
Abstract
Importin, a family of proteins essential for nuclear transport, specifically regulates the translocation of DNA repair enzymes, which execute a crucial process for maintaining genomic stability. KPNA2, a member of this family, is often overexpressed in cancerous tissues, a phenomenon that seems paradoxical given its critical role in supporting genomic stability. This study aimed to assess how missense mutations (SNPs) in the ARM domains of importin-α1 impact its structural integrity and cargo-binding ability, particularly with the double-stranded DNA repair enzyme NBS1. SNPs across a set of domains were analyzed using predictive scoring tools (e.g., CADD, PolyPhen), structural modelling (AlphaFold, PyMOL), and protein-protein docking tools (ClusPro, HADDOCK), to assess both affected binding affinity and structural conformation. A cancer genome analysis was also performed using GEPIA2. The majority of selected missense variants demonstrated high probabilities for severe structural deformation and stark disruptions in binding affinity. The results from this study demonstrated specific SNP-induced structural damages, as well as affected cargo docking abilities. These findings may reveal the nuanced relationship between SNP location and severity of missense mutations, as well as the unreliability of predictive measures compared to comprehensive biochemical and energetic analyses. These conclusions warrant and may later inspire deeper experimental studies on the mechanistic role of importin mutations in DNA repair efficiency and impairment.
Keywords: KPNA2, importin-α, karyopherin, missense SNPs, NBS1 nuclear localization, protein-protein docking
Introduction
Importin, a type of karyopherin, is responsible for the transportation of all NLS-bearing proteins from the cytoplasm to the nucleus of a cell1. Importin protein complexes bind to their cargo’s nuclear localization signal (NLS)—a specific stretch of amino acids that acts as a nuclear signal guide—and escort the cargo into the nucleus, facilitating their entry via the nuclear pore complex (NPC). Importin proteins are divided into two subclasses: importin-α and importin-β, which jointly form an importin heterodimer2. Importin-α, the smaller of the two karyopherins, is necessary for the direct binding of the importin heterodimer to its protein cargo, and consequently, must maintain a stable structure and function3. Functioning synergistically, importin-β facilitates the entry of the importin heterodimer into the NPC and there, disengages from its cellular cargo2,4.Various studies have shown instances where importin-β can act self-sufficiently as a monomer, performing the roles of both importin proteins without the need of importin-α, yet these are considered aberrant5; many of them occur when the cargo contains a non-classical NLS (ncNLS). Regardless of their class, importin transports a multitude of proteins with diverse characteristics, yet among the most crucial are DNA repair enzymes—enzymes that specifically act as the catalysts of DNA repair pathways, such as Base Excision Repair (BER) or Double-Stranded Break Repair (DSBR).
In mammalian cells, there exist six subunits of the importin-α protein class, each with distinct structural and functional characteristics. Often considered the main cNLS-binding importin-α protein is the importin-α1 protein, commonly referred to by its gene name KPNA2 (karyopherin subunit-alpha 1). The importin-α1 protein consists of three critical components: the importin-β binding domain (IBB), located at the N-terminus end of the protein, ten armadillo (ARM) domains, and the C-terminus end, which is necessary for cNLS-mediated protein transportation6. The IBB is the specific region of the protein where the importin-β binds to form the importin heterodimer complex, measuring approximately 58 amino acids in length (UniProt ID: P52292)7. The ARM domains are specific regions of the protein, each approximately 40 amino acids in length, which are repeating sequence motifs. The importin-α1 protein specifically contains ten ARM repeat regions, with ARM 1 beginning at residue 71 and ARM 10 ending at residue 496 (UniProt ID: P52292)7. In importin-α proteins, the ARM domains are divided into two regions classified by their cargo binding abilities: the major binding domain and the minor binding domain. For KPNA2, ARMS 2-4 are considered major binding sites, while ARMS 6-9 are classified as minor binding sites8. ARM 5, although not divided into a distinct binding domain, carries large structural and indirect effects on the rest of the importin; ARMs 1 and 10 are considered irrelevant in the context of the other armadillo domains.
Specific genetic mutations can be tracked across populations in studies known as Genome-Wide Association Studies (GWAS). GWAS measures the density and frequency of mutations known as single-nucleotide polymorphisms, commonly referred to as SNPs9. These mutations are particular, single-nucleotide mutations (SNVs) that exist in at least one percent of a species’ population. Although not all, many SNPs can also influence the phenotype of an organism, in a possibly immoderate manner10. Despite extensive study of importin pathways, the impact of specific KPNA2 variants on cargo binding and nuclear transport efficiency and functionality remains poorly understood. Our study investigates the structural and functional consequences of rare missense SNPs in the ARM domains, specifically the major and minor binding regions, of KPNA2, particularly focusing on their effects on DNA repair cargo docking11. Moreover, several data have shown how KPNA2 is largely overexpressed and dysregulated in many cancer tissue data sets, adding clinical significance to this study as well12,13,14. It is hypothesized that specific SNPs in key ARM regions will structurally and energetically disrupt KPNA2’s binding affinity with DNA repair cargo. Here, we present peculiar and nuanced relationships between genomic and proteomic variants, as well as other factors that may have an impact on mutation-induced structural and functional damage. Each of our chosen SNP-induced KPNA2 proteins underwent tests that intrinsically described the effects that their respective SNP had on their stability. Subsequently, they were analyzed to assess the possible severity of these effects on a widespread impairment of the DNA repair system.
Materials and Methods
SNP Selection and Identification
Data specifying the genomic and protein-level locations of KPNA2 SNPs were extracted from Ensembl (release 114) and imported into the Numbers application for organization and analysis15. For each ARM region (2–9), the SNP with the highest PolyPhen score was initially selected to ensure that each region was represented by a mutation with high predicted structural and/or functional disruption. This approach allowed us to explore the structural impact of mutations across the entire cargo-binding surface of KPNA2. These SNPs were then entered into ProtVar (v1.4) using their respective genomic variant IDs to obtain predicted values for AlphaMissense pathogenicity, CADD (Combined Annotation Dependent Depletion), and ΔΔG (change in Gibbs free energy)16,17. The aforementioned criteria were used as they established a wide field of structural, pathogenic, and energetic markers for our SNP selections. If a given SNP received uniformly low scores across these predictive measures, the SNP with the next-highest PolyPhen score from that region was selected and evaluated. This iterative process continued until seven distinct SNPs were identified, each demonstrating relatively strong scores across structural and pathogenicity prediction metrics. Final allele frequency data for each SNP were retrieved from GnomAD (v4.1.0)and dbSNP(Table 1)18,19. Note: although this study refers to these variants as SNPs for simplicity, all seven selected variants exhibit allele frequencies below 1%, and are therefore technically classified as SNVs (single-nucleotide variants) rather than SNPs. The number of SNPs within each ARM region (2–9), along with the distribution of corresponding CADD and PolyPhen scores, was individually graphed. Statistical significance for CADD and PolyPhen score differences across regions was assessed using Kruskal–Wallis testing (n=312/ARM; total n=2496). To analyze the ΔΔG score distribution, one SNP was randomly selected for approximately every 10 amino acids between residues 112–456—the residues between ARMS 2 and 9. The ΔΔG values of these randomly chosen SNPs were plotted alongside the ΔΔG values of the hand-selected SNPs on the same graph for comparison and added analysis. Statistical significance was calculated using Spearman correlation (n=42).
| # | SNP ID | Ref Nuc | Alt Nuc | AA Pos | ARM # | AA Change | ΔΔG | CADD | Alpha-Missense | Allele Freq. | PolyPhen |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs1059558 | G | A | 365 | 7–8 | Gly/Ser | 3.97 | 28.3 | 0.98 | A=0.000021 | 0.950 |
| 2 | rs11545989 | C | G | 165 | 3 | Pro/Arg | 2.14 | 26.7 | 0.54 | G=0.050257 | 0.987 |
| 3 | rs2071267327 | T | G | 116 | 2 | Ile/Ser | 2.91 | 28.8 | 1.00 | G=0.000007 | 0.998 |
| 4 | rs11545987 | C | A | 244 | 4 | Pro/His | 13.58 | 27.9 | 0.98 | A=0.00004 | 1.000 |
| 5 | rs1200044026 | C | T | 275 | 5 | Ile/Thr | 2.88 | 26.8 | 0.91 | C=0.000001 | 0.981 |
| 6 | rs1555705006 | T | C | 320 | 6 | Ile/Thr | 3.36 | 28.4 | 0.91 | C=0.000002 | 0.997 |
| 7 | rs538665386 | G | A | 446 | 9 | Asn/Asp | 3.72 | 27.2 | 0.94 | G=0.000005 | 0.927 |
Importin-α1 Modeling and SNP Mapping
The KPNA2 wild-type structure was obtained from the AlphaFold Protein Structure Database (v3)20,21. The model’s pLDDT scores exceeded 90 for the majority of residues, with an average pLDDT of 86.51, and alignment with RCSB PDB entry 3WPT showed a TM-score of 0.96 and RMSD of 1.48 Å, indicating high structural similarity. The AlphaFold-predicted structure of KPNA2 was used in place of the experimental structure from the RCSB PDB (PDB ID: 3WPT), as the AlphaFold model included an additional 94 amino acids absent from the crystal structure. The file was then uploaded to PyMOL (v3.1.6.1)22. In ribbon format, each ARM region within the major and minor binding domains, as well as ARM 5, was color-coded, and a polished KPNA2 model was created. Two surface models of KPNA2 were also generated, each distinctively highlighting either the major or minor binding domain of the protein. Following, a sphere model of KPNA2 was created that highlighted the residual locations of the seven chosen missense variants from Figure 1, as well as the ARM domain in which they were located. Finally, using the Wizard Mutagenesistool on PyMOL, the P244H SNP was chosen to be displayed using a ball-and-stick model to highlight the structural change between the wild and variant amino acids within the protein itself.
