
Abstract
Gefitinib resistance in EGFR-mutant non-small cell lung cancer (NSCLC) poses a critical challenge to effective treatment. We conducted transcriptomic analysis of a gefitinib-resistant HCC827R cell line to identify key molecular drivers of resistance and performed CRISPR-mediated gene editing and overexpression experiments to validate candidate genes. TRIM28 was significantly upregulated in resistant cells, promoting survival and proliferation via the PI3K/AKT/mTOR pathway. Functional studies confirmed that CRISPR-mediated TRIM28 knockout restored drug sensitivity, while overexpression conferred resistance. Rapamycin, an mTOR inhibitor, effectively reduced cell viability, impaired colony formation, and suppressed TRIM28 expression and pathway activation, demonstrating its potential to overcome resistance. Clinical data showed TRIM28 upregulation in lung adenocarcinoma (LUAD), correlating with advanced disease stages and poor prognosis. These findings establish TRIM28 as a critical mediator of gefitinib resistance and highlight targeting TRIM28 and the PI3K/AKT/mTOR pathway as a promising therapeutic strategy for improving outcomes in EGFR-mutant NSCLC.
Keywords: NSCLC, gefitinib resistance, TRIM28, PI3K/AKT/mTOR pathway, rapamycin.
Introduction
Non-small cell lung cancer (NSCLC) remains a leading cause of cancer-related mortality worldwide, with epidermal growth factor receptor (EGFR) mutations frequently identified as critical drivers of tumorigenesis1. EGFR-tyrosine kinase inhibitors (TKIs), such as gefitinib, have demonstrated significant clinical efficacy in patients harboring EGFR-activating mutations, including exon 19 deletions. However, acquired resistance to TKIs inevitably arises, limiting the long-term benefits of these targeted therapies2. Understanding the molecular mechanisms underlying resistance is essential to developing novel therapeutic strategies.
To investigate resistance mechanisms, we generated a gefitinib-resistant HCC827 cell line (HCC827R) by exposing parental HCC827 cells to escalating concentrations of gefitinib. RNA sequencing revealed transcriptomic reprogramming3 in HCC827R cells, compared to parental HCC827 cells, with significant upregulation of genes associated with survival and stress responses.
Among the differentially expressed genes, TRIM28 emerged as a particularly compelling candidate due to its marked upregulation and known roles in cellular stress adaptation, proliferation, and apoptosis4. While previous studies have explored various mechanisms of TKI resistance, the role of TRIM28 in this context has not been well characterized. This study addresses this gap by identifying and validating TRIM28 as a novel contributor to gefitinib resistance in EGFR-mutant NSCLC.
We further investigated the causal role of TRIM28 using CRISPR-Cas9-mediated gene editing and overexpression experiments in both resistant and sensitive cell lines. We hypothesized that TRIM28 promotes gefitinib resistance in EGFR-mutant NSCLC through activation of the PI3K/AKT/mTOR signaling pathway, and that targeting TRIM28 or its downstream effectors could restore drug sensitivity. To test this, we evaluated the effect of rapamycin, an mTOR inhibitor, as a potential therapeutic intervention5. Rapamycin treatment effectively suppressed cell viability and colony formation in HCC827R cells, accompanied by downregulation of TRIM28 and key components of the mTOR pathway.
These findings suggest that targeting TRIM28 and the PI3K/AKT/mTOR signaling axis may offer a promising strategy to overcome gefitinib resistance in EGFR-mutant NSCLC6.
This study aims to elucidate the molecular mechanisms underlying gefitinib resistance, clarify the role of TRIM28 in this process, and explore therapeutic approaches to improve outcomes in patients with EGFR-mutant NSCLC.
Results
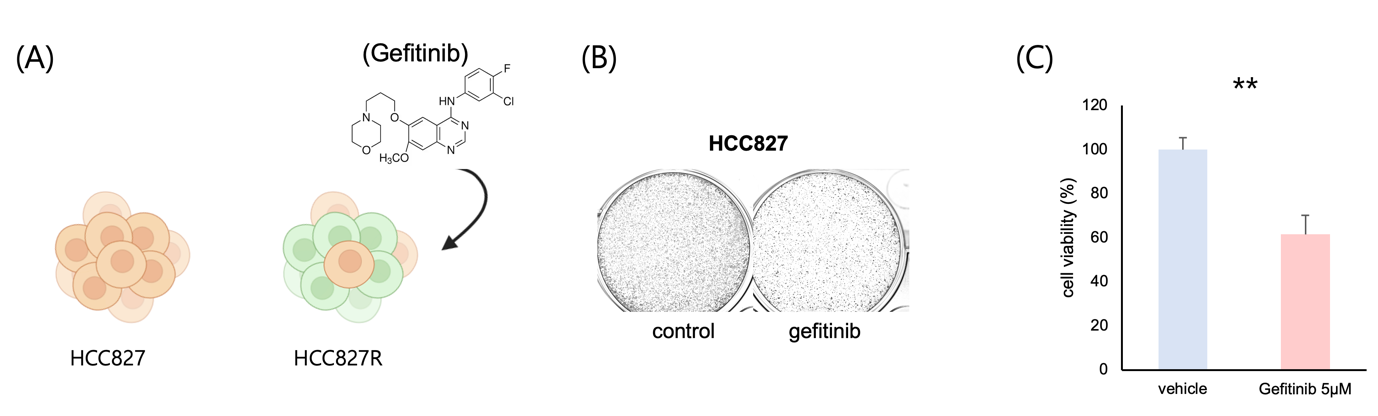
(A) A schematic diagram illustrates the development of gefitinib-resistant HCC827 cells. HCC827 parental cells were cultured and treated with escalating concentrations of gefitinib over eight weeks to generate a resistant cell line (HCC827R).
