
Abstract
This study focuses on diptericin, which is an antimicrobial peptide (AMP) involved in fly immune systems. It is primarily used against Gram-negative bacteria and its mode of action is to disrupt bacterial outer membranes. We investigated the molecular and structural evolution of Diptericin A across 51 different species in the genus Drosophila and related taxa, focusing on three paralogs: Diptericin A, B, and C. The goal of this paper is to characterize patterns of sequence divergence, gene duplication, and structural variation among Diptericin A, B, and C, and to evaluate whether these patterns are consistent with adaptive evolution. The protein sequences were obtained from a previous study and aligned, then phylogenetic gene trees were built for each paralog and for the entire Diptericin family. AlphaFold was used to predict 3D structures and map each protein. The 3D structures were used to identify structural components of each diptericin, such as the transmembrane pore, and the sequences encoding those components were used to build more focused phylogenetic trees. The Diptericin phylogenies were compared to a comprehensive species phylogeny, revealing that Diptericin B is the likely ancestor to all Diptericins, and that Diptericins A and C were created from an ancestral duplication of Diptericin B and subsequently diverged into what are now known as Diptericin A and Diptericin C. Diptericins A and C subsequently duplicated several more times in different lineages, whereas Diptericin B generally remained as a single copy on most taxa. These results reveal pronounced differences in evolutionary constraint among Diptericin paralogs and identify patterns of divergence and duplication that are consistent with adaptive evolution, generating testable hypotheses about functional diversification.
Keywords: AlphaFold, Antimicrobial peptides, Diptericin, Molecular evolution, Structural diversification.
Introduction
Insects lack adaptive immunity and rely entirely on innate immune defenses for protection against infection1,2. Antimicrobial peptides (AMPs) are a major part of the innate immune system in multicellular organisms. They play an essential role in defense against pathogens, rapidly deployed as biochemical defenses that directly target microbial membranes during early infection. Antimicrobial peptides evolve under pathogen-mediated selection and frequently undergo gene duplication and diversification3,4,5. AMP production and antimicrobial function does not depend on immunological memory or past infection experience. The effectiveness of AMPs is determined by their primary amino acid sequence, which is shaped by evolutionary pressures on the host lineage.
Upon infection, pathogen recognition molecules initiate intracellular signaling through the Toll and IMD (Immune Deficiency) pathways, activating transcription factors that drive expression of antimicrobial peptides. In contrast to the relative conservation of the signaling pathways, effector proteins like AMPs evolve much faster6,7. Previous studies have documented rapid sequence evolution, gene duplication and positive selection in AMP families, and some have examined either phylogenetic relationships or structural features of individual peptides7,8,9. Diptericin is a well-characterized AMP produced by Diptericin insects, comprising an evolutionarily diverse family that provides a useful system for studying how gene duplication and structural divergence shape immune effectors. Comparative genomics has revealed diversity in diptericin genes, including sequence divergence, gene duplication, and the recognition of three subtypes: diptericin A, diptericin B, and diptericin C10,11. Diptericin A has a well-established role in resistance against Providencia rettgeri12, while Diptericin B is more known to have a role in targeting the Acetobacter species in the gut8. Diptericin C is more divergent than Diptericin A and B, which could signal a more specialized function13.
Despite extensive work on antimicrobial peptides and their role in insect immunity, several key questions remain regarding how Diptericin has diversified across different species. The Drosophila clade contains hundreds of species, many of which are located in unique ecological niches with diverse challenges13. It is still unclear how patterns of duplication, sequence divergence and structural differentiation in Diptericin A, B and C manifest across these species. Specifically, most prior analyses have focused on individual diptericin paralogs, without systematically comparing paralogs across a broad phylogenetic scale. Thus, these studies do not distinguish how individual diptericin paralogs differ in evolutionary constraint across structurally defined domains.
Here, we address this gap by integrating comparative phylogenetic analyses, paralog-specific copy number mapping, and AlphaFold-based structural modeling across multiple fly species. Rather than relying solely on full-length sequence comparisons, we divided Diptericin sequences into structurally defined regions and examined their evolutionary patterns separately. This approach allows us to test explicit hypotheses about the ancestral status of Diptericin B, the derivation of Diptericin A and C through duplication, and the extent to which evolutionary divergence is concentrated in specific functional domains as might be predicted under adaptive evolution. By linking gene tree topologies, species relationships and structural features, this study provides a more detailed view of how diptericin paralogs have diversified in response to specific selective pressures.
We hypothesize that diptericin paralogs have experienced heterogeneous evolutionary pressures following gene duplication, resulting in lineage- and paralog-specific divergence patterns. We predict that species with multiple diptericin C copies will show either clusters of near-identical duplicates (consistent with recent duplication) or deeper divergence among retained copies (consistent with longer-term retention and functional differentiation).
This study focuses on Diptericin A, B, and C across Drosophila fly species using available sequence data that enables direct comparison of paralog distribution, copy number variation, and evolutionary divergence across lineages. Amino acid alignments were used to assess sequence divergence to assess sequence divergence, while phylogenetic trees were constructed for full-length proteins as well as for structurally defined regions identified through AlphaFold based modeling. We applied sequence alignment, phylogenetic tree construction, and AlphaFold-based structural modeling.
We only had access to the amino acid sequences so we could not do DNA-based analyses like the dN/dS test. We might also anticipate that synonymous sites could be saturated in comparing the Diptericins across the entire phylogeny, which would compromise the dN/dS test. However, a future study could use codon-based tests for adaptive evolution within specific clades.
Methodology
No human or animal subjects were involved in this study. Diptericin gene sequences were retrieved from a previous paper by Dhakad et al. (2025)14. Diptericin gene sequences obtained from 51 species were collected, including D. melanogaster, D. miranda, D. willistoni, D. suzukii, and others. While building the phylogenetic trees and comparing sequences, we removed one variant of D. suzukii due to its high level of divergence relative to other diptericin sequences. When placed on a gene tree, this variant produced a branch length much longer than any other sequence, indicating substantial divergence from both other species and even other D. suzukii paralogs. We modeled the sequence in AlphaFold and it did not create the open pore structure characteristic of Diptericins [Figure 1 compared to Figure 2]. Since Diptericin functions by destabilizing bacterial membranes, the absence of a pore was inconsistent with its expected activity. We therefore determined that this copy was likely to either be a nonfunctional pseudogene or to have a function that is highly divergent from other Diptericins and we excluded it from the study. All other Diptericin sequences modeled as trimers produced a pore structure. However, to avoid bias and to be transparent, we still show the sequence and its behavior directly in Figures 1 and 2 and discuss multiple explanations (pseudogene candidate vs. misannotation vs. highly diverged variant). We do not have access to raw genomic data for this locus, so we cannot definitively confirm pseudogenization (e.g., via frameshift mutation). We treat that as a limitation of our study.
Protein sequences were aligned using MAFFT and imported into AliView, where the “realign everything” option was applied using default settings15,16. Alignment quality control was limited to visual inspection in AliView to verify overall alignment consistency and correct placement of structural motifs. The alignments were then used to construct gene trees for all sequences of each Diptericin paralog, both for the entire protein sequence and for the pore region of the mature peptide. No automated trimming, masking of sites, or other exclusion of peptide regions was performed, and all aligned positions were retained for downstream analyses. As a result, the alignments contain gaps. Summary alignment statistics, including alignment length, proportion of gapped positions, number of variable sites, and number of parsimony-informative sites, were calculated using AMAS17. Maximum likelihood phylogenetic gene trees were constructed with IQ-TREE 3 using automated model selection and default inference options, including MixtureFinder, MAST, concordance factors, QMaker, CMAPLE, and IQ2MC18. Phylogenetic inference was performed using IQ-TREE 3 with default settings unless otherwise specified. Model selection was automated using MixtureFinder, which evaluates combinations of amino acid substitution models and rate heterogeneity schemes to identify the best-fitting model for each alignment. QMaker was used when appropriate to estimate empirical substitution models directly from the data rather than relying solely on predefined matrices. Multiple-tree mixture models (MAST) were enabled to account for potential heterogeneity in evolutionary histories across sites, particularly in datasets that include paralogs and lineage-specific duplications. Concordance factors were calculated to quantify the proportion of sites and gene histories supporting each internal branch, providing a measure of support that is less sensitive to alignment length than bootstrap values alone. CMAPLE and IQ2MC were used as part of the IQ-TREE 3 implementation to improve likelihood optimization and computational efficiency when analyzing moderately sized protein datasets. These options were chosen to allow data-driven model selection and to better accommodate heterogeneity across diptericin paralogs, rather than to test specific evolutionary models.
To build the trees, we used a GitHub implementation of IQ-TREE18. To run the program, the files “iqtree3” and “libiomp5md.dll” were copied into the working directory. The directory was opened in the command prompt using the cd command (e.g., cd “C:\Users\lisam\Downloads\iqtree-3.0.1-Windows\iqtree-3.0.1-Windows\new”). Once in the correct folder, the tree was generated with the command .\iqtree3.exe -s filename.phy -B 1000 –bnni -nt AUTO, where the input file contained aligned amino acid sequences in .phy format (e.g.,.\iqtree3.exe -s DiptericinA.phy -B 1000 –bnni -nt AUTO). The -s option specifies the input alignment file. Node reliability was assessed using ultrafast bootstrap analysis (-B) with 1000 bootstrap replicates. Branch-length optimization during bootstrap analysis was enabled using the –bnni option to improve the accuracy of support estimates. Computational resources were managed automatically using the -nt AUTO option, which allows IQ-TREE to determine the optimal number of CPU threads based on the system.