Protein-Protein Docking and KPNA2 Binding Affinity
Initial modeling aimed to evaluate the ARM repeat region of the protein in isolation. The KPNA2 PDB file was uploaded to PyMOL, where the PDB file was edited to only include ARM regions 2-9 of the protein22. The new, cut KPNA2 protein was uploaded to FoldX (v5.0), where it underwent computational energy minimization and stability control23. The cut, energy-minimized, and stabilized KPNA2 file was duplicated seven times, and each file was uploaded separately to PyMOL22. Each file was then mutated at a single, specific residue to mimic one of the seven chosen missense variants from Figure 1, totalling eight files: one wild-type (WT) KPNA2 and seven mutants22. Each PDB file was uploaded to ClusPro (v2.0) to undergo a protein-protein docking interaction between the importin-α1 and a DNA repair enzyme24. ClusPro is a protein–protein docking algorithm that generates and scores thousands of potential docking conformations based on rigid-body docking, energy minimization, and clustering of low-energy structures to predict the most likely binding orientations. For the study, Nibrin (NBS1), an essential repair enzyme in the DSBR pathway, which plays a necessary role in repairing cancerous tissues, was selected as the model DNA repair enzyme for our importin-cargo docking tests25,26. The NBS1 protein file (PDB) was likewise obtained from the AlphaFold Protein Structure Database (v3)20,21. Docking using ClusPro, however, yielded no results across all variants, including WT24. In response, we systematically tested four structure preparation combinations:
- Cut (ARM only) + FoldX
- Uncut (full protein) + FoldX
- Cut + No FoldX (raw)
- Uncut + No FoldX (raw)
Each ClusPro docking method was performed thrice. Variants and wild-type files across all preparation combinations were tested for quality assessments and structural integrity comparisons using MolProbity (v4.5.2) (Supplementary Table 2)27. Across all 3 replicates, docking outcomes and scores for all methods were parallel. Only method 4 (Uncut + No FoldX) produced valid ClusPro docking outputs for the WT. These findings (Supplementary Table 1) informed the decision to proceed with uncut, non-FoldX-treated structures for all ClusPro docking runs. Notably, one FoldX-minimized uncut structure did return valid results, though this was not consistent across all variants (specifically WT) and thus excluded in further analyses for uniformity.
Once method 4 was confirmed for pipeline use, full protein preparation pipeline methodology was repeated using method 4 and ClusPro docking tests were performed thrice again. For each mutant test, the returned cluster 0 was downloaded and loaded into PyMOL, along with the WT-KPNA2 and NBS1 docking file22. Central and lowest energy docking scores for cluster 0 were also downloaded from ClusPro and plotted; statistical significance was determined using pairwise Welch’s t-tests with applied Bonferroni correction. Downloaded cluster 0 models per variant were analyzed using MolProbity for post-docking quality assessments and ClusPro result verifications (Supplementary Table 3)27. To visualize structural differences, an overlay model was created, with each docking result shown in a different color to highlight any deviations. RMSD and RMSD_cur values were calculated for each WT-KPNA2 vs. mutant-type (MT) KPNA2 comparison. Each KPNA2 PDB file, attached with the NBS1 PDB file, was also uploaded to HADDOCK (v2.4), where it underwent a round of biochemical protein-protein docking28. HADDOCK is a protein–protein docking tool that uses biochemical and biophysical information—such as interface residues, mutational data, or NMR constraints—to guide the docking of biomolecular complexes. For all HADDOCKtests, active residues defined for KPNA2 were provided using a true-interface (TI) restraint file between WT-KPNA2 and NBS1, calculated via the haddock-restraints Command Line tool; cutoff distance for restraint generation was set at 5Å in accordance with reported optimal parameters28,29. NBS1 residues 401-520 were chosen as active after locating an approximate location for the NLS on the NBS1 using cNLS Mapperand PSORT II; passive residues for both KPNA2 and NBS1 were self-defined based on active residues. Surface area solvent accessibility (SASA) scorings per residue across all variants (WT and MT) were calculated and plotted using FreeSASA (v2.1.2) to determine whether our HADDOCK configuration should or should not exclude buried residues from KPNA2-NBS1 docking tests (Supplementary Figure 1)30; cutoff SASA threshold for buried residue exclusion was set at 20% in accordance with reported optical parameters31. Each variant was tested in triplicate, with metrics corresponding to the top 10 generated HADDOCK clusters (lowest Z-scores) being extracted (30 total scores per variant); only 10, rather than all, metrics were used for analysis and statistical testing per replicate due to the variance of total cluster quantities across variants. To ensure consistency, replicate-level means were initially computed and assessed via one-way Kruskal-Wallis; no significant intra-variant differences were found, so individual replicate means were not reported. For final visualization and statistical analysis, all 30 scores per variant were aggregated. Mean and standard deviation were calculated and visualized using bar plots. One-way Kruskall-Wallis followed by pairwise testing (Mann-Whitney U with Bonferroni correction) was used to statistically compare variants. Protein stability (ΔΔG) was then calculated via FoldX’s Stability command for each replicate of each variant24; final scorings per variant were calculated and graphed based on mean across replicates, and standard deviation nor intra-variant statistical analysis was reported due to negligible (<1.00) numeric differences across replicates.
As controls, we included a benign KPNA2 variant (p.Ser384=), docking with wild-type NBS1, and a scrambled NLS cargo negative control, docking with wild-type KPNA2. To generate the negative cargo control, the predicted NLS of the NBS1 protein was randomly shuffled, and used as a reference for manual amino acid rearrangement via PyMOL; the shuffled NLS was used as the active residue region in HADDOCK for the negative control docking. Each control was subject to the same pipeline as the pathogenic variants, and each was performed with equal replicates as described above. Numerical results, however, were not reported due to the results being consistent with standard expectations for positive and negative controls.
The complementary docking platforms ClusPro and HADDOCK take radically different approaches to protein–protein interactions. While HADDOCK incorporates user-defined restraints and evaluates models with energy terms and Root Mean Square Deviation (RMSD), a measure of atomic-level positional differences, ClusPro places more emphasis on large-scale sampling and clustering, evaluating models primarily through the Fraction of Common Contacts (FCC), which determines whether binding contacts are preserved despite slight conformational shifts. We ensured a more rigorous and balanced examination by utilizing both platforms, with HADDOCK offering energetic and structural validation and ClusPro emphasizing contact integrity.
KPNA2 Transcriptomic Analysis
Following the suit of other importin-focused computational studies, the findings of our study were correlated with insights into KPNA2 presence and function in cancerous tissues32.GEPIA2 was used to analyze KPNA2 gene expression across 33 human tissue types, comparing cancerous and non-cancerous samples33. GEPIA2, a large-scale gene expression analysis database for both control and tumor samples, obtains data from tumor and normal samples in both the TCGA and GTEx databases, respectively. A gene expression profile was generated to visualize quantitative differences between healthy and tumor tissues of the same type33. Seven diverse tissues were then selected for more detailed analysis using box-and-whisker plots33. Cancerous KPNA2 expression levels were further used to perform a Pearson correlation analysis with NBN (NBS1-coding gene)33. Lastly, Kaplan–Meier overall survival (OS) curves were generated to compare patient survival rates based on high versus low KPNA2 expression across 33 cancer types, and a Mantel-Cox test was conducted to determine individual p-values and overall statistical significance33.
