
Abstract
Progressive supranuclear palsy (PSP) is a rare neurodegenerative disorder characterized by impaired movement, balance, and cognitive function. Because many of its symptoms overlap with those of Parkinson’s disease, PSP is frequently misdiagnosed, making early and accurate detection a clinical challenge. Recent studies have identified the protein ATP6AP2 in the cerebrospinal fluid of PSP patients, suggesting that it may serve as a promising biomarker for the disease. Antibodies, which are produced by the immune system to recognize specific antigens, can be used as detection tools if they bind selectively to target proteins. In this research, computational simulations were employed to evaluate whether specific antibodies can effectively recognize and bind to ATP6AP2. The amino acid sequence of ATP6AP2 was first obtained and input into AlphaFold 3 to generate its three-dimensional structure. Antibody structures were collected from the Protein Data Bank (PDB). Molecular docking simulations were then performed to predict how ATP6AP2 interacts with each antibody and to analyze the stability and specificity of these interactions. To further validate the predicted binding sites, ScanNet, a deep learning–based binding site prediction tool, was used. Antibodies were evaluated based on three criteria: visual inspection of the binding interface, predicted binding energy, and hydrogen bond formation. Among all candidates, antibody 1B4J demonstrated the strongest potential, showing a favorable binding pose, strong affinity, and a high number of stabilizing interactions. This study provides a computational foundation for identifying antibodies capable of detecting ATP6AP2 and supports future experimental validation aimed at developing diagnostic tools for PSP.
Keywords: Progressive supranuclear palsy (PSP), ATP6AP2, Antibodies, Biomarker.
Introduction
Progressive supranuclear palsy (PSP) is a rare neurodegenerative disease characterized by impaired body movement, coordination, and cognitive decline1. It is caused by damage to nerve cells in the brain responsible for these functions2. PSP progresses over time, leading to severe complications such as pneumonia, choking, or injuries from falls. Some symptoms overlap with Parkinson’s disease1 however, PSP typically develops later, in the mid-to-late 60s. Key symptoms include balance difficulties, impaired eye movement (e.g., inability to look downward, blurriness, and double vision). Additional symptoms, which also worsen as the disease advances, include trouble swallowing, sleep disturbances, neck stiffness, impulsive behavior, depression, slowed or slurred speech, and loss of interest in activities. Currently, there are no treatments to slow or reverse the progression of PSP, nor are there medications that effectively treat its symptoms. While some individuals respond well to levodopa, which addresses stiffness, slowness, and balance issues, its effects are short-lived1. Levodopa is a medication that the brain converts into dopamine, commonly used to reduce movement-related symptoms in Parkinson’s disease and related disorders3. Botulinum toxin injections in the muscles around the eye can help with eye-closing, and some antidepressants not only treat depression but also the pain and drooling associated with PSP4. There are no brain imaging techniques or tests to diagnose PSP, but physicians will perform a physical and neurological exam after reviewing the patient’s medical history. Diagnostic imaging may be able to display shrinkage at the top of the brain stem. Identifying trouble with eye movement, walking, speech, and swallowing early is very important as it can help rule out similar diseases such as Parkinson’s1.
Diagnosing Progressive Supranuclear Palsy (PSP) is clinically challenging because its early symptoms closely resemble other movement disorders, especially Parkinson’s disease. Both PSP and Parkinson’s can present with balance problems, slow movement, stiffness, and frequent falls, making them difficult to distinguish during the initial stages. However, PSP has several unique features—such as difficulty moving the eyes vertically, early postural instability, and rapid progression—but these signs may not appear until the disease has advanced. As a result, many PSP patients are misdiagnosed for months or even years. Another challenge is that no definitive laboratory test or imaging biomarker currently exists for PSP, so clinicians must rely solely on physical examinations and patient history. This overlap with other neurological conditions, combined with the absence of reliable diagnostic tools, makes early and accurate PSP diagnosis extremely difficult.
The ATPase H+transporting lysosomal accessory protein, or ATP6AP2, is integral to vacuolar ATPase, playing a vital role for autophagy and lysosomal processes, Figure 1. It also plays a crucial role in neural and oligodendrocyte differentiation. Recently, scientists have discovered the protein ATP6AP2 to be present in the cerebrospinal fluid of PSP patients and have proposed that it could be a potential biomarker of the disease5. ATP6AP2 is essential for the growth and repair of brain cells. According to this study, it promotes the development of brain cells from stem cells (hADSCs). It functions via regulating lipid microdomains (CLR-Ms), which are microscopic cell components, and cell signals (Wnt and G-proteins). Brain cell proliferation is inhibited when ATP6AP2 is absent or diminished, which results in issues seen in conditions like PSP.

