocchi et al, 2021 |
| Early neurodegenerative stress, including Alzheimer’s disease | Cognitively intact aging human cortex; J20 and 3xTg Alzheimer’s disease mouse models | Adaptive induction at early pathology | Regulation of pathways linked to amyloid processing, tau-associated toxicity, and apoptosis | Delayed pathology and enhanced neuronal survival | Lu et al, 2014; Aron et al, 2023 |
| Huntington’s disease | Huntington’s disease cell models; Hdh knock-in and R6/2 mouse models; human postmortem HD brain | Pathological nuclear accumulation or dysregulation | Inappropriate repression of neuronal genes, including BDNF-related programs | Maladaptive transcriptional repression | Zuccato et al, 2003; Zuccato et al, 2007; Chen et al, 2017 |
| Acute ischemic injury | Adult rat global ischemia models; adult rat focal cerebral ischemia (MCAO); post-ischemic hippocampal neurons | Induced in vulnerable neurons | Repression of GluR2 and other plasticity-related genes; injury-linked chromatin remodeling | Enhanced neuronal injury and cell loss | Calderone et al, 2003; Formisano et al, 2007; Noh et al, 2012; Morris-Blanco et al, 2019 |
| Dopaminergic stress models | α-synuclein-overexpressing BAC-transgenic mice; human iPSC-derived dopaminergic neurons; dopaminergic REST-cKO mice exposed to manganese | Protective when maintained or restored | Support of mitochondrial function, oxidative stress control, mitophagy, and resistance to proteinopathy | Improved neuronal survival under stress | Ryan et al, 2021; Pajarillo et al, 2024 |
| Spinal cord injury and optic nerve injury | Adult mouse corticospinal tract injury model; adult mouse optic nerve crush model | Upregulated in injured CNS neurons | Suppression of Jun, STAT3, SOX11, SMAD1, ATF3, and broader regenerative gene modules | Inhibition of intrinsic axon regeneration programs | Cheng et al, 2022 |
| REST deletion or inhibition after CNS injury | Adult mouse corticospinal and optic nerve injury models | Functionally reduced in injured neurons | Reactivation of regeneration-associated transcription factor networks and growth-associated gene modules | Enhanced regenerative outcomes in preclinical models | Cheng et al, 2022 |
REST/NRSF exerts distinct effects depending on context, promoting neuroprotection in aging and neurodegenerative conditions while suppressing intrinsic axon regeneration after central nervous system injury.
Overall, confidence in the evidence is moderate for the conclusion that REST suppresses regeneration in preclinical CNS injury models, but lower for broader translational conclusions because the evidence base is heterogeneous, predominantly preclinical, and uneven across biological contexts.
Discussion
When the full literature is considered, REST should not be framed as a straightforward therapeutic target in the clinical sense, but as a preclinical, context-dependent regulatory node whose manipulation may be beneficial only under specific biological conditions23,33.
Accordingly, the available evidence supports a cautious therapeutic rationale. In mouse injury models, REST suppresses CNS regenerative transcription factor networks and limits axon growth, which makes REST modulation an attractive hypothesis-generating strategy for CNS repair21. At the same time, REST can preserve neuronal survival, while limiting misfolded protein pathology, autophagy, mitochondrial function, and stress resistance in aging and selected neurodegenerative contexts22,24,25,26,27. This duality means that the value of targeting REST depends not only on whether axon regeneration increases, but also on whether neuronal viability is maintained in the same tissue and time window23,33.
For that reason, claims about AAV-mediated gene therapy or CRISPR interference should be described as experimental strategies supported by animal and cell studies rather than clinical approaches. Any translational discussion should therefore remain provisional and explicitly address model dependence, off-target risk, delivery challenges, and the possibility that REST inhibition may worsen neurodegeneration or stress vulnerability in settings where REST is serving in a protective role. This caution is especially important because REST is beneficial in some aging and neurodegeneration models, harmful in ischemia, and inhibitory for axon regeneration after injury, which means that timing, tissue targeting, and disease contexts are likely to determine whether REST-directed interventions are restorative or detrimental21,22,24,30,38,40.