Statistical Analysis
All statistical analyses were performed using base R (v4.3.1) and were either integrated within visualization scripts or described in the corresponding figure captions. Both p-values and effect sizes were reported where applicable. Power sizes (when applicable) are reported in above sections of the methodology. T-tests, either Student’s or Welch’s, were applied to directly compare pairwise WT and variant data, while Kruskal-Wallis testing was used for dataset-wide statistical analysis, specifically for datasets without WT/variant conditions; Bonferroni FDR correction was applied to all pairwise statistical tests. Usage of Pearson vs Spearman testing was determined based on visual analysis of data distributions; normality was assumed for all datasets in our study. Statistical significance depicted as ns, non-significant; *P<0.05;**P<0.005. All plots were generated using ggplot2 (v4.0.0), and the majority of custom scripts are provided in Supplementary Document 1.
Computational predictions of importin function have previously been shown to align with experimental validations. For example, Riddick and Macara (2005) combined systems-level simulations with real-time nuclear import assays to validate the operation of computationally-modelled importin-α/β–mediated transport, while Panagiotopoulos et al. (2025) used bioinformatic tools to identify a novel NLS motif for importin-8 that was subsequently confirmed in vitro34,35. These notable studies provide evidence of experimental validation and the generation of similar findings to computational pipelines that investigate structural insights of various importin proteins. Although not directly replicating these studies, our computational procedure is derived from a combination of modeling and docking approaches, including ClusPro and HADDOCK, similar to those employed in importin-focused or conceptually related investigations32,36,37.
Comprehensive software details, custom (scripting) code, specific protein–protein docking parameters, and corresponding random seeds are provided in Supplementary Document 1.
Results
Characterization of the KPNA2 pathway and SNP Expression on Functional Impact Across ARM Repeats
Figure 1 explores how SNP density and predictive impact scores are distributed across the ARM domains of the KPNA2 protein. In Figure 1A, the diagram depicting the classical importin-α transport system accurately demonstrates the associated risks with missense variants in KPNA2 and other importin-α coding genes. Although SNPs of any gene can cause a range of catastrophic effects on an organism’s phenotype, the diagram emphasizes the direct correlation between SNPs in importin-coding genes and an elevated risk of pathway-wide DNA repair failure. SNPs, though, range in severity and biological significance, indicating that an ARM domain-wide analysis of trends in SNP characterization would be beneficial and can be used as additional context in further modelling and docking tests.

In examining the distribution of missense SNP counts across the ARM repeats (Figure 1B), an interesting pattern emerged: most ARM repeats (2-9) displayed relatively consistent SNP counts, ranging from 30 to 40. However, ARM 4 stood out, displaying a notably higher count of 51 SNPs. This suggests that ARM 4 may be more prone to variation compared to other regions. On the other hand, ARM repeats 3 and 5 had the fewest SNPs, each with just 35, indicating these regions may harbor fewer variants overall.
These findings prompted a deeper analysis to understand whether certain trends existed between SNP density, their severity, and their genomic locations. To investigate the potential impact of these SNPs, CADD (Combined Annotation Dependent Depletion) scoring was employed to model the average deleteriousness of SNPs within each ARM domain. When looking at the median CADD scores (Figure 1C), ARM 3 was an unexpected outlier, peaking at a score of 25, despite its relatively low SNP count. In contrast, ARM 5, with its similarly low SNP count, exhibited the lowest median CADD score of 21. However, it was notable that both ARM 3 and ARM 5 contained extreme outliers in their CADD scores, which were significantly higher than the median values. This indicates a clear inverse relationship between SNP density and the predicted deleteriousness of those variants across these regions.
PolyPhen scores (Figure 1D) reinforced this pattern. ARM 3, with its relatively low SNP count, still had the highest structural and functional disruption predictions, suggesting that the few variants present in this region could have a disproportionately high impact. ARM 5, conversely, showed lower predicted effects from the amino acid substitution despite its similarly low SNP count. These results highlight the intricacies of SNP distributions and their varying computationally backed impacts on protein function depending on the ARM repeat.
Finally, when examining the ΔΔG values for residues 112 to 456 (Figure 1E), no clear trends emerged in terms of position or domain, suggesting either an incompatibility between ΔΔG and other predictive metrics or a nonlinear basis for ΔΔG distribution. Overall, the analysis suggested important regional differences in SNP distributions, with ARM 3 and ARM 5 both presenting unique patterns in terms of both their SNP counts and their associated deleteriousness scores. These findings may imply that SNP density alone does not fully explain the functional consequences of genetic variation in these regions; yet, an isolated analysis of predictive metrics of a few SNPs also proves to be inaccurate for assuming protein-wide trends.
Structural Organization and Disruption of KPNA2 by Missense SNPs
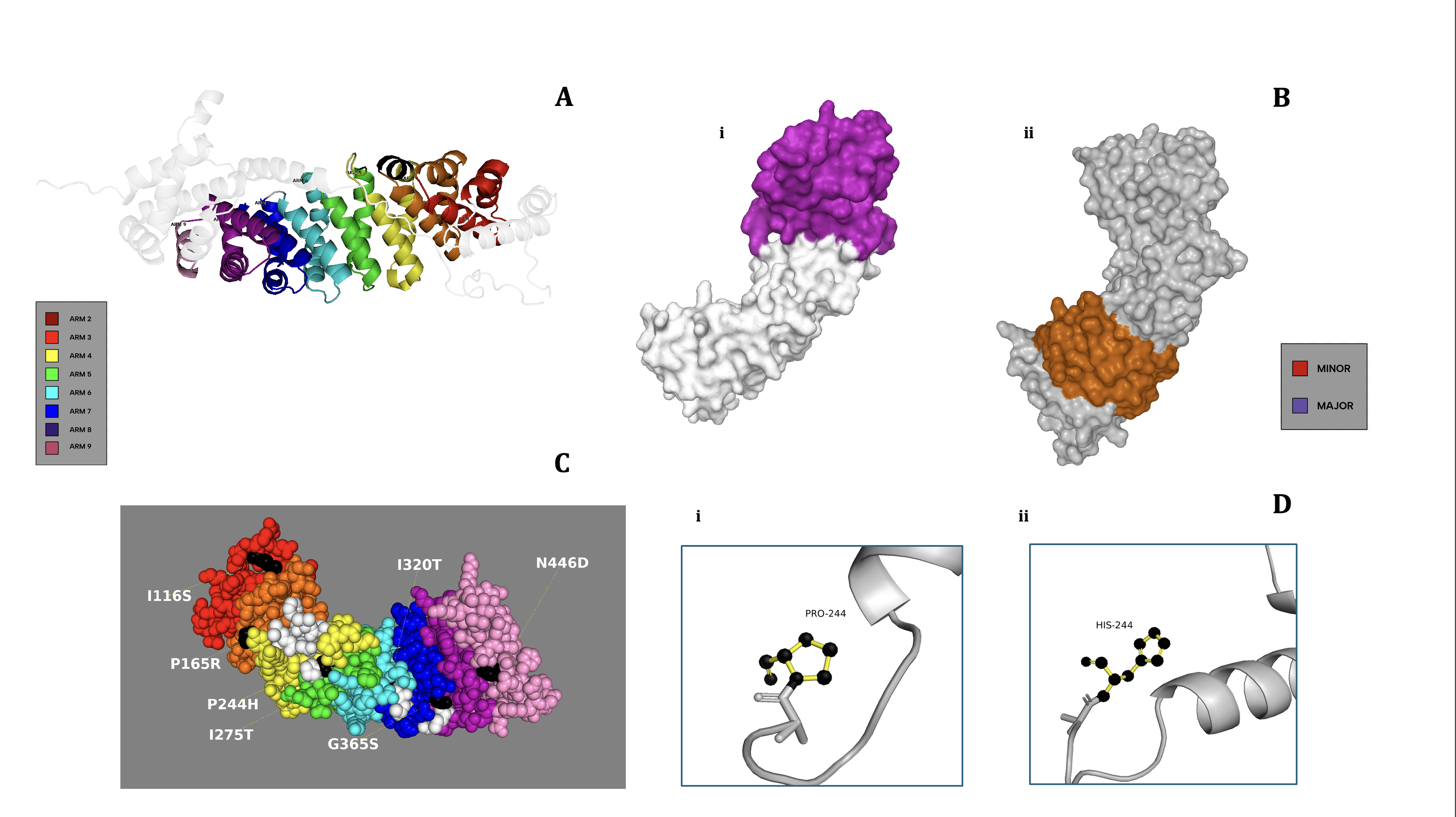
As previously discussed in Figure 1, clear relationships are apparent between SNP density within ARMs and the median structural and functional effects on KPNA2. However, these genomic trends do not always align with proteomic location. This acknowledgment prompted further exploration into the relationship between the genomic positioning of each SNP and its corresponding proteomic location. Domain-wide SNP analysis was not conducted on protein models, primarily due to logistical constraints and the assumption that significant findings would be unlikely. Instead, we focused on targeted structural analyses, providing a more detailed understanding of how specific missense variants may impact the KPNA2 protein.