Molecular docking is a computational technique utilized in drug discovery that predicts the interactions between biomolecules, such as proteins-proteins, and proteins-ligands6. It involves simulating these interactions to determine a prediction of the binding capacity of the molecule to the target7. Electrostatic interactions, including Van der Waal, hydrogen, and Coulombic bonds, are involved in the coupling between the two structures and can be considered in the simulation6. The bonding potential between the structures is estimated by a docking algorithm, which provides a docking score. It is a two-step process in which the algorithm first explores the conformational space and displays the possible places on the target where the molecule would bind. Then, it calculates the energy required for coupling in each potential coupling site6.
Antibodies are protective proteins the body’s immune system produces when harmful substances, called antigens, are detected8 Each type of antibody is unique and only defends against a specific type of antigen9. They bind to antigens such as bacteria, fungi, or viruses and destroy them directly or make it easier for other immune cells to do so.(Chiu, Goulet, Teplyakov, & Gilliland, 2019; Stanfield & Wilson, 2015) The Y-shape of antibodies consists of two identical “arms” with heavy and light chains. This Y-shape allows the “arms” that contain binding sites to attach specifically to antigens on the surface of pathogens10. When first exposed to an antigen, the body’s immune system creates antibodies specific to that organism. The immune system also remembers that first exposure and will reactivate the antibodies that stay in the body to destroy it if it enters the body again.
Methods
Protein Sequence Retrieval and Structural Modeling: The amino acid sequence of ATP6AP2 was obtained from the UniProt web server11. This sequence was used as input for de novo protein structure prediction using AlphaFold 3, which generates high-accuracy atomic models based on multimodal structural constraints12. The predicted 3D structure was imported into UCSF ChimeraX for preprocessing, including model refinement and conversion into PDB format for downstream computational analyses13.
Surface Characterization and Binding Site Prediction: To identify potential ligand-accessible regions, the ATP6AP2 structure was analyzed using P2Rank, an AI-based binding site prediction tool that evaluates surface geometry and physicochemical properties to locate probable interaction pockets14. Electrostatic surface potential (ESP) maps of ATP6AP2 were also generated in ChimeraX to visualize charge distribution patterns and assess compatibility with antibody complementarity-determining regions.
Antibody Acquisition and Structural Preparation: A set of ten antibody structures was downloaded from the Protein Data Bank15. Each antibody was preprocessed in ChimeraX to remove crystallographic artifacts, optimize side-chain conformations, and ensure structural compatibility with docking input requirements. The ATP6AP2 model and antibody files were then formatted appropriately for molecular docking.
Protein–Antibody Molecular Docking Simulations: Molecular docking simulations between ATP6AP2 and each antibody were conducted using the HDOCK server16. This platform integrates template-based modeling with ab initio docking to generate energetically favorable protein–antibody complexes. For each antibody, ten docking simulations were performed to ensure sufficient sampling of possible binding conformations. The highest-scoring complexes were selected based on docking metrics and interface quality.
Post-Docking Analysis and Complex Refinement: The top protein–antibody complexes were analyzed using ChimeraX, ScanNet, and P2Rank17. ScanNet was used to predict interaction hotspots and assess the plausibility of residue-level contacts. Detailed inspection of hydrogen-bond networks, hydrophobic interactions, and steric complementarity was performed in ChimeraX to evaluate interface stability and structural consistency.
Binding Affinity and Energetic Profiling: Binding energies for all ATP6AP2–antibody complexes were calculated using PRODIGY, which estimates binding affinity based on intermolecular contacts and structural features18. The predicted ΔG values and dissociation constants were used to rank antibodies based on their thermodynamic favorability and compatibility with the ATP6AP2 binding surface.
Results
In this research paper, we employed computational molecular docking simulations and identified antibody (PDB ID 1B4J) as a promising antibody candidate for ATP6AP2 for binding. This could be a potential biomarker for the early and accurate detection of PSP.
Binding Site Characterization: To identify the binding site on the protein surface, the machine learning-based method P2Rank was employed. The binding site is depicted in Figure 2(a) as the yellow region and in Figure 2(b) as the red region, boxed in white. The binding site and flexible regions were visualized using ScanNet and are depicted in Figure 2(b). This is an essential criterion for choosing an appropriate antibody that binds to the ATP protein, as the antibody exhibiting the strongest binding to the binding site, as shown through the docking results, will be the most suitable candidate for PSP detection. Lastly, the electrostatic surface potential (ESP) of ATP6AP2 was obtained with ChimeraX, with the charges displayed in Figure 2(c).

