
Abstract
Immune Checkpoint Inhibitors represent a paradigm shift in cancer therapy, providing significant treatment benefits for patients. These drugs target immune checkpoints, regulatory proteins that dampen T cell activity. Research has led to its various subtypes, with PD-1 and CTLA-4 blockades among the most prominent. However, clinical studies indicate their association with numerous immune-related adverse events, including autoimmunity, resistance to treatment, and tumor escape, affecting several organs and causing issues like diarrhea and pyrexia. Adoptive cellular therapy has also emerged as a key approach in cancer treatment, particularly for hematological malignancies, utilizing modified T cells such as T cell–receptor transgenic T cells and chimeric antigen receptor cells. However, barriers to its success remain, with the tumor microenvironment being a major factor responsible for inhibiting T cell activity and causing cellular exhaustion. This highlights the need for further research to understand the mechanisms behind the side effects of these novel approaches and explore new combination therapies. The synergy of immune checkpoint inhibitors and adoptive cell therapies, like CAR-T therapy with PD-1 drugs, has proven effective in addressing various adverse events, mainly due to personalized treatment strategies tailored for patients. This paper discusses the major advancements to Active and Passive Immunotherapy of Cancer, involving their mechanistic study, assessment of the U.S Food and Drug Administration (FDA) approved drugs, and effectiveness evaluation based on clinical trial response rates. Additionally, it offers a comprehensive review of the synergy between immune checkpoint inhibition and adoptive cell therapy, highlighting significantly enhanced response rates. This conclusion was based on an analysis of clinical trials involving 15 different cancer types. Lastly, the role of predictive biomarkers and gut microbiome modulation in addressing these side effects is explored, along with the potential of organoids and nanoparticles for future implications.
Introduction
Cancer stands among the most terminal diseases worldwide. With over 20 million new global cases annually according to GLOBOCAN, it is proving fatal for a significant portion of our population with a mortality rate of 9.6 million deaths per year. Cancer treatments are often systemic immunosuppressant which then increases a patient’s susceptibility to bacterial, viral and fungal infections; it often intensifies existing chronic conditions.1 Given its devastating consequences, effective remedies are crucial. Hence, extensive research aims to combat these cancerous effects. On this note, a primary strategy for the treatment of cancer is the surgical removal of tumors from the patient. However, as the tumor metastasis occurs before their detection, the surgical excision often requires the resection of the whole organ, a major complication. Other than surgery, chemotherapy is another widely used approach for cancer treatment. The mechanism for early therapeutic agents in this treatment, like antimetabolites and platinum compounds, involves directly targeting DNA strands, resulting in DNA damage and replication inhibition. While, the target for the later generation chemotherapeutic agents, such as camptothecin, vincristine and topoisomerase inhibitors (e.g., irinotecan, topotecan, etoposide, and doxorubicin) centered on inhibiting enzymes which were associated with DNA replication. Despite its efficacy, chemotherapy causes immunosuppression, leading to cytotoxic effects and a weakened anti-tumor response.2
In order to tackle the challenges in the pre-existing therapies, the modulation of the patient’s immune system to fight cancer (Immunotherapy) has emerged as a promising approach. Although enhancing the immune system to treat tumors is not new, it has proven effective for many cancer types, largely due to numerous treatment agents discovered by past research.3 This has led to the enhancement of clinical response rates, survival durations, personalized treatment strategies, and overcoming side effects associated with the other therapies mentioned prior.4 Hence, it is not much of a surprise to see that immunotherapy was recognized as the 2013 science breakthrough of the year, offering new hope to patients affected by the global epidemic of cancer.5 The broad spectrum of immunotherapeutic treatment is majorly classified into 3 main categories. Adjuvant therapies include drugs like cytokines which promote the proliferation of the immune cells such as dendritic cells, macrophages (a type of phagocyte derived from monocytes) and natural killer (NK) cells that work to destroy the cancer cells. Targeted therapies include monoclonal antibodies (mABs) that target specific antigens in cancer cells and inhibit signals promoting the tumor growth and invasion. This approach includes three key mechanisms: mABs that mark cancerous cells for destruction by immune cells, mABs that deliver therapeutic drugs or toxins directly to these cancer cells, and mABs that block tumor signaling networks, thereby inhibiting tumor growth and proliferation. Vaccines offer another promising treatment, as they contain cancer-specific antigens, which are injected into the body, allowing the immune system to recognize and target those antigens in the future and destroy them through antibodies.6Immune Checkpoint Inhibitors are a prominent strategy within this paradigm of therapeutic agents, and have proved to be the cornerstone of cancer immunotherapy.7Their function is to block T cell regulatory proteins (checkpoints) allowing the immune cells to attack the cancer cells. Adoptive cell therapy (ACT) is another forefront immunotherapy involving the collection and modification of a patient’s immune cells, typically T cells, outside the body and re-infusing them to fight cancer. This approach empowers the immune cells to better recognize and destroy cancer cells, offering tailored and targeted patient care.8
Despite their individual successes, there has been limited research into the mechanistic synergy between Immune Checkpoint Inhibitors (ICIs) and Adoptive Cell Therapy. This is mainly due to the complex interactions taking place in the Tumor Microenvironment (TME) and the absence of reliable representative models to mimic the TME. Additionally, tumor heterogeneity and variation in immune profiles of patients leads to diversity in patient responses to ICI and ACT combination treatment. This paper seeks to address these gaps by providing a detailed mechanistic analysis of ICI-ACT synergy and exploring engineered T-cells as Checkpoint Inhibitors. This is extremely important as lack of mechanistic insights limits rational drug designing and preparation of personalized treatment approaches. Additionally, robust strategies to implement the Checkpoint Inhibitor and Adoptive Cell Therapy synergy are discussed with a comparative analysis showing trends and limitations. Moreover, the role of predictive biomarkers is discussed in detail to offer personalized targeted therapies and tackle tumor heterogeneity. Hence by discussing these aspects, this paper aims to optimize cancer treatment strategies. For clarity, cancer refers to a general category of neoplastic growths and specific narrower types of cancer are addressed where relevant.
Methodology
Literature Search
An initial outline was formulated, which included all the key topics that were to be held under study during the review. A literature search was conducted specifically through Google Scholar and Pubmed search engines, chosen for their extensive access to peer reviewed biomedical literature. Rather than relying on predefined keywords, a flexible approach to search terms was employed, allowing adjustments based on emerging results during the search process.
Article Selection
Majority of the articles under review were from 2010-2024, to analyse uptodate research. However, some articles from the previous decades are also included in the review, which was done to trace the evolution of new treatment strategies as the time progressed. Article selection was carried out on the basis of relevance and validity, which was assessed through the peer reviewed status and the impact factor of the journals. Articles were included only if they are published in Journals with an impact factor of 3.0 or higher, based on the recent Clarivate Journal Citations Reports.
Data Extraction
Data from the selected studies was extracted and categorized into major findings, research designs used, clinical trial results, research gaps and future implications. A thematic analysis was conducted to identify recurring patterns and themes across the studies, such as treatment efficacy, immune-related adverse events, and challenges in the tumor microenvironment, in order to ensure credibility. These themes were then summarized and contextualized to provide a cohesive and comprehensive review.
Figure Creation
Fig1 and 3 were generated using LaTex Overleaf. Fig2 was designed on Microsoft PowerPoint (v16.0). Fig4 was generated using Matplotlib (Python 3.9).
Discussion
The emergence of several new immunotherapeutic agents has enhanced efficacy of the clinical outcomes in cancer treatment. The immunotherapy of cancer is implemented in two different modes: active immunotherapy and passive immunotherapy.9
Active Immunotherapy
Active immunotherapy involves manipulation of the patient’s immune system to recognize and destroy the tumor cells and other pathogens. The effective eradication of tumor cells achieved through this therapy makes it a novel approach in cancer treatment. Without reliance on external agents, it provides prolonged protection due to immunological memory, thus reducing the risk of tumor relapse.
Cancer Vaccine Therapy
Cancer vaccines which primarily target the tumor associated antigens (TAAs) have been used to destroy the malignant tumor cells expressing such antigens.10An exciting strategy cultured the immune cells (extracted through leukapheresis) with a protein complex incorporating Prostatic Acid Phosphatase and granulocyte-macrophage colony-stimulating factor (GM-CSF), which was then reinfused back to the patient. This vaccine, called Sipuleucel-T (Provenge), was consequently granted clearance by the U.S Food and Drug Administration (FDA) as the first cancer vaccine for prostate cancer.11
Oncolytic Virus Therapy
Some viruses, referred to as oncolytic viruses (OVs) selectively replicate within cancer cells inducing tumor lysis. For example, Talimogene laherparepvec (T-VEC), which is designed to produce the human granulocyte-macrophage colony-stimulating factor (GM-CSF), has gained FDA approval for IIIB/C–IVM1a melanoma.12 ‘13 However, the complex tumor microenvironment (TME) contributing to increased acidity, hypoxia and tumor heterogeneity reduces the efficacy of oncolytic viruses (OVs). Therefore, researchers have manipulated the genomes of the OVs through genetic engineering to overcome these limiting factors, utilizing diverse techniques. One of them was demonstrated by Ju et al. who made use of OVs expressing PD-1 inhibitors, resulting in increased T-cell proliferation in mice.14
Cytokine Therapy
Cytokines are a large group of signaling proteins which have exhibited a dual nature in the TME, either inhibiting or promoting the tumors. On the basis of their structures, they have been divided into 6 classes. Chemokines guide the cell migration during the immune responses and the growth-promoting factors promote cell proliferation and tissue repair. Interleukins (ILs) are responsible for regulating the communication between the cells and interferons (IFNs) are pivotal for defence against viruses. Tumor necrosis factors (TNFs) and colony-stimulating factors (CSFs) are responsible for inflammation and production of blood cells respectively. In 1986, Interferon alpha received approval by the FDA for hairy cell leukemia (HCL) making it the first cytokine therapy to be approved for cancer. Moreover, many other cytokines have gained approval, including metastatic renal cell carcinoma, metastatic melanoma, chronic myelogenous leukemia (CML), Hodgkin Lymphoma, and Kaposi’s sarcoma.15
Limitations and Ethical Concerns
Although Active Immunotherapy has shown some therapeutic potential as shown by Fig 1, there are certain limitations to its clinical effectiveness. According to a study by Micheal A Morse et al. , many cancer vaccine trials utilized target antigens that lacked the necessary immunogenicity to provoke effective clinical responses.16 Likewise, limited efficacy was reported in oncolytic virus therapy as mentioned in a study by Shang Jiang et al. and the overall survival varied significantly from 3.25 to 20.2 months. Moreover, the challenges of delivering oncolytic viruses across the blood-brain barrier limited their potential. Trials involving oncolytic viruses present challenges in obtaining informed consent due the uncertain nature of the outcome of this therapy.17 Similarly, a study by Yi Qiu et al. analysed 2630 clinical trials on application of cytokines in cancer treatment. They highlighted that the short half-life of cytokines in the bloodstream necessitates frequent dosing which can lead to greater patient risks.18 These limitations are significant research gaps in the current landscape if Cancer Immunotherapy.