The ribbon model (Figure 2A) of the entire KPNA2 protein illustrates the overall curvature and highlights the location of each ARM repeat along the binding groove and structural contour. This model offers a general overview of the protein, but for a deeper focus on the ARM domains, Figure 2B provides a surface-rendered model. This representation distinguishes between the major binding region (ARMs 2–4, Figure 2B(i)) and the minor binding region (ARMs 6–9, Figure 2B(ii)). According to Table 1, variants I116S, P165R, and P244H are located within the major binding region, whereas I320T, G365S, and N446D reside in the minor region. Notably, despite the greater functional relevance of the major domain, predictive scoring suggests that SNPs in the minor region may exert comparable functional and structural disruptions; this implies that both domains are functionally significant and potentially vulnerable to disruption. The models also show that these regions extend into the protein’s binding groove, reinforcing the idea that mutations in either domain could impair the cargo transport capabilities of KPNA2.
To further explore the structural and functional nuances of this observation, we modelled the 7 missense variants on the WT-KPNA2 protein (Figure 2C) for visual analysis. Among the variants mapped, variant I116S is the only mutation embedded within the central binding groove, while the remaining variants reside on the protein’s periphery or internal core. Aside from this distinction, the model reveals few consistent structural trends; yet, the structural distinctiveness of SNP I116S was recognized, and it was used to better understand any further abnormalities it might introduce into KPNA2’s structure.
While each SNP was modeled on the protein for visual analysis, we focused on performing a chemical visual analysis of one specific SNP to predict and assess potential major residue-level structural damage before conducting docking tests. Due to its extreme scoring metrics (Table 1), SNP P244H was chosen to be modelled. The histidine residue modelled (Figure 2D(i)) is noticeably bulkier, with 10 atoms and bonds compared to proline’s 7 (Figure 2D(ii)), and it extends further outward, potentially clashing with the adjacent residues. Notably, the proline points into the page while histidine projects outward, suggesting a directional clash that could destabilize the protein’s folding. These details suggest the high possibility of steric hindrance introduced by this SNP, explaining the high predictive scores from Table 1, including the modelled SNP’s extreme ΔΔG of 13.58 kcal/mol. Moreover, this visualization also supports the idea that the most structurally damaging variants may involve two structurally divergent amino acids. Before any dynamic testing, we hypothesized that SNP P244H is the most structurally destabilizing SNP in the KPNA2 ARM domains. However, dynamic testing is essential to confirm or challenge our hypothesis, as it will provide insights into the energetic and structural feasibility of the P244H mutation in the context of KPNA2’s function.
Structural and Biochemical Impact of KPNA2 SNP Variants on Cargo Docking
Figure 3 compares modelling, scoring, and graphical analysis of KPNA2’s cargo docking abilities and binding affinity under different genomic contexts. Figures 3A, 3B, and 3C exclude data from variants P165R and P244H, as those SNP-induced KPNA2 proteins failed to return any docking results from ClusPro; thus, no models or docking scores were outputted. These results, independently, may indicate severe structural deformation caused by both variants, which likely destabilized the protein structure incredibly, inhibiting any form of plausible, structural docking with the NBS1 enzyme. SNP 244H, based on Figure 2C, is located in the core of KPNA2, suggesting that core-centered or near missense variants could have larger structural effects on the protein compared to others. Due to their complete docking failures in ClusPro, our study considered P244H and P165R as the most damage-inducing missense variants in the ARM domains of KPNA2, as they originally had the highest PolyPhen metrics from their respective ARM domain in Ensembl. As a result, these tests are not used for the large majority of the analyses completed in Figure 3, as docking structures or scores could not be computed by ClusPro.
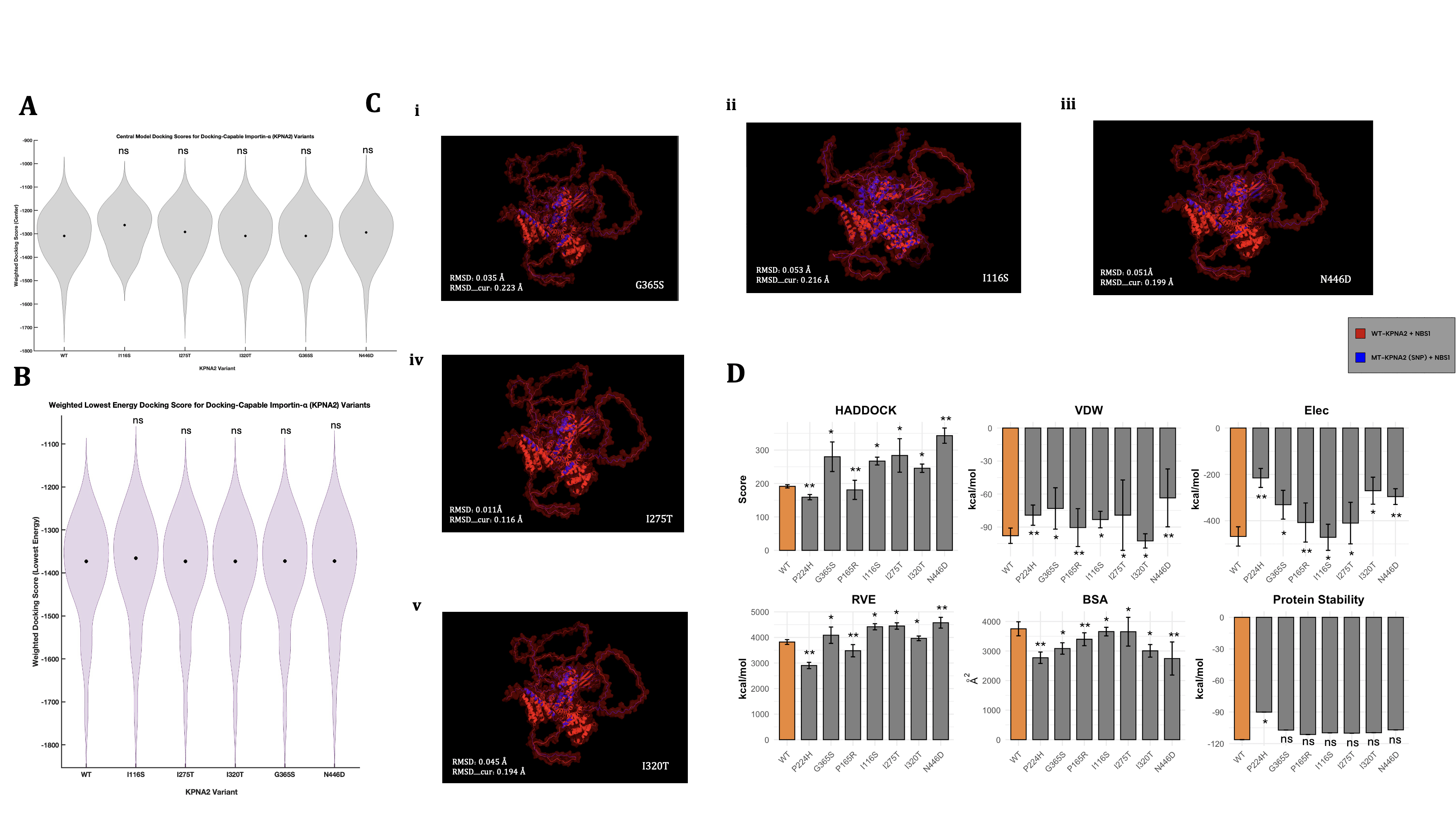
In order to quantify successful structural dockings from ClusPro, we can analyze the relative trends between outputted docking scores—scores that describe the overall predicted binding affinity across many test runs. The graph depicting the central version of these scores (Figure 3A) showed that all variant tests exhibited analogous scoring (approx 1300), except I116S, which resulted in a score of approximately -1275. Despite minor fluctuations, I116S was the only variant to exhibit a central docking score greater than the wild-type docking test, possibly signifying weaker binding between the KPNA2 protein and NBS1 when residue 116 contains an isoleucine-serine missense mutation. The I116S test also showed a shorter range of docking scoring (min -1575), whereas all other tests resulted in minimum values of approximately -1750. Moreover, I116S displayed a differing graphical structure than the other tests, with slightly higher scoring density at two other instances (-1200 and -1450). Statistical testing, however, revealed no major differences between the scoring means across any of the variants, including I116S. Alongside central docking scores—which depict the most common and centralized docking prediction—we also assessed the lowest docking score predictions (Figure 3B), which are estimated based on the most energetically favorable (highest binding affinity) docking test across all models. Similarly to Figure 3A, variant I116S showed an elevated median (-1350), while the other tests’ medians hovered around -1375. However, the graphical structure and density of the I116S variant were extremely similar to those of the other tests, contrasting the results from Panel 3A. These results, coupled with the location of I116S (Figure 2C), may imply that groove-localized variants could be the largest destabilizers to the importin’s cargo binding abilities. Overall, these biophysically derived scores indicated a limited range of results, especially when aiming to answer specific questions around the nuances of each individual docking test. Moreover, for the tests that did not generate plausible docking structures (P244H and P165R), ClusPro similarly failed to produce docking scores, indicating an incomplete scope of results from these two figures alone. Nevertheless, Figures 3A and 3B did indicate a peculiarity within the I116S-KPNA2 docking tests, which supports previous findings in the visual analysis of SNP mapping on WT-KPNA2.
