
Abstract
Background: Parkinson’s disease is a degenerative disorder defined by the loss of dopaminergic cells within the substantia nigra. The degeneration is associated with Lewy bodies, aggregates of the α-synuclein protein. Research suggests that metals and other environmental toxins affect α-synuclein aggregation; however, the underlying mechanisms remain poorly understood. This review aims to summarize current evidence on how environmental toxins influence α-synuclein aggregation and identify directions for future research.
Methods: A systematic review following PRISMA guidelines was conducted on PubMed using predefined keywords related to Parkinson’s disease, α-synuclein, and environmental exposures. After screening 553 studies, five primary research articles were selected for analysis.
Results: Three included papers focused on copper (Cu²⁺ and Cu⁺), two on iron (Fe³⁺), and one on zinc (Zn²⁺). Cu²⁺ bound with high affinity to non-acetylated wild-type α-synuclein and accelerated aggregation, but N-terminal acetylation, the modification present in the human form, significantly reduced this effect. Cu⁺ binds through a different mechanism and, under membrane-like conditions, protects against oxidative damage. Fe³⁺ showed concentration- and variant-dependent effects: it accelerated aggregation of non-acetylated wild-type at low concentrations but slowed it in the A53T familial mutation. Zn²⁺ had only modest effects.
Conclusion: This systematic review clarifies the mechanistic interplay between metal exposure and α-synuclein aggregation, providing insights into how metals bind and influence this process. It also highlights the importance of acetylation and mutations in metal-protein binding. These findings offer valuable insights into the role of metal ions in α-synuclein aggregation, potentially guiding future research and therapeutic strategies for Parkinson’s disease.
Keywords: α-synuclein, environmental toxins, Lewy bodies, metal ions, protein aggregation
Introduction
As described by James Parkinson in his 1817 publication, “Essay on the Shaking Palsy”1, Parkinson’s disease is a neurodegenerative disorder of middle and late life. Motor symptoms for Parkinson’s include bradykinesia (slowness of movement), tremors, involuntary movements, rigidity, walking difficulties, and impaired balance. Additional neurological symptoms include multiple non-motor symptoms such as cognitive impairment, mental health disorders, sleep problems, and sensory disturbances2.
Parkinson’s disease is characterized by the loss of dopaminergic cells within the substantia nigra pars compacta2, linked to the accumulation of Lewy bodies – abnormal aggregates primarily consisting of misfolded α-synuclein proteins2,3. α-Synuclein is a small 140-amino-acid protein with three distinct regions: a lipid-binding amino-terminal region, a central hydrophobic domain involved in oligomerization, and an acidic carboxy-terminal segment4. These are referred to as the N-terminal, NAC region, and C-terminal. Approximately 85% of Parkinson’s cases are idiopathic (with no known cause)4. However, evidence suggests a positive correlation between industrialization and Parkinson’s disease cases. This trend is partly attributed to increased exposure to heavy metals commonly used in industrial processes, which may contribute to the disease’s prevalence5.
Candelise et al. published a review6 about the role of environmental factors in α-synuclein aggregation. Metal ions were found to induce a more aggregation-prone structure of α-synuclein by neutralizing charge repulsion, with positively charged ions reducing electrostatic repulsion by binding to α-synuclein, specifically at carboxylate groups. Similarly, polyamines such as putrescine, spermine, and spermidine, which are involved in cellular toxicity in Parkinson’s disease, enhance α-synuclein aggregation. The previous review highlighted the different toxins that affect α-synuclein aggregation; however, the mechanistic functions of how each toxin interacts with the protein remain unclear.
The objective of this systematic review is to build on the literature following Candelise et al.’s 2020 publication6 by examining recent evidence on how specific metal ions bind to α-synuclein and mechanistically drive or inhibit its aggregation. Metal ions, particularly Cu²⁺, Cu⁺, Fe³⁺, and Zn²⁺, are consistently elevated in the substantia nigra of Parkinson’s disease patients, yet the molecular mechanisms by which they influence protein misfolding remain incompletely understood.7,8,9 By clarifying these mechanisms, this review aims to identify gaps in the current literature and suggest directions for future research into Parkinson’s disease biomarkers and therapeutic targets.
This review is organized as follows. The Methods section explains how studies were identified and selected. The Results section summarizes the five included papers, organized by metal ion, covering binding locations, effects on aggregation, and structural consequences. The Discussion section synthesizes these findings into a broader picture of how metal exposure may contribute to Parkinson’s disease, and acknowledges important limitations of comparing results across studies conducted under different laboratory conditions. Figure 1 presents a conceptual overview of the mechanistic pathways reviewed in this study. This review is a mechanistic, narrative synthesis of in vitro studies examining how selected metal ions bind to human α-synuclein and alter its aggregation behavior. It does not attempt a clinical or therapeutic efficacy review, and downstream pathological consequences are discussed only insofar as they are directly supported by the included studies.
Methods
Protocol and search strategy
A systematic approach was employed during the screening of 553 papers to minimize bias, in accordance with PRISMA guidelines10. The database used for the search was PubMed. Initial scoping reviews were performed to survey data on current literature and determine the most appropriate search method. The following MeSH (Medical Subject Headings) terms were used for environmental toxins: “Occupational Exposure/adverse effects”; “Metals, Heavy”; “Air Pollution”; and “Agrochemicals”. Then “alpha-Synuclein”, “Lewy Bodies”, “Parkinson Disease”, and “Lewy Body Disease” were added to the search. The method was finalized as  “Occupational Exposure/adverse effects”[Mesh]) OR (“Metals, Heavy”[Mesh])) OR (“Air Pollution”[Mesh])) OR (“Agrochemicals”[Mesh])) AND “Lewy Bodies”[Mesh]) OR (“alpha-Synuclein”[Mesh])) OR (“Parkinson Disease”[Mesh])) OR (“Lewy Body Disease”[Mesh])).