Limitations
This review has several limitations. First, the evidence base is predominantly preclinical and includes heterogeneous in vivo and in vitro models, limiting direct comparison across studies. As a result, the literature does not yet provide a sufficiently consistent evidence base to support broad translational conclusions, and findings should instead be interpreted primarily as mechanistic and hypothesis-generating rather than directly predictive of human therapeutic outcomes. Second, the literature specifically addressing REST in spinal cord injury remains smaller than the literature on REST in aging, ischemia, and neurodegeneration. This relative imbalance means that conclusions about REST as a regulator of axon regeneration after spinal cord injury, although supported by important preclinical studies, currently rest on a narrower evidence base than conclusions drawn in other biological contexts. Third, limitations of the review process itself should be acknowledged: screening, study selection, data extraction, and qualitative methodological assessment were conducted by a single author, although a second-pass audit using prespecified criteria was used to improve consistency. No prospectively registered protocol or formal inter-reviewer procedures were used, and single-reviewer decisions may therefore have increased the risk of selection or interpretation bias. Finally, some mechanistic interpretation of regeneration-associated transcription factor networks relied on contextual non-REST primary studies that were not part of the systematic evidence set6,7,8,9,10,11,12,13,14. This means that some broader network-level interpretations should be understood as contextual extensions of the direct REST literature rather than conclusions derived solely from the formally included studies.
More specifically, this evidence base relies heavily on preclinical rodent and cell-based systems, including mouse optic nerve crush, complete spinal cord injury, global and focal ischemia, BAC-transgenic alpha-synuclein models, Alzheimer’s disease mouse models, and mutant huntingtin cell models; variability in cell type, injury paradigm, and REST-manipulation strategy across these systems further limits direct generalization to humans21,24,25,26,27,30,37,40. These limitations reflect the current state of the field, in which mechanistic interrogation of REST has depended largely on manipulable preclinical systems, while direct human experimental studies are constrained by tissue access, disease heterogeneity, and the context-dependent dual role of REST across regeneration, ischemia, aging, and neurodegeneration1,23,33. Model variability also likely contributes to inconsistent outcomes because transcription factor interventions can produce different effects across neuronal subtypes and endpoints13,14,21.
Overall, the current evidence suggests that REST should presently be understood as a promising context-dependent preclinical target, and it underscores the need for more standardized study designs, broader replication across injury models, and carefully controlled human-relevant systems in future work.
Conclusion
Overall, the literature supports a more precise understanding of REST: it is neither a transcriptional silencer that should simply be removed nor a universally protective factor that should be preserved in every setting. Instead, REST is a context-dependent transcriptional regulator whose effects diverge across multiple contexts23,29,33. In CNS injury models, REST represses a defined pro-regenerative transcription factor network and limits axon growth, whereas in aging and selected neurodegenerative settings, it supports neuronal stress resistance and homeostasis21,22,24,25,26,27. Accordingly, the significance of REST modulation depends on when, where, and in which neural context it is applied. Future work should therefore focus on temporally restricted and spatially targeted preclinical strategies to determine whether REST can be modulated to improve regeneration without compromising the protective functions it serves in other neural contexts21,23,33.