Although docking scores represent a summarized ranking of each docking test, further analyses must be completed to fully understand the docking relationships between each MT-KPNA2 and NBS1; such methods include the use of root mean square deviation (RMSD) to measure the physical divergence between wild-type vs mutant-type importin-ɑ1 and NBS1 enzyme docking. The manually overlaid docking structure configurations (Figure 3C) visually appeared relatively similar across all tests, with a consistent ratio of blue (MT-KPNA2) vs red (WT-KPNA2) structures. Yet again, the outlier of this trend is SNP I116S (Figure 3Cii), which appeared to visually display more of the blue structure overlaid on the red structure. This distinction, although not quantitative or extremely informative, does imply that the I116S-KPNA2 and NBS1 faced larger structural disruptions compared to the other variants. This structural change, however, did not prevent docking, again suggesting that I116S primarily reduces binding affinity rather than fully disrupting the docking interface, such as missense variants P244H and P165R.
SNP N446D (Figure 3C(iii)) also displayed a slight increase in blue appearance, yet not nearly as evident as the I116S-KPNA2 docking model, and therefore, insignificant. For quantitative analysis of the same structural differences, we used RMSD to pinpoint the exact severity of the structural separations. The RMSD (avg=0.039 Å) and RMSD_cur (avg=0.189 Å) scores remained low and consistent across all variant testing, with the only slight outlier being SNP I116S, as expected, resulting in the highest of both types of RMSD scoring. Mirroring Figures 3A, 3B, and 3C, the RMSD docking simulations indicated consistent structural divergencies across all tests, except I116S, which again showed higher deformations of some sort. It is important to note that the conclusions that arise from the visual analyses executed in Figure 3C are relative, as a control relationship between blue and red cannot be accurately measured without proper experimental testing and is solely based on comparisons to a wild-type computational test.
Alongside the usage of structural docking predictions from ClusPro, our study executed parallel biochemical and energetically-focused docking tests in HADDOCK; this choice aimed to identify key incongruities between varying docking methods, especially two that use contrasting scientific procedures. To evaluate how each missense mutation impacted KPNA2’s interaction with NBS1, a range of biochemical and structural metrics from HADDOCK were analyzed and visualized (Figure 3D). Most variants weakened the interaction, as indicated by higher HADDOCK scores—reflecting less favorable binding energies—compared to the wild-type. The most extreme disruption came from N446D, which consistently produced the weakest performance across all key categories, including HADDOCK score, Van der Waals energy, electrostatics, restraint violation energy, and buried surface area. These results could strongly suggest a broad and severe destabilizing effect of N446D on the KPNA2-NBS1 complex, as well as the possibility of it being classified as an allosteric destabilizer.
In contrast, the findings from the P165R and P244H were incredibly discernible. While all other variants resulted in weaker biochemical interactions, these two were the only variants to produce lower HADDOCK scores than the wild-type, suggesting improved binding affinity of a moderate amount. Both also showed lower restraint violation energy (RVE), indicating fewer structural or energetic conflicts in the predicted docking. However, when compared alongside the ClusPro results, where P165R and P244H were the only variants that failed to bind to NBS1 at all, the conclusions become quite complex. This contradiction—enhanced biochemical scores in HADDOCK but no physical binding observed in ClusPro—could imply that while these mutations may favorably alter the overall energy of the docking interaction, they could simultaneously heavily interfere with docking plausibility in three-dimensional space, ultimately preventing a physical complex from being computed. Otherwise, both variants still demonstrated reduced BSA, VDW, protein stability, and electrostatic energy compared to the wild-type, following the general trend of more solvent-exposed and potentially destabilized mutations.
Specifically across other variants, these trends demonstrated less variability. For instance, I320T uniquely exhibited improved van der Waals energy—suggesting tighter atomic packing—but was not distinguishable in any other metric. Meanwhile, FoldX-predicted stabilities remained similar to the wild-type for most variants, except P244H, which again deviated with a significant decrease in folding energy, reinforcing the notion that its effects are distinct from the other SNP tests; this finding is similar to the predictive metrics that were obtained from ProtVar (Table 1). Taken together, these results point toward a broader pattern of weakened KPNA2-NBS1 interaction across most variants, with most metrics having indicated specific missense variants that markedly trump over others, in terms of severity and conformational impact. Moreover, when considered as a whole, Figure 3 highlights P165R and P244H as uniquely contradictory cases, as they both exhibit signs of energetic favorability, yet ultimately fail to bind in structural docking simulations. Contextualizing these findings, these two variants are located nearby and in the same binding region (Figures 1, 2), suggesting a possible locational linkage between the two variants and inherently providing a reason for the similar effects they have on KPNA2.
To validate our docking pipeline, we included both a benign KPNA2 variant and a scrambled NLS sequence as controls. The benign variant yielded docking energies and structural quality scores that closely paralleled wild-type across ClusPro, MolProbity, and HADDOCK, consistent with its expected non-pathogenic behavior. In contrast, the scrambled NLS produced no stable complexes in ClusPro and returned non-physical or unrealistic values in HADDOCK, confirming that the pipeline does not generate plausible results from biologically meaningless inputs. Together, these controls support the specificity and reliability of the docking analyses.
Transcriptomic Landscape of KPNA2 and Genomic Analysis in Cancerous and Non-Cancerous Tissues
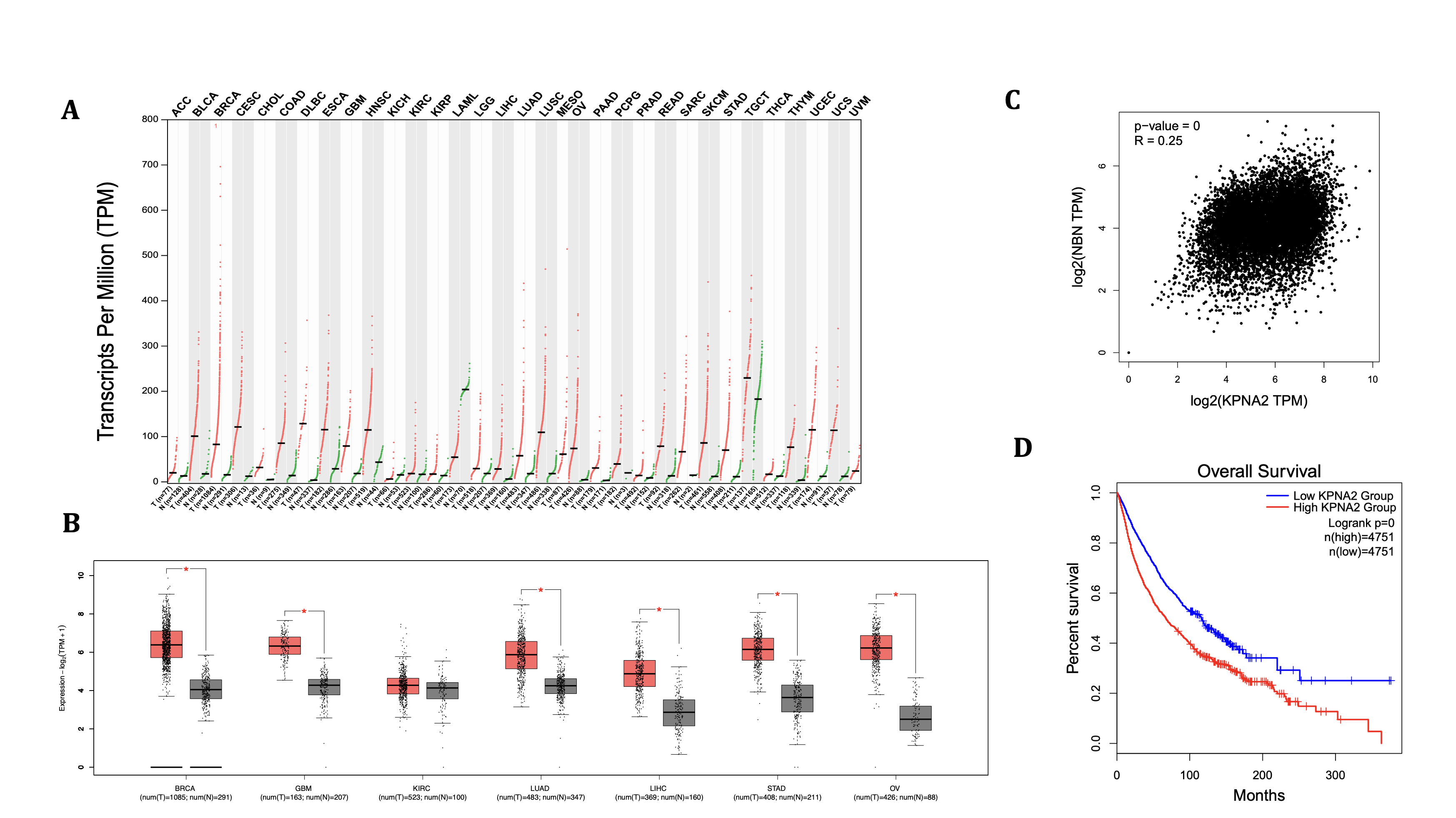
Although missense mutations in KPNA2 can be detrimental regardless of the cargo used for binding, NBS1 was specifically chosen due to its critical role in double-stranded break repair (DSBR). The usage of a DNA repair enzyme adds biological significance, as missense mutations in KPNA2 will have a larger amplification consequence if the cargo being bound is a repair enzyme. Furthermore, correlations could also be created to link specific SNPs to possible cell-wide DNA repair failure. Regardless of SNP mutations, however, KPNA2 expression, correlation, and impacts on survival levels in cancers can help identify oncogenic linkage between importin-ɑ production and cellular responses (Figure 4). Moreover, the gene’s expression can help solidify the exact consequential effects that damaging missense mutations will have on the cell. Across the tissue profile (Figure 4A)—which consists of 33 various tissue and cancer types—KPNA2 gene expression in each tissue is elevated in the cancerous samples compared to their respective non-cancerous tissues. Kidney chromophobe (KICH) and acute myeloid leukemia (LAML) are the only instances, however, where the median gene expression (TTM) of the non-cancerous tissue (KICH=15.37, LAML=203.97) is greater than that of the cancerous tissue (KICH=6.22, LAML=54.44), indicating severe genetic downregulations. In comparing the median gene expression scores, the cancerous tissue that ranked the highest was TGCT (229.51), with the highest median for non-cancerous tissue being the aforementioned LAML test (203.97). Spreads of expression levels do vary across the KPNA2 profile; yet, BRCA had the highest range, with its highest (cancerous) expression value at approximately 690, indicating possible heterogeneity within the collected data.