Each analysis produced multiple output files, including a maximum-likelihood tree file (.treefile) containing bootstrap support values mapped to internal nodes. These tree files were visualized using the Interactive Tree of Life (iTOL) with all trees displayed in rectangular format for consistency across figures19. For visualization clarity, bootstrap support labels were displayed on trees for nodes with UFBoot ≥70. For the Diptericin A with the omitted suzukii comparison tree, a lower display threshold (41%) was used to illustrate instability in weakly supported regions.
Separate phylogenetic trees were generated for full-length amino acid sequences and individual structural domains (pores). These were contrasted to a published species tree based on complete mitochondrial genomes4. The number of Diptericin A, B, and C paralogs was also mapped onto the species tree to estimate the number of duplications and losses and to clarify how the three paralogs are related.
Diptericin sequences were submitted to AlphaFold version 2 (publicly available implementation as of August 2025) to generate three-dimensional structural models. All predictions were performed using default parameters, with no manual parameter tuning, template enforcement, or custom restraints applied. Structural confidence was assessed using AlphaFold’s per-residue confidence metric (pLDDT), which was used qualitatively to verify that identified structural elements were supported by high-confidence predictions. Structural sites were identified and used to separate amino acid sequences into sections corresponding to each structural domain. The results of the phylogenetic reconstructions and structural mapping were then integrated to assess the evolutionary history of diptericin.
The following procedure was applied to identify structural domains inferred from AlphaFold structural predictions of Diptericin trimers (pore, helices, string).
- Generate trimer models: For each Diptericin amino acid sequence, we generated a trimer structure prediction in AlphaFold (same settings across all sequences; default parameters). Each sequence was modeled three independent times to check consistency.
- Open model & standardize viewing: we viewed each trimer model in the AlphaFold structure viewer in cartoon/ribbon mode with secondary structure displayed (β-sheets vs helices vs coils). We also inspected pLDDT confidence coloring to flag low-confidence segments. In Alphafold and in the corresponding figures in this paper, confidence is shown using color. Cooler colors like blue represent more confidence and orange and yellow represent less confidence
– In this paper, the “pore region” is defined as the β-sheet barrel-like segment that forms the central cavity when the three peptide copies assemble (Figure 1). This pore is predicted to span the bacterial membrane and act as an open channel, destabilizing bacterial cell integrity and leading to lysis and death. In the present study, the pore region is predicted computationally and is not directly functionally measured.
– “P-domain” definition: P-domain is used as a descriptive label for the flexible, low-structure proline-rich motif that is between the pore and the signal peptide. This domain is predicted to be removed from Diptericin B by furin cleavage, but loss of the furin cleavage site means that the P-domain is predicted to remain attached to the pore in Diptericins A and C (Figure 1). We do not know whether it extends into the bacterial cytoplasm or into the extracellular space. There is no confident prediction of the secondary structure of the P-domain. - Mark pore start: Starting at the N-terminus, we move residue-by-residue along the chain (mouse-over residue labeling) until reaching the first residue that is part of the contiguous β-sheet barrel contributing to the central cavity. That index was recorded as pore start.
– If the transition fell inside a low-confidence segment, we still recorded the boundary but flagged it as low-confidence (pLDDT low) in my notes. - Mark pore end: Continuing along the sequence, we recorded the last residue that still belongs to the β-sheet barrel before the structure transitions into a non–β-barrel region (coil/flexible linker, i.e., the “string,” or other non-barrel structure). That residue index was recorded as pore end.
- Consensus across 3 runs: we repeated steps 2–5 for all three AlphaFold trimer runs per sequence. Final pore boundaries were defined using the consensus across runs (the mode start/end positions; if all three differed, we used the rounded mean).
- Documentation: For transparency, we included an annotated screenshot showing an example of pore start identification (mouse-over residue label at the β-barrel transition)
To evaluate these predictions, we focused on a small set of comparative analyses that directly reflect the structure and duplication patterns of diptericin. First, we quantified sequence divergence across diptericin A alignments and compared variability between the full-length peptide and the predicted pore/interface region to test whether divergence is concentrated in functionally exposed domains. Second, for species with multiple diptericin C copies, we examined pairwise amino-acid similarity and phylogenetic clustering among paralogs to determine whether duplicates represent recent expansions or deeper, retained divergence.
Results
Our AlphaFold modeling of Diptericins A, B, and C indicated that each Diptericin is likely to form a trimer. Three copies of the Diptericin peptide assemble into a barrel (Figure 1) that we predict would insert into the membrane of a Gram-negative bacterium, acting as an open pore that would result in bacterial lysis. This hypothesized mode of action is consistent with published reports that Diptericin disrupts bacterial membranes20. We do not know whether Diptericin B can heterodimerize with Diptericin A or C to make chimeric pores. In our dataset there are no genomes that contain both Diptericin A and C (Table 1).
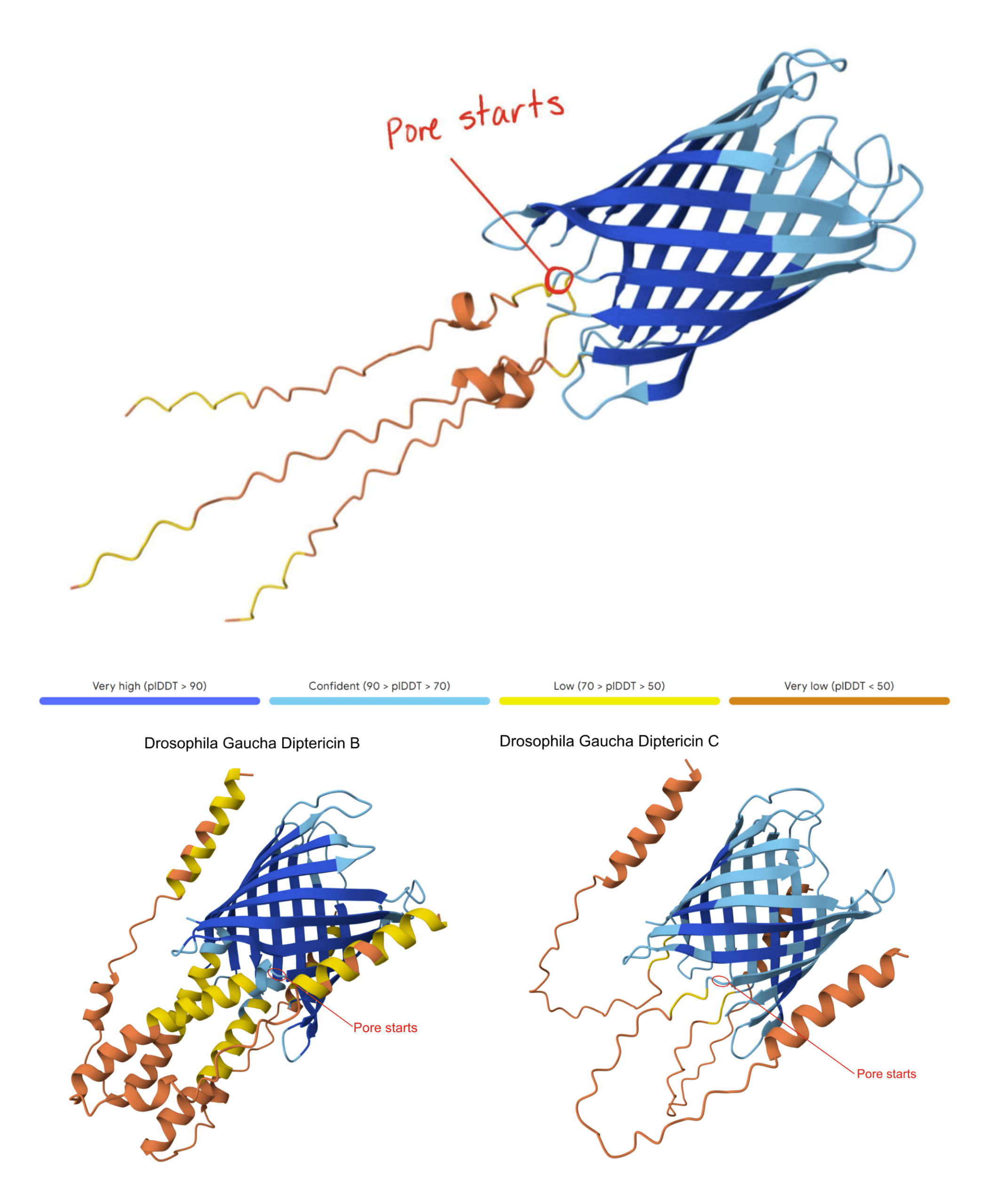
This AlphaFold model shows the modeled Diptericin A peptide from D. setifemur and the Diptericin B and C peptides from D. gaucha with the pore start region indicated in red. To locate the pore, we generated an AlphaFold model of each gene using three copies and identified the consistent region where the β-sheet barrel transitions into the P-domain. We then averaged the amino acid base positions across these models to generalize the pore start for Diptericin sequences. This figure is provided as an example; across species, Diptericin A, B, and C models showed the same overall architecture and a comparable β-sheet barrel–to–P transition. The pore initiation site is defined as the transition between the β-sheet barrel and the P-region (see Methods). Note that the illustrated alpha helices in Diptericin B may be removed from the mature peptide, as there is a furin cleavage site near the start of the pore in Diptericin B. There is no furin cleavage site in Diptericins A or C.