(B) Representative images of colony formation in HCC827 cells with and without gefitinib treatment. Untreated control cells (left) formed a significantly higher number of colonies, while gefitinib-treated cells (right) exhibited markedly reduced colony numbers and sizes, indicating suppressed proliferation.
(C) Quantitative analysis of colony numbers. Bar graphs represent the mean colony counts from three independent experiments, showing a significant reduction in colony formation in gefitinib-treated HCC827 cells compared to untreated controls. Error bars indicate the standard error of the mean (SEM). Statistical significance was determined using a two-tailed t-test (**p < 0.01).
To investigate mechanisms of acquired resistance to EGFR tyrosine kinase inhibitors (TKIs), we established a gefitinib-resistant HCC827 cell line (HCC827R) by chronically treating parental HCC827 cells with escalating concentrations of gefitinib over an eight-week period. Parental HCC827 cells, which are EGFR mutation-positive and highly sensitive to gefitinib, gradually acquired resistance under prolonged exposure to the drug, as illustrated schematically (Fig. 1A).
First, to evaluate the effect of gefitinib, an EGFR tyrosine kinase inhibitor (TKI)7, on the proliferation of HCC827 cells, we treated the cells with gefitinib and assessed their growth using a colony formation assay. HCC827 cells, which harbor activating EGFR mutations (exon 19 deletion), are highly sensitive to gefitinib, making them an ideal model to study TKI efficacy.
Colony formation assays revealed a significant suppression of cell growth upon gefitinib treatment. As shown in the representative images (Fig. 1B), the number and size of colonies formed by HCC827 cells were substantially reduced when gefitinib was present, compared to untreated control cells. This indicates that gefitinib effectively inhibits the proliferative capacity of HCC827 cells under these experimental conditions.
Quantitative analysis of colony numbers further confirmed this observation (Fig. 1C). Gefitinib-treated HCC827 cells exhibited a significant decrease in colony formation compared to untreated controls, with a reduction of approximately 50% (p < 0.01). These data highlight the potent anti-proliferative effect of gefitinib in this EGFR-mutant NSCLC cell line, aligning with its clinical role as a targeted therapy for tumors driven by EGFR signaling.
Overall, these findings demonstrate that gefitinib effectively suppresses the growth of EGFR-mutant HCC827 cells, validating its use in targeted therapy for non-small cell lung cancer (NSCLC). This model serves as a robust platform for further investigating resistance mechanisms and optimizing treatment strategies for EGFR-TKI-sensitive cancers.
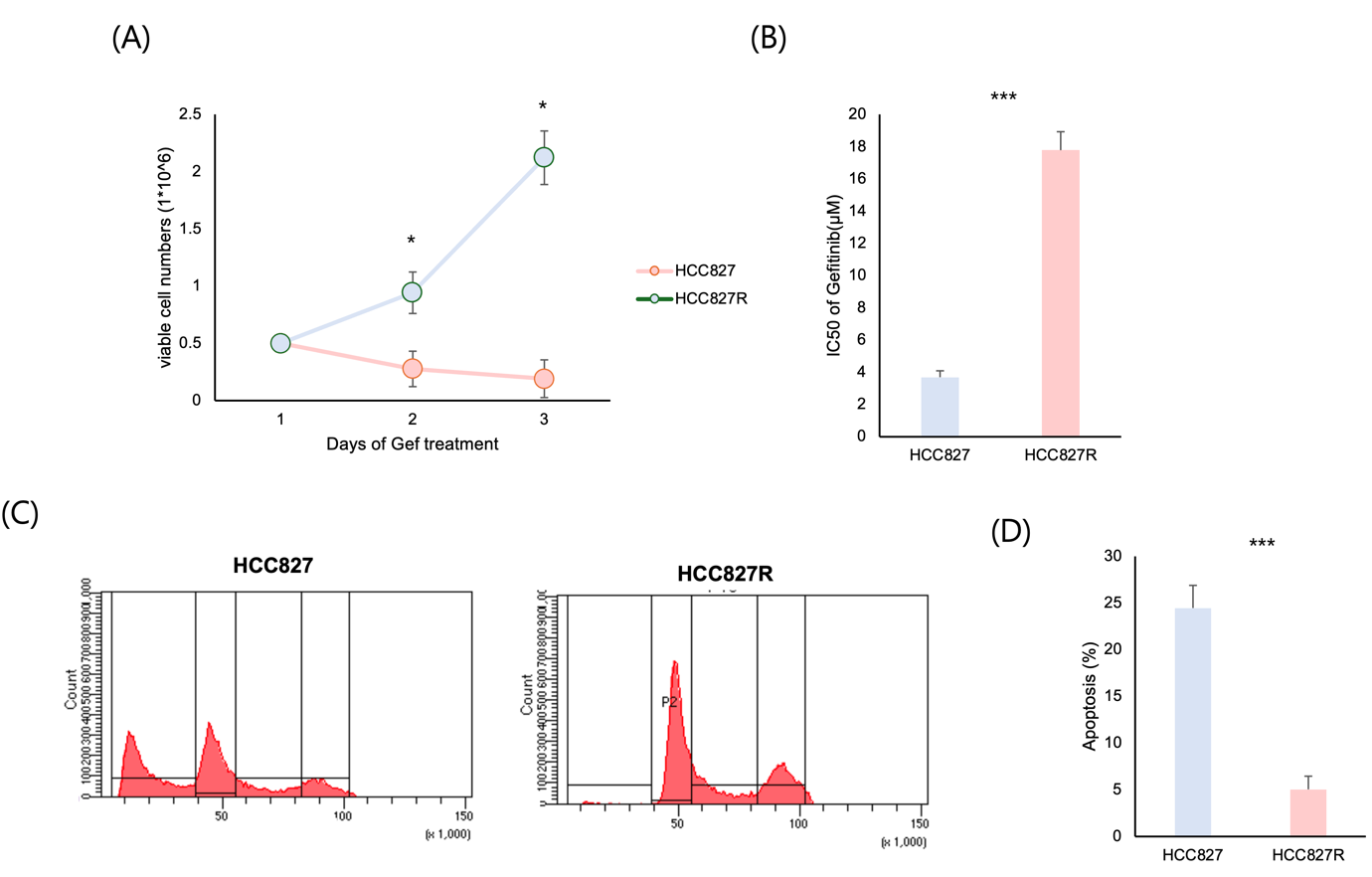
(A) HCC827 parental cells and gefitinib-resistant HCC827R cells were treated with gefitinib for 1–3 days. Viable cell numbers were quantified using a cell viability assay. HCC827R cells exhibited 10-fold higher viability compared to HCC827 cells under gefitinib treatment (*p < 0.05).