While the expression profile revealed broad overexpression of KPNA2 across cancerous tissues, the box plot (Panel 4B) offers a refined view of seven representative tissue types. In stomach adenocarcinoma (STAD) and ovarian cancer (OV), for instance, cancerous samples (STAD=70.07, OV=73.81) showed markedly elevated KPNA2 levels compared to their non-cancerous counterparts (STAD=11.47, OV=4.68), with relatively tighter distributions, suggesting consistent overexpression. In contrast, kidney renal clear cell carcinoma (KIRC) displayed minimal differential expression (T=18.4, N=16.61), implying a more scarce role of KPNA2 in this tissue. Despite the size of their separations, all the randomly selected tissues in Figure 4B showed higher expression levels in cancerous tissues, implying that a consistent and predominant role of KPNA2 expression exists in most cancerous tissues. This correlation exists (to our knowledge) without the addition of majorly deforming SNPs in KPNA2, signifying that the total linkage between the KPNA2 protein and DNA repair integrity is substantial.
Cross-expression—the instance where two genes are co-regulated in a way where their expressions are biologically corresponding—can also be analyzed between two genes’ expressions in cancerous tissues. To further explore the biological relationship between KPNA2 and NBN, beyond the fact that they encode physically interacting proteins, a Pearson correlation plot was used (Figure 4C) to display the genes’ relationships within both cancerous and non-cancerous tissue variations. The correlation coefficient (R) for the plot was 0.25, demonstrating a positive yet mildly strong correlation between the two genes’ expressions, regardless of the tested tissue’s malignancy. Despite the relatively weak correlation, the p-value (<0.0001) does suggest an extreme likelihood of achieving the observed correlation coefficient of 0.25 and indicates the relationship as statistically significant; yet, the biological significance is insufficient to make any formal conclusions.
As a final analysis of the genomic landscape of KPNA2, we aimed to connect our study to clinical data and explore whether any trends exist between KPNA2 expression and the overall survival (OS) rates of patients with varying KPNA2 expressions (Figure 4). Kaplan-Meier OS curves, which compare patients with low (Group A) vs high (Group B) KPNA2 expression across 33 tumor types (based only on tumor expression levels), demonstrated lower percent survival in Group A across the entire span of the plot, with some minor fluctuations in differences between the two groups. The identical p-value (< 0.0001) as Figure 4C indicates strong statistical significance, supporting the association between KPNA2 overexpression and poor cancer prognosis. As previously found, KPNA2 appears to have clear biological effects and connections to cancer development, regardless of the SNPs expressed in the gene, as its expression skyrocketed drastically, and patient survival in the high-expression group declined exponentially over 400 months. Therefore, these SNP mutations appear to induce biologically and clinically significant disruptions in both in vitro and in vivo cellular environments.
Discussion
Domain-wide SNP analysis (Figures 1A, 1C) across the ARM repeats of KPNA2 revealed intriguing patterns in the relationship between SNP density and predicted impact. Although the correlation—where the most severe SNPs are located in the least dense regions of the gene—does not follow a linear or easily modeled pattern, it does reflect unique sensitivity. This insight could form the basis of a broader hypothesis regarding ARM-specific vulnerability to missense mutations, specifically the relationship between density and severity, and supports further investigation across larger SNP datasets. Furthermore, the lack of clustering in the ΔΔG analysis (Figure 1E) suggests that SNPs across any residue of the protein could cause high pathogenicity and structural damage, challenging the common assumption of extremely preeminent vulnerability in only certain KPNA2 domains or regions (e.g., the IBB domain). In prior studies, even a single amino acid deletion within the IBB domain was shown to reduce nuclear import efficiency by ~50%, indicating domain-specific sensitivity38. Our study emphasizes that this severe vulnerability extends across numerous regions, especially ARM domains, of KPNA2. These findings are based on a dataset of 35 SNPs; however, broader generalizations would require a full-domain-wide analysis to be validated.
Structural modeling of KPNA2 in various formats (Figure 2) revealed key nuances that may affect its function and binding affinity. When comparing the proteomic location and predictive metrics of each SNP (Figure 2C), our findings hinted that residues closer to the center of the protein may have a higher susceptibility to damaging missense mutations. Although the dataset we analyzed lacks sufficient depth to fully validate this hypothesis, an independent study should be conducted to determine whether SNPs in the center or embedded in the binding groove have more multifaceted effects on the protein. Future studies should also consider the biochemical divergence between wild-type and mutant amino acids—known as a radical replacement—as a key predictor of structural impairment39.
The central and lowest energy docking scores—obtained from ClusPro—for mutant KPNA2 proteins and NBS1 indicated a consistent value across all tests (Figures 3A, 3B); however, it is important to note that central docking scores are generally more stable and representative of a larger range of data compared to the lowest docking scores, which could often be outliers.Notably, two previous hypotheses were made (Figure 3): I116S is predicted to be the most damaging to the protein’s binding affinity—supporting the idea that groove-localized missense variants are particularly disruptive—while P244H is predicted to be the most damaging to the protein’s overall structure, highlighting the potential impact of core-localized variants. These hypotheses are based on distinct lines of evidence and are not mutually exclusive.
The results from the RMSD alignment tests (Figure 3C) suggest that missense-induced docking damages can vary in manner (e.g., structural, functional) and severity (I116S vs P244H and P165R). Additionally, the predicted RMSD values observed in all of the tests could have arisen as early as KPNA2 folding or as late as KPNA2-NBS1 docking—this could not be specified through visual or statistical analysis. Wet-lab validation would be required to specify and characterize the precise structural mutation timeline of each missense variant.