Unlike most diptericin homologs modeled here, this sequence does not yield a pore-like trimeric assembly under the same modeling procedure. This result is consistent with either a non-canonical structural variant or annotation-related issues. Therefore, the sequence is treated as an outlier and is excluded from primary comparative pore analyses.
| Species | A | B | C |
|---|---|---|---|
| Acommunis | 1 | ||
| Ccostata | 4 | ||
| Cfuscimana | 4 | ||
| Dalanassae | 1 | ||
| Dananassae | 1 | 1 | |
| Dalbomicans | 1 | 1 | |
| Dbipectinata | 1 | 1 | |
| Dbusckii | 5 | 8 | |
| Delegans | 1 | 1 | |
| Dflavopinic | 1 | 2 | |
| Dfunebris | 1 | 1 | |
| Dgaucha | 1 | 5 | |
| Dgrimshawi | 1 | 2 | |
| Dhydei | 1 | 3 | |
| Dimmigrans | 1 | 1 | |
| Dinnubila | 1 | 1 | |
| Dkikkawai | 1 | ||
| Dmaculinota | 1 | 2 | |
| Dmelanogaster | 1 | 1 | |
| Dmimica | 1 | 2 | |
| Dmiranda | 4 | 1 | |
| Dmojavensis | 1 | 3 | |
| Dneotestace | 1 | ||
| Dobscura | 1 | 1 | |
| Dpallidipen | 1 | ||
| Dparamelani | 1 | 1 | |
| Dpseudotala | 3 | ||
| Dpruinosa | 1 | 1 | |
| Drepleta | 1 | 4 | |
| Drepletoide | 1 | 1 | |
| Dsetifemur | 3 | 1 | |
| Dsturtevant | 1 | 1 | |
| Dsuzukii | 2 | 1 | |
| Dvirilis | 1 | 1 | |
| Dwillistoni | 1 | 2 | |
| Hcam | 2 | ||
| Hconf | 1 | 3 | |
| Hhistrioide | 1 | 4 | |
| Hduncani | 2 | ||
| Htrivittata | 2 | ||
| Landalusiac | 3 | 1 | |
| Lfenestraru | 3 | 1 | |
| rna-DptB | 1 | ||
| Sdef-DptB | 1 | ||
| Shsui | 1 | 2 | |
| Slebanonens | 1 | ||
| Slatifascia | 1 | ||
| Stumidula | 1 | 2 | |
| Zbogoriensi | 1 | 1 | |
| Zdavidi | 1 | 1 | |
| Ztuberculat | 1 |
This table provides a comprehensive list of which species have diptericin A, B, or C, with copy counts included. It reveals clear lineage-specific patterns. Diptericin B is the most widely conserved, as it is present in nearly all species, often as a single copy. However, there are exceptions, such as D. busckii (5 copies) and C. costata (4 copies). Diptericin A is more variably distributed, with most species having one copy but with significant duplications in D. setifemur (3 copies) and D. miranda (4 copies). Diptericin C shows the greatest duplication in species like D. busckii (8 copies) and D. gaucha (5 copies), while being absent from others. The variation in copy number across species indicates lineage-specific duplication/retention dynamics. These patterns are consistent with ecological or pathogen-mediated selection in some lineages, but other explanations (neutral copy-number change and assembly/annotation artifacts) cannot be excluded given reliance on published genome annotations.
| Alignment | Taxa | Length (AA) | % Missing | % Variable sites | % Parsimony-Informative |
|---|---|---|---|---|---|
| Diptericin A | 22 | 145 | 27.4 | 63.4 | 45.5 |
| Diptericin B | 56 | 204 | 40.2 | 60.8 | 49 |
| Diptericin C | 61 | 204 | 48.6 | 46.1 | 42.6 |
| AC Pore | 83 | 70 | 7.5 | 85.7 | 78.6 |
| ABC Pore | 139 | 71 | 9.4 | 87.3 | 78.9 |
Full-length Diptericin A alignments contained moderate missing data (27–49%) and 46–63% variable sites (42–49% parsimony-informative). In contrast, pore-region alignments showed very low missing data (<10%) and a high proportion of variable and parsimony-informative sites (86–87% variable; ~79% parsimony-informative), indicating that the pore region is information-dense across taxa and provides substantial phylogenetic signal despite its shorter length. Because “variable sites” are counted when any species differs at a position, these values reflect the breadth of substitutions across the dataset rather than average pairwise divergence.
Table 2 displays the alignment statistics for full-length amino acid sequences of Diptericin A, Diptericin B, and Diptericin C, as well as pore-region alignments for Diptericin A & Diptericin C and for the pore region across Diptericin A&B&C [Table 2]. Across full-length alignments, 27–49% of the sites are coded as “missing data” because they fall in alignment gaps, whereas less than 10% of the aligned sites are gapped in the sequence encoding the pore. This indicates that the pore is more consistently alignable than the remainder of the protein, and that this region should give the alignment with the greatest confidence. Consistent with that, 78% of the aligned sites in the pore region are parsimony informative compared to 42-49% across the whole peptide [Table 2]. Diptericin A shows substantial sequence variation despite its smaller size, and its relatively high proportions of variable and informative sites indicate meaningful evolutionary signal rather than noise; missing data are present but not excessive given limited species representation [Table 2]. Despite higher missing data, Diptericin B retains strong phylogenetic signal and its high proportion of parsimony-informative sites supports reliable inference and reflects broad but structured variation across taxa [Table 2]. Diptericin C exhibits substantial missing data consistent with lineage-specific duplication and incomplete annotation across species, yet nearly half the alignment remains variable and informative, supporting real evolutionary divergence rather than an alignment artifact [Table 2]. In contrast to full-length alignments, pore-region alignments show very low missing data (<10%) and exceptionally high variability (>85% variable sites), meaning the most intense divergence is concentrated in this functional domain rather than spread evenly across the peptide [Table 2]. This same pattern holds when pore residues are considered across paralogs (Diptericin A&B&C): pore sequences remain exceptionally variable and informative while maintaining low missingness, which is the qualitative pattern expected if diversifying selection is acting on a functional AMP [Table 2].
We mapped Diptericin paralogs onto a previously published species tree based on mitochondrial genome sequence4. Across the Drosophila phylogeny, mapping paralog presence onto a published species tree shows that diptericin B is conserved in most lineages, while diptericin A and diptericin C occur in more restricted clades [Figure 3]. Species typically retain diptericin B and tend to contain either diptericin A or diptericin C (or neither), but diptericin A and diptericin C do not co-occur within the same species, producing a clear pattern of mutual exclusivity at the species level [Figure 3]. This distribution is consistent with the hypothesis that A and C were derived from ancestral B copies and subsequently evolved along distinct trajectories [Figure 3]. When copy counts are considered in the same framework, some species show strong lineage-specific expansion (e.g., D. busckii, D. miranda, D. repleta), whereas other species maintain only a single copy (e.g., D. sturtevantii, D. willistoni), suggesting that selection may influence not only amino acid divergence but also the number of diptericin gene copies retained in a lineage [Table 1, Figure 3]. Outcomes following duplication are also heterogeneous across species: in some lineages, multiple copies are nearly identical, consistent with recent duplication events, whereas in other lineages, paralogs are deeply divergent and occupy distinct positions in gene trees, consistent with longer-term retention rather than rapid loss [Figure 5, 7]. This within-species contrast matters because it separates “many copies because of very recent expansion” from “many copies because divergent duplicates were retained,” and both patterns show up in the trees [Figure 4, 5, 6]. In the Diptericin C tree, D. gaucha is a good example of the “recent expansion / low divergence” pattern. Its multiple Diptericin C copies are nearly identical to each other, with very short branch lengths and little visible separation, which is what would be expected if the copies duplicated recently and have not had much time to diverge [Figure 6]. In contrast, D. pseudomantica shows the opposite pattern. Its Diptericin C copies are not grouped together as one tight cluster. Instead, they are split across different parts of the tree, which is more consistent with older duplication events followed by long-term retention of divergent duplicates rather than a single recent burst [Figure 6].
Diptericin A shows a similar pattern. In the Diptericin A tree, D. andalusiaca and D. fenestrarum do not form clean within-species clusters. Their copies are interleaved, with sequences from the two species alternating across the same part of the tree, which suggests these duplicates are not just species-specific “extra copies” that arose yesterday. Instead, it fits better with duplication predating the species split (or with recent gene conversion / strong constraint keeping copies similar), and it shows why copy number alone is not enough to interpret duplication as either purely recent expansion or long-term retention [Figure 4].