Registration and Protocol
This systematic review was not prospectively registered, and no formal review protocol was prepared. Accordingly, no protocol amendments apply.
Support
This review received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Competing Interests
The author declares no competing interests.
Availability of Data, Code, and Other Materials
No analytic code was generated for this review. Full search strategies are provided in Supplementary Table S1.
Supplementary information
References
- Y. Fu, X. Rui, S. Zhu, C. Guo, H. Li, Z. Pan, X. Wu, W. He. Research status of regenerative difficulties after central nervous system injury. Regenerative Therapy. Vol. 29, pg. 493–498, 2025, https://doi.org/10.1016/j.reth.2025.04.011. [↩] [↩] [↩] [↩] [↩]
- P. Li, Z.-Q. Teng, C.-M. Liu. Extrinsic and intrinsic regulation of axon regeneration by microRNAs after spinal cord injury. Neural Plasticity. Vol. 2016, pg. 1279051, 2016, https://doi.org/10.1155/2016/1279051. [↩] [↩] [↩]
- P. Li, Z.-Q. Teng, C.-M. Liu. Extrinsic and intrinsic regulation of axon regeneration by microRNAs after spinal cord injury. Neural Plasticity. Vol. 2016, pg. 1279051, 2016, https://doi.org/10.1155/2016/1279051. [↩]
- S. Neumann, C. J. Woolf. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. Vol. 23, pg. 83–91, 1999, https://doi.org/10.1016/S0896-6273(00)80755-2. [↩]
- S. Neumann, C. J. Woolf. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. Vol. 23, pg. 83–91, 1999, https://doi.org/10.1016/S0896-6273(00)80755-2. [↩]
- J. Qiu, W. B. J. Cafferty, S. B. McMahon, S. W. N. Thompson. Conditioning injury-induced spinal axon regeneration requires STAT3 activation. Journal of Neuroscience. Vol. 25, pg. 1645–1653, 2005, https://doi.org/10.1523/JNEUROSCI.3269-04.2005. [↩] [↩] [↩] [↩] [↩] [↩]
- P. Parikh, Y. Hao, M. Hosseinkhani, S. B. Patil, G. W. Huntley, M. Tessier-Lavigne, H. Zou. Regeneration of axons in injured spinal cord by activation of SMAD1 signaling. Proceedings of the National Academy of Sciences of the United States of America. Vol. 108, pg. E99–E107, 2011, https://doi.org/10.1073/pnas.1100426108. [↩] [↩] [↩] [↩] [↩] [↩]
- S. T. Mehta, X. Luo, K. K. Park, J. L. Bixby, V. P. Lemmon. Hyperactivated STAT3 boosts axon regeneration in the CNS. Experimental Neurology. Vol. 280, pg. 115–120, 2016, https://doi.org/10.1016/j.expneurol.2016.03.004. [↩] [↩] [↩] [↩] [↩] [↩]
- P. D. Smith, F. Sun, K. K. Park, B. Cai, C. Wang, K. Kuwako, I. Martinez-Carrasco, L. Connolly, Z. He. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. Vol. 64, pg. 617–623, 2009, https://doi.org/10.1016/j.neuron.2009.11.021. [↩] [↩] [↩] [↩] [↩]
- K. Liu, Y. Lu, J. K. Lee, R. Samara, R. Willenberg, I. Sears-Kraxberger, A. Tedeschi, K. K. Park, D. Jin, B. Cai, B. Xu, L. Connolly, O. Steward, B. Zheng, Z. He. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nature Neuroscience. Vol. 13, pg. 1075–1081, 2010, https://doi.org/10.1038/nn.2603. [↩] [↩] [↩] [↩] [↩]
- F. Sun, K. K. Park, S. Belin, D. Wang, T. Lu, G. Chen, K. Zhang, C. Yeung, G. Feng, B. A. Yankner, Z. He. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. Vol. 480, pg. 372–375, 2011, https://doi.org/10.1038/nature10594. [↩] [↩] [↩] [↩] [↩]
- F. M. Nathan, Y. Ohtake, S. Wang, X. Jiang, A. Sami, H. Guo, F.-Q. Zhou, S. Li. Upregulating Lin28a promotes axon regeneration in adult mice with optic nerve and spinal cord injury. Molecular Therapy. Vol. 28, pg. 1902–1917, 2020, https://doi.org/10.1016/j.ymthe.2020.04.010. [↩] [↩] [↩]
- M. W. Norsworthy, F. Bei, R. Kawaguchi, Q. Wang, N. M. Tran, Y. Li, B. Brommer, Y. Zhang, C. Wang, J. R. Sanes, G. Coppola, Z. He. SOX11 expression promotes regeneration of some retinal ganglion cell types but kills others. Neuron. Vol. 94, pg. 1112–1120, 2017, https://doi.org/10.1016/j.neuron.2017.05.035. [↩] [↩] [↩] [↩] [↩] [↩]
- T. Noro, S. H. Shah, Y. Yin, R. Kawaguchi, S. Yokota, K.-C. Chang, A. Madaan, C. Sun, G. Coppola, D. Geschwind, L. I. Benowitz, J. L. Goldberg. Elk-1 regulates retinal ganglion cell axon regeneration after injury. Scientific Reports. Vol. 12, pg. 17446, 2022, https://doi.org/10.1038/s41598-022-21767-3. [↩] [↩] [↩] [↩] [↩] [↩]