The variants examined through HADDOCK metrics revealed many vital insights into the relationship between location, predictability, and amino acid biochemistry. Interestingly, variants P244H and P165R were the only variants to exhibit lower HADDOCK and RVE scores than the wild-type test did, and yet, they were the only ones to fail in ClusPro. These variants were also the only ones located in the direct core of the major binding region, suggesting a possible link between residual location and structural vulnerability. Although the existence of this duality is plausible, a clear answer to why this is true is currently unknown, and future research into a clear breakdown between missense variant-induced structural and biochemical damage is necessary. Accordingly, the large trend of inconsistency between protein software is vivid, such as docking failures in ClusPro but strong HADDOCK scores, or ProtVar-predicted ΔΔG values being moderately distant from their FoldX-ΔΔG values. Interestingly, our original FoldX-based preprocessing pipeline for ClusPro docking consistently failed. Further investigation showed that MolProbity scoring revealed reduced structural integrity after energy minimization, an ironic outcome given that FoldX is designed to improve stability. This consistent observation highlights an important caveat in computational biology and underscores the broader lesson that careful, context-specific software choices are critical. Moreover, we also visualized that the missense variants with the greatest predictive metrics (ΔΔG, CADD, PolyPhen, AlphaMissense) were not the most structurally, pathogenically, or functionally disruptive, indicating a lack of consistency and validity in Ensembl- and ProtVar-based scoring metrics. A deeper exploration and understanding of the mathematical and computational tools used to predict those metrics should be undertaken to improve the reliability and accuracy of these tools. Moreover, while our study used a TI-restraint file for active residues in HADDOCK, further studies with similar intentions should also explore the use of location-relevant active residues per KPNA2 variant (ex. major binding residues for major binding site variants); our study, however, prioritized unbiased restraints to mimic biological accuracy to the highest possible degree.
GEPIA2 was used to analyze KPNA2 gene expression across a variety of cancer types, including non-cancerous tissue counterparts. The profile generated from the data signifies a vast trend of overexpression in cancerous tissues, with a wide majority of tests indicating severe overexpression (Figure 4A, 4B). This conclusion alone indicates a deeper consequence of the results outlined in Figures A-C, as KPNA2 missense variants within cancerswill have larger cellular effects compared to the same mutations in non-cancerous tissues. Interestingly, two issues showed the opposite: downregulation of the gene in certain cancers. These outliers may warrant a peculiar relationship between KPNA2 in certain cancers and illustrate a possible future research question in the overall mechanism of the gene’s expression and consequent dysregulation. Due to the in silico nature of this study, correlation studies are limited in detail, and thus, a reason for our contradictory findings between biological and statistical significance (Figure 4C); this calls for the need for a nuanced wet-lab comparative analysis of the two genes’ expression levels in varying tissues, as genetic co-regulation could be apparent. Finally, since the datasets used for the high vs low survival plots were of equal sample size (Figure 4D), it is possible to imply that the group with higher KPNA2 expression is the group with cancerous tissues rather than non-cancerous alternatives, thus supporting our previous findings in Figure 4.
Besides minimal outliers, the multipurpose data from GEPIA2 signifies a trend of KPNA2 being upregulated in patients with cancerous tissues, as well as a decrease in their chances of survival over a profound period. Severe mutations, such as P244H, P165R, and N446D, may not only inhibit the DNA repair system but also may enhance cancer development or exhibit other major effects on cellular functions. Moreover, the direct link between importin-ɑ and its exact effects on cancer development and growth is currently underexplored by researchers across all scientific fields. Based on the totality of conclusions from this study, we believe that this mechanistic link is the most vital, and further experimental and computational analysis is highly encouraged to explore novel cancer insights and hypothetical KPNA2-based treatments.
Conclusion
The purpose of this study was to model and evaluate the effect of missense mutations in the KPNA2 gene on the structure and function of the importin-α1 protein in cargo transportation. By examining the protein’s predicted ability to transport NBS1, or any DNA repair enzyme, we aimed to see if single-nucleotide mutations, dependent or independent of genomic location, could affect KPNA2’s role critically. The deeper goal of the study was to explore the nuanced differences between effects on protein binding affinity versus structural disruption, and how each depends on a myriad of factors. Our results first indicated that a direct correlation most likely does not exist between SNP density in an ARM region and mutational severity, as many SNPs that were predicted to cause major structural and functional damage were located in relatively unmutated ARM regions. Our results also indicated that SNPs located in the binding groove of the importin do not necessarily have a larger impact on the protein structure, but possibly rather binding affinity, whereas SNPs located closer to the core of the protein may have induced higher structural deformation. This theory contradicts previous claims of generally higher impact mutations being found in only the binding groove and/or the major binding region. Synthesizing all results, a confident claim was concluded: all seven of our chosen missense variants indicated some form of weakened binding affinity or structural instability; however, the quantitative severity of their disruptions was not proportional to the predictive metrics used in Table 1, signifying an incompatibility between energetic and structural bioinformatic metrics and varying protein-protein docking platforms. Additionally, peculiar conclusions were developed involving results from HADDOCK and ClusPro, demonstrating a complex but biological disconnect between structural and biochemical bioinformatic tools. Finally, analyzing KPNA2 expression in cancer datasets illustrated an evident trend of genetic overexpression in almost 32 cancerous tissues, which proves clinically significant in itself. Despite the abundance of tissues with KPNA2 overexpression, however, two tissues displayed downregulation of the gene, an important and rare nuance to further explore. On the contrary, the survival analysis graph supports the major trend of KPNA2 dysregulation in cancers, as patients with higher KPNA2 expression faced a consistently reduced survival compared to those with a relatively lower expression rate. Moreover, the correlation plot between KPNA2 and the gene producing one of its cargo DNA repair enzymes, NBN, demonstrated low to moderate but statistically significant correlation between the genes’ expressions, suggesting possible co-regulation or functional linkage, supporting the biological relevance of the NBS1 docking models.
Although this study primarily examined an isolated system of genomic variants and their effects on protein stability, the intentional inclusion of DNA repair enzymes as cargo in docking simulations could not be understated. We originally hypothesized that at least one chosen variant would lead to critical effects in the importin’s overall stability, and thus, may lead to impacts on the entire transportation system of cargo into the nucleus. Mutations of adequate severity would exhibit a likely chance of risking the stability of the DNA repair process, surpassing isolated effects within one protein, one enzyme, or even a singular DNA repair pathway; the rippled effects would be catastrophic. Based on the results mentioned above, at least 3-4 of the tested missense variants demonstrate extremely weakened binding affinity and/or structural impairment to suggest it as a possible systematic missense impairment. More specifically, the DNA repair enzyme chosen to be docked with our tests, NBS1, has been previously discovered to play a crucial role in DSBR, which in turn, plays an even greater role in repairing DNA in budding cancerous cells25,26. Coupled with the nuanced analysis of KPNA2 overexpression and dysregulation in a wide majority of cancer types, and a decently positive correlation with NBN, incredibly destabilizing missense variants in KPNA2 may inevitably lead to an elevated, multifaceted risk in clinical tumor development. We hope that researchers will use our research as a basis for novel cancer prognosis, biomarkers, or therapeutic targets in the future, aimed at exposing the pathogenic severity of importin-based mutations that compromise DNA repair integrity and endanger cellular homeostasis.
Despite the important conclusions and revelations that this study discusses, limitations did exist when designing and executing this experiment. By nature of the in silico and bioinformatic-based methodology of this study, certain results and hypotheses are strictly hypothetical and may be false due to errors in the virtual tools that were used; these errors could stem from either machine-based inaccuracies or implausible input parameters. Additionally, this study solely employed seven missense variants for its principal tests, signifying that particular conclusions may be inaccurate without a full understanding and analysis of every missense variant located within the ARM regions of the protein. Moreover, the usage of varying docking platforms could result in conflicting results, which may be falsely interpreted and misunderstood if one platform is comparatively more accurate than the other. For future research and experiments within the same scientific field, a corresponding wet-lab validation is vital for confirmation of several results demonstrated in this study, as well as further analyses that were unable to be completed through in silico methodologies; such methods could include: site-directed mutagenesis mediated by CRISPR-Cas9, several binding assays between importin-ɑ/β, and DNA repair enzymes, or Western blotting with nuclear fractionation. Further bioinformatic or experimental studies should use larger-scale quantities for all methodologies, including a minimum of 100 tested missense variants and/or a multitude of varying DNA repair enzymes. Finally, employing a higher quantity of nuanced genomic and proteomic methods, such as molecular dynamics (MD) simulations or patient-specific cancerous tissue datasets, would prove to be useful and likely result in more robust and translational conclusions. In all, our study successfully emphasizes the significance of KPNA2-associated mutations, which can consequently lead to tumor development, and provides an introductory stepping stone for exploration within this scientific niche, experimentally and computationally.