“Occupational Exposure/adverse effects”[Mesh]) OR (“Metals, Heavy”[Mesh])) OR (“Air Pollution”[Mesh])) OR (“Agrochemicals”[Mesh])) AND “Lewy Bodies”[Mesh]) OR (“alpha-Synuclein”[Mesh])) OR (“Parkinson Disease”[Mesh])) OR (“Lewy Body Disease”[Mesh])).
A filter was added to include literature published after 2020 to find data published later than the paper by Candelise et al6. The final search was conducted on April 13, 2025.
Study Inclusion and Exclusion Criteria
As this review focuses on human α-synuclein, only experimental studies testing the defined toxins on human α-synuclein expressed in microorganisms were included. This focus was intended to ensure a high-quality review of how these toxins mechanistically affect human α-synuclein and contribute to Lewy body-related diseases such as Parkinson’s, diseases that affect humans and would present differently in non-human species. Environmental toxins were defined as inorganic substances commonly encountered through environmental exposure, such as in air, water, or soil, rather than through ingestion. To evaluate the extent of new research conducted and identify significant developments, only studies published after Candelise et al.6 were included. Review papers and studies in languages other than English were excluded. Table 1 presents the eligibility criteria for studies included in the final review sample.
| Criteria | Inclusion | Exclusion |
|---|---|---|
| Population | Human α-synuclein | Non-human α-synuclein |
| Intervention / Exposure | Metals Air pollution Pesticides Agrochemicals | UV radiation Electromagnetic radiation Diet Smoking Caffeine Viruses Bacteria |
| Comparator | None | — |
| Outcome | Mechanism explaining α-synuclein aggregation | — |
| Study Characteristics | Any | Non-English Article prior to 1/1/2020 Review Paper |
Study Selection
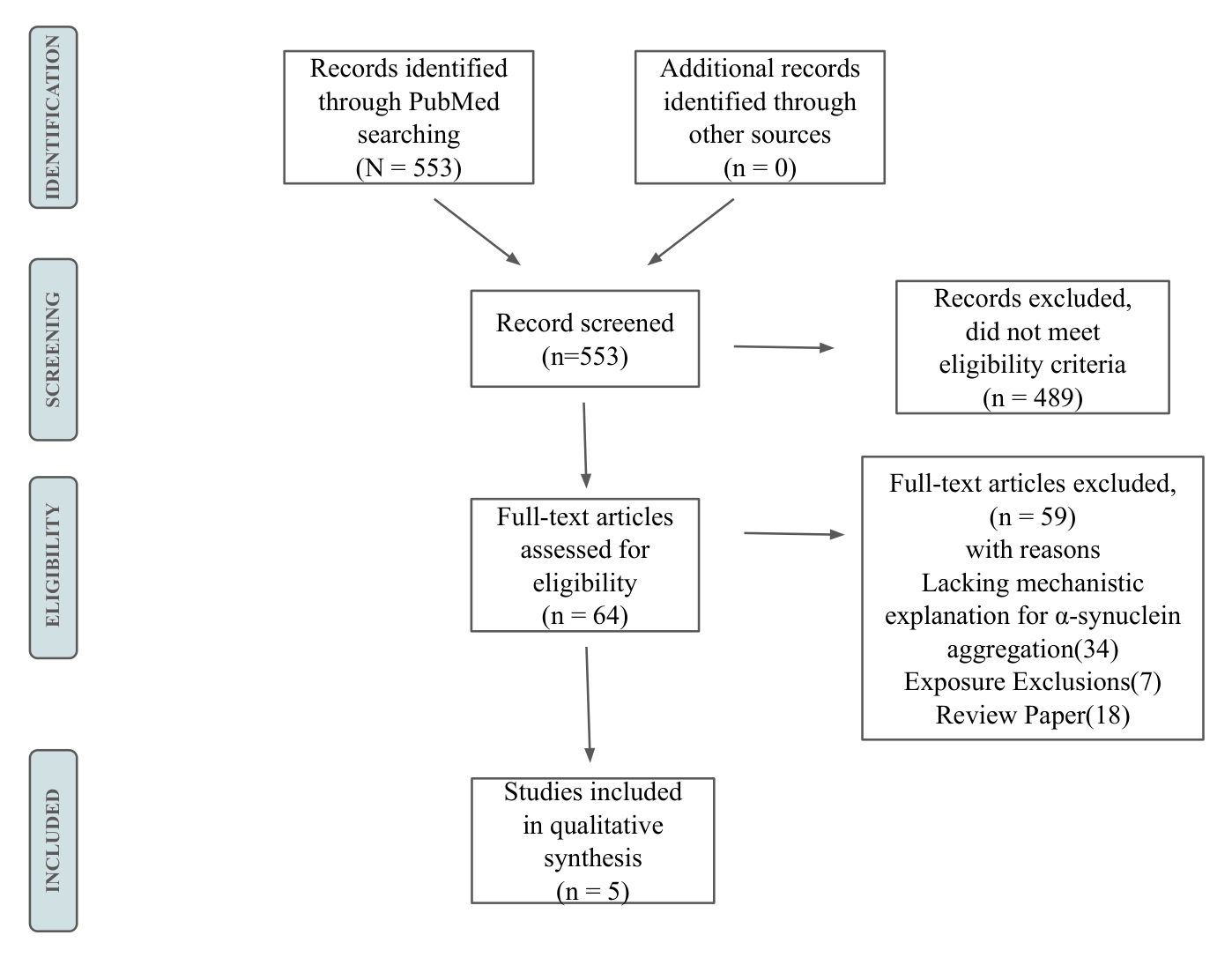
A total of 553 references were exported from PubMed into the systematic review management software Covidence11 to facilitate the screening process. Titles and abstracts were screened based on the inclusion criteria. A second reviewer cross-checked, and discrepancies or papers not clearly meeting criteria were resolved through discussion. Articles included after screening were then exported to the reference management software Zotero for full-text screening. Sixty-four full-text articles were then screened, with reasons noted for each exclusion. Ultimately, five articles were included in this review. Figure 2 presents a PRISMA flowchart illustrating the search and screening process, including reasons for exclusions from the full-text screening.