Passive Immunotherapy of Cancer
While Active Immunotherapy has shown promising results in some cases, its difficulty in generating robust immune responses underscore the need of complementary therapeutic strategies such as passive Immunotherapy. Other than stimulation of the immune system, researchers have successfully implemented the strategy of providing patients with pre modified immune components to fight infections.19 This has opened the doors to more personalized treatment strategies, along with enabling researchers to implement diverse techniques. 19 But, unlike active immunotherapy, this will result in exclusion of immunological memory, posing risks of tumor relapse.20 Later sections of this paper will explain these side effects and ongoing research towards their management.
Monoclonal Antibody Therapy
Monoclonal antibodies (mAbs) are a class of immunoglobulins, cloned by a single B cell. They work through recognizing unique molecules for example, HER2, CD20, or PD-L1. Afterwards, the mABs bind to specific sections on these antigens (epitopes) and block the binding function of the target molecules, leading to its neutralization. Researchers utilized biotechnological methods e.g (Hybridoma Technology) to produce lab-made mABs, which resulted in its large-scale manufacturing, along with specificity to alter different molecular pathways.21 This accuracy makes mAbs a form of targeted therapy, mitigating damage to surrounding healthy tissues which often result in chemo and radiation therapy that rapidly kill all dividing cells.22 ‘23
Mechanistic study
Growth factor receptors are proteins located on cell surfaces, which upon binding to their ligands trigger initiate a cascade of intracellular signals. Blockage of growth factor receptor signaling is the primary mode of action by which tumor cell death is stimulated by antibodies. But mutations in the growth factor receptors or their overexpression contributes to uncontrolled cell signaling leading to a multitude of cell divisions, more than required (metastasis) leading to cancer. The monoclonal antibodies bind to these growth factor receptors and alter the activation sites, prohibiting the uncontrolled tumor development.24
Clinical Applications
As of 2024, over 30 mABs have gained approval by the FDA for a range of cancers. Rituximab, a genetically engineered chimeric (composed of both mouse and human components) monoclonal antibody, targets CD 20 protein on B cell surfaces. Upon this binding an intracellular signaling cascade is triggered, by rituximab which kills the targeted B cell. Moreover, Hervé T. S. Cartron et al. in their study introduced a technique, called Antibody-dependent Cellular Cytotoxicity, that made use of mABs as markers for recognition by immunological cells. It was demonstrated that the Fc region of rituximab binds to certain immune cells, such as the Natural killer (NK), which then destroys the target antigens bound to rituximab. Following the success of this clinical study, it has been approved by the FDA for patients with relapsed low-grade or follicular B-cell non-Hodgkin’s lymphoma (NHL), and relapsed stage III/IV follicular lymphoma.25 This ADCC implies that rituximab can also play a complementary role in combination with other direct cancer cell killing treatments e.g chemotherapy and radiotherapy and enhance the therapeutic efficacy26 . A phase II clinical trial of rituximab in 166 patients with non-hodgkin’s lymphoma resulted in a 48% objective response rate.27
Trastuzumab is another FDA approved mAB which inhibits human epidermal growth factor receptor 2 (HER 2) and is a vital component for cancer treatment. The function of trastuzumab is to bind to the HER 2 on the cell surface of the tumor, thereby preventing cancer cells from proliferating. This is achieved through the induction of antibody-dependent cell-mediated immune response.28
Limitations
However, the exact mechanisms behind trastuzumab-induced cytotoxicity remain poorly understood, with oxidative stress being a significant factor leading to cellular damage for HER2-amplified breast cancer.29
Adoptive Cell Therapy
Mechanistic Study
Adoptive cell therapy utilizes T lymphocytes (with antitumor activity) derived from the patient, which are then modified in vitro through gene engineering and reinfused into the cancer patient. It encompasses various immunotherapy strategies aimed at harnessing cytotoxic T cells, but only two will be discussed in this review. T cells are core agents of adaptive immune systems and induce a cytotoxic response against viral infections and diseases. Each T cell is equipped with a protein complex called T cell receptor, which recognizes foreign agents loaded onto the Major Histocompatibility Complex of the Antigen Presenting Cells (APCs).30 Due to some intracellular infections like a virus, peptides of viral proteins are displayed by the class I of Major Histocompatibility Complex (MHC). This presentation enables the cytotoxic CD8+ T cells to recognize and attack these abnormal proteins. However, extracellular peptides require the Major Histocompatibility Complex to be expressed on antigen presenting cells (APCs), which classifies the MHC as MHC class II. These antigen presenting cells activate the CD4+ helper T cells, which further secrete cytokines and trigger B cells.8
Clinical Effectiveness
This therapy has shown great efficacy in the treatment of metastatic melanoma. According to a clinical trial mentioned by Yinqi li et al. approximately 50% of the patients showed an objective response (OR) whereas 15% of the patients obtained a durable complete response (CR) through adoptive cell therapy.31
Adoptive Cell Therapy using TCR-T cells
Mechanistic Study
A major breakthrough in immunology came through the discovery of alpha and beta chain heterodimer on T cell receptors, by Bojan Dembić et al. in the 1980s.32 The alpha and beta chains each are composed of a variable and a constant region, where the variable region (CD23) forms the antigen–binding site. However, due to a short intracellular domain (part of a protein receptor inside the cell), the TCR lacks the signaling capacity to activate the T cells. To tackle this, the conformational change in the shape of the receptor upon binding the antigen–MHC complex activities CD3 (which is a protein in the intracellular domain of the CAR that initiates T cell activation) subunits which are complexed with the TCR. The CD3 subunits contain immunoreceptor tyrosine-based activation motifs (ITAMs) which are short amino acid sequences found in the cytoplasmic tails of certain cell surface receptors. There are tyrosine residues present in these ITAMs, which become phosphorylated upon the engagement with TCRs. This phosphorylation creates binding sites for ZAP-70 kinase, which triggers all the major signaling pathways i.e MAPK Pathway and AP-1 Activation, Calcium Influx and NFAT Activation. The MAPK pathway, AP-1, and NFAT collectively coordinate cellular responses to external stimuli, such as activation signals in immune cells. The MAPK pathway transduces signals that lead to the activation of AP-1, a transcription factor that regulates genes involved in cell proliferation and differentiation. Concurrently, NFAT (Nuclear Factor of Activated T-cells) is activated in response to calcium signaling and works together with AP-1 to regulate genes critical for immune cell activation, cytokine production, and T cell differentiation. This collective signaling network plays a central role in immune responses, particularly in T cell activation and function. As a result of this process, T cell function is boosted in three primary ways; T cells proliferate rapidly through multiple clonal expansions. Moreover, these T cells differentiate into two further classes; cytotoxic T lymphocytes (CD8+) which kill the antigen binded to the MHC by releasing toxins, and helper T (CD4+) cells which aid the process through secreting cytokines.33
Clinical Applications
Naturally present T cell receptors face significant limitations, including weak affinity to bind with the tumor antigens and their dependence on presentation by the Major Histocompatibility Complex.28To address this, researchers made use of genetic engineering to further modify these T cell receptors in vitro (experiments done in a lab), enhancing its binding affinity with antigens. Clinical studies have revealed positive results of this treatment modality, which can be evaluated by a phase II clinical trial conducted by Robbins et al. in which TCR expressing T cells targeting the New York esophageal squamous cell carcinoma (NY-ESO)-1 antigen in patients through a retrovirus vector. The objective survival rate in response (ORR) was 57.9%, including 5 complete and 17 partial responses. Additionally, no adverse effects were reported from the trial which showcase an effective and on target immune response.34 Numerous clinical trials showcased the therapeutic effectiveness of TCR–T cell based ACT across multiple solid tumor types. The melanoma-associated antigen (MAGE) gene expressed on melanoma cells encodes cancer testis antigens (CTA), a group of tumor associated antigen expressed in many cancers i.e breast cancer, ovarian cancer, melanoma. A clinical study by Rosenburg et al. focused on treating patients with metastatic cancers expressing the MAGE-A3 antigen. The treatment involved a lymphodepleting conditioning regimen followed by HLA-DP 0401/0402-restricted-anti-MAGE-A3 T cell receptor along with aldesleukin admininstration. Two partial responses and a single complete response was observed during the study.35
Limitations and Future Perspectives
From the results, we can deduce that although the therapy has shown mild efficacy, there is a need for further strategies to improve its functionality and accuracy. Additionally, focus is being laid to make a bank of healthy donor cells which can be engineered to express the efficient T cell receptors, which will reduce the extensive time taken by taking cells from the patient itself. Currently ongoing trials include the TCRs targeting MAGE-A3/A6, MAGE-C2, MAGEA4/8 among others.31
Chimeric Antigen Receptor-T cell therapy (CAR-T)
Mechanistic Study
Chimeric antigen receptor T cell therapy also involves the genetic modification of a patients’ T cells, but this time to express the chimeric antigen receptor (CAR). It is different from the TCR therapy as it induces tumor lysis (tumor destruction) independent of the MHC, and is for hematological malignancies (blood cancers).33 In similarity with the TCR-T cell therapy, the patients’ T cells are separated from their blood through leukapheresis (A procedure to separate white blood cells from the patients’ blood). Afterwards they are engineered genetically in the lab to express the chimeric antigen receptor (CAR) and then reinfused to the patient through viral vectors (modified viruses to deliver genetic material into T cells). The structure of this chimeric antigen receptor consists of three primary sections. The first is the extracellular domain, encompassing an antigen binding scaffold which consists of a single chain variable fragment (ScFv) which is a fusion protein of the variable regions of both the heavy (VH) and light chains (VL) of immunoglobulins, connected with a short linker peptide of ten to about 25 amino acids. This ScFv aids the binding of the CAR–T cells to the targeted antigens, offering two benefits; each ScFv is engineered to recognize a specific epitope (sites recognized by antibodies), enabling specificity in antigen recognition. The other is versatility, which is achieved by a flexible segment called the hinge region which is a flexible segment that connects the antigen-binding domain to the transmembrane domain, allowing better orientation of the CAR to target epitopes on tumors. Longer hinges provide greater flexibility, as it allows better orientation of the CAR to target the epitopes on tumors.36 The next is the transmembrane domain which is characterized by a single traversal of the membrane, meaning that it spans the membrane only once, forming an alpha-helical structure that is lipophilic. It enables CARs to be attached onto the plasma membrane of T cells. Furthermore, it ensures correct positioning of the extracellular domain ( part of the CAR that extends out of the T cell for antigen binding). In the interior of CARs lies the intracellular signaling domain, which integrates signals to activate the T cells upon antigen–binding. This process is triggered by a CD3ζ chain co-receptor. Afterwards the intracellular signalling pathways are initiated as detailed in the prior section.