(B) The half-maximal inhibitory concentration (IC50) of gefitinib was 5-fold higher in HCC827R cells compared to HCC827 cells, indicating acquired resistance to the drug. Data represent mean ± SEM from three independent experiments (***p < 0.001).
(C) Flow cytometry analysis revealed cell cycle profiles of HCC827 and HCC827R cells after gefitinib treatment. HCC827 cells displayed a marked G1 phase arrest, while HCC827R cells bypassed the G1 checkpoint with increased S and G2/M phase populations.
(D) The percentage of apoptotic cells (Annexin V-positive) was reduced by ¼ in HCC827R cells compared to HCC827 cells following gefitinib treatment, highlighting the reduced apoptotic response in resistant cells (***p < 0.001).
To investigate the mechanisms of resistance to EGFR tyrosine kinase inhibitors (TKIs), gefitinib-resistant HCC827R cells were developed by continuously exposing HCC827 parental cells to increasing concentrations of gefitinib. These resistant cells exhibited distinct growth characteristics, survival, and cell cycle profiles compared to parental cells, highlighting their resistance to gefitinib.
HCC827R cells maintained robust proliferation under high gefitinib concentrations, unlike parental cells, which showed significant growth inhibition (Fig. 2A). MTT assays revealed that resistant cells formed more colonies under gefitinib treatment than parental cells (Fig. 2B). Flow cytometry showed that gefitinib induced G1 phase arrest in parental cells, while HCC827R cells bypassed the G1 checkpoint, with increased S and G2/M phase populations (Fig. 2C). Apoptotic response analysis confirmed reduced annexin V-positive cells in HCC827R compared to parental cells, indicating attenuated cell death under gefitinib treatment (Fig. 2D).
These findings demonstrate that chronic gefitinib exposure induces a resistant subpopulation in HCC827 cells, characterized by sustained proliferation, altered cell cycle dynamics, and diminished apoptosis. The HCC827R model is a valuable tool for understanding EGFR-TKI resistance and developing strategies to overcome it in EGFR-mutant NSCLC.
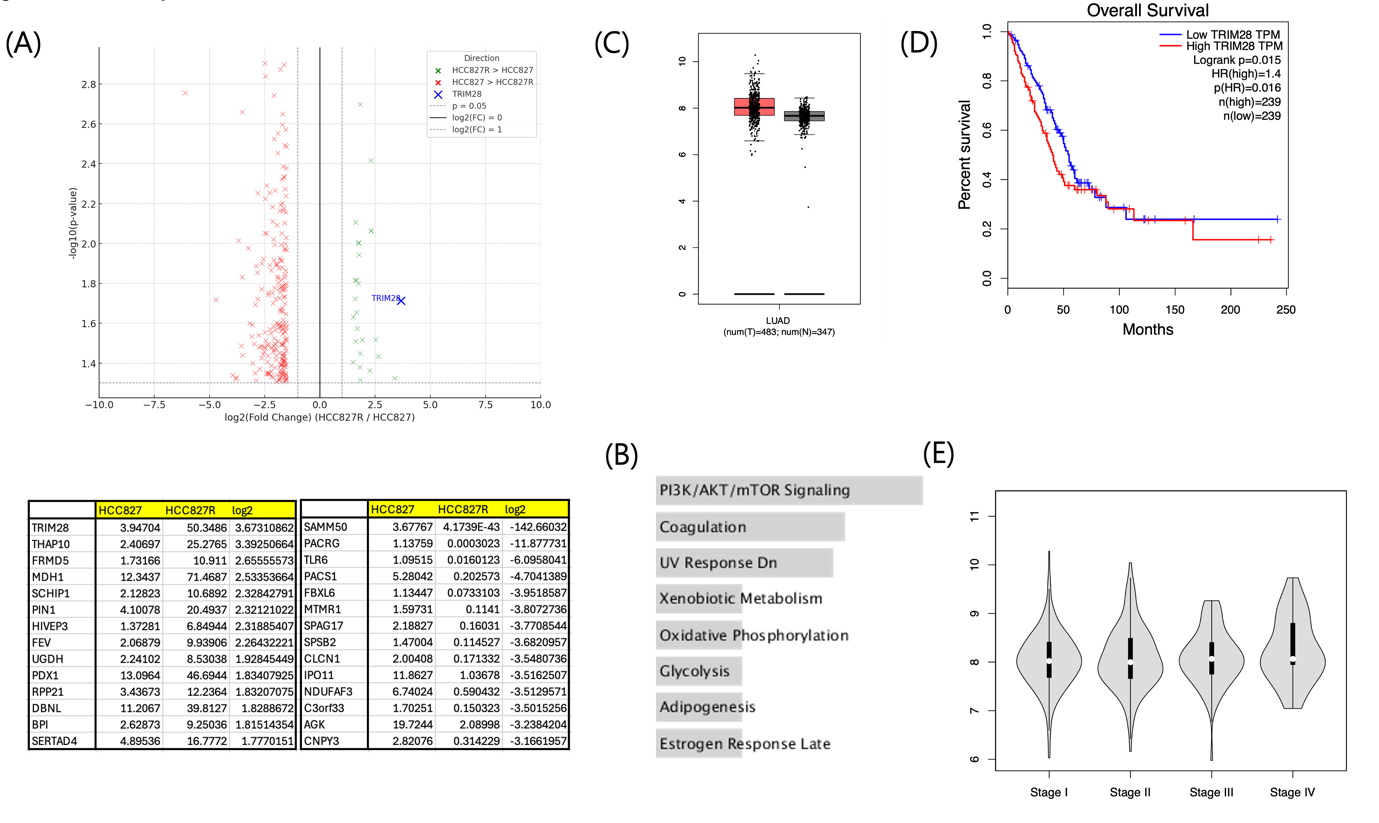
(A) Volcano plot of RNA sequencing analysis (top) comparing HCC827R and HCC827 cells revealed significant changes in gene expression. Table of top 15 genes (bottom) lists genes that show either increased or decreased expression in HCC827R cells compared to HCC827, with TRIM28 exhibiting the highest expression increase in HCC827R cells, with a log2 fold change > 2 and an adjusted p-value < 0.05.