Data extraction
Data on the binding of metals to α-synuclein were manually extracted from each of the five papers. All data on the effects of toxins on aggregation for individual variants of α-synuclein were extracted. The extracted data were synthesized thematically by grouping findings based on the specific metal ion studied, the α-synuclein variant used, and the resulting impact on protein aggregation.
Meta-analysis consideration
The feasibility of a quantitative data synthesis was evaluated prior to analysis. Formal meta-analysis was determined to be inappropriate due to substantial methodological differences across the five included studies: the studies used different α-synuclein variants, buffer compositions, metal ion concentrations spanning several orders of magnitude, and distinct biophysical assays (ThT fluorescence, AFM, FTIR, NMR, and EPR) with non-interchangeable metrics. Accordingly, a narrative synthesis was employed, organizing findings thematically by metal ion and protein variant rather than pooling effect estimates.
Bias Assessment
To assess potential methodological and interpretive bias across the included studies, a hybrid risk-of-bias framework was constructed by combining elements from SYRCLE’s12 and ROBINS-I’s13 risk-of-bias tools. Since neither tool was directly suited to in vitro biophysical investigations, the bias domains were adapted to address protein chemistry experiments. The final tool incorporated domains for selection bias, performance bias, detection bias, reporting bias, and confounding bias. Each domain was reframed with context-specific guiding questions. Each paper was evaluated against these domains and assigned a rating (low, moderate, or high risk of bias).
| Domain | Lorentzon et al. (2020) | Teng et al. (2021) | Li et al. (2022) | Bacchella et al. (2023) | Walke et al. (2024) |
|---|---|---|---|---|---|
| Selection Bias | Low | Low | Low | Low | Low |
| Performance Bias | Low | Low | Low | Unclear | Low |
| Detection Bias | Unclear | Unclear | Unclear | Low | Low |
| Reporting Bias | Low | Low | Low | Low | Low |
| Confounding | Low | Low | Low | Low | Low |
Results
Study characteristics
The final systematic review comprised five articles14,15,16,17,18. Table 3 presents the characteristics of each article. Each paper investigated one to three toxins using various α-synuclein variants. Four of the five papers provided information about the specific locations of binding for the tested toxins, as shown in Table 4.
| Study | Date of Publication | Primary Objective | α-Synuclein Variants Studied | Toxin Studied |
|---|---|---|---|---|
| Copper Binding and Redox Activity of α-Synuclein in Membrane-Like Environment | 03 February 2023 | Investigate properties of copper-αSyn binding and its redox activity in membrane-like environments | Wild-type αSyn (WT); N-terminal αSyn1–15 peptide; αSyn45–55 peptide; N-terminally acetylated WT αSyn (Ac-WT) | Copper(II) (Cu²⁺); Copper(I) (Cu⁺); Silver(I) (Ag⁺) as a Cu⁺ probe |
| Copper ion incorporation in α-synuclein amyloids | 19 February 2024 | Investigate how copper ions (Cu(II)) interact with αSyn amyloid fibers, specifically their incorporation during aggregation or binding to pre-formed amyloids | Wild-type αSyn (WT); N-terminally acetylated WT αSyn (Ac-WT); αSyn variant His50Ala (H50A); N-terminally acetylated His50Ala αSyn (Ac-H50A) | Copper(II) (Cu²⁺) |
| Differential effects of Cu²⁺ and Fe³⁺ ions on in vitro amyloid formation of biologically-relevant α-synuclein variants | 13 March 2020 | Investigate and compare the effects of Cu²⁺ and Fe³⁺ ions on amyloid formation in specific AS variants | Wild-type αSyn (WT); N-terminally acetylated WT αSyn (Ac-WT); Ala53Thr αSyn (A53T); N-terminally acetylated Ala53Thr αSyn; truncated αSyn (residues 1–97) | Copper(II) (Cu²⁺); Iron(III) (Fe³⁺) |
| Acetylation Rather than H50Q Mutation Impacts the Kinetics of Cu(II) Binding to α-Synuclein | 07 October 2021 | To investigate the effects of the H50Q mutation and N-terminal acetylation on the kinetics of Cu²⁺ binding to αSyn | Wild-type αSyn (WT-αSyn); H50Q αSyn (H50Q WT-αSyn); N-terminally acetylated αSyn; H50Q NAc-αSyn | Copper(II) (Cu²⁺); Copper(I) (Cu⁺) |
| Modulation Effects of Fe³⁺, Zn²⁺, and Cu²⁺ Ions on the Amyloid Fibrillation of α-Synuclein: Insights from a FTIR Investigation | 01 December 2022 | Investigate modulation effects of Fe³⁺, Zn²⁺, and Cu²⁺ ions on the amyloid fibrillation of α-synuclein at both the secondary and quaternary structural levels | Wild-type α-synuclein | Copper(II) (Cu²⁺); Zinc(II) (Zn²⁺); Iron(III) (Fe³⁺) |
Review of α-Synuclein Variants and Toxin Effects
Before examining each metal individually, it is important to clarify how Cu²⁺, Cu⁺, Fe³⁺, and Zn²⁺ differ chemically, because these differences directly determine how each metal behaves when it encounters α-synuclein. Cu²⁺ and Fe³⁺ are both redox-active, meaning they can gain or lose electrons and, in doing so, generate reactive oxygen species (ROS), which are chemically unstable molecules that can damage proteins and other cellular components. Cu⁺ is also redox-active in free solution, but under certain conditions its behavior is very different, as described below. Zn²⁺, by contrast, is redox-inactive and cannot participate in these damaging reactions. Each metal also binds to a different region of α-synuclein: Cu²⁺ targets the N-terminal (beginning) region of the protein; Cu⁺ also uses the N-terminal region but binds through different atoms; Fe³⁺ binds to the C-terminal (end) region; and Zn²⁺ interacts with negatively charged side chains spread across the protein. These chemical and structural differences mean that each metal influences aggregation through a distinct pathway, and results from one metal cannot be assumed to apply to the others.14,15,16,17,18