Generational modifications to CAR-T cell therapy
Multiple modifications have been introduced to this therapy, leading to what we now classify as the generations of CAR-T cell therapy. The first generation of CAR-T therapy was introduced in the early 1990s which involved the incorporation of the CD3ζ into the CAR construct. But a single CD3ζ is not efficient in creating a robust immune response, as this therapy showed limited efficacy in clinical trial testing.37 This was because T cells required a second signal (co simulation) to become fully optimized rather than only a primary signal received by binding to the antigen. This signal is provided by the co-stimulatory molecules (molecules on the T cell surface that provide additional signals to enhance T cell activation and persistence) on the T cell surface by binding to their ligands on the APCs (e.g B7-1). Hence, the second generation of CAR-T cell therapy incorporated these co-stimulatory molecules such as CD28 and 4-1BB(CD137). Aaron J. Harrison et al. in their review mention a clinical trial of a patient of advanced follicular lymphoma, whose cancerous B cells were destroyed through the CAR with CD28 (co stimulatory molecule), making its efficacy evident.38 Researchers are currently working to integrate other co-stimulatory domains, which has led to further generations of the CAR-T cell therapy. In 2012, the third generation of the CAR-T therapy was introduced, which involved the combination of CD3ζ and two co-stimulatory molecules (CMs). This led to the development of various CAR-T cell constructs, including combinations of CD3ζ with costimulatory molecules such as OX40, CD27, ICOS, and 4-1BB, to optimize T cell activation and persistence in immunotherapeutic applications. The third generational CAR therapy involved a combination of two co-stimulatory molecules in addition to the primary CD3ζ signaling domain. The inclusion of two co-stimulatory domains promotes additional signaling which enhances T cell expansion. For instance, pairing of CD28 with OX40 or 4-1BB with ICOS ( examples of co-stimulatory molecules) creates a dual signaling mechanism that generates a more robust immune response. Theoretically, the third generational CAR therapy should have superior efficacy to the first. 38 However, a contrast is seen in the studies of Ramos et al. and Abate-Daga D et al. who have demonstrated a clearly greater and a variable immune response respectively upon the third generational CAR treatment.39 ‘40 The heterogeneity observed in these results could be largely due to the difference in study design and focus. Ramos et al. focused on hematological malignancies, which have a less immunosuppressive environment compared to the solid tumors studied by Abate-Daga D et al. In the fourth generational CAR-T cell therapy also known as universal cytokine mediated killing (TRUCK) or universal CAR-T (UniCAR-T), the nuclear factor of activated T cells (NFAT) is incorporated through genetic engineering to enhance functionality. It is translocated on T cell surfaces and initiates the gene transcription for the production of cytokines. This mechanism enables precise control over cytokine release, ensuring that they will only be released when the T cell has binded to the antigen. The UniCAR-T cells have led to enhanced T cell expansion and effective anti tumor activity by the production of these cytokines, but have shown less efficacy against solid tumors.38
Clinical applications
Additionally, the CD-19 targeted autologous (involving extraction of the patient’s own cells) CAR-T cell therapies have manifested great efficacy in treatment of B–cell malignancies which can be seen in the following clinical results. In an ELARA phase II global trial Tisagenlecleucel was administered in 97 patients with Refractory Lymphoma41. The results demonstrated an objective response rate (ORR) of 86% and a complete response rate (CRR) of 69%. Moreover, Axicabtagene ciloleucel (Yescarta) was approved by the FDA in October 2017, for the treatment of refractory large B-cell lymphoma (DLBCL). Furthermore, it was approved for refractory follicular lymphoma in 2021 based on the positive results on the ZUMA-5 clinical trial.33 Although the immense potential of the CAR-T cell therapy is demonstrated from the above results, this treatment comes with several limitations including the cytokine release syndrome leading to systemic inflammation, and tumor antigen escape leading to loss of recognition by the CAR-T cells. Additionally, the environment surrounding a tumor, known as immunosuppressive tumor microenvironment (TME) includes factors like checkpoints, regulatory T cells, myeloid-derived suppressor cells which actively inhibit the immune response. As a result of this CAR-T cells may face difficulty infiltrating tumors. Hence, overcoming tumor heterogeneity (diverse genetic, phenotypic, and molecular differences) and reducing tumor suppression may lead to a significantly enhanced CAR-T cell therapy response.
Future directions
Contemporary studies on CAR-T cell therapy are focused on its 5th generation, making use of more advanced and sophisticated techniques. One of them is to make use of cytokine receptors (cell surface proteins for binding to cytokines e.g IL-2R chain fragment) to incorporate multiple intracellular signal domains. These are designed to simultaneously attack multiple antigens on tumor cells, reducing the risk of tumor escape (ability of cancer cells to evade destruction). Another exciting strategy to enhance the CAR cell precision is to use CRISPR/Cas9 that allows precise modification of DNA within living organisms.40
Immune checkpoint Inhibitors
Among all the treatment strategies mentioned earlier, Immune checkpoint Inhibitors (ICIs) stand among the most significant advancements in cancer immunotherapy. Over the previous decade, the number of FDA approvals for these checkpoint blockade drugs has increased significantly, revolutionizing the landscape of cancer therapy.
Mechanistic detail of Immune checkpoints
As discussed in detail above, the activation of T cells and their complete proliferation requires a costimulatory or a two-signal process. When the pathogen enters the body, the dendritic cells engulf it through phagocytosis, in a process known as antigen capturing. The antigen is then broken down into fragments and loaded onto the major histocompatibility complex (MHC) for antigen presentation. The variable region of the TCR recognizes and binds to the MHC originating the first signal. The second signal comes from the binding of the co-stimulatory molecule (mainly CD28) to the B-7 ligand on the APCs (CD80,CD86). This binding to the ligand invokes a signaling relay which leads to complete T cell expansion. However, excessive increase of these T cells may result in autoimmune disorders such as tissue damage and inflammation, thyroiditis, hypophysitis. Hence, to regulate this excessive T cell expansion, inhibitory molecules known as immune checkpoints found on T cell surfaces downregulate the activation signals. Upon their interaction with their specific ligands, these immune checkpoints deliver an inhibitory signal (also referred to as negative signal) that suppresses the T cell activation. Hence they play a vital role in the prevention of autoimmune disorders. Despite this, these checkpoints have consequences as far as cancer is concerned, as the tumors escape the T cell attack. ICIs are a class of monoclonal antibodies that block these inhibitory signaling pathways and reinvigorate the immune-mediated response.42 The below section contains a review of two novel immune checkpoint inhibition therapies, restricting the cytotoxic t-Lymphocyte antigen-4, and programmed cell death-1 checkpoints.
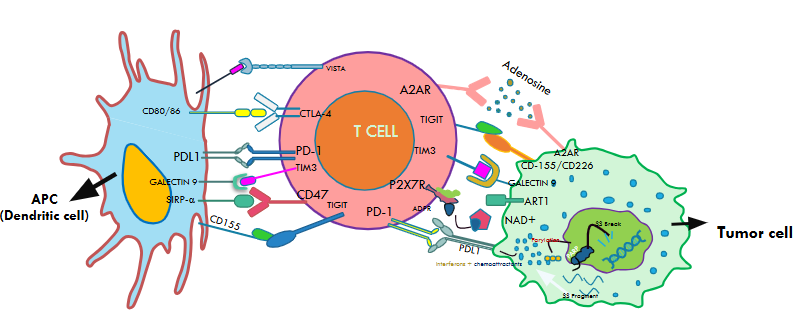
Cytotoxic T Lymphocyte Associated Antigen-4
Historical Context
Brunet et al. in 1987 discovered the cytotoxic T lymphocyte antigen-4 (CTLA-4) during his functional studies in mice, which was then cloned next year. .42 Subsequently, a landmark research was carried out by James Allison et al. who discovered that CTLA-4 is a co–inhibitory receptor that competes with CD28 on binding to the co-stimulatory ligands (i.e B7-1 and B7-2) won the antigen presenting cells.