(B) Pathways enriched in HCC827R cells include PI3K/AKT/mTOR signaling, oxidative phosphorylation, and glycolysis, which support cell survival and metabolic adaptation.
(C) Boxplot analysis of publicly available datasets shows that TRIM28 expression is significantly upregulated in LUAD tumor samples compared to normal tissues (*p < 0.05).
(D) LUAD patients with high TRIM28 expression exhibit significantly poorer overall survival compared to those with low TRIM28 expression (p = 0.015, hazard ratio [HR] = 1.4), emphasizing its potential as a prognostic marker.
(E) Violin plot analysis shows a progressive increase in TRIM28 expression with advancing LUAD stages, further supporting its role in tumor progression.
To investigate the molecular mechanisms underlying gefitinib resistance in HCC827R cells8, RNA sequencing (RNA-seq) was performed on both HCC827 parental and HCC827R cells, revealing significant changes in gene expression profiles. Volcano plot analysis identified numerous genes significantly upregulated or downregulated in HCC827R cells (log2 fold change > 2 or < -2, adjusted p-value < 0.05), with TRIM28 notably upregulated (Fig. 3A), suggesting its potential role in resistance through survival and stress-related processes.
Gene set enrichment analysis (GSEA) showed enrichment of pathways such as PI3K/AKT/mTOR signaling, oxidative phosphorylation, and glycolysis in HCC827R cells, supporting cellular proliferation and metabolic adaptation, while pathways related to apoptosis and UV response were downregulated (Fig. 3B), consistent with reduced sensitivity to gefitinib-induced apoptosis.
To evaluate TRIM28’s clinical relevance, its expression in lung adenocarcinoma (LUAD) samples was analyzed using publicly available datasets. TRIM28 was significantly upregulated in tumor tissues compared to normal tissues (Fig. 3C), and Kaplan-Meier survival analysis revealed that high TRIM28 expression was associated with poor overall survival (p = 0.015, HR = 1.4; Fig. 3D). Violin plot analysis showed a trend of increasing TRIM28 expression with advancing cancer stages (Fig. 3E), indicating its progressive upregulation during tumor progression.
These findings highlight transcriptomic reprogramming in HCC827R cells, emphasizing TRIM28‘s role in resistance through survival-related pathways, including PI3K/AKT/mTOR signaling. The association of TRIM28 with poor LUAD patient outcomes suggests its potential as a biomarker and therapeutic target in EGFR-mutant NSCLC.

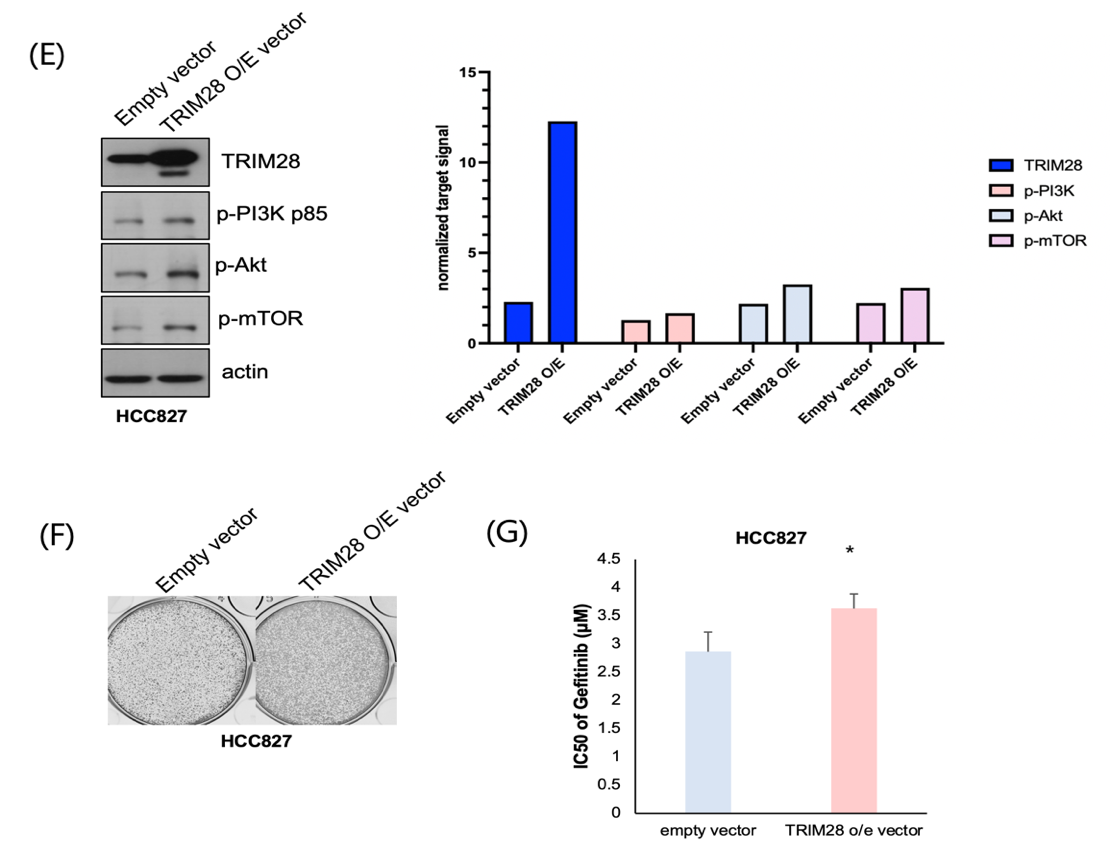
(A) Relative mRNA expression of TRIM28 was 5-fold higher in HCC827R cells compared to HCC827 cells, as measured by RT-qPCR. Data are presented as mean ± SEM from three independent experiments. (***p<0.001)
(B) Western blot analysis and quantification confirmed that CRISPR-mediated knockout (KO) of TRIM28 in HCC827R cells led to a significant reduction in TRIM28 protein levels along with phosphorylated PI3K (p-PI3K p85), phosphorylated AKT (p-AKT), and phosphorylated mTOR (p-mTOR) levels, confirming TRIM28’s role in activating this pathway.