Cu²⁺ (Oxidized Copper)
Three of the five included studies examined how Cu²⁺ binds to α-synuclein, and all three agreed on the key binding region. Using NMR spectroscopy, a technique that detects changes in the magnetic signals of specific atoms when a metal binds nearby, Bacchella et al.14 showed that Cu²⁺ attaches to the very beginning of the protein at the nitrogen atom of the first amino acid (Met1) and the nearby Asp2 residue. Additional measurements by electron paramagnetic resonance (EPR) spectroscopy, which detects unpaired electrons in metals, confirmed this location. Walke et al.15 independently confirmed binding in the same N-terminal region and additionally found that a second site, the amino acid histidine at position 50 (His50), is required for copper to become fully trapped in protein fibrils once they form. Teng et al.17 measured exactly how fast and how tightly Cu²⁺ binds using rapid-mixing experiments. In non-acetylated wild-type (WT) α-synuclein, the dissociation constant (Kd), a standard measure of binding strength where a lower value indicates tighter binding, was approximately 3 nanomolar (nM), confirming this is an extremely strong interaction. However, in the physiological human form of the protein, which carries a chemical tag called N-terminal acetylation (NAc) on its first amino acid, the N-terminal binding site is blocked. As a result, Cu²⁺ shifts to binding at His50 instead, the binding strength drops dramatically to a Kd of approximately 23 micromolar (μM), nearly 8,000-fold weaker, and the rate at which Cu²⁺ first attaches falls roughly 1,300-fold.17
The effect of Cu²⁺ on aggregation depends heavily on which form of α-synuclein is present. This was tested by monitoring protein aggregation with Thioflavin T (ThT) fluorescence, a dye that gives off a brighter signal as more amyloid fibrils form, allowing researchers to track the speed of aggregation over time. Walke et al.15 showed that Cu²⁺ accelerates aggregation of non-acetylated WT α-synuclein and of the H50A mutant (a modified version where histidine at position 50 is replaced with alanine), but has no accelerating effect on the acetylated NAc-WT form – the form found in humans. Lorentzon et al.16 further showed that the Parkinson’s-associated A53T mutation, in which threonine replaces alanine at position 53, also showed no increase in aggregation rate when Cu²⁺ was added. The likely explanation is that the A53T mutation already makes the protein more prone to misfolding on its own, so adding Cu²⁺ does not produce any additional effect. Together, these results show that Cu²⁺ only accelerates aggregation in protein forms that have a free, accessible N-terminal binding site, reinforcing that binding location matters, not just binding itself.
When α-synuclein forms fibrils in the presence of Cu²⁺, the metal actually becomes incorporated into the fibril structure itself, with important consequences. Walke et al.15 quantified how much copper was trapped in the fibrils using a colorimetric assay (a color-change test sensitive to copper concentration) and found that the ratio of copper to protein in NAc-WT fibrils reached as high as 1.9 copper ions per protein molecule. This is higher than the simple 1:1 ratio seen in free solution, which suggests that the cross-linked, stacked architecture of a fibril creates new metal-binding opportunities that do not exist in the individual protein. Atomic force microscopy (AFM), which uses a tiny physical probe to image surfaces at the nanometer scale, confirmed that Cu²⁺ also changes the physical structure of the fibril: the repeating twist pattern (pitch) of WT fibrils shifted from roughly 70-80 nm to about 100-110 nm. Electrochemical measurements showed that Cu²⁺ bound inside a fibril is harder to chemically reduce than Cu²⁺ bound to a single protein molecule, meaning the fibril structure stabilizes the copper and reduces its reactivity. Accordingly, the ability of copper to generate damaging radicals followed this order: free copper > copper bound to a single protein > copper trapped in a fibril.15 Infrared spectroscopy (FTIR) performed by Li et al.18 further showed that Cu²⁺ causes a modest increase in the proportion of the protein locked into a tightly packed β-sheet structure, from a baseline of about 39.5% to 43.6% at the highest concentration tested, confirming that Cu²⁺ influences the protein’s internal architecture even after fibril formation.
In addition to driving aggregation, Cu²⁺ can directly damage the α-synuclein protein through oxidative chemistry. Copper can cycle between its two charged forms, Cu²⁺ and Cu⁺, and each cycle through this reaction, known as Fenton-like chemistry, releases reactive oxygen species (ROS) such as hydroxyl radicals (•OH) and hydrogen peroxide (H₂O₂). These ROS can then chemically modify the protein itself. Bacchella et al.14 tracked these modifications using mass spectrometry (a technique that weighs molecules and fragments to identify chemical changes) and found that after 30 minutes, the sulfur-containing amino acids Met5 (85% modified) and Met1 (38% modified) were the most heavily damaged. Over four hours, additional chemical modifications appeared at His50. These oxidative modifications occur at the very sites where copper binds, meaning that the same interaction that promotes aggregation also progressively chemically damages the protein. This damage could further destabilize the protein’s normal structure and contribute to a self-reinforcing cycle of misfolding and aggregation.