Mechanism of action
CTLA-4 and CD28 are both homodimers (two identical protein subunits joined), but bind to the ligands with different affinities. CTLA-4 having greater affinity to bind with B7-1 and B7-2, sequesters the ligand towards itself, binds to it, and dampens the T cell activation as shown in Fig2. Moreover, the engagement of CTLA-4 reduces interleukin-2 (IL2) production, which is critical for an effective T cell function.43 A groundbreaking discovery which truly revolutionized cancer research came through the research of Allison et al. which showed that blocking the interaction of CTLA-4 with the ligands (B7-1 and B7-2) resulted in a significantly enhanced immune response. Other extensive studies of CTLA-4 blockade conducted in animal-based models, showed enhanced clinical responses to prostrate, breast, lymphoma and other cancers.44
Clinical application
Results of a phase III clinical trial demonstrated that patients receiving Ipilimumab (anti CTLA–4 mAB) had a median overall survival of 10.1 months (ORR of 10.3%). Hence, Ipilimumab was granted clearance by the FDA as an igG1 mAB for the blockade of CTLA-4.45
Programmed Cell Death Protein-1 (PD-1)
Historical Context
Programmed Cell Death Protein-1 (PD-1), discovered by Honjo et al. at Kyoto University in 1992, is classified within the same structural family of proteins as the CD28 and CTLA-4 immune checkpoints.46 PD-1, just like CTLA-4, is a receptor present on various immune cell surfaces such as those of T cells and myeloid cells as depicted in Fig2.
Mechanistic study
In the Tumor Microenvironment, PD-L1, a ligand expressed on tumor cells and antigen presenting cells binds to the PD-1 receptor on T cells as shown in Fig2. This interaction suppresses the T cell activity, which allows the tumors to evade immune surveillance. Upon this binding, the intracellular domain of PD–1 becomes tyrosine phosphorylated. This phosphorylation occurs in specific regions of its intracellular domain, including immunoreceptor tyrosine-based inhibitory motifs (ITIM) and immunoreceptor tyrosine-based switch motifs (ITSM). The phosphate group added to the tyrosine residues is critical in activating the signaling pathways as we saw in earlier sections. However, binding of SHP-1 to the ITSM leads to the dephosphorylation which inhibits all the necessary signaling pathways, including PI3K/Akt pathway and the stimulation of PTEN phosphatase activity. The suppression of these crucial signaling pathways leads to decrease in the T cell proliferation and expansion. Immune checkpoint inhibition drugs inhibiting the PD-1 and PDL-1 interactions have been at the forefront of cancer immunotherapy.
Clinical applications
PD-1 inhibition has shown efficient immune responses to melanoma, colon cancer, non–small lung cancer, Hodgkin lymphoma, and many other cancer types.47 Nivolumab, an igG4 mAB, was the first PD-1 inhibitor was tested in clinical trials for refractory solid tumors, and subsequently gained approval by the FDA for metastatic melanoma in December 2014. A landmark clinical trial (CheckMate 037) demonstrated nivolumab’s greater efficiency, than the chemotherapy (dacarbazine orcarboplatin) as nivolumab showed an objective response rate of 31.7%, which was higher than that of dacarbazine orcarboplatin which was 10.6%. This study laid the foundation for further research on PD-1, leading to its FDA approvals for 12 different types of cancers.48 Pembrolizumab is another anti PD-1 humanized igG4 mAB, which was approved by the FDA in September 2014 to treat metastatic melanoma. Clinical studies of pembrolizumab demonstrated an ORR of greater than 30% in the patients with advanced melanoma.49 It has been approved by the FDA for Melanoma, MSI-H solid tumors, urothelial carcinoma where immune checkpoint pathways play a crucial role. Cemiplimab, another igG4 mAB, was approved by the FDA in 2018 for the treatment of cutaneous squamous cell carcinoma (CSCC) due to its ability to counteract PD-1 mediated immune suppression. Additionally, Atezolizumab, Durvalumab, along with Avelumab have also received approval by the FDA as PD-L1 inhibitors.
Challenges and Limitations
The extensive range of the FDA approved checkpoint blockade drugs mentioned above showcases the success of the therapy, but comes with certain limitations which are being explored by current research. In this view, a prominent study by Filleron et al. demonstrated that only 20-30% patients showed a clinical response after receiving ICI therapy, through their flexible parametric cure model trial.50 Immune checkpoints as discussed above play a crucial role in immune tolerance, however their blockage may result in overactivation of the immune system. This leads to a number of immune-mediated toxicities, jointly referred to as immune related adverse events (irAEs). These may occur through the following mode of actions: Autoreactive CD4+ T cells, Cytotoxic CD8+T cells, release of self antigens, autoreactive B cells, and pro-inflammatory mediators like cytokines.51 A recent retrospective study conducted by Guihong Wan et al. showed the statistics of the patients receiving checkpoint blockade therapy. According to it, immune related adverse events were present in 37.7% of the 15,246 recipients in the MGBD cohort and 30.5% of the 50,503 recipients in the TriNetX network. Additionally, seven patient clusters were identified based on organ-specific irAEs, including those affecting the endocrine system, skin, respiratory tract, gastrointestinal tract, musculoskeletal system, liver and neurological system.52 Along with it, there was a lack of response to ICI therapy in glioblastoma, pancreatic cancer, ovarian cancer.53 Another major problem faced in this view was the complexity of the Tumor Microenvironment (TME) which not only suppressed immune cell infiltration, but their heterogeneity makes the testing and understanding of the mechanisms complicated. In the light of these challenges, careful monitoring of these side effects in patients has become a dire need.
Predictive biomarker development
Predictive biomarker development is of immense importance to minimize these side effects in the early and later stages of ICI therapy. In this regard, PDL-1 expression is a promising biomarker which is assessed in patients through histochemical staining. Studies demonstrate a correlation of PDL-1 expression and enhanced response to ICI therapy. However the sensitivity and specificity of PDL-1 expression vary significantly across cancer types and testing platforms.54 Tumor Mutational Burden is the number of mutations per megabase in a tumor, which has a direct correlation with the number of neoantigens. Hence, high TMB has been associated as a predictive biomarker, producing more neoantigens which can be recognized by the immune system. However, TMB lacks thresholds for defining high mutational burden across tumor types and sequencing methods affecting its predictive accuracy. Microsatellite instability (MSI), another biomarker produced by the defective DNA mismatch repair (dMMP) increases the immune cell infiltration, enhancing the susceptibility of tumors to the ICIs. The FDA has approved several therapies incorporating MSI as a response biomarker, including Pembrolizumab for MSI-H colorectal cancer in 2017 and Dostarlimab for MSI-Endometrial cancer in 2021. Yet, MSI is rare in many cancer types limiting its broader application as a predictive biomarker.55
Gut Microbiome Modulation
The modulation of immunotherapy through the gut microbiome has also been persistently linked with better clinical outcomes associated with checkpoint blockade therapy. The fundamental clinical model for the implication of this strategy makes use of metabolites which spread out from the gut and influences the working of ICIs, thereby increasing their efficiency.56However, in their systematic review, Anjali Bhatt et al. demonstrated a lack of significant experimental evidence for the microbiome suggesting the need for better-designed and more randomized clinical trials.57 Although these approaches have resulted in significant increase in efficacy, further understanding of the mechanisms underlying these adverse effects and the complexities of the TME is the need of the hour for better therapeutic regimens.
Synergistic Combination of Immune Checkpoint Inhibition and Adoptive Cell Therapy
Combination therapies exploit the complementary mechanisms of action of diverse agents to enhance anti-tumor efficacy, mitigate resistance, and improve clinical outcomes, thereby representing a transformative approach in cancer management.58 This can be judged by a landmark clinical trial Check-Mate 67 (NCT01844505) which demonstrated that ipilimumab (originally having a 5-year OS rate) showed an OS-rate of 46% in combination with nivolumab.59 Synergy is a type of combination therapy in which the interaction of two or multiple drugs yields better efficacy compared to their individual outcomes. The synergistic combination of adoptive cell therapy and Checkpoint Blockade is a novel therapeutic with multiple clinical benefits. This section will present the two novel approaches for implementing this synergy to address the challenges of their monotherapies such as irAes, T cell exhaustion, tumor escape, cytokine release syndrome (associated with CAR-T therapy).