- J. A. Chong, J. Tapia-Ramírez, S. Kim, J. J. Toledo-Aral, Y. Zheng, M. C. Boutros, Y. M. Altshuller, M. A. Frohman, S. D. Kraner, G. Mandel. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. Vol. 80, pg. 949–957, 1995, https://doi.org/10.1016/0092-8674(95)90298-8. [↩] [↩] [↩] [↩] [↩] [↩]
- C. J. Schoenherr, D. J. Anderson. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. Vol. 267, pg. 1360–1363, 1995, https://doi.org/10.1126/science.7871435. [↩] [↩] [↩] [↩] [↩] [↩]
- M. E. Andrés, C. Burger, M. J. Peral-Rubio, E. Battaglioli, M. E. Anderson, J. Grimes, J. Dallman, N. Ballas, G. Mandel. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proceedings of the National Academy of Sciences of the United States of America. Vol. 96, pg. 9873–9878, 1999, https://doi.org/10.1073/pnas.96.17.9873. [↩] [↩] [↩]
- J. A. Grimes, S. J. Nielsen, E. Battaglioli, E. A. Miska, J. C. Speh, D. L. Berry, F. Atouf, B. C. Holdener, G. Mandel, T. Kouzarides. The co-repressor mSin3A is a functional component of the REST-CoREST repressor complex. Journal of Biological Chemistry. Vol. 275, pg. 9461–9467, 2000, https://doi.org/10.1074/jbc.275.13.9461. [↩] [↩] [↩]
- A. Roopra, R. Qazi, B. Schoenike, T. J. Daley, J. F. Morrison. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Molecular Cell. Vol. 14, pg. 727–738, 2004, https://doi.org/10.1016/j.molcel.2004.05.026. [↩] [↩]
- P. Mulligan, T. F. Westbrook, M. Ottinger, N. Pavlova, B. Chang, E. Macia, Y.-J. Shi, J. Barretina, J. Liu, P. M. Howley, S. J. Elledge, Y. Shi. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Molecular Cell. Vol. 32, pg. 718–726, 2008, https://doi.org/10.1016/j.molcel.2008.10.025. [↩] [↩]
- Y. Cheng, Y. Yin, A. Zhang, A. M. Bernstein, R. Kawaguchi, K. Gao, K. Potter, H.-Y. Gilbert, Y. Ao, J. Ou, C. J. Fricano-Kugler, J. L. Goldberg, Z. He, C. J. Woolf, M. V. Sofroniew, L. I. Benowitz, D. H. Geschwind. Transcription factor network analysis identifies REST/NRSF as an intrinsic regulator of CNS regeneration in mice. Nature Communications. Vol. 13, pg. 4418, 2022, https://doi.org/10.1038/s41467-022-31960-7. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- T. Lu, L. Aron, J. Zullo, Y. Pan, H. Kim, Y. Chen, T.-H. Yang, H.-M. Kim, D. Drake, X. S. Liu, D. A. Bennett, M. P. Colaiácovo, B. A. Yankner. REST and stress resistance in ageing and Alzheimer’s disease. Nature. Vol. 507, pg. 448–454, 2014, https://doi.org/10.1038/nature13163. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- J.-Y. Hwang, R. S. Zukin. REST, a master transcriptional regulator in neurodegenerative disease. Current Opinion in Neurobiology. Vol. 48, pg. 193–200, 2018, https://doi.org/10.1016/j.conb.2017.12.008. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- L. Aron, C. Qiu, Z. K. Ngian, M. Liang, D. Drake, J. Choi, M. A. Fernandez, P. Roche, E. L. Bunting, E. K. Lacey, S. E. Hamplova, M. Yuan, M. S. Wolfe, D. A. Bennett, E. A. Lee, B. A. Yankner. A neurodegeneration checkpoint mediated by REST protects against the onset of Alzheimer’s disease. Nature Communications. Vol. 14, pg. 7030, 2023, https://doi.org/10.1038/s41467-023-42704-6. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- A. Rocchi, E. Carminati, A. De Fusco, J. A. Kowalska, T. Floss, F. Benfenati. REST/NRSF deficiency impairs autophagy and leads to cellular senescence in neurons. Aging Cell. Vol. 20, pg. e13471, 2021, https://doi.org/10.1111/acel.13471. [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- B. J. Ryan, N. Bengoa-Vergniory, M. Williamson, E. Kirkiz, R. Roberts, G. Corda, M. Sloan, S. Saqlain, M. Cherubini, J. Poppinga, H. Bogetofte, M. Cioroch, S. Hester, R. Wade-Martins. REST protects dopaminergic neurons from mitochondrial and α-synuclein oligomer pathology in an α-synuclein-overexpressing BAC-transgenic mouse model. Journal of Neuroscience. Vol. 41, pg. 3731–3746, 2021, https://doi.org/10.1523/JNEUROSCI.1478-20.2021. [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- E. Pajarillo, S. Kim, A. Digman, I. Ajayi, I. Nyarko-Danquah, D.-S. Son, M. Aschner, E. Lee. Dopaminergic REST/NRSF is protective against manganese-induced neurotoxicity in mice. Journal of Biological Chemistry. Vol. 300, pg. 107707, 2024, https://doi.org/10.1016/j.jbc.2024.107707. [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- A. W. Bruce, I. J. Donaldson, I. C. Wood, S. A. Yerbury, M. I. Sadowski, M. Chapman, B. Göttgens, N. J. Buckley. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proceedings of the National Academy of Sciences of the United States of America. Vol. 101, pg. 10458–10463, 2004, https://doi.org/10.1073/pnas.0401827101. [↩] [↩] [↩]