Cu⁺ (Reduced Copper)
The reduced form of copper, Cu⁺, binds α-synuclein through an entirely different mechanism and has different functional effects. Bacchella et al.14 used X-ray absorption spectroscopy (XAS), a technique that identifies how a metal is connected to surrounding atoms by measuring which energies of X-rays it absorbs, to characterize this binding. The data showed that one Cu⁺ ion simultaneously bridges two α-synuclein molecules by attaching to the sulfur atoms of Met1 and Met5 from each protein chain, forming a symmetrical, four-sulfur complex (CuS₄). This structure only forms stably when the two protein molecules are held in close proximity, which naturally occurs when α-synuclein is anchored to a cell membrane. Because all four attachment points on copper are occupied by sulfur atoms, copper is fully shielded from contact with oxygen molecules in the cell. When Bacchella et al. measured radical-generating activity using a fluorescence-based test, they found that this membrane-bound complex produced almost no ROS, in sharp contrast to Cu⁺ in free solution, which readily reacts with oxygen to generate harmful radicals.14
Because Cu⁺’s tendency to undergo redox reactions can complicate certain measurements, Bacchella et al.14 used silver(I) ions (Ag⁺) as a stand-in to cross-check their structural conclusions. Ag⁺ has a similar preference for sulfur-containing binding sites as Cu⁺ but does not undergo redox cycling under these conditions, making it an easier and cleaner experimental tool. When they compared NMR chemical shift patterns, which indicate which parts of the protein are affected by metal binding, between Ag⁺ and Cu⁺, the patterns were essentially identical. This confirms that the structural model derived from the Ag⁺ experiments accurately reflects how Cu⁺ binds, strengthening confidence in the CuS₄ bis-complex model.
Teng et al.17 showed that the human (N-terminally acetylated) form of α-synuclein binds Cu⁺ with moderate affinity (Kd ≈ 12 μM) and proposed an interesting protective role for this interaction. In neurons, copper ions are briefly released at synapses during neurotransmission. The authors propose that NAc-α-synuclein could act as a temporary copper buffer, picking up free Cu⁺ and handing it off to dedicated copper-transport proteins before it can cause oxidative damage or trigger aggregation. This proposed function depends on the N-terminal acetylation modification, which is the predominant form in human neurons, making this mechanism potentially relevant to how the brain normally manages copper. This also means that earlier laboratory studies using non-acetylated protein may have overestimated copper’s role in promoting aggregation in real human cells.17
Fe³⁺ (Oxidized Iron)
Unlike Cu²⁺, which targets the beginning of α-synuclein, Fe³⁺ binds at the opposite end of the protein, at the C-terminal region (specifically residues 119-124). Lorentzon et al.16 identified this binding site using near-UV circular dichroism (CD) spectroscopy, which detects small changes in protein shape caused by metal binding. The clearest evidence came from testing a truncated version of α-synuclein that is missing these C-terminal residues (residues 1-97): this shortened form showed no response to Fe³⁺ at all, confirming the C-terminus is required for binding. Because N-terminal acetylation does not affect the C-terminal region, both the acetylated and non-acetylated forms of the protein bind Fe³⁺ at this site.16
The way Fe³⁺ affects aggregation is more complex than Cu²⁺ as it depends on both the concentration used and the specific protein variant. Interestingly, in non-acetylated WT α-synuclein, low concentrations of Fe³⁺ (25-50 μM) slightly sped up aggregation, while high concentrations (200-400 μM) slowed it down.16 In the A53T mutation, Fe³⁺ slowed aggregation at all concentrations tested, with the strongest inhibition seen in the acetylated A53T form. To understand why, Lorentzon et al.16 used seeding experiments, where pre-formed fibril fragments are added to accelerate aggregation, and showed that the inhibitory effect of Fe³⁺ applies only to the very first step of aggregation (nucleation: when the first small clusters of misfolded protein form), not to the elongation step (when fibrils grow by adding more protein). This is mechanistically opposite to Cu²⁺, which promotes aggregation by making the early conformational steps easier. The contrast between Fe³⁺ and Cu²⁺ illustrates why these two metals, despite both being found in Parkinson’s disease brain tissue and both capable of generating ROS, cannot be treated as equivalent.
At the level of fibril structure, Fe³⁺ has the most dramatic effects of the three metals studied by Li et al.18 Using FTIR spectroscopy (infrared spectroscopy that reveals protein structure by measuring how the protein absorbs different frequencies of light), they found that Fe³⁺ increased the proportion of the protein locked into the tightly packed β-sheet structure from 39.5% to 49.0% at 4.0 mM Fe³⁺ – a 24% relative increase, larger than the increases caused by Zn²⁺ or Cu²⁺ at comparable concentrations. The spectroscopy also revealed that Fe³⁺ interacts with two distinct types of carboxylate (negatively charged) side chains within the fibril, while Zn²⁺ and Cu²⁺ only interact with one type. This difference likely reflects the stronger electrostatic attraction of the +3 charged Fe³⁺ compared to the +2 charge of the other metals. AFM imaging showed that Fe³⁺ changes fibril shape most dramatically: instead of smooth, uniform fibrils about 5 nm tall, Fe³⁺ produced irregular, worm-like fibrils with height variations between 4 and 10 nm.18
Zn²⁺ (Zinc)
Zn²⁺ is redox-inactive under physiological conditions, meaning it cannot generate reactive oxygen species on its own, and its effects on α-synuclein are the most modest of the four metals examined. Li et al.18 used FTIR spectroscopy and AFM to characterize Zn²⁺’s effects on fibrils. FTIR showed that Zn²⁺ interacts primarily with the negatively charged (carboxylate) side chains of the protein and causes a moderate, concentration-dependent increase in parallel β-sheet content less than that caused by Fe³⁺ or Cu²⁺ at the same concentrations. AFM showed that Zn²⁺ did not substantially change the overall fibril architecture as fibrils were slightly taller and more rigid than those formed without metal, but their basic structure remained intact. Because α-synuclein is an intrinsically disordered protein (IDP), meaning it does not fold into a fixed stable shape under normal conditions but instead exists as a flexible ensemble of conformations, it is naturally sensitive to the chemical environment around it. However, in the case of Zn²⁺, this sensitivity results in only modest structural changes. The absence of redox activity means Zn²⁺’s contribution to Parkinson’s pathology, if any, is likely through physical crowding or charge shielding effects rather than oxidative damage.18