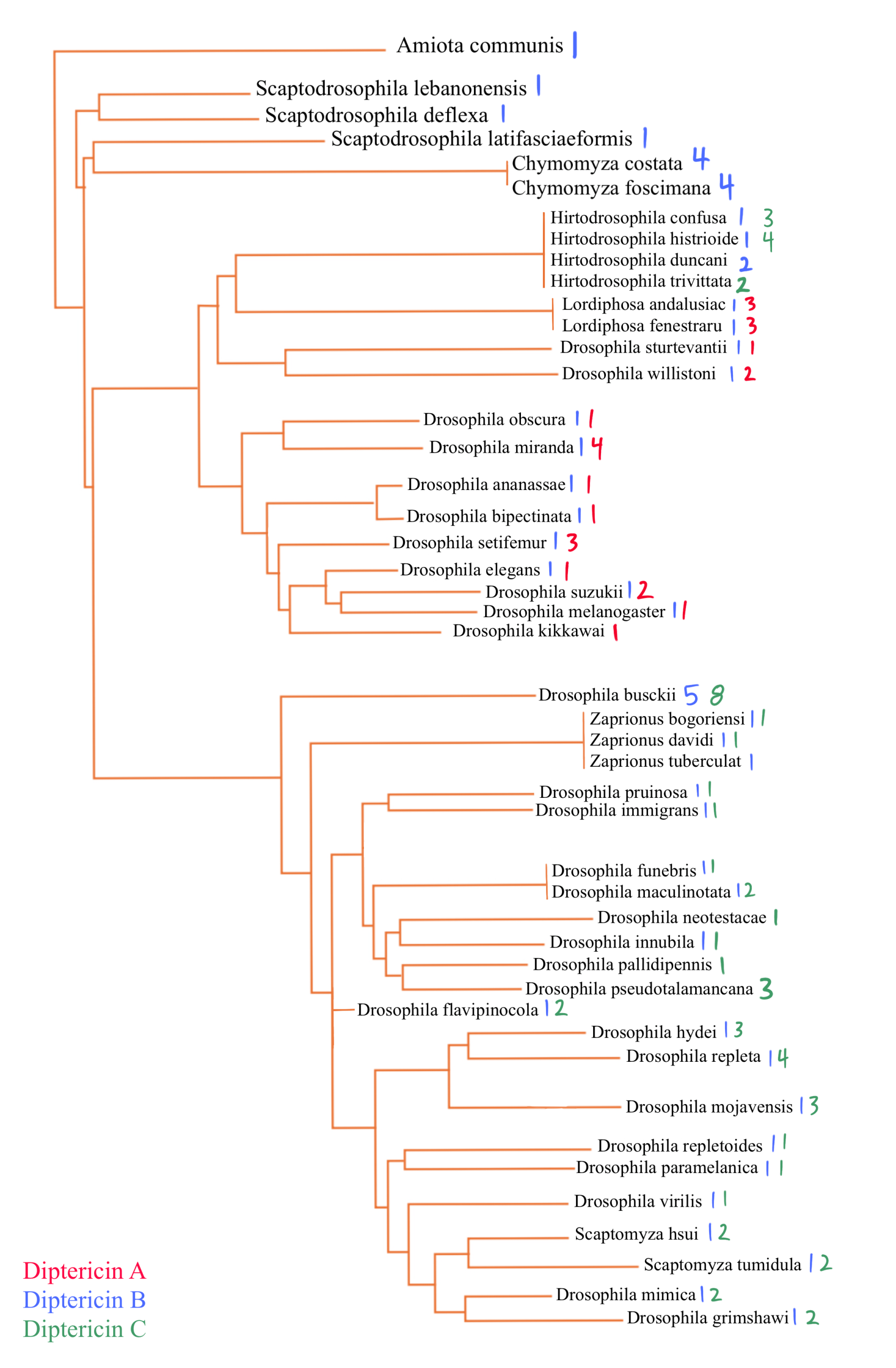
This figure is the comprehensive species phylogeny of all the flies in this paper based on a previously published mitochondrial phylogeny4, with species arranged to reflect their evolutionary relationships. Diptericin A is red, Diptericin B is blue, and Diptericin C is green. Diptericin subtypes were also mapped onto the tree. There is a consistent pattern of diptericin B having one copy in most species. Each species either has diptericin A or diptericin C, or neither. diptericin A and diptericin C never appear together in the same species.
The paralog counts reinforce the same story from a different angle. Across 51 species, diptericin B is the most widespread, present in 44/51 species, while diptericin C is present in 27 species and diptericin A in only 13 species, supporting the hypothesis that B is ancestral and broadly conserved whereas A and C are more restricted [Table 3]. Figure 3 shows that the paralog combinations are not randomly scattered across taxa. The earliest-branching lineages in this dataset mostly show diptericin B alone (for example Amiota, Scaptodrosophila, and Chymomyza), which fits the idea that B represents the ancestral background state before the duplication events that generated the derived paralogs [Figure 3]. Moving up the tree, diptericin A appears in the Sophophora part of the phylogeny where it is consistently found alongside B, while diptericin C appears in the subgenus Drosophila where it is also almost always found alongside B and often shows additional within-lineage copy expansion (for example the high copy number in D. busckii) [Figure 3, Table 1]. This phylogenetic structure supports a simple evolutionary model where diptericin B is the stable and ancestral core paralog and the A/C branch reflects an early duplication of B followed by lineage-specific divergence. Under that model, the duplicated copy evolved into diptericin A in Sophophora and into diptericin C in the subgenus Drosophila, with repeated secondary duplications contributing to the higher copy numbers seen in some lineages [Figure 3, Table 1]. The Hirtodrosophila placements look “off” in that they map near the A-bearing side of the tree but show sequences classified as C, but that is not necessarily biologically meaningful because A and C are both derived from the same original duplication and the label can be ambiguous at deep divergence [Figure 3].
Taken together, the frequency and combination patterns show diptericin B as the stable background subtype across lineages, while diptericin A and diptericin C are less commonly retained by themselves and more often appear in the presence of B, consistent with more specialized, lineage-specific evolutionary trajectories for A and C [Figure 3]. Copy-number summary statistics match this pattern. Diptericin B, which represents the ancestral form in this dataset, is most often retained as a single-copy gene. Consistent with that, diptericin B has the lowest mean copy number (1.27) and is typically single-copy, although a few lineages reach higher counts (max 5) [Table 3]. Diptericin A shows intermediate copy-number expansion (mean 1.77, max 4) [Table 3]. Diptericin C shows the strongest expansion overall, with the highest mean copy number (2.22) and the highest maximum (8 copies), consistent with repeated duplication in many species [Table 3]. These distributions support subtype-specific evolutionary trajectories where B behaves like a conserved core locus while A and especially C show greater copy-number flexibility, consistent with lineage-specific duplication or retention (while still acknowledging that neutral copy-number dynamics and genome assembly/annotation artifacts can contribute when relying on published annotations) [Table 3].
| Statistic | Diptericin A | Diptericin B | Diptericin C |
|---|---|---|---|
| Average | 1.769230769 | 1.272727273 | 2.222222222 |
| Max | 4 | 5 | 8 |
| Min | 1 | 1 | 1 |
| Median | 1 | 1 | 2 |
This table shows the statistics for diptericin A, B, and C copy numbers across the analyzed fly species. diptericin C has the highest average copy number (2.22) as well as the highest maximum, with it being present up to 8 times in some species. diptericin C shows the highest mean and maximum copy number across species (Table 3), indicating more frequent duplication and/or retention in some lineages. diptericin A averages 1.77 copies with a maximum of 4, while diptericin B is the most conserved, with the lowest average (1.27) and a maximum of 5 copies. The median values indicate that most species maintain one copy for diptericin A and diptericin B and two copies for diptericin C, reflecting its broader variation and possible adaptive diversification. High copy number is consistent with lineage-specific expansion that could be shaped by pathogen-mediated selection, but it may also arise through neutral duplication processes, copy-number variation dynamics, or assembly/annotation artifacts. Therefore, copy-number patterns are interpreted here as hypothesis-generating rather than definitive evidence of adaptation.
Across maximum-likelihood phylogenies, many shallow clades show high bootstrap support (often >95%), indicating confident clustering of closely related sequences and duplicated copies within lineages, whereas deeper backbone relationships include multiple moderately supported nodes (70–90%) and, when displayed, some weakly supported nodes (<70%), so the precise relationships in the deep branches are interpreted cautiously [Figure 4, 5, 6]. This is also seen in the pore-only tree [Figure 8]. Although pore alignments are shorter, they are also cleaner and less gappy than full-length sequences, so pore-only trees can show equal or stronger support for some relationships by avoiding poorly alignable regions of the full peptide. Because several internal nodes exhibit low support, apparent discordance between gene trees and the species phylogeny is treated as unresolved rather than definitive evidence of duplication or loss in cases where the relevant nodes are weakly supported [Figure 1, 4, 5, 6].
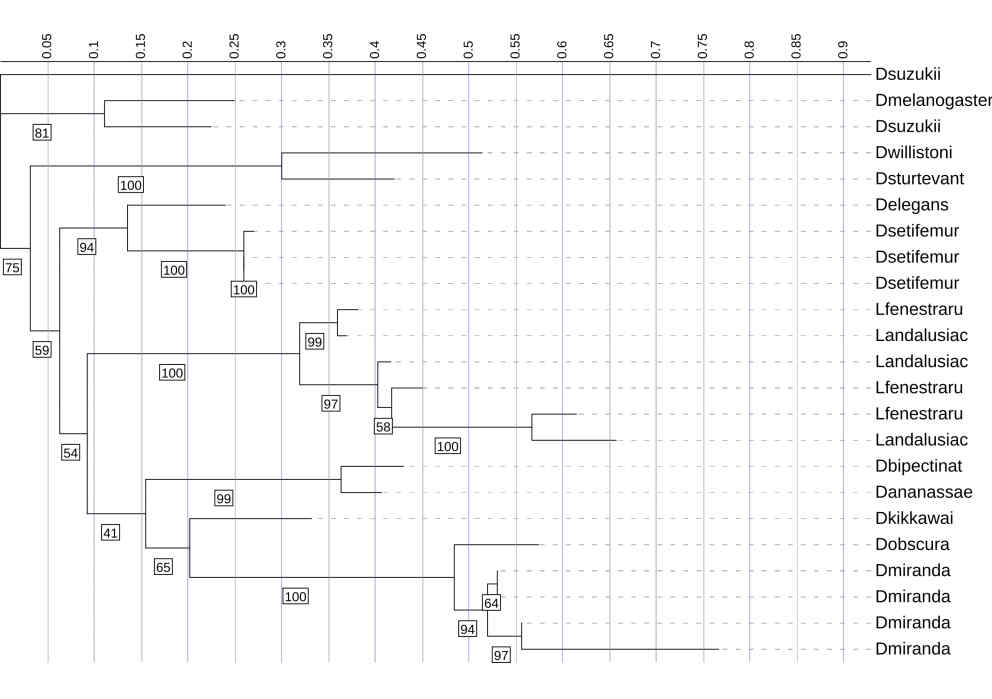
This is a phylogenetic tree made using Diptericin A sequences from multiple fly species. The tree was constructed using maximum likelihood analysis based on full-length amino acid sequences. Branch lengths indicate relative sequence divergence, with longer branches representing greater evolutionary change. There is clustering within species such as D. setifemur and D. miranda. Diptericin A copies from other species, such as L. andalusiaca and L. fenestratum, are distributed across the tree. Overall node support was high for Diptericin A (median UFBoot = 96), but several deeper internal nodes fell below 70, so the branching order among major lineages should be interpreted cautiously. Node labels indicate bootstrap support (1000 replicates); values below 70 are not shown.