Molecular docking simulations: To further validate these interactions, we conducted molecular docking simulations using HDOCK, a computational technique that models the binding of antibodies to ATP6AP2. This approach allowed us to assess the strength and stability of these interactions, as illustrated in Figure 3, and make a transition from the binding site to molecular docking.Molecular docking is a computational technique that simulates the interactions between molecules, such as proteins and ligands. This technique was conducted using the software HDOCK to evaluate the interactions between ATP6AP2 and the antibodies used in this research. Figure 3 illustrates the binding interactions of antibodies with AT6AP2. In this figure, the protein is represented in purple, and the antibody is depicted in pink and grey. In this image, the protein is oriented in the same orientation to show the binding of the antibody to the predicted binding site.


Molecular docking analysis: After molecular docking simulations, the antibodies were selected based on the following criteria. First, through visual inspection using the ChimeraX software, the protein-antibody structures were analyzed and compared with the predicted binding sites obtained from ScanNet and P2Rank. ScanNet and P2Rank are AI-based techniques used to identify the binding site on the surface of a protein. It displays the amino acids that have a higher probability of binding with biomolecules. These structures are shown in Figure 3. The antibodies that did not bind to the specific binding site shown in Figures 2(a) and 2(b) were rejected, and only the antibodies that bonded to the predicted site were selected for the subsequent analysis, which involved binding energy calculation. Based on the study, the following antibodies were selected: (PDB ID: 1A5F, 1B4J, 1FL5, 1IL1, and 2HRP). The second selection criterion was to compute the binding energy between the protein and antibodies. Binding energy is the energy required to break apart a molecule, atom, or nucleus and is measured in kcal/mol. Binding energy is always measured in negative values, and the most negative value indicates the highest binding affinity. Notably in Table 1, antibody 1F8T has the strongest binding affinity. Antibodies 1NLB and 1B4J follow close behind 1F8T. However, 1B4J was selected based on the previous criteria– visual inspection. 1NLB and 1F8T did not satisfy the first criteria and, therefore, were not selected. Finally, the last criterion is the number of hydrogen bonds formed between ATP6AP2 and the antibodies. These hydrogen bonds are crucial for interacting with the two biomolecules and were calculated using a Python programming script. The number of hydrogen bonds between each antibody and AT6AP2 are displayed in Figure 4. Based on this data, antibody 1B4J (the purple bar) has the greatest number of hydrogen bonds and, therefore, the strongest hydrogen bond interaction. If only binding energy were considered a criterion, then 1F8T would be the winner. However, 1F8T did not pass the visual inspection nor the hydrogen bond criteria, as it had the lowest number of bonds.
| S.No. | PDB ID | Binding Energy (Set 1) | Binding Energy (Set 2) | Average |
| 1 | 1A5F | -14.1 | -22.8 | -18.45 |
| 2 | 4A6Y | -12.3 | -17.1 | -14.70 |
| 3 | 2UYL | -14.3 | -11.7 | -13.00 |
| 4 | 1NLB | -13.6 | -24.4 | -19.00 |
| 5 | 1B4J | -22.3 | -24.1 | -23.20 |
| 6 | 2HRP | -15.1 | -17.9 | -16.50 |
| 7 | 1IL1 | -18.1 | -11.4 | -14.75 |
| 8 | 1FL5 | -12.1 | -21.0 | -16.55 |