Rationale for Synergism of CAR-T therapy with Checkpoint Blockade
The administration of genetically engineered T cells like CAR-T cells followed by the infusion of checkpoint blockade antibodies (cell-extrinsic strategy) has shown promising results in clinical studies. A prerequisite for the effective functioning of ICIs is a hot tumor microenvironment (TME) (infiltrated by a significant number of immune cells). In contrast, a cold TME lacking effector T cells leads to several immune evasion mechanisms. One of them is the reduced expression of tumor associated antigens, which limits the immune system’s ability to recognize the cancer cells. Moreover, the MHC downregulation occurs (which hampers the antigen presentation) along with resistance to IFN-γ signaling (which is an essential signaling pathway to activate the immune system).60 The CAR-T cells combined with the checkpoint inhibitors can provide such T cell infiltrates for the immune-inactive tumors, regulating T cell proliferation. In parallel, persistent exposure to antigen presenting cells leads to functional impairment of T cells in the CAR-T therapy, which is also referred to as T cell exhaustion. When PD-1 binds to its ligand PD-L1, SHP-2 Phosphatase is recruited which leads to dephosphorylation of ZAP-70 and CD3ζ, which reduces the T cell receptor signalling. PD-1 inhibition reverses T cell exhaustion by inhibiting the PD1/PDL-1 signaling pathways, which typically transmits negative signals through the SHP-2 Phosphatase. By blocking the PD-1, inhibition on TCR signalling is lifted, which then reactivates T cell proliferation. Additionally, a study by Songnan Sui et al. revealed that T cell exhaustion exists with distinct subpopulations with a varying extent of dysfunction. Notably, the progenitor exhausted CD8+ T cells expressing markers such as TC57 and IL7R, retain responsiveness to PD-1 inhibition. This finding also suggests ICIs can partially reverse the T cell exhaustion.61 Moreover, the addition of ICIs help to inhibit excessive cytokine release, thereby tackling the challenge of cytokine release syndrome (CRS) which is commonly faced in CAR-T therapy.62
Comparative Insights from key studies
In this view, a prominent investigation was done by Chong EA et al. on the effects of pembrolizumab administered 26 days post-infusion of CD19 targeted CAR-T cells. This was carried out in a patient with diffuse large B-cell lymphoma (DLBCL). Positive results were extracted from the study including an enhanced number of CAR-T cells, along with elevated serum IL-6 concentrations.62 ‘63 Another study in a breast cancer model by John et al. in 2013 investigated the synergistic combination of HER-2 targeted CAR-T cells and PD-1 blockade. This combination therapy enhanced CAR-T cell functionality effectively, through increase in IFN-γ production, along with granzyme B expression.64 Both studies highlight the critical role that PD-1 inhibition plays in improving CAR-T therapy. However, according to the emphasis by James Allison and Padmanee Sharma, this combination should involve the targeting of multiple inhibitors so that the resistive effects to a single ICI can be overcomed.65 Ongoing trials are exploring the synergistic effect of CAR-T cells and other checkpoint inhibitors such as LAG-3 and TIM-3, which can lead to further optimization of this therapy.
Rationale for Engineering T cells as Intrinsic Checkpoint Blockade agents
Long-term use of many pharmacological ICIs can result in immune related adverse events (irAes) which lead to long term complications and increased healthcare costs. To overcome this challenge, T cells (CAR-T or TCR-T) are engineered to downregulate the inhibitory receptor signaling, thus, functioning as a checkpoint inhibitor.60 This further engineering of T cells to provide the patient with a continuous source of ICI is known as a cell-intrinsic model.66
Comparative analysis of mechanistic approaches
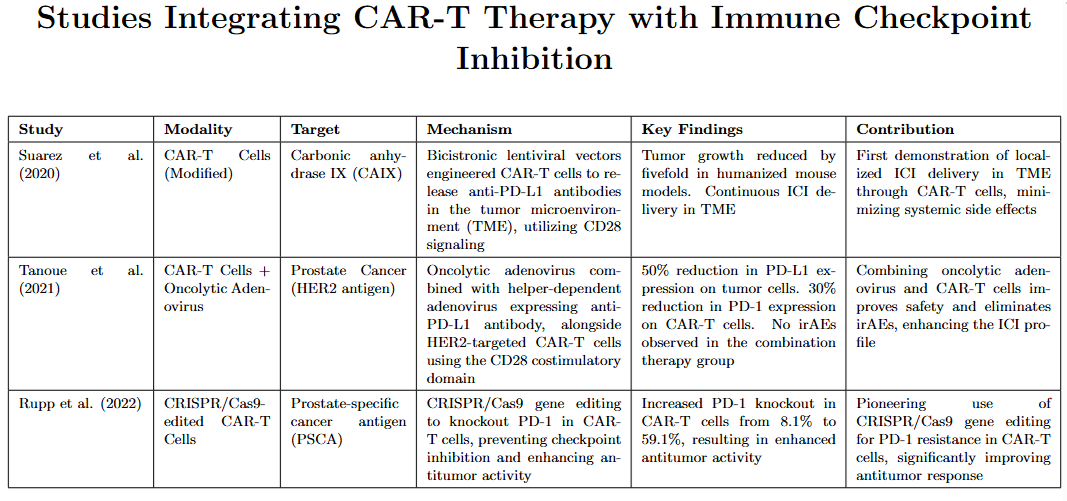
The studies of Suarez et al. (2020), Tanoue et al. (2021), and (Rupp et al. 2022) all highlight strategies to enhance the efficacy of CAR-T cell therapies by targeting immune checkpoints. In their study Suarez et al. designed a novel strategy by using CAR-T cells to target carbonic anhydrase IX (CAIX) through CD28 signaling. The modification of these CAR-T cells was done through a bicistronic lentiviral vector. This enabled them to release anti PD-L1 antibodies in the TME. According to clinical studies investigating the effect of these antibodies in humanized mice models, tumor growth was reduced by five times.67 Building on this concept of targeted ICI delivery, Tanoue et al. introduced an innovative approach for treating prostate cancer that combines an oncolytic adenovirus (Onc.Ad), which was engineered to replicate and lyse the cancer cells, and a helper-dependent adenovirus (HDAd), which was specifically engineered to express a single-domain antibody that blocks PD-L1. This was administered alongside intravenous HER2-targeted CAR-T cells utilizing the CD28 costimulatory domain.60 The results showed that tumor cells had 50% lower PD-L1 expression, whereas a 30% reduced PD-1 expression on CAR-T cells was reported. Diarrhea was reported in some mice which were infused with anti-PD-L1 IgG. However, no irAe was reported in the ones infused with CAR-T cells supplemented by oncolytic adenovirus and a helper-dependent viral vector.64 Hence this approach focused more on safety, highlighting a shift towards tolerability of the therapy alongside its effectiveness. Another exciting approach presented by Rupp et al. is to use gene editing methods such as CRISPR/Cas9 to resist PD-1 inhibition (also known as PD-1 knockout). Ren et al. investigated the antitumor activity in prostate-specific cancer antigen (PSCA) provoked by using modified CAR-T cells through CRISPR/Cas9 to deactivate PD-1. This gene editing approach resulted in an increase in the fraction of CAR-T cells lacking PD-1 expression, rising from 8.1% in the control group to 59.1%. Across these studies, it was found that the engineering of CAR-T cells to function as Checkpoint Inhibitors is crucial for improving the safety and efficiency of cancer Immunotherapy.68 These innovative approaches not only present effective solutions, but also set the ground for future research in immunotherapy.
Clinical trial analysis
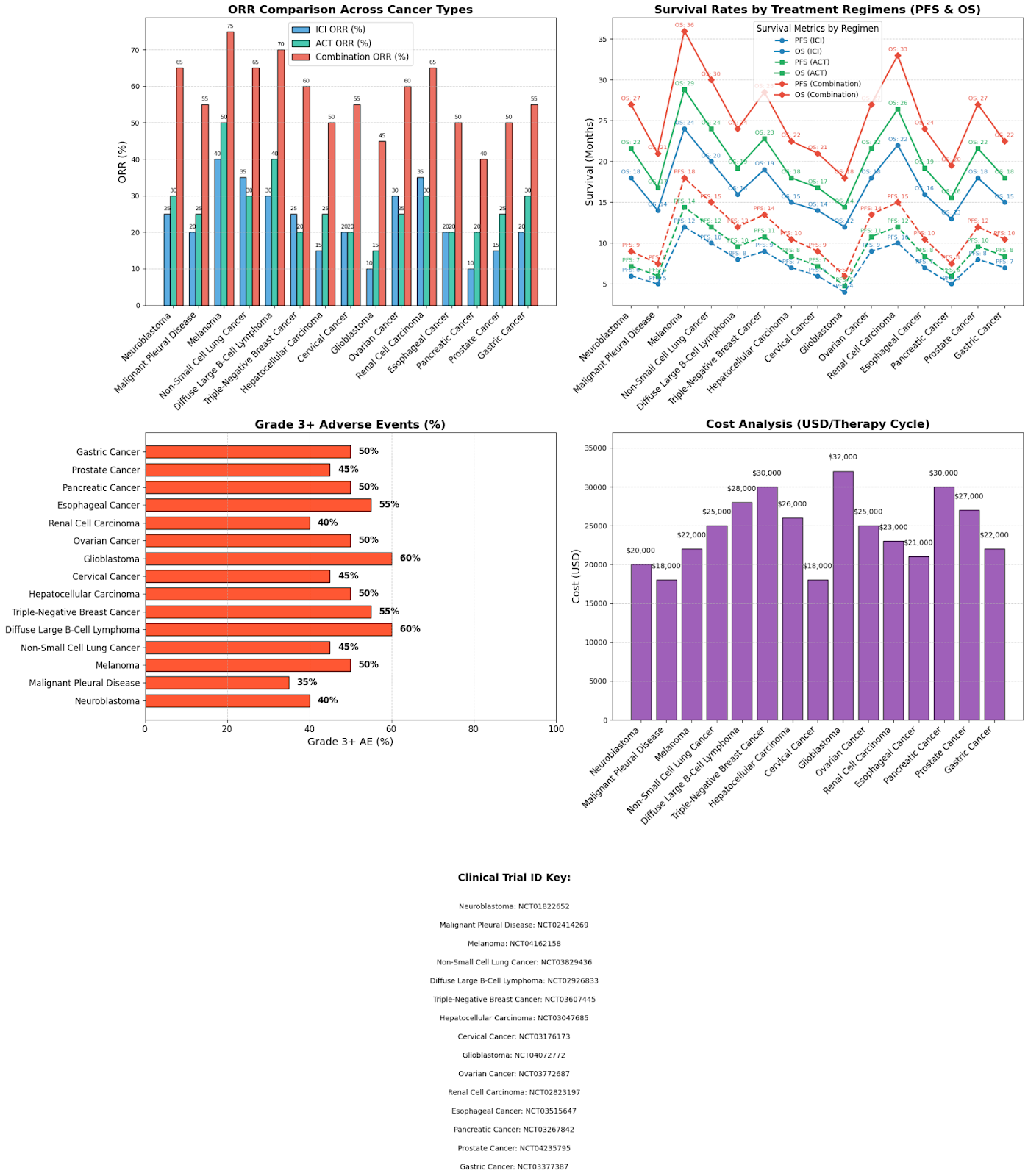
This clinical trial analysis shown in Fig4 compares the response and survival rates of the monotherapies of ICI and the ACT to their combination therapies. Clinical trial studies of 15 cancers were assessed in the study on the basis of their objective response rates, survival rates, and the percentage of grade 3+ adverse events after receiving the treatment regimen. A broad variety of combination treatments were used in these clinical trials which have been broadly classified as adoptive cell therapy and Immune checkpoint Inhibitor therapy for easy understanding. The study results revealed a substantial increase in objective response rate for all the 15 cancers upon the ICI and ACT combination therapy, compared to their monotherapies. For instance, the objective response rate in neuroblastoma rose from 25% and 30% in ICI and ACT therapy respectively to 65% in the combination therapy. Additionally, the patients receiving the combination therapy showed remarkably increased progression-free survival and objective survival durations, which is indicative that the mechanism is effective in dealing with the associated adverse events. These results show the effectiveness of this synergistic approach across multiple tumor types.