This phylogenetic tree is made from Diptericin B sequences. It was constructed using maximum likelihood analysis of full-length amino acid sequences, with longer branches indicating greater evolutionary divergence. There is some duplication evident within this tree, such as in D. busckii, but overall, there are fewer duplicates than for Diptericins A and C. There are also many clusters of species with low divergence. Many shallow clades were strongly supported, but nearly half of internal nodes had bootstrap support lower than 70%, limiting confidence in deeper branching relationships. Node labels indicate bootstrap support (1000 replicates); values below 70 are not shown.
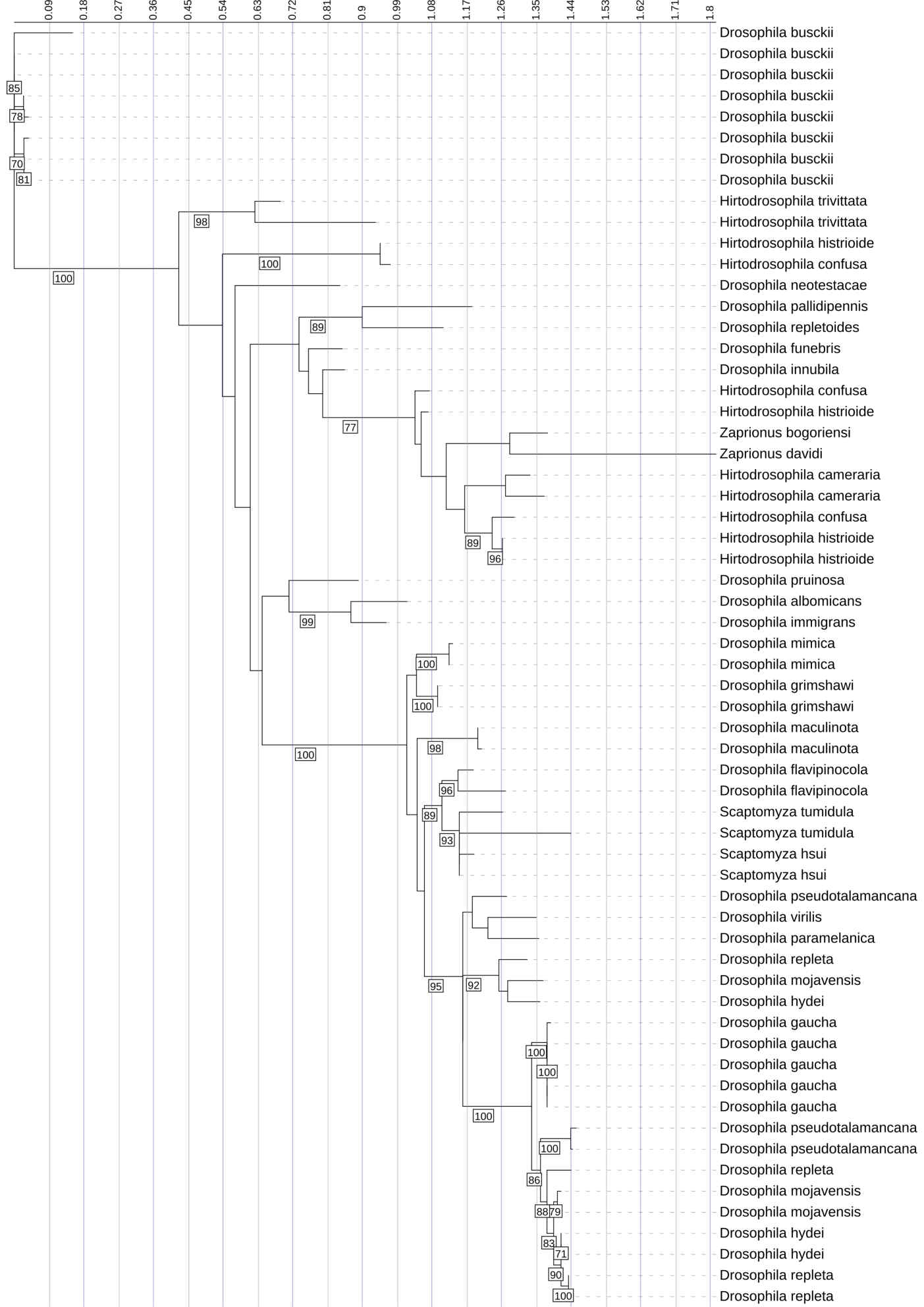
This phylogenetic tree represents the evolutionary relationships among Diptericin C sequences across diverse fly species. It was constructed using maximum likelihood analysis of full-length amino acid sequences, with longer branches indicating more extensive evolutionary change. There are large clades with high copy numbers in certain species, such as D. busckii and D. gaucha. These species show greater variability and diversity, as reflected by longer branches and multiple distinct clusters. This may reflect recent duplication events (tight clusters of closely related copies) and/or longer-term retention of divergent paralogs (multiple distinct clusters within a species). Since duplication can also reflect neutral processes or assembly/annotation artifacts, we interpret high copy number as consistent with lineage-specific expansion rather than as definitive evidence of pathogen-driven adaptation. Diptericin C similarly contained both strongly supported local clades and weaker backbone support, indicating that some higher-level relationships are not robustly resolved. Node labels indicate bootstrap support (1000 replicates); values below 70 are not shown.
Within this overall support context, full-length trees show subtype-specific patterns that match the copy-number and frequency results. Diptericin A full-length phylogenies show many strong terminal clusters and within-species groupings. Biologically, this clustering means that multiple sequences from the same species form a tight clade with short internal branch lengths and strong bootstrap support, consistent with recent duplication followed by limited divergence among paralogs [Figure 4]. For example, the D. miranda copies form a tight, strongly supported clade consistent with recent duplication, while several Lordiphosa sequences are distributed across longer branches and do not consistently cluster by species with strong support, suggesting substantial divergence or unresolved deeper relationships, especially where backbone support is moderate [Figure 4]. Diptericin B sequences generally show shorter branch lengths and many compact clades, consistent with comparative conservation, with strong support for many shallow groupings but only moderate/weak support on several deeper nodes; D. mojavensis is a notable long-branch exception consistent with elevated divergence, while recognizing that branch length alone does not specify evolutionary mechanism (relaxed constraint vs adaptive change) [Figure 5]. Diptericin C shows frequent lineage-specific duplication and diversification, with multiple clades containing tight, strongly supported clusters of closely related paralogs (including clusters in D. busckii and D. gaucha) consistent with recent expansion and limited divergence among copies, alongside other taxa with fewer copies and longer internal branches consistent with deeper divergence or more conserved copy number [Figure 6]. Across these trees, strong shallow support supports recent duplication and clustering patterns, while reduced deep-node support limits confidence in the precise ordering of older splits [Figure 4, 5, 6].