(C) CRISPR-mediated TRIM28 knockout reduced colony formation in HCC827R cells compared to scrambled control, indicating restored sensitivity to gefitinib.
(D) TRIM28 knockout significantly reduced the IC50 of gefitinib in HCC827R cells, restoring drug sensitivity (*p < 0.05).
(E) Western blot analysis and quantification confirmed that lentiviral overexpression of TRIM28 (TRIM28 OE) in HCC827 cells led to increased TRIM28 protein levels and upregulated phosphorylated PI3K, AKT, and mTOR, confirming TRIM28’s role in promoting this signaling pathway.
(F) TRIM28 overexpression in HCC827 cells enhanced colony formation under gefitinib treatment compared to the empty vector control, indicating induced resistance.
(G) Overexpression of TRIM28 increased the IC50 of gefitinib in HCC827 cells, conferring resistance (*p < 0.05).
To investigate the functional role of TRIM28 in gefitinib resistance, we employed CRISPR-Cas9 technology to knockout TRIM28 in HCC827R cells9 and conducted overexpression experiments in parental HCC827 cells. These experiments aimed to validate TRIM28’s role in mediating resistance and assess its potential as a therapeutic target.
RT-qPCR analysis confirmed elevated TRIM28 mRNA levels in HCC827R cells compared to parental HCC827 cells, consistent with RNA-seq results (Fig. 4A). CRISPR-mediated TRIM28 knockout (TRIM28-KO) in HCC827R cells successfully eliminated TRIM28 expression, as validated by Western blot (Fig. 4B), and restored sensitivity to gefitinib. Functional assays revealed that TRIM28-KO cells exhibited significantly reduced proliferation and survival under gefitinib treatment. Colony formation assays demonstrated a marked reduction in both the number and size of colonies in TRIM28-KO cells compared to control cells transduced with a non-targeting guide RNA (Fig. 4C). Similarly, MTT assays showed a significant decrease in cell viability in TRIM28-KO cells treated with gefitinib (p < 0.05, Fig. 4D).
In contrast, TRIM28 overexpression in parental HCC827 cells conferred resistance to gefitinib. Lentiviral transduction of TRIM28-expressing vectors resulted in a substantial increase in TRIM28 protein levels, confirmed by Western blot (Fig. 4E). Functional assays indicated that TRIM28-overexpressing cells displayed enhanced survival and proliferation in the presence of gefitinib compared to vector-only controls. Colony formation assays showed an increase in both the number and size of colonies in TRIM28-overexpressing cells (Fig. 4F), while MTT assays revealed over a 20% increase in cell viability under gefitinib treatment (p < 0.01, Fig. 4G).
These findings establish TRIM28 as a critical regulator of gefitinib resistance in EGFR-mutant NSCLC. CRISPR-mediated TRIM28 knockout restores drug sensitivity by impairing survival and proliferation, whereas TRIM28 overexpression induces resistance by enhancing these processes. This underscores the therapeutic potential of targeting TRIM28 to overcome gefitinib resistance, offering valuable insights into resistance mechanisms and guiding the development of novel strategies for EGFR-mutant NSCLC treatment.
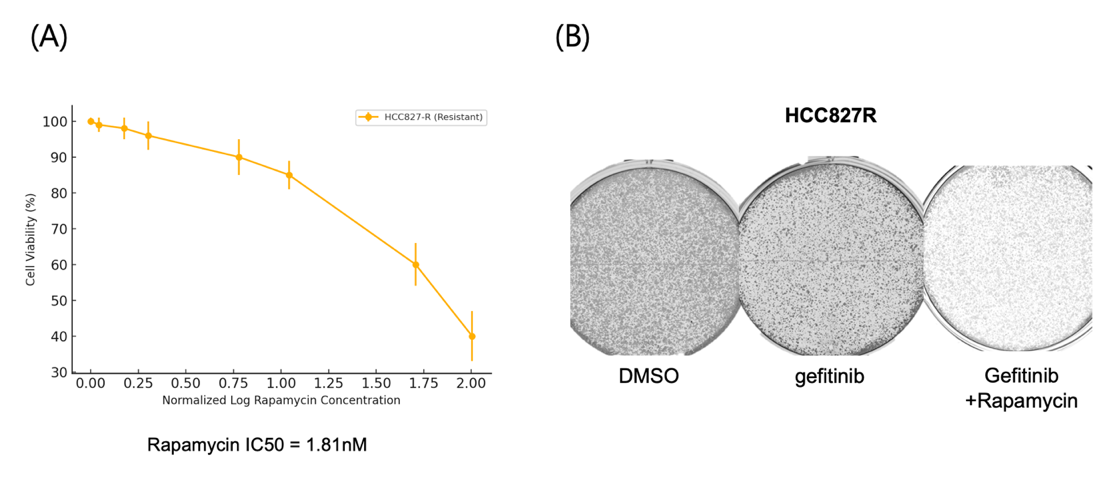
(A) Cell viability was assessed using the MTT assay after treatment with increasing concentrations of rapamycin for 72 hours. Rapamycin reduced cell viability in a dose-dependent manner, with an IC50 of 1.81 nM, indicating its effectiveness in targeting resistant cells.