| 9 | 1IQW | -12.2 | -12.2 | -12.20 |
| 10 | 1F8T | -14.3 | -26.1 | -20.20 |

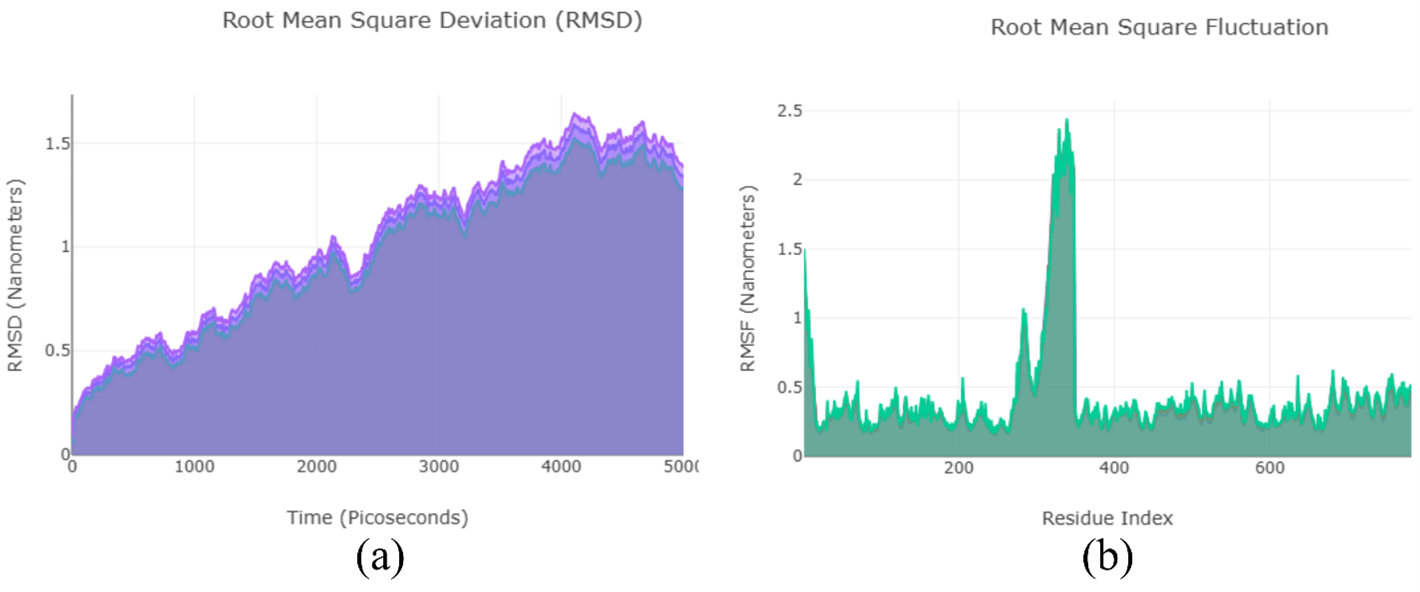
Molecular Dynamics Simulations: To further validate the findings from the molecular docking analysis, we also performed molecular dynamics simulations, and the results are shown in Figure 6. Throughout the entire MD simulation, the antibodies remained bound to the active site of the protein. This provides additional confirmation of the docking results and strengthens the overall validity of the study.
Discussion
Previous studies described ATP6AP2 as a multifunctional protein contributing to autophagy, lysosomal maintenance, and neuroinflammatory signaling. Its dysregulation has been linked to Alzheimer’s disease as well as neuronal development disorders5. Our findings are consistent with these earlier studies in demonstrating that ATP6AP2 contains stable extracellular regions that may be accessible for molecular recognition in the CSF. However, unlike existing research that examines ATP6AP2’s biological function, our work provides the first structural docking-based evidence that ATP6AP2’s ectodomain can interact with antibodies in a predictable and energetically favorable manner. This contrast highlights the novelty of the current research: while earlier studies focus on ATP6AP2 as a functional protein, our work positions ATP6AP2 as a structurally targetable biomarker.
Current Diagnostic Methods for PSP: Although there are no brain imaging techniques or tests specifically designed to detect PSP, physical and neurological exams can be conducted after reviewing the patient’s medical history. Diagnostic imaging may also reveal shrinkage at the top of the brainstem. Identifying trouble with eye movement, walking, speech, and swallowing early is crucial, as it can help rule out similar disorders, such as Parkinson’s. However, these methods are often ineffective because diagnostic imaging and physical problems are not always definitive in diagnosing PSP.
Role of ATP6AP2 Protein in Neurodegenerative Diseases: The ATP6AP2 protein is primarily located on the surface of the cell membrane; therefore, its binding site is located on the ectodomain of the protein. This protein is also present on the plasma membrane of intracellular organelles. The protein plays a vital role in increasing the acidity within intracellular components of the cell. Additionally, the protein regulates blood pressure and maintains fluid and electrolyte balance within the body. Ultimately, the protein regulates cell multiplication, inflammation, and fibrosis. The protein is crucial in neurodegenerative diseases, such as Alzheimer’s disease. Recently, this protein has been identified as a potential biomarker in the cerebrospinal fluid of patients with supranuclear palsy. Since no non-invasive detection techniques are available, this research aims to provide a novel technique. The proposed method will focus solely on detecting the protein in the cerebrospinal fluid of patients for disease identification and analysis.
Potential Benefits of Using ATP6AP2 as a Biomarker: This research has significant applications in the field of science, particularly in the development of diagnostic tools. The antibody can detect the ATP6AP2 protein in patients suspected of having Progressive Supranuclear Palsy (PSP). A biomarker will be used to detect the presence of AT6AP2 in this antibody. The antibody can detect the ATP6AP2 protein in patients suspected of having progressive supranuclear palsy (PSP), Figure 7.