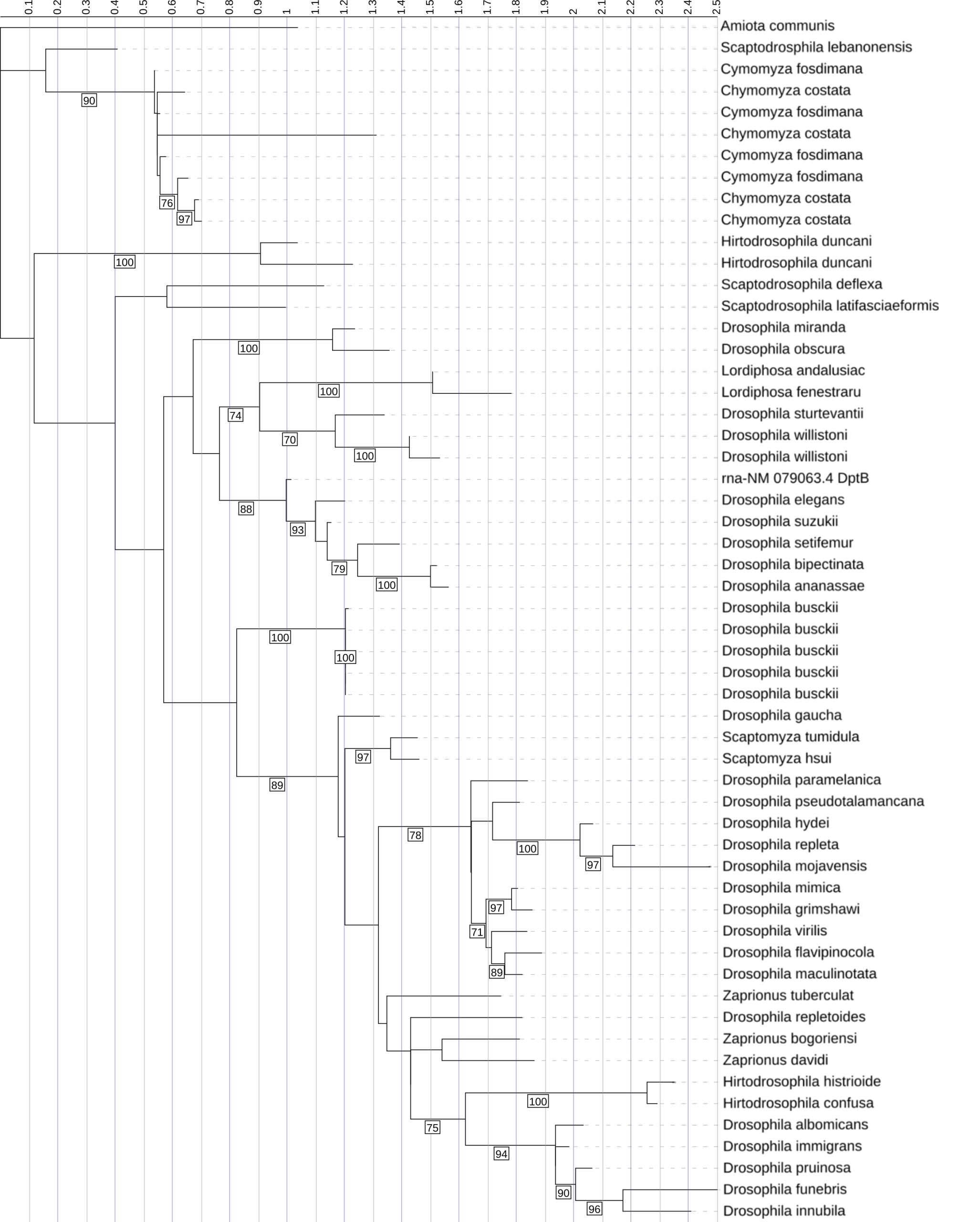
Structural mapping and pore-only phylogenies show that the pore domain is a focal point of conservation and that these domain-based trees capture patterns not obvious from full-length sequences alone. Pore residues were defined using AlphaFold trimer modeling, where the predicted pore initiation site is identified at the transition between the β-sheet barrel (blue) and the adjacent low-structure “string” region (orange/yellow), and pore boundary positions were averaged across three independently generated trimer models to standardize pore extraction across sequences [Figure 1]. Using these pore boundaries, the Diptericin A & Diptericin C pore phylogeny recovers multiple well-supported clusters within each subtype, and Diptericin C pore sequences show several clades with substantial divergence and high copy number in certain taxa, consistent with repeated duplication and diversification of Diptericin C, whereas Diptericin A pore sequences more often form tighter clusters with shorter branch lengths consistent with comparatively lower divergence in this domain in many taxa [Figure 7, 8]. Extending to all three paralogs, the ABC pore tree shows Diptericin B pore sequences forming compact clusters with shorter branch lengths (comparative conservation) while Diptericin A and Diptericin C pore sequences show greater divergence overall; several taxa (e.g., D. busckii, D. gaucha) contain multiple closely related pore variants that cluster together, consistent with recent duplication and retention of similar copies [Figure 8]. However, deeper relationships among the A, B, and C clades depend on support at the internal nodes connecting major subtype groups, so any inferred sister relationship (e.g., A and C vs B and C) is treated as tentative unless connecting support is strong (≥70), and cases where Diptericin A shows an unstable or weakly supported placement relative to the B and C clades are interpreted cautiously, since this pattern can reflect long-branch behavior, elevated divergence, or limited phylogenetic signal rather than a definitive statement of ancestry [Figure 8].
One D. suzukii Diptericin A–like sequence is highly diverged and behaves atypically on the phylogeny. Another copy of Diptericin A in D. suzukii is more similar to the Diptericin A sequences from the remaining species [Figure 2, 9]. The D. suzukii outlier paralog falls on an extremely long branch with weakly supported placement in the diptericin A tree ( bootstrap <41), distorting overall scaling and reducing interpretability of other relationships [Figure 9]. AlphaFold modeling supports the hypothesis that this paralog is either highly functionally divergent or is a nonfunctional pseudogene. This paralog failed to produce a pore-like structure in trimeric assembly [Figure 2]. This sequence was excluded from the results shown above.
This phylogenetic tree represents the evolutionary relationships inferred from the pore sequences from Diptericin A and C across diverse Drosophilid species. It was constructed using maximum likelihood analysis of full-length amino acid sequences, with longer branches indicating more extensive evolutionary change. Diptericin A is shown in red and Diptericin C in green. We determined which amino acids were part of the pore by modeling three copies in AlphaFold and averaging the pore sequence positions. The tree reveals several large clades with high sequence variation. The Diptericin paralogs formed their own clades. Support values were lower overall in pore-only trees than full-length trees, consistent with reduced phylogenetic signal in shorter, rapidly evolving domains. Node labels indicate bootstrap support (1000 replicates); values below 70 are not shown.

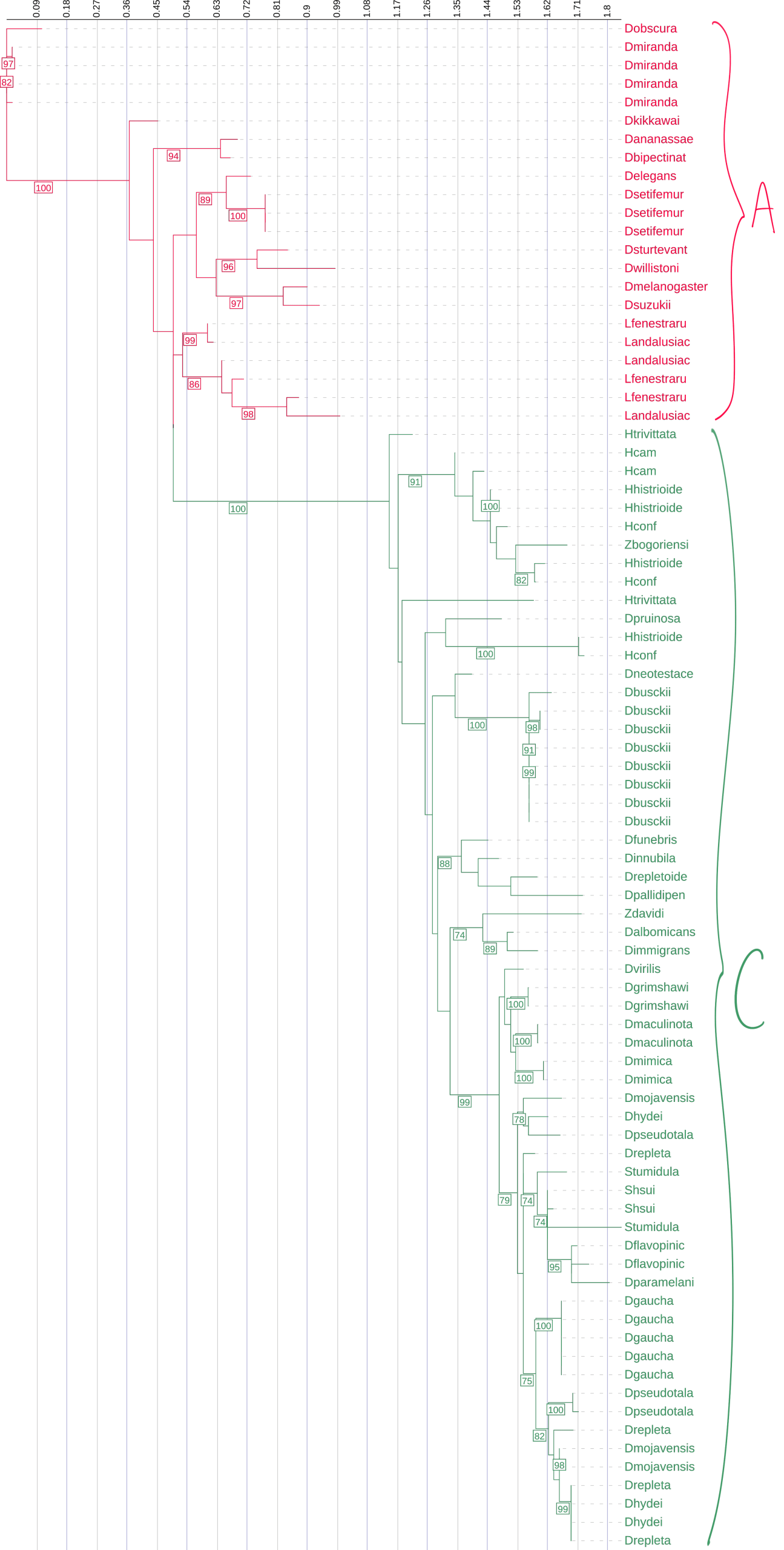
This phylogenetic tree represents the evolutionary relationships among pore-region sequences from Diptericin A, B, and C across diverse fly species. It was constructed using maximum likelihood analysis of full-length amino acid sequences, with longer branches indicating more extensive evolutionary change. Diptericin A is shown in red, Diptericin B in blue, and Diptericin C in green. We determined which amino acids were part of the pore by modeling three copies in AlphaFold and averaging the pore sequence positions. This tree shows that the paralogs form their own clades. Diptericin A is positioned closer to the root, while Diptericin B and Diptericin C share a common ancestor before sharing one with Diptericin A. The ABC pore tree showed generally low backbone support (majority of nodes with bootstrap support <70%), but the clustering of Diptericin B sequences was strongly supported (98% bootstrap support), and B and C grouped together relative to A with strong support (100% bootstrap support). Node labels indicate ultrafast support (1000 replicates); values below 70 are not shown.