- G. Thiel, M. Ekici, O. G. Rössler. RE-1 silencing transcription factor (REST): a regulator of neuronal development and neuronal/endocrine function. Cell and Tissue Research. Vol. 359, pg. 99–109, 2015, https://doi.org/10.1007/s00441-014-1963-0. [↩] [↩] [↩] [↩] [↩] [↩]
- K.-M. Noh, J.-Y. Hwang, A. Follenzi, R. Athanasiadou, T. Miyawaki, J. M. Greally, M. V. L. Bennett, R. S. Zukin. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proceedings of the National Academy of Sciences of the United States of America. Vol. 109, pg. E962–E971, 2012, https://doi.org/10.1073/pnas.1121568109. [↩] [↩] [↩] [↩] [↩] [↩]
- N. Ballas, C. Grunseich, D. D. Lu, J. C. Speh, G. Mandel. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. Vol. 121, pg. 645–657, 2005, https://doi.org/10.1016/j.cell.2005.03.013. [↩]
- A. Perera, D. Eisen, M. Wagner, S. K. Laube, A. F. Künzel, S. Koch, J. Steinbacher, E. Schulze, V. Splith, N. Mittermeier, M. Müller, M. Biel, T. Carell, S. Michalakis. TET3 is recruited by REST for context-specific hydroxymethylation and induction of gene expression. Cell Reports. Vol. 11, pg. 283–294, 2015, https://doi.org/10.1016/j.celrep.2015.03.020. [↩] [↩]
- L. Jin, Y. Liu, Y. Wu, Y. Huang, D. Zhang. REST is not resting: REST/NRSF in health and disease. Biomolecules. Vol. 13, pg. 1477, 2023, https://doi.org/10.3390/biom13101477. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- T. Soga, S. Nakajima, M. Kawaguchi, I. S. Parhar. Repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) in social stress and depression. Progress in Neuro-Psychopharmacology and Biological Psychiatry. Vol. 104, pg. 110053, 2021, https://doi.org/10.1016/j.pnpbp.2020.110053. [↩]
- C. Zuccato, M. Tartari, A. Crotti, D. Goffredo, M. Valenza, L. Conti, T. Cataudella, B. R. Leavitt, M. R. Hayden, T. Timmusk, D. Rigamonti, E. Cattaneo. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nature Genetics. Vol. 35, pg. 76–83, 2003, https://doi.org/10.1038/ng1219. [↩]
- C. Zuccato, N. Belyaev, P. Conforti, L. Ooi, M. Tartari, E. Papadimou, M. MacDonald, E. Fossale, S. Zeitlin, N. Buckley, E. Cattaneo. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. Journal of Neuroscience. Vol. 27, pg. 6972–6983, 2007, https://doi.org/10.1523/JNEUROSCI.4278-06.2007. [↩]
- G. L. Chen, Q. Ma, D. Goswami, J. Shang, G. M. Miller. Modulation of nuclear REST by alternative splicing: a potential therapeutic target for Huntington’s disease. Journal of Cellular and Molecular Medicine. Vol. 21, pg. 2974–2984, 2017, https://doi.org/10.1111/jcmm.13209. [↩] [↩]
- A. Calderone, T. Jover, K. M. Noh, H. Tanaka, H. Yokota, Y. Lin, S. Y. Grooms, R. Regis, M. V. L. Bennett, R. S. Zukin. Ischemic insults derepress the gene silencer REST in neurons destined to die. Journal of Neuroscience. Vol. 23, pg. 2112–2121, 2003, https://doi.org/10.1523/JNEUROSCI.23-06-02112.2003. [↩] [↩] [↩]
- L. Formisano, K. M. Noh, T. Miyawaki, T. Mashiko, M. V. L. Bennett, R. S. Zukin. Ischemic insults promote epigenetic reprogramming of μ opioid receptor expression in hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America. Vol. 104, pg. 4170–4175, 2007, https://doi.org/10.1073/pnas.0611704104. [↩] [↩] [↩]
- K. C. Morris-Blanco, T. Kim, M. J. Bertogliat, S. L. Mehta, A. K. Chokkalla, R. Vemuganti. Inhibition of the epigenetic regulator REST ameliorates ischemic brain injury. Molecular Neurobiology. Vol. 56, pg. 2542–2550, 2019, https://doi.org/10.1007/s12035-018-1254-y. [↩] [↩] [↩] [↩] [↩]



{kind=link}