(B) Representative images show that rapamycin treatment significantly impaired colony formation in HCC827R cells under gefitinib treatment compared to gefitinib alone or the DMSO control, highlighting its ability to suppress long-term proliferation.
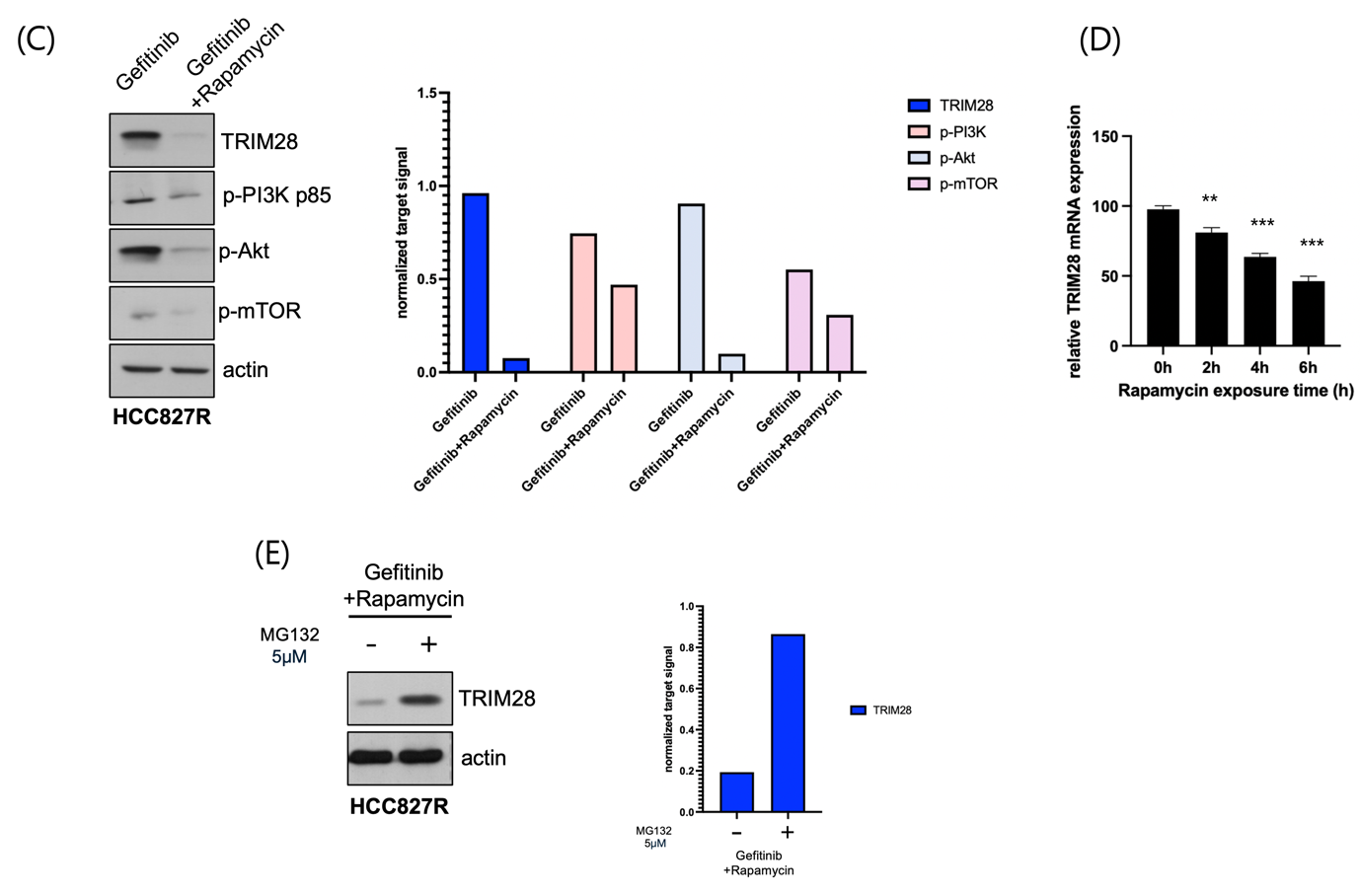
(C) Western blot analysis and quantification of rapamycin treatment in gefitinib-resistant HCC827R cells reduced the expression of TRIM28 and key phosphorylated signaling components of the PI3K/AKT/mTOR pathway, including p-PI3K p85, p-AKT, and p-mTOR, indicating that rapamycin inhibits TRIM28-mediated pathway activation.
(D) Relative TRIM28 mRNA expression after rapamycin exposure at different time points. Data represent the mean ± SD of three independent experiments. Statistically significant differences compared to the 0h time point are indicated by **p < 0.01 and ***p < 0.001 as shown.
(E) Western blot analysis and quantification of TRIM28 expression in gefitinib-resistant HCC827R cells treated with 5 μM MG132 for 6 hours showed an increase in TRIM28 levels. The treatment was applied in the context of a combination of gefitinib and rapamycin, suggesting that MG132 exposure may influence the stability of TRIM28 in this specific treatment condition.