Confounding factors and inter-study heterogeneity:
Despite the promising survival outcomes in the 15 referenced trials, several confounding factors and inter-study heterogeneity limit the interpretation of efficacy. Variability was seen in tumor types, for example the NCT04162158 trial on melanoma and the NCT02823197 trial on renal cell carcinoma involved tumors with high baseline immunogenicity, which is more likely to respond to ICI and ACT therapy. In contrast, the NCT03267842 trial on pancreatic cancer targets cold tumors which are typically more resistant to therapy. Additionally, diversity was seen in trial design and ACT engineering approaches.
Adverse event analysis
Across the referenced trials in Fig 4, combination therapies generally showed higher rates of Grade ≥ 3 adverse events compared to the monotherapies. These adverse events included numerous toxicities such as cytokine release syndrome, neurotoxicity, auto-immune related colitis, pneumonitis, and endocrinopathies. Management strategies reported in the literature, including corticosteroids and ICU support for cytokine release syndrome, anti-leptics for neurotoxicity, and dosing modifications and premedication as preventive measures. While the ICI and ACT combination therapy enhances anti tumor activity, it comes with a greater overall percentage of adverse events compared to the monotherapies. This highlights the importance of improved biomarker-based patient selection and early adverse event prediction models.
Conclusion and Future Perspectives
This paper provided a comprehensive review of the evolving landscape of cancer Immunotherapy by highlighting the mechanisms, clinical applications, and comparative effectiveness of key treatment strategies. The review bridges the mechanistic understanding of ACT/ICI synergy with clinical response data across diverse cancer types – a perspective lacking in current literature. Through the analysis of 15 clinical trials it was demonstrated that ACT and ICI combination led to markedly improved progression-free survival and objective response rates. Although this synergistic approach of ICI and ACT has shown great efficacy in cancer therapy, challenges such as resistance to treatment and less response rates in patients remain. On this note, there is robust ongoing research and new experimental-designs are being evaluated. However, the complexity of the TME involving innate myeloid and lymphoid cells, cancer-associated fibroblasts, and the tumor vasculature (contributing to tumor escape) as discussed above, pose a challenge in the efficient testing for these experimental-models. This indicates the need for better clinical models to recapitulate the TME, other than the pre-existing mouse models. In this view, the use of 3D organoid cultures which mimic the structure and function of organs is a very promising approach. These organoid cultures are capable of an enhanced replication of the TME as they maintain the diversity of including malignant cells, stromal cells (like cancer-associated fibroblasts), endothelial cells, and other immune cells. Moreover, their 3-D structures allow better understanding of the spatial interactions between different cells in the TME. Other than that, the organoids derived from human cells avoid the species-related discrepancies, which are often found in mouse models. Recent studies have investigated the combination of organoid cultures with organ-on-a-chip models for better functionality of such laboratory models. The organ-on-a-chip models make use of microfluidics to mimic the fluid flow in organs and make spatial gradients, for example, a hypoxic gradient (lower oxygen levels in specific areas) can be created to simulate the conditions in a tumor’s core.69 Moreover, these organ-on-a-chip models can involve transparent materials for real-time imaging, leading to effective TME monitoring. In addition to the precision-oncology achieved due to these factors, organoids can lead to more personalized therapeutic regimens for patients through their biopsies.70 Along with it, a novel and effective delivery approach of the ICIs to the infected site is essential for accurate tumor targeting. Nanoparticles have emerged as effective delivery agents of the ICIs, resulting in their targeted delivery and overall modulation of the immune response. Study conducted by Song W et al. showed that the use of lipid-protamine-DNA (LPD) nanoparticles (carrying PD-L1 plasmid) to disrupt PD-L1 signaling works synergistically to inhibit tumor growth, and reduce toxicity.71 Further research on the customization and scalability of nanoparticles could enable a more tailored immune response to tumors. Moreover, the utilization of machine learning and deep learning methods can result in robust Artificial Intelligence models. These models can increase our understanding of immune related adverse events through the simulation of tumor-immune interactions. Additionally, certain AI algorithms can mine genomic, transcriptomic, and proteomic data to predict the response rate of patients with ICI and ACT therapies.
References
- 1) Rui, R., Zhou, L., & He, S. (2023). Cancer immunotherapies: Advances and bottlenecks. Frontiers in Immunology, 14, 1212476. https://doi.org/10.3389/fimmu.2023.1212476 [↩]
- Dai, M., Liu, D., Liu, M., Zhou, F., Li, G., Chen, Z., Zhang, Z., You, H., Wu, M., Zheng, Q., Xiong, Y., Xiong, H., Wang, C., Chen, C., Xiong, F., Zhang, Y., Peng, Y., Ge, S., Zhen, B., … Cai, H. (2020). Patients with cancer appear more vulnerable to sars-cov-2: A multicenter study during the covid-19 outbreak. Cancer Discovery, 10(6), 783–791. https://doi.org/10.1158/2159-8290.CD-20-0422 [↩]
- Faculty of Biological Sciences Kampala International University Uganda, & Nsubuga, S. N. (2024). The role of immunotherapy in cancer treatment: Mechanisms, efficacy, and future directions. Newport International Journal Of Public Health And Pharmacy, 5(2), 57–61. https://doi.org/10.59298/NIJPP/2024/523945761 [↩]
- Chu, X., Tian, W., Wang, Z., Zhang, J., & Zhou, R. (2023). Co-inhibition of tigit and pd-1/pd-l1 in cancer immunotherapy: Mechanisms and clinical trials. Molecular Cancer, 22(1), 93. https://doi.org/10.1186/s12943-023-01800-3 [↩]
- Vanneman, M., & Dranoff, G. (2012). Combining immunotherapy and targeted therapies in cancer treatment. Nature Reviews Cancer, 12(4), 237–251. https://doi.org/10.1038/nrc3237 [↩]
- Vanneman, M., & Dranoff, G. (2012). Combining immunotherapy and targeted therapies in cancer treatment. Nature Reviews Cancer, 12(4), 237–251. https://doi.org/10.1038/nrc3237 [↩]
- Chu, X., Tian, W., Wang, Z., Zhang, J., & Zhou, R. (2023). Co-inhibition of tigit and pd-1/pd-l1 in cancer immunotherapy: Mechanisms and clinical trials. Molecular Cancer, 22(1), 93. https://doi.org/10.1186/s12943-023-01800-3 [↩]
- Verdegaal, E. M. (2016). Adoptive cell therapy: A highly successful individualized therapy for melanoma with great potential for other malignancies. Current Opinion in Immunology, 39, 90–95. https://doi.org/10.1016/j.coi.2016.01.004 [↩] [↩]
- Immunotherapy | Seer training. (n.d.). Retrieved January 13, 2025, from https://training.seer.cancer.gov/treatment/biotherapy/immunotherapy.html#:~:text=Active%20immunotherapy%20involves%20setting%20an,can%20be%20specified%20 or%20 [↩]
- Miao, L., Zhang, Y., & Huang, L. (2021). Mrna vaccine for cancer immunotherapy. Molecular Cancer, 20(1), 41. https://doi.org/10.1186/s12943-021-01335-5 [↩]
- 10) Zhou, J. (2014). Advances and prospects in cancer immunotherapy. New Journal of Science, 2014, 1–13. https://doi.org/10.1155/2014/745808 [↩]
- Volovat, S. R., Scripcariu, D. V., Vasilache, I. A., Stolniceanu, C. R., Volovat, C., Augustin, I. G., Volovat, C. C., Ostafe, M.-R., Andreea-Voichița, S.-G., Bejusca-Vieriu, T., Lungulescu, C. V., Sur, D., & Boboc, D. (2024). Oncolytic virotherapy: A new paradigm in cancer immunotherapy. International Journal of Molecular Sciences, 25(2), 1180. https://doi.org/10.3390/ijms25021180 [↩]
- Rosen, A. (2023). Oncolytic virus: A promising immunotherapy to treat tumors. Science Insights, 42(2), 801–805. https://doi.org/10.15354/si.23.ps021 [↩]