| α-Synuclein Variant | Cu⁺ (reduced copper) redox-inert in bis-complex | Cu²⁺ (oxidized copper) redox-active | Fe³⁺ (oxidized iron) redox-active | Zn²⁺ (zinc) redox-inactive |
|---|---|---|---|---|
| Wild-type (WT) | Binding:Met1, Met5 (1:2 Cu⁺:αSyn bis-complex)14 No aggregation acceleration; Cu⁺ redox-inert within complex, suppresses ROS generation14,16 | Binding: Met1, Asp2 (Kd ≈ 3 nM); His50 required for fibril trapping14,15,17 Strongly accelerates aggregation; promotes β-sheet structure (+4.1%)14,15,18 | Binding: C-terminal residues 119–12416 Slight acceleration at low [Fe³⁺] (25–50 µM); ↓ slows at high [Fe³⁺] (200-400 µM)16 | Binding: Carboxylate side chains (COO⁻)18 Minimal effect; fibrils slightly taller and more rigid; β-sheet +3.7%18 |
| N-terminally acetylated WT (NAc-WT) | Binding: Met1, Met5 (same as WT; Kd ≈ 12 µM)14,17 No acceleration; proposed copper-buffering function – transfers Cu⁺ to dedicated transporters17 | Binding: N-terminal site blocked; shifts to His50/Asp121 (Kd ≈ 23 µM; ~8,000× weaker)15,17 No acceleration; acetylation largely eliminates Cu²⁺-driven aggregation15,17 | Binding: C-terminal residues 119–124 (acetylation does not affect this region)16 Accelerates at low [Fe³⁺] (25–50 µM); ↓ inhibits at high [Fe³⁺] (200–400 µM)16 | NR |
| A53T (familial Parkinson’s mutation) | Binding: Met1, Asp2 (same affinity as WT)16 No effect on aggregation (variant already predisposed to rapid misfolding)16 | NR | Binding: C-terminal residues 119–12416 Inhibits aggregation at all concentrations tested16 | NR |
| N-terminally acetylated A53T | No binding detected16 No effect on aggregation16 | NR | Binding: C-terminal residues 119–12416 Strongly inhibits aggregation16 | NR |
| His50Ala (H50A) | NR | Binding: N-terminal region intact; still binds Cu²⁺15 Moderate acceleration of aggregation (less than WT, confirming His50 role in fibril trapping)15 | NR | NR |
| N-terminally acetylated H50A | NR | Both key sites disrupted (N-term blocked by acetylation; His50 mutated)15 No binding; no effect on aggregation15 | NR | NR |
| H50Q WT | Binding: Met1, Met517 Aggregation effect NR | Binding: Met1–Asp2; no change in affinity vs. WT17 Aggregation effect NR | NR | NR |
| H50Q NAc | Binding: Met1, Met517 Aggregation effect NR | Binding: Asp121 only; weakest binding of all variants tested17 Aggregation effect NR | NR | NR |
| Truncated αSyn (residues 1–97) | NR | NR | NR |
* Ag⁺ was used by Bacchella et al.14 as a redox-inactive structural probe for Cu⁺, forming similar bis-complexes at Met1/Met5. NMR shift patterns for Ag⁺ were identical to Cu⁺ across all variants tested, validating the Cu⁺ structural model.
** αSyn1–15 and αSyn45–55 peptide fragments were used as methodological controls to confirm binding site assignments (Met1/Met5 and His50, respectively) and are not included as independent variants above.
NR = not reported in the included studies.
Discussion
This review synthesizes findings from five studies published since 2020 on how Cu²⁺, Cu⁺, Fe³⁺, and Zn²⁺ interact with α-synuclein and influence its aggregation. Taken together, these results support a stepwise model of metal-driven aggregation: metal ions bind to specific sites on the protein, this binding changes the protein’s shape, and those shape changes alter the probability that the protein will begin to misfold and form toxic aggregates. The most critical upstream step appears to be metal binding to the N-terminal region, which in non-acetylated WT α-synuclein reduces electrostatic repulsion between neighboring protein molecules, making it easier for them to cluster together and form the seed structures needed to initiate fibril growth. Cu²⁺ is particularly effective at this in non-acetylated protein, consistent with the 3 nM Kd reported by Teng et al.17 However, in the physiologically relevant acetylated form, this binding site is blocked and the Cu²⁺ effect on aggregation largely disappears, a critical nuance for interpreting the disease relevance of in vitro copper data.17
Beyond just the rate of aggregation, the studies reviewed here show that metal ions change the physical character of the fibrils themselves. Cu²⁺ incorporation into fibrils, as shown by Walke et al.,15 alters fibril twist geometry and reduces copper’s ability to generate radicals, suggesting that fibril formation may partly reduce copper toxicity in this regard. Fe³⁺ produces the most dramatic fibril changes: larger increases in β-sheet content and irregular, worm-like morphology by AFM, likely because its +3 charge creates stronger electrostatic interactions than the +2 divalent metals.18 Different fibril structures (polymorphs) are thought to have different spreading and toxicity properties in the brain, so these morphological differences may have direct relevance to disease progression, though this link was not directly tested in any of the included studies.
A second major theme is oxidative stress. Both Cu²⁺ and Fe³⁺ can cycle between oxidation states in a Fenton-like reaction, generating reactive oxygen species (ROS) that damage the protein and surrounding cellular components. However, the included studies also describe three mechanisms that may counter this damage. First, Cu⁺ bound at the membrane in the CuS₄ bis-complex generates almost no ROS, suggesting that membrane-bound α-synuclein may serve a protective copper-detoxifying function.14 Second, N-terminal acetylation reduces Cu²⁺ binding affinity by nearly four orders of magnitude, dramatically limiting the amount of copper that can bind to α-synuclein and trigger redox cycling in human neurons.17 Third, Cu²⁺ incorporated into mature fibrils is redox-dampened relative to free or monomer-bound copper,15 indicating that the protein architecture of the fibril itself partially neutralizes copper’s oxidative potential.