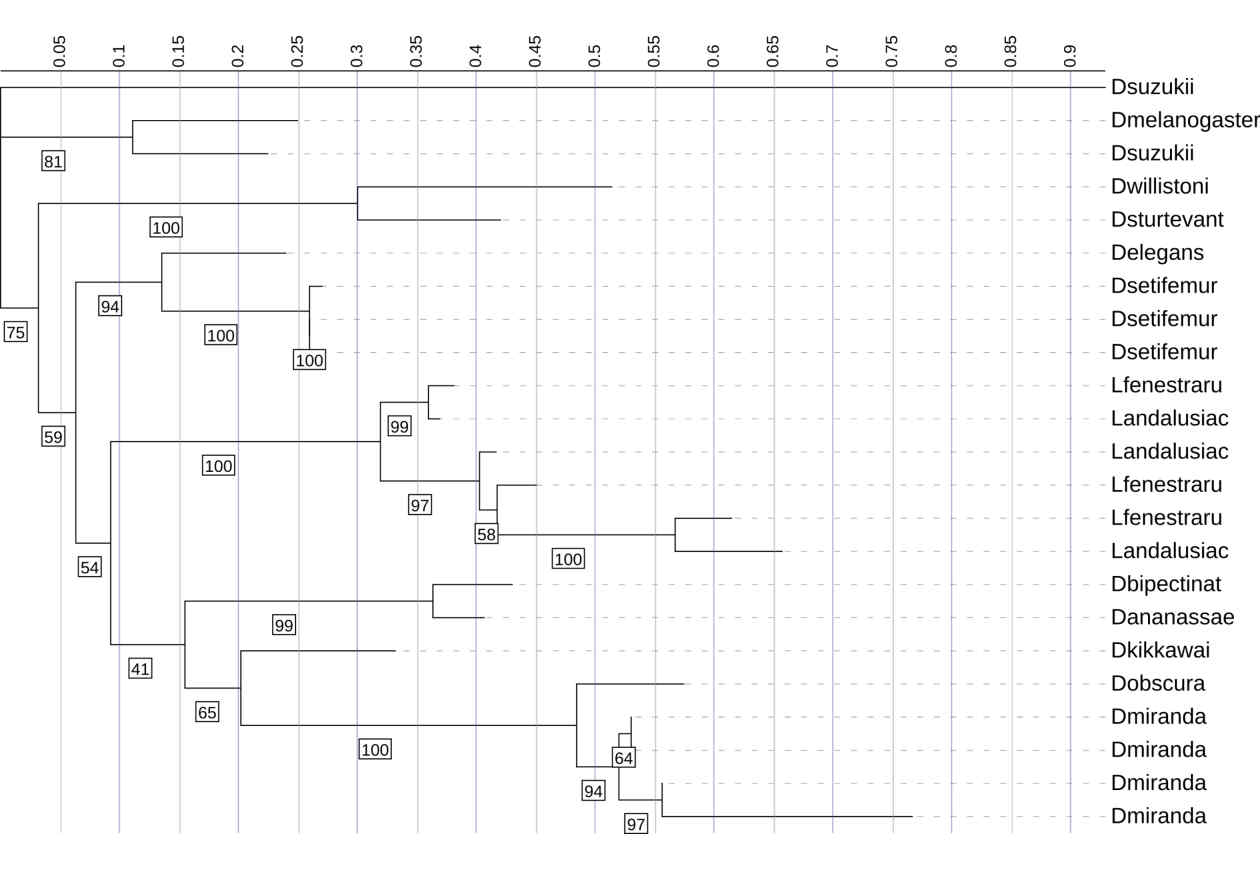
This phylogenetic tree shows the relationships among Diptericin A sequences across multiple fly species. It was constructed using maximum likelihood analysis. Most of the sequences cluster into lineage-specific clades with relatively short branch lengths, indicating moderate levels of divergence. In contrast, the D. suzukii sequence fell on an abnormally long branch length, indicating extreme divergence, and the placement had low bootstrap support (41%). Therefore, it was omitted from the primary analyses presented in this paper. Node labels indicate bootstrap support (1000 replicates); values below 41 are not shown.
Discussion
Diptericin is a major IMD-regulated antimicrobial peptide in Drosophila, and comparative genomics shows that diptericin genes diversify through sequence divergence, gene duplication, and the emergence of three subtypes: Diptericin A, Diptericin B, and Diptericin C. Even within that basic framework, the paralogs often duplicate within species. In this study, the goal was to use phylogenies, copy-number patterns, and AlphaFold-based structural models together to describe how Diptericin has diversified across fly species.
Across the dataset, Diptericin B is the most broadly retained subtype, while Diptericin A and Diptericin C are more lineage-associated and show more copy-number variability. Mapping presence and copy counts onto the species phylogeny supports an early split from a Diptericin B-like ancestral state, followed by divergence along different lineage trajectories where A occurs in Sophophora and C occurs in the subgenus Drosophila, with repeated subsequent duplication within species lineages. Phylogenetic trees generated from the full-length peptide sequence or the more conserved pore region only show the same major subtype structure, revealing specific lineages and domains where duplication and sequence divergence have accumulated.
The evolutionary patterns discovered in this study provide new insights into how immune effectors like AMPs diversify in response to microbial and pathogenic pressure. Across the fly phylogeny, the diptericin gene family showcases evolutionary patterns that probably reflect lineage-specific ecological pressures. In Table II, the presence of duplicated copies of diptericin paralogs in some species suggests that these AMPs may be especially important in those lineages.
One limitation of this study is that our conclusions are based on sequence evolution, phylogenetic patterns, and predicted structure. We do not have experimental data for any of the Diptericin variants analyzed here, so we cannot determine whether duplicated or diverged Diptericin paralogs and subgroups have specialized antimicrobial functions differences in activity. However, we can clearly conclude that Diptericin A and C show evolutionary and structural divergence consistent with functional divergence, and our results generate hypotheses that should be tested experimentally. These predictions could be validated with heterologous expression of representative Diptericin A, B, and C variants (including highly diverged copies) followed by antimicrobial activity assays against Gram-negative bacteria to determine minimum inhibitory concentration (MIC) or bactericidal activity21. The diptericin mode of action is thought to be membrane disruption, so complementary membrane-permeabilization assays (e.g., dye uptake or leakage assays using bacterial membranes or model liposomes) could test whether structural differences correspond to changes in pore-forming efficacy21,22. In addition, expression studies in vivo (constitutively or or in a controlled infection setup) could test whether Diptericin A, B, and C differ in induction across pathogens or tissues, which would support regulatory specialization4,23. Finally, genetic tests such as knockouts or targeted knockdowns followed by transgenic rescue with specific paralogs would provide the strongest evidence for functional differences among A, B, and C24.
Because we only had access to protein sequences, we were unable to perform DNA-based analyses such as dN/dS tests, which limits our ability to conclusively detect positive selection. Instead, we focus on identifying evolutionary patterns such as lineage-specific gene duplication, elevated amino acid divergence, and concentrated variability in functional domains that are consistent with adaptive evolution and motivate future hypothesis-driven testing. Formal gene tree–species tree reconciliation analyses were not performed; therefore, interpretations based on topological comparisons are presented as hypotheses rather than explicit inferences of duplication, loss, or deep coalescence. When coding sequences are available, future work could apply codon-based evolutionary models (e.g., tests for episodic positive selection or relaxed purifying selection) to better distinguish adaptive diversification from rate relaxation. Because the analyses in this study are largely based on amino-acid sequences, results are interpreted as patterns of rate heterogeneity and domain-specific divergence rather than definitive evidence of positive selection.
Because model selection and likelihood optimization were automated, this study does not systematically evaluate how alternative substitution models or inference settings would affect tree topology. As a result, interpretations are based on consistent patterns observed across multiple trees and domains rather than on the robustness of any single model choice. Future work could explicitly test the sensitivity of these results to alternative phylogenetic models.
Multiple lines of evidence indicate that Diptericin B is the ancestral Diptericin paralog, and that is duplicated and subsequently diverged to generate Diptericin A in the subgenus Sophophora and diptericin C is only present in the subgenus Drosophila. The conservation of diptericin B while diptericins A and C recurrently duplicated suggests that ancestral Diptericin B may have a distinct ancestral function, while A and C may have adapted to more specialized functions. The duplication patterns observed in certain clades, including D. busckii, D. miranda, and D. repleta [Figure 4, 5, 6], indicate lineage-specific expansion rather than uniform duplication across all species. These lineages have sustained multiple duplications but this study does not test what ecological or genomic factors drive those expansions.
Previous functional studies of diptericin provide useful context for interpreting the evolutionary patterns observed here, but they do not allow direct functional conclusions to be drawn from our data alone6,7. In Drosophila melanogaster, Diptericin A contributes to resistance against Providencia rettgeri during systemic infection, while Diptericin B is required for defense against Acetobacter in the gut7,8,24. Naturally occurring variation in Diptericin A can also have large and pathogen-dependent effects on resistance. The S69R polymorphism is strongly associated with bacterial load and survival after Providencia infection, while showing little to no effect for several other pathogens25,26. Together, these results support the broader premise that diptericin paralogs and variants can differ in biological context and importance, which motivates the hypothesis that divergence among Diptericin A, B, and C across species may reflect differences in function or regulation8.
Most of the species retained a single copy of Diptericin B, suggesting it performs a conserved, essential function such as regulating bacteria such as Acetobacter in the gut. However, some species, such as D. busckii and D. gaucha, show lineage-specific expansions of Diptericin B23. This could be a way to expand antimicrobial coverage without fundamentally altering Diptericin B’s ancestral function. In contrast, a few species have a complete absence of B. This may reflect either gene loss following the functional replacement of B by other diptericins (A or C) or shifts in that fly’s ecological niche that reduced the selective advantage of keeping it22,26. Alternatively, those absences may reflect incomplete annotation of those species’ genomes. Future work could validate diptericin copy numbers by checking read-depth and assembly quality metrics or by confirming paralog copy numbers through targeted sequencing26.