Future Directions for Research: This study has several limitations, one of which is that computational simulations, such as those employed in this study, lack experimental validation. Since this research is not conducted within a physical lab, definite experimental data cannot be obtained. Experimental methods, such as Surface Plasmon Resonance (SPR), validate the results—especially Isothermal Calorimetry (ITC), which measures binding energy in the laboratory. Additionally, computational simulations are currently limited by the availability of a small number of antibodies for testing; therefore, expanding computational resources in the future would likely improve results.
Conclusion
In this research, computational simulations were employed to develop a novel detection technique for PSP. Various computational simulations were utilized to determine the most suitable antibody capable of binding to ATP6AP2. First, antibody-protein interactions were computed. Next, the most suitable antibodies were selected based on the predicted binding site, binding energy, and hydrogen bond analysis. The results indicate that antibody 1B4J was the optimal candidate and was, therefore, selected. In future research, experimental validations such as Surface Plasmon Resonance and Isothermal Calorimetry will be used to confirm these findings further. This study provides an initial computational framework that may support the development of a non-invasive detection approach for PSP.
References
- Williams, D. R., & Lees, A. J. (2009). Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. The Lancet Neurology, 8(3), 270-279 [↩] [↩] [↩] [↩]
- Dickson, D. W., Rademakers, R., & Hutton, M. L. (2007). Progressive Supranuclear Palsy: Pathology and Genetics. Brain Pathology, 17(1), 74-82 [↩]
- Stamelou, M., & Hoeglinger, G. U. (2013). Atypical parkinsonism: an update. Curr Opin Neurol, 26(4), 401-405 [↩]
- Litvan, I., Mangone, C. A., McKee, A., Verny, M., Parsa, A., Jellinger, K., . . . Pearce, R. K. (1996). Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry, 60(6), 615-620 [↩]
- Jang, Y., Oh, S., Hall, A. J., Zhang, Z., Tropea, T. F., Chen-Plotkin, A., . . . Pantelyat, A. Y. (2024). Biomarker discovery in progressive supranuclear palsy from human cerebrospinal fluid. Clin Proteomics, 21(1), 56 [↩] [↩]
- Fan, J., Fu, A., & Zhang, L. (2019). Progress in molecular docking. Quantitative Biology, 7(2), 83-89 [↩] [↩] [↩]
- Ghasemi, J. B., Abdolmaleki, A., & Shiri, F. (2017). Molecular Docking Challenges and Limitations. In I. R. Management Association (Ed.), Pharmaceutical Sciences: Breakthroughs in Research and Practice (pp. 770-794). Hershey, PA, USA: IGI Global [↩]
- Tsuchikama, K., Anami, Y., Ha, S. Y. Y., & Yamazaki, C. M. (2024). Exploring the next generation of antibody–drug conjugates. Nature Reviews Clinical Oncology, 21(3), 203-223 [↩]
- Stanfield, R. L., & Wilson, I. A. (2015). Antibody Structure. In Antibodies for Infectious Diseases (pp. 49-62). [↩]
- Chiu, M. L., Goulet, D. R., Teplyakov, A., & Gilliland, G. L. (2019). Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies, 8(4), 55 [↩]
- Consortium, T. U. (2014). UniProt: a hub for protein information. Nucleic acids research, 43(D1), D204-D212 [↩]
- Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., . . . Jumper, J. M. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630(8016), 493-500 [↩]
- Meng, C. E., Goddard, D. T., Pettersen, F. E., Couch, S. G., Pearson, J. Z., Morris, H. J., & Ferrin, E. T. (2023). ChimeraX: Tools for structure building and analysis. Protein science, 32(11). [↩]
- Krivák, R., & Hoksza, D. (2018). P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of Cheminformatics, 10(1), 39 [↩]
- Burley, S. K., Berman, H. M., Kleywegt, G. J., Markley, J. L., Nakamura, H., & Velankar, S. (2017). Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. In A. Wlodawer, Z. Dauter, & M. Jaskolski (Eds.), Protein Crystallography: Methods and Protocols (pp. 627-641). New York, NY: Springer New York [↩]
- Yan, Y., Zhang, D., Zhou, P., Li, B., & Huang, S. Y. (2017). HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res, 45(W1), W365-w373 [↩]
- Tubiana, J., Schneidman-Duhovny, D., & Wolfson, H. J. (2022). ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nature Methods, 19(6), 730-739 [↩]
- Vangone, A., & Bonvin, A. (2017). PRODIGY: A Contact-based Predictor of Binding Affinity in Protein-protein Complexes. Bio Protoc, 7(3), e2124 [↩]



{kind=link}