The mechanisms addressed in this review – metal binding, redox cycling, and β-sheet stabilization – represent upstream molecular events in the pathogenesis of Parkinson’s disease. Downstream consequences such as mitochondrial dysfunction, membrane disruption, and impaired proteostasis, which are also implicated in metal-driven neurodegeneration, fall outside the scope of this review because none of the five included studies examined these pathways. Future mechanistic reviews that incorporate cell-based and animal model studies will be necessary to map how the binding events described here connect causally to these broader pathological processes.
Compared to the 2020 review by Candelise et al.,6 which established that metal ions generally enhance aggregation by neutralizing charge repulsion at the protein surface, the present review adds several important layers of specificity. Candelise et al.6 did not distinguish between different protein variants or post-translational modifications; this review demonstrates that N-terminal acetylation, the standard human form, fundamentally changes how copper interacts with the protein. The earlier review also grouped metals together as aggregation enhancers, whereas this review shows that Fe³⁺ can inhibit aggregation in A53T α-synuclein and that Cu⁺ under membrane conditions is protective rather than harmful. These distinctions matter because they determine whether a given metal-protein interaction is likely to contribute to, or protect against, neurodegeneration in a physiologically realistic context.
While the present review is limited to in vitro mechanistic studies, the findings are consistent with in vivo observations. Post-mortem analyses have demonstrated significantly elevated iron concentrations in the substantia nigra of Parkinson’s disease patients, with increases of up to 176% in total iron and 255% in Fe³⁺ compared to age-matched controls.19 Subcellular fractionation studies have further shown that the Parkinson’s disease substantia nigra exhibits both elevated iron and decreased copper specifically in the soluble cellular fraction, directly demonstrating metal dysregulation at the level relevant to protein-metal interactions.20 These observations suggest that the metal-α-synuclein interactions characterized in the in vitro studies reviewed here may be operative in the disease context, though direct confirmation of the specific binding and aggregation mechanisms described here requires further investigation in cell-based and animal model systems.
An important limitation of all five included studies is that they were conducted in vitro, that is, in laboratory conditions using purified protein in solution or on artificial membranes, not in living cells or animal models. In vitro conditions differ from the brain environment in several important ways. Intracellular environments contain many other proteins, lipids, and metabolites that could compete with α-synuclein for metal binding or alter its folding landscape. Real neurons also maintain tightly regulated metal homeostasis through chaperone proteins and transport systems, meaning the effective free metal concentrations available to α-synuclein in vivo are likely much lower than those used in most experiments. For reference, Cu²⁺ concentrations used in the included studies ranged from approximately 1 μM to 200 μM, while the free copper concentration in healthy neurons is estimated to be in the picomolar range. Fe³⁺ concentrations of up to 4 mM were used in FTIR experiments,18 far above physiological levels. These comparisons do not invalidate the mechanistic findings, but they do caution against directly extrapolating in vitro aggregation rates or metal effects to disease conditions without further in vivo validation.
Comparing findings across the five included studies is also complicated by methodological variability. The studies used different buffer systems, protein concentrations, agitation methods, pH conditions, and temperature protocols, all of which are known to influence α-synuclein aggregation kinetics. For example, Li et al.18 used very high metal concentrations (up to 4 mM) to detect FTIR signal changes, while Walke et al.15 and Lorentzon et al.16 worked in the low micromolar range. Aggregation assays were performed using Thioflavin T (ThT) fluorescence in some studies and AFM imaging in others, and not all studies used the same variants of α-synuclein. As a result, direct quantitative comparisons, such as which metal accelerates aggregation most, should be made cautiously. The main conclusions drawn here are qualitative and mechanistic rather than quantitative rankings of metal toxicity.
Limitations
Several limitations should be considered when interpreting this review. First, only five articles met the inclusion criteria, all focused exclusively on metal ions. Although studies on pesticides and other environmental toxins were identified during screening, they were excluded because they did not provide mechanistic data on protein-toxin binding at the molecular level, meaning this review reflects a narrow slice of the full environmental toxin landscape relevant to Parkinson’s disease. Second, this review was conducted using PubMed as the sole database. While databases such as EMBASE, Web of Science, and Scopus would ordinarily be included in a comprehensive systematic search, preliminary scoping confirmed that the relevant primary biophysical literature on metal ion–α-synuclein interactions is concentrated in journals well-indexed by PubMed. Nevertheless, the exclusive use of PubMed represents a limitation, as studies indexed only in other databases may have been missed, and future reviews on this topic should employ a multi-database search strategy.10
Third, the included studies varied substantially in their experimental conditions, including metal concentrations, buffer composition, agitation protocols, and α-synuclein variants used. This makes it difficult to directly compare quantitative findings across papers. This variability also means that a formal meta-analysis, which would pool numerical data from multiple studies to produce a combined statistical estimate, was not appropriate for this review. A meta-analysis would require a sufficient number of studies using sufficiently similar methods; future work that standardizes experimental protocols across laboratories would better enable this type of analysis. Fourth, all included studies were conducted in vitro, limiting the direct applicability of these findings to in vivo or clinical contexts, as discussed above. Future research should prioritize cell-based and animal model studies to test whether the mechanistic relationships identified here hold under biologically realistic conditions.
Conclusion
In conclusion, this systematic review demonstrates that copper and other heavy metals are associated with increased rates of α-synuclein aggregation in non-acetylated WT and H50A αSyn, and A53T αSyn, confirming the general conclusions made in the 2020 review. In contrast, in other variants, aggregation was slower or even inhibited. The findings emphasize the significance of post-translational modifications, such as N-terminal acetylation, and highlight how specific mutations, such as A53T, can alter metal-induced aggregation behavior. These insights into metal binding could inform targeted therapeutic strategies as well as preventative measures for individuals at higher risk of toxin exposure. Future research should investigate the role of other environmental toxins such as pesticides and herbicides and the associated mechanisms leading to α-synuclein aggregation.
Acknowledgments
I want to thank Dr. Anna J. Dreyer for her mentorship and insightful discussions on protein aggregation mechanisms and environmental toxicity and for aiding in the screening process. I am also grateful to Rakhee Sohan and to Dr. Asha Jose for providing feedback on the final draft of this paper.