Differences between gene trees and the species phylogeny can come from several evolutionary processes, including gene duplication and loss, incomplete lineage sorting among closely related taxa, or uncertainty in gene tree reconstruction. Horizontal gene transfer is considered unlikely for nuclear-encoded antimicrobial peptides in insects and is not supported by the broader phylogenetic context of this dataset. Importantly, many of the apparent mismatches observed here involve nodes with low bootstrap support, limiting confidence in the inferred gene tree topology. Thus, these patterns are best interpreted as unresolved relationships rather than strong evidence for specific evolutionary events.
The findings of this study align with prior studies. There is rapid diversification of AMP families under pathogen-mediated selection1,2, but our paper extends this conclusion by linking gene duplication to structural diversification. Importantly, the observed divergence between gene and species phylogenies suggests that the evolution of diptericin is shaped not only by ancestry but also by duplication and adaptive evolution. There are limitations to this paper. Genome sequences vary in quality, as shown with the D. suzukii sequence being omitted in Diptericin A.
We observed one D. suzukii sequence that was highly divergent by sequence and structure. This could reflect strong, recent accelerated evolution for a new function. Alternatively, the divergence might result from relaxed selection if that specific diptericin A became functionally redundant due to the presence of other AMPs, more specifically the other D. suzukii diptericin A. Or this gene copy may have sustained a mutation that rendered it nonfunctional so it became a pseudogene. Either of these latter two would lead to accelerated mutation accumulation since the redundant Diptericin A would not have any functional constraint. Since this sequence also failed to produce the pore-like trimeric assembly observed for other diptericin homologs under the same AlphaFold procedure, we infer that it has substantially altered function or no function, so we excluded it from our primary analyses.
Overall, the results presented in this paper support the view that diversifying selection and gene duplication are important forces shaping diptericin. Diptericin B is the most conserved and ancestral, and Diptericin A and C are likely to be derived lineages that arose from the duplication of an ancestral diptericin B before undergoing stronger adaptive pressure. This work highlights the evolutionary relationships between gene family, protein structure, and immune function. It also provides a foundation for studies aimed at testing the functional consequences of diptericin diversification. Future studies could build on these findings by testing the antimicrobial activity of divergent Diptericin A and C variants. They could also extend similar evolutionary and structural analyses to other AMP gene families. That research could help provide context and more information for the evolutionary patterns identified here and provide a broader framework for understanding how immune effectors diversify across insect lineages.
Acknowledgements
Thank you for the guidance of Dr. Brian Lazzaro from Cornell University in the development of this research paper. We thank Dr. Pankaj Dhakad and Dr. Darren Obbard for sharing unpublished Diptericin sequences with us.
Data Availability
All Diptericin sequences analyzed in this study were obtained from the primary publication Dhakad et al., 2025. Species presence/absence counts and frequency calculations were performed manually in Google Sheets using these published data; no custom scripts or computational pipelines were used.
Supplementary Materials
Phylogenetic trees were inferred using IQ-TREE 3 (version 3.0.1). Alignments were provided in PHYLIP format. Trees were constructed using automated model selection and maximum likelihood inference with the following command structure: iqtree3.exe -s DiptericinA.phy -m MFP -B 1000. Equivalent commands were used for Diptericin B, Diptericin C, and pore-only alignments, with the input file name changed accordingly. The -m MFP option enabled automated model selection using MixtureFinder. Ultrafast bootstrap support was calculated with 1000 replicates using -B 1000. Concordance factors were computed using default IQ-TREE 3 settings. Output tree files were visualized using iTOL.
References
- J. Jumper, R. Evans, A. Pritzel, et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). doi:10.1038/s41586-021-03819-2. PMID:34265844 [↩] [↩]
- M. A. Hanson, B. Lemaitre, R. L. Unckless. Dynamic evolution of antimicrobial peptides underscores trade-offs between immunity and ecological fitness. Front. Immunol. 10, 2620 (2019). doi:10.3389/fimmu.2019.02620. PMID:31781114 [↩] [↩]
- H. E. S. Matthews, J. H. Jiggins, F. M. Jiggins. Adaptive evolution and gene family expansions in insect immunity. Philos. Trans. R. Soc. B. 374, 20190085 (2019). [↩]
- R. DeSalle, S. Oppenheim, P. M. O’Grady. Whole mitochondrial genome phylogeny of Drosophilidae. Mitochondrial DNA A DNA Mapp. Seq. Anal. 33, 1–9 (2024). doi:10.1080/24701394.2023.2295247. PMID:38269531 [↩] [↩] [↩] [↩] [↩]
- A. H. Benfield, S. T. Henriques. Mode-of-action of antimicrobial peptides: membrane disruption vs intracellular mechanisms. Front. Med. Technol. 2, 610997 (2020). doi:10.3389/fmedt.2020.610997. PMID:35047892 [↩]
- P. Bulet, R. Stöcklin, L. Menin. Antimicrobial peptides in insects: structure and function. Dev. Comp. Immunol. 23, 329–344 (1999). doi:10.1016/S0145-305X(99)00017-9. PMID:10426426 [↩] [↩]
- D. J. Obbard, J. J. Welch, K.-W. Kim, F. M. Jiggins. Quantifying adaptive evolution in the Drosophila immune system. PLoS Genet. 5, e1000698 (2009). doi:10.1371/journal.pgen.1000698. PMID:19851448 [↩] [↩] [↩] [↩]
- R. L. Unckless, V. M. Howick, B. P. Lazzaro. Convergent balancing selection on an antimicrobial peptide in Drosophila. Curr. Biol. 26, 257–262 (2016). doi:10.1016/j.cub.2015.11.063. PMID:26776733 [↩] [↩] [↩] [↩]
- T. B. Sackton, B. P. Lazzaro, T. A. Schlenke, et al. Dynamic evolution of the innate immune system in Drosophila. Nat. Genet. 39, 1461–1468 (2007). doi:10.1038/ng.2007.60. PMID:17987029 [↩]
- J. Li, J. J. Koh, S. Liu, et al. Membrane active antimicrobial peptides: translating mechanistic insights to design. Front. Neurosci. 11, 294 (2017). doi:10.3389/fnins.2017.00294. PMID:28659875 [↩]
- M. A. Hanson, L. Grollmus, B. Lemaitre. Ecology-relevant bacteria drive the evolution of host antimicrobial peptides in Drosophila. Science 381, eadg5725 (2023). doi:10.1126/science.adg5725. PMID:37471548 [↩]
- E. De Gregorio, P. T. Spellman, P. Tzou, G. M. Rubin, B. Lemaitre. The Toll and Imd pathways are major regulators of the immune response in Drosophila. EMBO J. 21, 2568–2579 (2002). doi:10.1093/emboj/21.11.2568. PMID:12032070 [↩]
- B. Lemaitre, E. Nicolas, L. Michaut, J. M. Reichhart, J. A. Hoffmann. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983 (1996). doi:10.1016/S0092-8674(00)80172-5. PMID:8808632 [↩] [↩]
- P. Dhakad, et al. Transcriptomic analysis of non-model Drosophilidae reveals novel antimicrobial peptide candidates. bioRxiv (preprint) (2025). doi:10.1101/2025.06.06.658223 [↩]
- K. Katoh, D. M. Standley. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). doi:10.1093/molbev/mst010. PMID:23329690 [↩]
- A. Larsson. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278 (2014). doi:10.1093/bioinformatics/btu531. PMID:25095880 [↩]
- M. L. Borowiec. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4, e1660 (2016). doi:10.7717/peerj.1660. PMID:26835189 [↩]
- T. K. F. Wong, et al. IQ-TREE 3: phylogenomic inference software using complex evolutionary models. EcoEvoRxiv (preprint) (2025). doi:10.32942/X2P62N [↩]
- I. Letunic, P. Bork. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024). doi:10.1093/nar/gkae268. PMID:38613393 [↩]
- K. A. Winans, D. S. King, V. R. Rao, C. R. Bertozzi. A chemically synthesized version of the insect antibacterial glycopeptide, diptericin, disrupts bacterial membrane integrity. Biochemistry 38, 11700–11710 (1999). doi:10.1021/bi991247f. PMID:10512626 [↩]
- B. Lemaitre, J. A. Hoffmann. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697–743 (2007). doi:10.1146/annurev.immunol.25.022106.141615. PMID:17201680 [↩]
- M. Lynch, A. Force. The probability of duplicate gene preservation by subfunctionalization. Genetics 154, 459–473 (2000). doi:10.1093/genetics/154.1.459. PMID:10629003 [↩] [↩]
- P. Laštovka, J. Máca. European species of the Drosophila subgenus Lordiphosa. Acta Entomol. Bohemoslov. 75, 404–420 (1978). doi:NA. PMID:NA [↩] [↩]
- J. O. Wertheim, B. Murrell, M. D. Smith, S. L. Kosakovsky Pond, K. Scheffler. RELAX: detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 32, 820–832 (2015). doi:10.1093/molbev/msu400. PMID:25540451 [↩] [↩]
- L. Viljakainen. Evolutionary genetics of insect innate immunity. Brief Funct. Genomics 14, 407–412 (2015). doi:10.1093/bfgp/elv002. PMID:25750410 [↩]
- B. Murrell, J. O. Wertheim, S. Moola, T. Weighill, K. Scheffler, S. L. Kosakovsky Pond. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8, e1002764 (2012). doi:10.1371/journal.pgen.1002764. PMID:22807683 [↩] [↩] [↩]



{kind=link}