The rationale for evaluating rapamycin as a therapeutic agent stems from the transcriptomic analysis (Figure 3), which revealed significant enrichment of the PI3K/AKT/mTOR signaling pathway in gefitinib-resistant HCC827R cells. This pathway, heavily implicated in promoting survival, proliferation, and metabolic adaptation, presents a compelling target for overcoming gefitinib resistance.
To assess the potential of rapamycin, a well-known mTOR inhibitor5, in counteracting this resistance, HCC827R cells were treated with increasing concentrations of rapamycin for 72 hours, and viability was measured using the MTT assay. A dose-dependent reduction in cell viability was observed, and the IC50 value was calculated to quantify rapamycin’s inhibitory effect (Fig. 5A). These results confirm that rapamycin effectively reduces the survival of resistant cells by targeting the mTOR pathway.
A colony formation assay was conducted to evaluate the long-term impact of rapamycin on the proliferative capacity of HCC827R cells. Rapamycin-treated cells exhibited a marked reduction in both colony number and size compared to untreated controls, demonstrating a significant impairment in the cells’ ability to sustain growth (Fig. 5B). Representative images of the assay further highlight the pronounced reduction in colony density, emphasizing rapamycin’s potential as a therapeutic strategy.
To elucidate the molecular mechanisms underlying rapamycin’s effect, Western blot analysis was performed to measure key components of the PI3K/AKT/mTOR pathway. Rapamycin treatment resulted in a marked decrease in the expression of TRIM28, a regulator implicated in gefitinib resistance, as well as phosphorylated mTOR (p-mTOR) and phosphorylated AKT (p-AKT) (Fig. 5C). These findings align with the pathway enrichment data (Fig. 3), further substantiating the role of PI3K/AKT/mTOR signaling in gefitinib resistance and its suppression by rapamycin.
In an effort to investigate the sequence of events following rapamycin treatment and determine the kinetics of TRIM28 mRNA regulation, time-course experiments were performed (Fig. 5D). These experiments suggest that rapamycin-mediated inhibition of TRIM28 expression occurs over time, providing insight into the temporal dynamics of gene suppression and potential feedback mechanisms. Although the inhibition of TRIM28 by rapamycin (Fig. 5C) is intriguing, the underlying mechanism remains unclear. It is uncertain whether TRIM28 is a direct transcriptional target of mTOR or if rapamycin influences its stability through an indirect mechanism. Further experiments involving proteasome inhibition or mRNA stability analysis will be necessary to differentiate between these possibilities.
Finally, treatment with MG132, a proteasome inhibitor10, in combination with rapamycin further restored TRIM28 expression (Fig. 5E), suggesting that mTOR pathway may modulate TRIM28 stability through proteasomal degradation. This provides additional insight into the mechanisms underlying rapamycin’s regulatory effect on TRIM28.
Discussion
Non-small cell lung cancer (NSCLC) driven by EGFR mutations poses a significant clinical challenge due to the inevitable emergence of resistance to EGFR-tyrosine kinase inhibitors (TKIs) like gefitinib11. This study highlights TRIM28 as a key regulator of gefitinib resistance and demonstrates the therapeutic potential of targeting the PI3K/AKT/mTOR pathway with rapamycin to overcome resistance in gefitinib-resistant HCC827R cells.
RNA sequencing and functional assays revealed that TRIM28 is significantly upregulated in gefitinib-resistant HCC827R cells, driving resistance through enhanced survival and proliferation. CRISPR-Cas9-mediated TRIM28 knockout9 restored drug sensitivity, while TRIM28 overexpression conferred resistance in parental HCC827 cells. Potential off-target effects of CRISPR-Cas9 were considered, and consistent results with two sgRNAs support the specificity of TRIM28 knockout. These findings identify TRIM28 as a critical therapeutic target in overcoming TKI resistance.
In addition to TRIM28, several other mechanisms of resistance to EGFR-TKIs have been reported, including secondary EGFR mutations (e.g., T790M), MET amplification, epithelial-mesenchymal transition (EMT), and activation of bypass signaling pathways such as AXL or IGF-1R12. While these mechanisms have been well-characterized, our findings suggest that TRIM28 represents an alternative and potentially complementary resistance pathway. Notably, TRIM28 may function in concert with these known mechanisms by modulating survival signaling and chromatin regulation, which warrants further investigation.
Transcriptomic analysis showed significant enrichment of the PI3K/AKT/mTOR signaling pathway in HCC827R cells, which supports survival and metabolic adaptation in resistant cells. This pathway’s activation provides a strong rationale for therapeutic targeting.
Rapamycin, an mTOR inhibitor13, effectively reduced cell viability and impaired the proliferative capacity of HCC827R cells. It also suppressed TRIM28 expression and key mTOR pathway components (p-mTOR and p-AKT), demonstrating its ability to overcome resistance by targeting this pathway. Although the precise mechanism by which rapamycin suppresses TRIM28 remains to be fully elucidated, it is plausible that mTOR pathway inhibition indirectly regulates TRIM28 expression through downstream transcriptional or epigenetic mechanisms. Further investigation is warranted to clarify this relationship. Additionally, future studies should consider potential challenges related to TRIM28-targeted drug delivery, treatment-associated toxicity, and the emergence of resistance to rapamycin.
TRIM28 upregulation correlates with poor prognosis in lung adenocarcinoma (LUAD), highlighting its potential as both a prognostic biomarker and a therapeutic target. While TRIM28 has been implicated in various cancer types, its function appears to be context-dependent. In particular, its role in EGFR-mutant NSCLC requires further investigation to determine whether its regulatory mechanisms are unique to this subtype. Understanding how TRIM28 contributes to other resistance mechanisms may further support its value in therapy development.