References
- J. Parkinson. An essay on shaking palsy. Sherwood, Neely, and Jones, London (1817). [↩]
- D. K. Simon, C. M. Tanner, P. Brundin. Parkinson disease: Epidemiology, pathology, genetics and pathophysiology. Clin Geriatr Med, 36, 1-12 (2020). doi:10.1016/j.cger.2019.08.002. [↩] [↩] [↩]
- V. W. Sung, A. P. Nicholas. Nonmotor symptoms in Parkinson’s disease. Neurol Clin. 31, S1-S16 (2013). doi:10.1016/j.ncl.2013.04.013. [↩]
- L. Amaral, M. Martins, M. Côrte-Real, T. F. Outeiro, S. R. Chaves, A. Rego. The neurotoxicity of pesticides: implications for Parkinson’s disease. Chemosphere. 377, 144348 (2025). doi:10.1016/j.chemosphere.2025.144348. [↩] [↩]
- O. Myhre, H. Utkilen, O. Duale, G. Brunborg, J. Hofer. Metal dyshomeostasis and inflammation in Alzheimer’s and Parkinson’s diseases. Oxid Med Cell Longev. 2013, 726954 (2013). doi:10.1155/2013/726954. [↩]
- N. Candelise, M. Schmitz, K. Thüne, M. Cramm, A. Rabano, S. Zafar, E. Stoops, H. Vanderstichele, A. Villar-Pique, F. Llorens, I. Zerr. Effect of the micro-environment on α-synuclein conversion. Transl Neurodegener. 9 (2020). doi:10.1186/s40035-019-0181-9. [↩] [↩] [↩] [↩] [↩] [↩]
- C. R. Fields, N. Bengoa-Vergniory, R. Wade-Martins. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front Mol Neurosci, 12, 299 (2019). doi:10.3389/fnmol.2019.00299. [↩]
- A. Aravindan, M. E. Newell, R. U. Halden. Literature review and meta-analysis of environmental toxins associated with increased risk of Parkinson’s disease. Sci Total Environ, 931, 172838 (2024). doi:10.1016/j.scitotenv.2024.172838. [↩]
- L. Shan, H. J. Heusinkveld, K. C. Paul, S. Hughes, S. K. L. Darweesh, B. R. Bloem, J. R. Homberg. Towards improved screening of toxins for Parkinson’s risk. NPJ Parkinsons Dis, 9, 169 (2023). doi:10.1038/s41531-023-00615-9. [↩]
- M.J. Page, J.E. McKenzie, P.M. Bossuyt, I. Boutron, T.C. Hoffmann, C.D. Mulrow, L. Shamseer, J.M. Tetzlaff, E.A. Akl, S.E. Brennan, R. Chou, J. Glanville, J.M. Grimshaw, A. Hróbjartsson, M.M. Lalu, T. Li, E.W. Loder, E. Mayo-Wilson, S. McDonald, L.A. McGuinness, L.A. Stewart, J. Thomas, A.C. Tricco, V.A. Welch, P. Whiting, D. Moher. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ, 372, n71 (2021). [↩] [↩]
- Covidence. Covidence systematic review software. https://www.covidence.org. [↩]
- A. Hooijmans, R. Tillema, M. Leenaars, M. Ritskes-Hoitinga. SYRCLE’s risk of bias tool for animal studies. BMC Med Res Methodol, 14, 43 (2014). doi:10.1186/1471-2288-14-43. [↩]
- J. P. Sterne, J. Hernán, M. Reeves, R. Savović, A. Berkman, N. Viswanathan, K. Henry, C. Altman, J. Ansari, A. Boutron, D. Carpenter, P. Chan, J. Cochrane, L. Deeks, T. Duval, J. Higgins, T. Holroyd-Leduc, S. Ioannidis, C. M. Khalid, A. Meerpohl, T. Rind, R. S. Shrier, S. H. Sterne, L. Tugwell, D. A. Wells. ROBINS-I: A tool for assessing risk of bias in non-randomized studies of interventions. BMJ, 355, i4919 (2016). doi:10.1136/bmj.i4919. [↩]
- C. Bacchella, F. Camponeschi, G. Furlan, S. Gaggelli, M. L. Bini, S. Spreti, M. Remelli, S. Valensin. Copper binding and redox activity of α-synuclein in membrane-like environment. Biomolecules. 13, 287 (2023). doi:10.3390/biom13020287. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- G. Walke, R. Kumar, P. Wittung-Stafshede. Copper ion incorporation in α-synuclein amyloids. Protein Sci. 33, e4956 (2024). doi:10.1002/pro.4956. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- E. Lorentzon, R. Kumar, I. Horvath, P. Wittung-Stafshede. Differential effects of Cu²⁺ and Fe³⁺ ions on amyloid formation of α-synuclein variants. Biometals. 33, 97-106 (2020). doi:10.1007/s10534-020-00234-4. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- X. Teng, A. Sheveleva, F. Tuna, K. R. Willison, L. Ying. Acetylation rather than H50Q mutation impacts Cu(II) binding to α-synuclein. ChemPhysChem. 22, 2413-2419 (2021). doi:10.1002/cphc.202100651. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Y. Li, Y. Yu, G. Ma. Modulation effects of Fe³⁺, Zn²⁺, and Cu²⁺ on α-synuclein fibrillation: insights from FTIR. Molecules. 27, 8383 (2022). doi:10.3390/molecules27238383. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- E. Sofic, P. Riederer, H. Heinsen, H. Beckmann, G.P. Reynolds, G. Hebenstreit, M.B.H. Youdim. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Transm. 74, 199-205 (1988). doi:10.1007/BF01244786. [↩]
- S. Genoud, B.R. Roberts, A.P. Gunn, G.M. Halliday, S.J.G. Lewis, H.J. Ball, D.J. Hare, K.L. Double. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics. 9, 1447-1455 (2017). doi:10.1039/C7MT00244K. [↩]



{kind=link}