Acknowledgments
I would like to sincerely appreciate Dr. Corrado Mazzaglia (Italian Institute of Technology) for his invaluable mentorship and guidance throughout this study.
References
- B. Di Ventura, B. Kuhlman. Go in! Go out! Inducible control of nuclear localization. Curr. Opin. Chem. Biol. 34, 62–71 (2016). [↩]
- R. A. Pumroy, G. Cingolani. Diversification of importin-alpha isoforms in cellular trafficking and disease states. Biochem. J. 466, 13–28 (2015). [↩] [↩]
- D. Görlich, S. Prehn, R. A. Laskey, E. Hartmann. Isolation of a protein that is essential for the first step of nuclear protein import. Cell. 79, 767–778 (1994). [↩]
- C. Enenkel, G. Blobel, M. Rexach. Identification of a yeast karyopherin heterodimer that targets import substrate to mammalian nuclear pore complexes. J. Biol. Chem. 270, 16499–16502 (1995). [↩]
- D. Palmeri, M. H. Malim. Importin beta can mediate the nuclear import of an arginine-rich nuclear localization signal in the absence of importin alpha. Mol. Cell. Biol. 19, 1218–1225 (1999). [↩]
- J. Lu, T. Wu, B. Zhang, S. Liu, W. Song, J. Qiao, H. Ruan. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal. 19, 60 (2021). [↩]
- The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 53, D609–D617 (2025). [↩] [↩]
- S. Kosugi, M. Hasebe, N. Matsumura, H. Takashima, E. Miyamoto-Sato, M. Tomita, H. Yanagawa. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J. Biol. Chem. 284, 478–485 (2009). [↩]
- E. Uffelmann, Q. Q. Huang, N. S. Munung, J. de Vries, Y. Okada, A. R. Martin, H. C. Martin, T. Lappalainen, D. Posthuma. Genome-wide association studies. Nat. Rev. Methods Primers. 1, 1–21 (2021). [↩]
- U.S. National Library of Medicine. What are single nucleotide polymorphisms (SNPs)? MedlinePlus. https://medlineplus.gov/genetics/understanding/genomicresearch/snp/ (2025). [↩]
- T. Ulrich. Rare genetic variants can reveal much about disease biology. Broad Institute. https://www.broadinstitute.org/news/rare-genetic-variants-can-reveal-much-about-disease-biology (2023). [↩]
- L. Alnoumas, L. van den Driest, Z. Apczynski, A. Lannigan, C. H. Johnson, N. J. W. Rattray, Z. Rattray. Evaluation of the role of KPNA2 mutations in breast cancer prognosis using bioinformatics datasets. BMC Cancer. 22, 874 (2022). [↩]
- Y. Han, X. Wang. The emerging roles of KPNA2 in cancer. Life Sci. 241, 117140 (2020). [↩]
- L. Huang, H.-Y. Wang, J.-D. Li, J.-H. Wang, Y. Zhou, R.-Z. Luo, J.-P. Yun, Y. Zhang, W.-H. Jia, M. Zheng. KPNA2 promotes cell proliferation and tumorigenicity in epithelial ovarian carcinoma through upregulation of c-Myc and downregulation of FOXO3a. Cell Death Dis. 4, e745 (2013). [↩]
- S. C. Dyer, O. Austine-Orimoloye, A. G. Azov, M. Barba, I. Barnes, V. P. Barrera-Enriquez, A. Becker, R. Bennett, M. Beracochea, A. Berry, et al. Ensembl 2025. Nucleic Acids Res. 53, D948–D957 (2025). [↩]
- J. D. Stephenson, P. Totoo, D. F. Burke, J. Jänes, P. Beltrao, M. J. Martin. ProtVar: Mapping and contextualizing human missense variation. Nucleic Acids Res. 52, W140–W147 (2024). [↩]
- J. Cheng, G. Novati, J. Pan, C. Bycroft, A. Žemgulytė, T. Applebaum, A. Pritzel, L. H. Wong, M. Zielinski, T. Sargeant, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 381, eadg7492 (2023). [↩]
- K. J. Karczewski, L. C. Francioli, G. Tiao, B. B. Cummings, J. Alföldi, Q. Wang, R. L. Collins, K. M. Laricchia, A. Ganna, D. P. Birnbaum, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020). [↩]
- S. T. Sherry, M. Ward, K. Sirotkin. dbSNP—database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9, 677–679 (1999). [↩]
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 596, 583–589 (2021). [↩] [↩]
- M. Varadi, D. Bertoni, P. Magana, U. Paramval, I. Pidruchna, M. Radhakrishnan, M. Tsenkov, S. Nair, M. Mirdita, J. Yeo, et al. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 52, D368–D375 (2024). [↩] [↩]
- The PyMOL Molecular Graphics System, Version 3.1. Schrödinger, LLC. [↩] [↩] [↩] [↩] [↩]
- D. Kozakov, D. R. Hall, B. Xia, K. A. Porter, D. Padhorny, C. Yueh, D. Beglov, S. Vajda. The ClusPro web server for protein–protein docking. Nat. Protoc. 12, 255–278 (2017). [↩]
- J. Delgado, A. Radusky, D. Cianferoni, L. Serrano. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics. 35, 4168–4169 (2019). [↩] [↩] [↩]
- S.-F. Tseng, C.-Y. Chang, K.-J. Wu, S.-C. Teng. Importin KPNA2 is required for proper nuclear localization and multiple functions of NBS1. J. Biol. Chem. 280, 39594–39600 (2005). [↩] [↩]
- X. Liu, F. Li, Q. Huang, Z. Zhang, L. Zhou, Y. Deng, M. Zhou, D. E. Fleenor, H. Wang, M. B. Kastan, et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res. 27, 764–783 (2017). [↩] [↩]
- I. W. Davis, L. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. Arendall, J. Snoeyink, J. S. Richardson, D. C. Richardson. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007). [↩] [↩]
- R. V. Honorato, M. E. Trellet, B. Jiménez-García, J. J. Schaarschmidt, M. Giulini, V. Reys, P. I. Koukos, J. P. G. L. M. Rodrigues, E. Karaca, G. C. P. van Zundert, et al. The HADDOCK2.4 web server for integrative modeling of biomolecular complexes. Nat. Protoc. 19, 3219–3241 (2024). [↩] [↩]
- J. Salamanca Viloria, S. Allega, G. A. Colombo, A. Milanetti, L. P. Niccolai, G. Micheletti. An optimal distance cutoff for contact-based protein structure networks using side-chain centers of mass. Sci. Rep. 7, 2838 (2017). [↩]
- S. Mitternacht. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Research. 5, 189 (2016). [↩]
- C. Savojardo, G. Fariselli, P. Lió, R. Casadio. Solvent accessibility of residues undergoing pathogenic variations in humans: from protein structures to protein sequences. Front. Mol. Biosci. 7, 626363 (2020). [↩]
- M. T. Zimmermann, K. A. Porter, R. A. Pumroy, J. B. Dunbrack, G. Cingolani. Modeling post-translational modifications and cancer-associated mutations that impact the heterochromatin protein 1α–importin-α heterodimers. Proteins. 87, 904–916 (2019). [↩] [↩]
- Z. Tang, B. Kang, C. Li, T. Chen, Z. Zhang. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560 (2019). [↩] [↩] [↩] [↩] [↩]
- G. Riddick, I. G. Macara. A systems analysis of importin-alpha–beta mediated nuclear protein import. J. Cell Biol. 168, 1027–1038 (2005). [↩]
- A. A. Panagiotopoulos, G. F. Sykiotis, P. Deloukas, M. D. Kapodistria. The sequence [RRKLPVGRS] is a nuclear localization signal for importin 8 binding (NLS8): A chemical biology and bioinformatics study. Int. J. Mol. Sci. 26, 2814 (2025). [↩]
- S. B. Rathod. Decoding nonspecific interactions between human nuclear transport proteins: A computational study. bioRxiv. doi:10.1101/2021.03.22.436462 (2021). [↩]
- M. Jadidi, A. A. Miryounesi, A. Vakili, H. Mirfakhraie, M. Kahrizi, H. Parsimehr, R. Darvish. Identification of a rare variant in TNNT3 responsible for familial dilated cardiomyopathy through whole-exome sequencing and in silico analysis. Eur. J. Med. Res. 30, 424 (2025). [↩]
- D. Görlich, P. Henklein, R. A. Laskey, E. Hartmann. A 41 amino acid motif in importin-alpha confers binding to importin-beta and hence transit into the nucleus. EMBO J. 15, 1810–1817 (1996). [↩]
- J. Majewski, J. Ott. Amino acid substitutions in the human genome: evolutionary implications of single nucleotide polymorphisms. Gene. 305, 167–173 (2003). [↩]



{kind=link}