- Ju, F., Luo, Y., Lin, C., Jia, X., Xu, Z., Tian, R., Lin, Y., Zhao, M., Chang, Y., Huang, X., Li, S., Ren, W., Qin, Y., Yu, M., Jia, J., Han, J., Luo, W., Zhang, J., Fu, G., … Xia, N. (2022). Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade. Journal for ImmunoTherapy of Cancer, 10(6), e004762. https://doi.org/10.1136/jitc-2022-004762 [↩]
- Kelly, E., & Russell, S. J. (2007). History of oncolytic viruses: Genesis to genetic engineering. Molecular Therapy, 15(4), 651–659. https://doi.org/10.1038/sj.mt.6300108 [↩]
- Morse, M. A., Gwin, W. R., & Mitchell, D. A. (2021). Vaccine therapies for cancer: Then and now. Targeted Oncology, 16(2), 121–152. https://doi.org/10.1007/s11523-020-00788-w [↩]
- Jiang, S., Chai, H., Tang, Q., Shi, Z., & Zhou, L. (2023). Clinical advances in oncolytic virus therapy for malignant glioma: A systematic review. Discover Oncology, 14(1), 183. https://doi.org/10.1007/s12672-023-00769-1 [↩]
- Qiu, Y., Su, M., Liu, L., Tang, Y., Pan, Y., & Sun, J. (2021). Clinical application of cytokines in cancer immunotherapy. Drug Design, Development and Therapy, 15, 2269–2287. https://doi.org/10.2147/DDDT.S308578 [↩]
- Guo, X., Yan, L., Zhang, D., & Zhao, Y. (2024). Passive immunotherapy for Alzheimer’s disease. Ageing Research Reviews, 94, 102192. https://doi.org/10.1016/j.arr.2024.102192 [↩]
- Diaz-Cano, I., Paz-Ares, L., & Otano, I. (2022). Adoptive tumor infiltrating lymphocyte transfer as personalized immunotherapy. In International Review of Cell and Molecular Biology, 370, 163–192. Elsevier. https://doi.org/10.1016/bs.ircmb.2022.04.003 [↩]
- Mitra, S., & Tomar, P. C. (2021). Hybridoma technology; advancements, clinical significance, and future aspects. Journal of Genetic Engineering and Biotechnology, 19(1), 159. https://doi.org/10.1186/s43141-021-00264-6 [↩]
- Zahavi, D., & Weiner, L. (2020). Monoclonal antibodies in cancer therapy. Antibodies, 9(3), 34. https://doi.org/10.3390/antib9030034 [↩]
- Jin, S., Sun, Y., Liang, X., Gu, X., Ning, J., Xu, Y., Chen, S., & Pan, L. (2022). Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduction and Targeted Therapy, 7(1), 39. https://doi.org/10.1038/s41392-021-00868-x [↩]
- Vega, M. I., Huerta-Yepez, S., Martinez-Paniagua, M., Martinez-Miguel, B., Hernandez-Pando, R., González-Bonilla, C. R., Chinn, P., Hanna, N., Hariharan, K., Jazirehi, A. R., & Bonavida, B. (2009). Rituximab-mediated cell signaling and chemo/immuno-sensitization of drug-resistant b-nhl is independent of its fc functions. Clinical Cancer Research, 15(21), 6582–6594. https://doi.org/10.1158/1078-0432.CCR-09-1234 [↩]
- Mileski, W., Rothlien, R., & Lipsky, P. (1994). Interference with the function of leukocyte adhesion molecules by monoclonal antibodies: A new approach to burn injury. European Journal of Pediatric Surgery, 4(04), 225–230. https://doi.org/10.1055/s-2008-1066110 [↩]
- Shiah, J.-G., Sun, Y., Kopečková, P., Peterson, C. M., Straight, R. C., & Kopeček, J. (2001). Combination chemotherapy and photodynamic therapy of targetable N-(2-hydroxypropyl)methacrylamide copolymer–doxorubicin/meso chlorin e6-OV-TL 16 antibody immunoconjugates. Journal of Controlled Release, 74(1–3), 249–253. https://doi.org/10.1016/S0168-3659(01)00325-X [↩]
- McLaughlin, P., Grillo-López, A. J., Link, B. K., Levy, R., Czuczman, M. S., Williams, M. E., Heyman, M. R., Bence-Bruckler, I., White, C. A., Cabanillas, F., Jain, V., Ho, A. D., Lister, J., Wey, K., Shen, D., & Dallaire, B. K. (1998). Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: Half of patients respond to a four-dose treatment program. Journal of Clinical Oncology, 16(8), 2825–2833. https://doi.org/10.1200/JCO.1998.16.8.2825 [↩]
- Ferrer, G., Álvarez-Errico, D., & Esteller, M. (2022). Biological and molecular factors predicting response to adoptive cell therapies in cancer. JNCI: Journal of the National Cancer Institute, 114(7), 930–939. https://doi.org/10.1093/jnci/djac088 [↩] [↩]
- Vu, T., & Claret, F. X. (2012). Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Frontiers in Oncology, 2. https://doi.org/10.3389/fonc.2012.00062 [↩]
- Karahan, Z. S., Aras, M., & Sütlü, T. (2023). Tcr-nk cells: A novel source for adoptive immunotherapy of cancer. Turkish Journal of Hematology. https://doi.org/10.4274/tjh.galenos.2022.2022.0534 [↩]
- Li, Y., Zheng, Y., Liu, T., Liao, C., Shen, G., & He, Z. (2024). The potential and promise for clinical application of adoptive T cell therapy in cancer. Journal of Translational Medicine, 22(1), 413. https://doi.org/10.1186/s12967-024-05206-7 [↩] [↩]
- DembiĆ, Z., Haas, W., Weiss, S., McCubrey, J., Kiefer, H., Von Boehmer, H., & Steinmetz, M. (1986). Transfer of specificity by murine α and β T-cell receptor genes. Nature, 320(6059), 232–238. https://doi.org/10.1038/320232a0 [↩]
- Li, Y., Zheng, Y., Liu, T., Liao, C., Shen, G., & He, Z. (2024). The potential and promise for clinical application of adoptive T cell therapy in cancer. Journal of Translational Medicine, 22(1), 413. https://doi.org/10.1186/s12967-024-05206-7 [↩] [↩] [↩]
- Albarrán, V., San Román, M., Pozas, J., Chamorro, J., Rosero, D. I., Guerrero, P., Calvo, J. C., González, C., García de Quevedo, C., Pérez de Aguado, P., Moreno, J., Cortés, A., & Soria, A. (2024). Adoptive T cell therapy for solid tumors: Current landscape and future challenges. Frontiers in Immunology, 15. https://doi.org/10.3389/fimmu.2024.1352805 [↩]
- Lu, Y.-C., Parker, L. L., Lu, T., Zheng, Z., Toomey, M. A., White, D. E., Yao, X., Li, Y. F., Robbins, P. F., Feldman, S. A., Van Der Bruggen, P., Klebanoff, C. A., Goff, S. L., Sherry, R. M., Kammula, U. S., Yang, J. C., & Rosenberg, S. A. (2017). Treatment of patients with metastatic cancer using a major histocompatibility complex class ii–restricted t-cell receptor targeting the cancer germline antigen mage-a3. Journal of Clinical Oncology, 35(29), 3322–3329. https://doi.org/10.1200/JCO.2017.74.5463 [↩]
- Testa, U., Castelli, G., Leone, G., Pelosi, E., Castelli, G., & Hohaus, S. (2023). Car-t cell therapy in large b cell lymphoma: Car-t; large b cell lymphoma; salvage therapy,. Mediterranean Journal of Hematology and Infectious Diseases, 15(1), e2023066. https://doi.org/10.4084/MJHID.2023.066 [↩]
- Evernden, C., Dowhan, M., Dabas, R., Chaudhry, A., Kalra, A., Dharmani-Khan, P., Gregson, D., Johnson, A., Jupp, J., Jimenez-Zepeda, V., Jamani, K., Duggan, P., Tay, J., Khan, F., Daly, A., & Storek, J. (2020). High incidence of Pneumocystis jirovecii pneumonia in allogeneic hematopoietic cell transplant recipients in the modern era. Cytotherapy, 22(1), 27–34. https://doi.org/10.1016/j.jcyt.2019.11.002 [↩]
- Asmamaw Dejenie, T., Tiruneh G/Medhin, M., Dessie Terefe, G., Tadele Admasu, F., Wale Tesega, W., & Chekol Abebe, E. (2022). Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Human Vaccines & Immunotherapeutics, 18(6), 2114254. https://doi.org/10.1080/21645515.2022.2114254 [↩] [↩]
- Abate-Daga, D., & Davila, M. L. (2016). CAR models: Next-generation CAR modifications for enhanced T-cell function. Molecular Therapy – Oncolytics, 3, 16014. https://doi.org/10.1038/mto.2016.14 [↩]
- Ramos, C. A., Royce, R., Robertson, C. S., Reyna, A., Narala, N., Vyas, G., Mehta, B., Zhang, H., Dakhova, O., Carrum, G., Kamble, R. T., Gee, A. P., Mei, Z., Wu, M.-F., Liu, H., Grilley, B., Rooney, C. M., Heslop, H. E., Brenner, M. K., … Dotti, G. (2018). In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Molecular Therapy, 26(12), 2727–2737. https://doi.org/10.1016/j.ymthe.2018.09.009 [↩] [↩]
- Harrison, A. J., Du, X., Von Scheidt, B., Kershaw, M. H., & Slaney, C. Y. (2021). Enhancing co-stimulation of CAR T cells to improve treatment outcomes in solid cancers. Immunotherapy Advances, 1(1), tb016. https://doi.org/10.1093/immadv/ltab016 [↩]
- Schuster, S. J., Dickinson, M. J., Dreyling, M. H., Martínez, J., Kolstad, A., Butler, J. P., Ghosh, M., Popplewell, L., Chavez, J. C., Bachy, E., Kato, K., Harigae, H., Kersten, M. J., ipoAndreadis, C., Riedell, P. A., Abdelhady, A. M., Zia, A., Morisse, M. C., Fowler, N. H., & Thieblemont, C. (2021). Efficacy and safety of tisagenlecleucel (Tisa-cel) in adult patients (Pts) with relapsed/refractory follicular lymphoma (R/r fl): Primary analysis of the phase 2 Elara trial. Journal of Clinical Oncology, 39(15_suppl), 7508–7508. https://doi.org/10.1200/JCO.2021.39.15_suppl.75 [↩] [↩]