In summary, this study establishes TRIM28 as a critical driver of gefitinib resistance in EGFR-mutant NSCLC and demonstrates that targeting the PI3K/AKT/mTOR pathway with rapamycin is a promising strategy to overcome this resistance, offering insights for improving patient outcomes. However, this study is based on a single gefitinib-resistant cell line (HCC827R), which may reflect cell line–specific adaptations rather than clinically prevalent mechanisms such as T790M or MET amplification. Further validation in diverse models, including patient-derived samples, is needed to confirm the generalizability of TRIM28’s role in EGFR-TKI resistance.
Materials and Methods
1. Cell Lines and Culture Conditions
HCC827 parental cells (EGFR exon 19 deletion, gefitinib-sensitive) were obtained from the Korea Cell Line Bank. Cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a 5% CO₂ atmosphere.
2. Establishment of Gefitinib-Resistant Cells
To generate gefitinib-resistant cells, HCC827 cells were exposed to gradually increasing concentrations of gefitinib (0.01 µM to 1 µM) over an eight-week period. The resulting resistant line (HCC827R) was maintained under 1 µM gefitinib and validated for resistance through viability and colony formation assays.
3. Drug Treatment and Viability Assays
To evaluate the sensitivity of HCC827 and HCC827R cells to gefitinib and rapamycin, we conducted viability and colony formation assays.
3.1. MTT Assay
Cells (5,000 cells/well) were seeded in 96-well plates and treated with gefitinib, rapamycin, or their combination for 72 hours. Cell viability was assessed using MTT reagent (5 mg/mL, Sigma-Aldrich), and absorbance was measured at 570 nm. IC₅₀ values were calculated using GraphPad Prism.
3.2. Colony Formation Assay
Cells (500 cells/well) were seeded in 6-well plates and treated with drugs for 10–14 days. Colonies were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet, and manually counted.
4. Genetic Manipulation of TRIM28
To assess the functional role of TRIM28 in drug resistance, we performed both knockout and overexpression experiments.
4.1. CRISPR-Cas9-Mediated Knockout
Guide RNAs targeting TRIM28 were cloned into the lentiCRISPRv2 vector (provided by Dr. Kim, Chromogen). Lentiviral particles were produced in HEK293T cells and used to transduce HCC827R cells, followed by puromycin selection (2 µg/mL). Knockout efficiency was confirmed by Western blot and RT-qPCR.
4.2. TRIM28 Overexpression
Full-length TRIM28 cDNA was cloned into the pCDH-CMV-MCS-EF1-Puro vector (Dr. Kim, Chromogen). Lentiviral transduction of HCC827 cells was followed by puromycin selection (2 µg/mL). Overexpression was verified by Western blot and RT-qPCR.
5. RNA Sequencing and Bioinformatics Analysis
Total RNA was extracted using TRIzol reagent and sequenced using the Illumina NovaSeq 6000 platform. Differential gene expression was analyzed using DESeq2, and pathway enrichment analysis was performed using GSEA (Gene Set Enrichment Analysis).
6. Western Blot Analysis
Protein expression and signaling pathways were assessed via Western blotting. Cells were lysed using RIPA buffer with protease and phosphatase inhibitors. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and probed with antibodies against TRIM28(Invitrogen, MA1-2023), p-mTOR(cell signaling technology, #2971), p-AKT(cell signaling technology, #9271), and β-actin(cell signaling technology, #4967). Bands were visualized using an ECL detection system.
7. Flow Cytometry
Flow cytometry was used to evaluate cell cycle distribution and apoptosis.
7.1. Cell Cycle Analysis
Cells were fixed in ethanol, stained with propidium iodide (PI), and analyzed using a flow cytometer.
7.2. Apoptosis Assay
Apoptotic cells were identified using Annexin V/PI staining and quantified by flow cytometry.
8. Statistical Analysis
All experiments were conducted in triplicate. Data are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using two-tailed Student’s t-tests or one-way ANOVA with Tukey’s post hoc test (GraphPad Prism). A p-value < 0.05 was considered statistically significant.
References
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 366:2–16 (2006). [↩]
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 361:947–57(2009), Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3:75ra26–75 (2011). [↩]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10(1):57-63 (2009). [↩]
- Li K, Wang H, Jiang B, Jin X. TRIM28 in cancer and cancer therapy. Front Genet. 19;15:1431564 (2024). [↩]
- Bouyahya A, El Allam A, Aboulaghras S, Bakrim S, El Menyiy N, Alshahrani MM, Al Awadh AA, Benali T, Lee LH, El Omari N, Goh KW, Ming LC, Mubarak MS. Targeting mTOR as a Cancer Therapy: Recent Advances in Natural Bioactive Compounds and Immunotherapy. Cancers (Basel).14(2022). [↩] [↩]
- Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, Lee EY, Weiss HL, O’Connor KL, Gao T, Evers BM. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 71(9):3246-56(2011). [↩]
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 361:947–57(2009). [↩]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10(1):57-63 (2009). [↩]
- Housden BE, Perrimon N. Comparing CRISPR and RNAi-based screening technologies. Nat Biotechnol. 34(6):621-3 (2016). [↩] [↩]
- Zanotto-Filho, A., Braganhol, E., Battastini, A. M. & Moreira, J. C. Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFkappaB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling. Invest New Drugs. 30, 2252-2262 (2012). [↩]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3:75ra26–75 (2011). [↩]
- Takamizawa S, Okuma Y, Kato Y, Hakozaki T, Kitagawa S, Zenke Y. First-line osimertinib in EGFR mutation-positive non-small cell lung cancer patients with poor performance status. Future Oncol. 18, 291-300 (2022). [↩]
- Lamming DW. Inhibition of the Mechanistic Target of Rapamycin (mTOR)-Rapamycin and Beyond. Cold Spring Harb Perspect Med. 6(5):a025924 (2016). [↩]



{kind=link}