- Wang, Y., Yang, S., Wan, L., Ling, W., Chen, H., & Wang, J. (2023). New developments in the mechanism and application of immune checkpoint inhibitors in cancer therapy (Review). International Journal of Oncology, 63(1), 86. https://doi.org/10.3892/ijo.2023.5534 [↩]
- Basudan, A. M. (2022). The role of immune checkpoint inhibitors in cancer therapy. Clinics and Practice, 13(1), 22–40. https://doi.org/10.3390/clinpract13010003 [↩]
- Chowdhury, F., Johnson, P. W., Glennie, M. J., & Williams, A. P. (2014). Ex vivo assays of dendritic cell activation and cytokine profiles as predictors of in vivo effects in an anti-human cd40 monoclonal antibody chilob 7/4 phase i trial. Cancer Immunology Research, 2(3), 229–240. https://doi.org/10.1158/2326-6066.CIR-13-0070 [↩]
- Hodi, F. S., O’Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., Gonzalez, R., Robert, C., Schadendorf, D., Hassel, J. C., Akerley, W., Van Den Eertwegh, A. J. M., Lutzky, J., Lorigan, P., Vaubel, J. M., Linette, G. P., Hogg, D., Ottensmeier, C. H., Lebbé, C., … Urba, W. J. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. New England Journal of Medicine, 363(8), 711–723. https://doi.org/10.1056/NEJMoa1003466 [↩]
- Wang, Y., Yang, S., Wan, L., Ling, W., Chen, H., & Wang, J. (2023). New developments in the mechanism and application of immune checkpoint inhibitors in cancer therapy (Review). International Journal of Oncology, 63(1), 86. https://doi.org/10.3892/ijo.2023.5534 [↩]
- Barrueto, L., Caminero, F., Cash, L., Makris, C., Lamichhane, P., & Deshmukh, R. R. (2020). Resistance to checkpoint inhibition in cancer immunotherapy. Translational Oncology, 13(3), 100738. https://doi.org/10.1016/j.tranon.2019.12.010 [↩]
- Robert, C., Long, G. V., Brady, B., Dutriaux, C., Maio, M., Mortier, L., Hassel, J. C., Rutkowski, P., McNeil, C., Kalinka-Warzocha, E., Savage, K. J., Hernberg, M. M., Lebbé, C., Charles, J., Mihalcioiu, C., Chiarion-Sileni, V., Mauch, C., Cognetti, F., Arance, A., … Ascierto, P. A. (2015). Nivolumab in previously untreated melanoma without braf mutation. New England Journal of Medicine, 372(4), 320–330. https://doi.org/10.1056/NEJMoa1412082 [↩]
- Robert, C., Schachter, J., Long, G. V., Arance, A., Grob, J. J., Mortier, L., Daud, A., Carlino, M. S., McNeil, C., Lotem, M., Larkin, J., Lorigan, P., Neyns, B., Blank, C. U., Hamid, O., Mateus, C., Shapira-Frommer, R., Kosh, M., Zhou, H., … Ribas, A. (2015). Pembrolizumab versus ipilimumab in advanced melanoma. New England Journal of Medicine, 372(26), 2521–2532. https://doi.org/10.1056/NEJMoa1503093 [↩]
- Filleron, T., Bachelier, M., Mazieres, J., Pérol, M., Meyer, N., Martin, E., Mathevet, F., Dauxois, J.-Y., Porcher, R., & Delord, J.-P. (2021). Assessment of treatment effects and long-term benefits in immune checkpoint inhibitor trials using the flexible parametric cure model: A systematic review. JAMA Network Open, 4(12), e2139573. https://doi.org/10.1001/jamanetworkopen.2021.39573 [↩]
- Casagrande, S., Sospetto, G. B., Bertalot, G., Bortolotti, R., Racanelli, V., Caffo, O., Giometto, B., Berti, A., & Veccia, A. (2024). Immune-related adverse events due to cancer immunotherapy: Immune mechanisms and clinical manifestations. Cancers, 16(7), 1440. https://doi.org/10.3390/cancers16071440 [↩]
- Wan, G., Chen, W., Khattab, S., Roster, K., Nguyen, N., Yan, B., Rajeh, A., Seo, J., Rashdan, H., Zubiri, L., Hadfield, M. J., Demehri, S., Yu, K.-H., Lotter, W., Gusev, A., LeBoeuf, N. R., Reynolds, K. L., Kwatra, S. G., & Semenov, Y. R. (2024). Multi-organ immune-related adverse events from immune checkpoint inhibitors and their downstream implications: A retrospective multicohort study. The Lancet Oncology, 25(8), 1053–1069. https://doi.org/10.1016/S1470-2045(24)00278-X [↩]
- Morad, G., Helmink, B. A., Sharma, P., & Wargo, J. A. (2021). Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell, 184(21), 5309–5337. https://doi.org/10.1016/j.cell.2021.09.020 [↩]
- Bai, R., Lv, Z., Xu, D., & Cui, J. (2020). Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomarker Research, 8(1), 34. https://doi.org/10.1186/s40364-020-00209-0 [↩]
- Yamaguchi, H., Hsu, J.-M., Sun, L., Wang, S.-C., & Hung, M.-C. (2024). Advances and prospects of biomarkers for immune checkpoint inhibitors. Cell Reports Medicine, 5(7), 101621. https://doi.org/10.1016/j.xcrm.2024.101621 [↩]
- Lu, Y., Yuan, X., Wang, M., He, Z., Li, H., Wang, J., & Li, Q. (2022). Gut microbiota influence immunotherapy responses: Mechanisms and therapeutic strategies. Journal of Hematology & Oncology, 15(1), 47. https://doi.org/10.1186/s13045-022-01273-9 [↩]
- Bhatt, A., Haslam, A., & Prasad, V. (2023). The effect of gastrointestinal microbiome supplementation on immune checkpoint inhibitor immunotherapy: A systematic review. Journal of Cancer Research and Clinical Oncology, 149(10), 7355–7362. https://doi.org/10.1007/s00432-023-04656-8 [↩]
- Schmidt, E. V., Sun, L. Z., Palmer, A. C., & Chen, C. (2023). Rationales for combining therapies to treat cancer: Independent action, response correlation, and collateral sensitivity versus synergy. Annual Review of Cancer Biology, 7(1), 247–263. https://doi.org/10.1146/annurev-cancerbio-061421-020411 [↩]
- Ramteke, A. S. (2023). Dual immune checkpoints inhibition: Cancer treatment and immunological modes of action. Journal of Drug Delivery and Therapeutics, 13(6), 175–187. https://doi.org/10.22270/jddt.v13i6.5880 [↩] [↩] [↩]
- Sui, S., Tian, Y., Wang, X., Zeng, C., Luo, O. J., & Li, Y. (2024). Single‐cell RNA sequencing gene signatures for classifying and scoring exhausted CD8+ T cells in B‐cell acute lymphoblastic leukaemia. Cell Proliferation, 57(3), e13583. https://doi.org/10.1111/cpr.13583 [↩]
- Rossetti, R., Brand, H., Lima, S. C. G., Furtado, I. P., Silveira, R. M., Fantacini, D. M. C., Covas, D. T., & De Souza, L. E. B. (2022). Combination of genetically engineered T cells and immune checkpoint blockade for the treatment of cancer. Immunotherapy Advances, 2(1), ltac005. https://doi.org/10.1093/immadv/ltac005 [↩] [↩]
- Chong, E. A., Alanio, C., Svoboda, J., Nasta, S. D., Landsburg, D. J., Lacey, S. F., Ruella, M., Bhattacharyya, S., Wherry, E. J., & Schuster, S. J. (2022). Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood, 139(7), 1026–1038. https://doi.org/10.1182/blood.2021012634 [↩]
- Ramteke, A. S. (2023). Dual immune checkpoints inhibition: Cancer treatment and immunological modes of action. Journal of Drug Delivery and Therapeutics, 13(6), 175–187. https://doi.org/10.22270/jddt.v13i6.5880 [↩] [↩]
- Sharma, P., Siddiqui, B. A., Anandhan, S., Yadav, S. S., Subudhi, S. K., Gao, J., Goswami, S., & Allison, J. P. (2021). The next decade of immune checkpoint therapy. Cancer Discovery, 11(4), 838–857. https://doi.org/10.1158/2159-8290.CD-20-1680 [↩]
- Rossetti, R., Brand, H., Lima, S. C. G., Furtado, I. P., Silveira, R. M., Fantacini, D. M. C., Covas, D. T., & De Souza, L. E. B. (2022). Combination of genetically engineered T cells and immune checkpoint blockade for the treatment of cancer. Immunotherapy Advances, 2(1), ltac005. https://doi.org/10.1093/immadv/ltac005 [↩]
- Suarez, E. R., Chang, D.-K., Sun, J., Sui, J., Freeman, G. J., Signoretti, S., Zhu, Q., & Marasco, W. A. (2016). Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget, 7(23), 34341–34355. https://doi.org/10.18632/oncotarget.9114 [↩]
- Yoon, D., Osborn, M., Tolar, J., & Kim, C. (2018). Incorporation of immune checkpoint blockade into chimeric antigen receptor t cells (Car-ts): Combination or built-in car-t. International Journal of Molecular Sciences, 19(2), 340. https://doi.org/10.3390/ijms19020340 [↩]
- Shin, W., & Kim, H. J. (2022). 3D in vitro morphogenesis of human intestinal epithelium in a gut-on-a-chip or a hybrid chip with a cell culture insert. Nature Protocols, 17(3), 910–939. https://doi.org/10.1038/s41596-021-00674-3
[↩]
- Yang, S., Hu, H., Kung, H., Zou, R., Dai, Y., Hu, Y., Wang, T., Lv, T., Yu, J., & Li, F. (2023). Organoids: The current status and biomedical applications. MedComm, 4(3), e274. https://doi.org/10.1002/mco2.274 [↩]
- Song, W., Shen, L., Wang, Y., Liu, Q., Goodwin, T. J., Li, J., Dorosheva, O., Liu, T., Liu, R., & Huang, L. (2018). Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nature Communications, 9(1), 2237. https://doi.org/10.1038/s41467-018-04605-x [↩]



{kind=link}