
Abstract
DNA repair pathways are essential mechanisms to ensure the stability of cellular processes, as they counteract mutations, which, if left unrepaired, result in damage to the genome. This paper aims to summarize information from literature addressing the following question: how do faulty repair mechanisms affect proliferating versus post-mitotic cells? Information was accessed mainly through reliable scholarly databases such as Google Scholar or PubMed. Keywords were used and sources were assessed on the basis of their citation index and their relevance before being included. Previous research was compiled into a comprehensive collection of multiple studies that correlate factors across the literature into a cohesive picture of the two diseases researched, xeroderma pigmentosum and Alzheimer’s Disease. The results of the studies described showed a correlation between errors in cellular DNA repair pathways and the symptoms or causation of the diseases. Nucleotide-excision repair was correlated with xeroderma pigmentosum and homologous recombination and nonhomologous end joining was correlated with Alzheimer’s disease.
Keywords: DNA Repair, Xeroderma Pigmentosum, Alzheimer’s Disease
Introduction
The information required for an organism’s survival and proliferation is all encoded within the genome, the complete genetic material of the organism, which, in the case of most organisms, including humans, consists of deoxyribonucleic acid (DNA)1. DNA is the molecule that carries the genetic information essential to the functioning and development of many organisms, and to ensure that all the cells of an organism contain an accurate copy of the genome, this is ensured through the process of DNA replication2.
The DNA replication process itself is not without fault. Errors can occur during the various steps of their molecular pathways as a result of external and internal mutagens. These errors are gene and chromosomal mutations that can occur during cell division, DNA replication, or other metabolic processes such as transcription. Errors can range from single point mutations to deletions or insertion errors where genes or chromosomal sections are either removed or inserted aberrantly.
Sometimes these mutations are not significant enough to make an impact. For example, when a mutation within a coding sequence is synonymous, the sequence of the protein produced remains unaltered, and therefore the error does not manifest3. Similarly, if the mutation occurs in a non-coding region with no functional significance, such as a repeating sequence, the effect is again silent4. However, other times they can lead to drastic changes and disrupt important processes. For example, if a missense or nonsense mutation occurs within a coding region, it can result in a change of a functionally important amino acid of a protein or premature termination of protein production. Additionally, if major DNA deletion or insertion occurs, it can cause severe chromosomal disorders, an example of which is Down’s syndrome resulting from an extra copy of chromosome 215.
To protect the genetic material from harmful mutations and ensure its survival and proliferation, the cell has developed specialized mechanisms of DNA repair, which will be the main focus of this review. Importantly, DNA repair pathways fall under different categories, each with a specific timing and site of action, namely pre-replicative repair pathways, post-replicative repair pathways, and transcription-coupled repair pathways.
Proliferating vs post-mitotic cells
DNA repair is an essential process to ensure that the integrity of the genome is maintained. Different types of cells may have unique needs for the maintenance of the genomic information they contain and therefore may have different preferred DNA repair pathways. Specifically, a major distinction that can have an impact on a cell’s preferred DNA repair pathway is whether the cell is proliferating or post-mitotic.
Cell division is an essential process for the growth and development of a cell, as well as to replenish dead cells. Multicellular organisms go through an essential process called the cell cycle. The stages of the cell cycle are Growth Phase 1, Synthesis, Growth Phase 2, and finally Mitosis or Meiosis when the cell divides6. Proliferating cells progress through the stages of the cell cycle numerous times throughout their life span. On the other hand, post-mitotic cells no longer divide; instead instead the majority of these cells permanently rest in G0, also known as the resting phase7.
Proliferating cells are cells that continue to traverse through the cell cycle, actively growing and producing more cells, with examples including skin cells and epithelial cells8. On the other hand, post-mitotic cells are no longer capable of dividing, so their proliferation is halted9. A notable example of post-mitotic cells is mature neurons in the central nervous system, loss of these cells results in neurodegeneration associated with various disorders, including Alzheimer’s disease, and a decrease in cognitive ability8.
Mitotic cells are susceptible to tumorigenic transformation, where as post-mitotic cells are not, because that post-mitotic cells cannot be stimulated to proliferate by any physiological or non-physiological stimulus such as carcinogens, irradiation, or oncogene expression8.
Proliferating cells still undergo the process of DNA replication prior to their division and can therefore rely on all processes of DNA repair, including pre-replicative repair pathways, replication-coupled repair, post-replicative repair pathways, and transcription-coupled repair pathways. On the other hand, since post-mitotic cells are arrested within the resting phase of the cell cycle, these types of cells tend to rely more heavily on post-replicative DNA repair pathways. Thus, DNA damage can be far more dangerous.
In xeroderma pigmentosum, the afflicted cells are typically skin cells; thus, the disease affects proliferating cells and the repair mechanisms that are correlated with the disease are primarily pre-replicative repair or replication-coupled repair. In Alzheimer’s disease, the afflicted cells are typically neurons; thus, the disease affects post-mitotic cells and the repair mechanisms that are correlated with the disease are primarily post-replicative repair.
DNA Repair
Pre-replicative DNA repair pathways
Pre-replicative DNA repair pathways occur before the replication process has concluded. There are numerous pre-replicative DNA repair mechanisms that can broadly be separated into three different categories: direct reversal, excision repair, and double-strand break repair.
The purpose of the first mechanism, known as direct reversal, is reversing specific chemical damage10. One cause of DNA mutations most commonly resolved by direct reversal is DNA alkylating agents which are typically involved in chemotherapy with anticancer drugs11. Repair is carried out by two pathways involving O6-methylguanine-DNA methyltransferase (MGMT) and the alkylated DNA repair protein B (AlkB) homologs11.
The second mechanism, excision repair, is responsible for removing the specific nucleotide bases that are damaged and replacing them with newly synthesized, undamaged bases10. There are three forms of excision repair: base-excision repair, nucleotide-excision repair, and mismatch repair (Cleaver et al.).
Base-excision repair is when a single damaged base is recognized, excised from the rest of the strand, and then replaced with a newly synthesized, accurate base with gaps filled by ligase, as seen in Figure 110. This process is especially important in the cell’s defense against oxidative damage. Oxidative damage can be caused by external factors such as chemicals or radiation, but the most common cause is through the byproducts of normal oxygen metabolism in ordinary metabolism and cellular respiration. These two functions are essential to the body, and the body has therefore evolved to produce antioxidant defense pathways. Typically, the body uses base-excision repair or direct reversal with scavenging molecules to reverse the damage.

Another example is nucleotide-excision repair (NER) which is demonstrated in Figure 2, where a section of damaged bases is recognized and a section of nucleotides that contains the damaged bases is excised. Then, these excised nucleotides are replaced with a new section of undamaged bases10. This is mainly used by mammals to remove bulky DNA lesions caused by UV radiation or environmental mutagens.

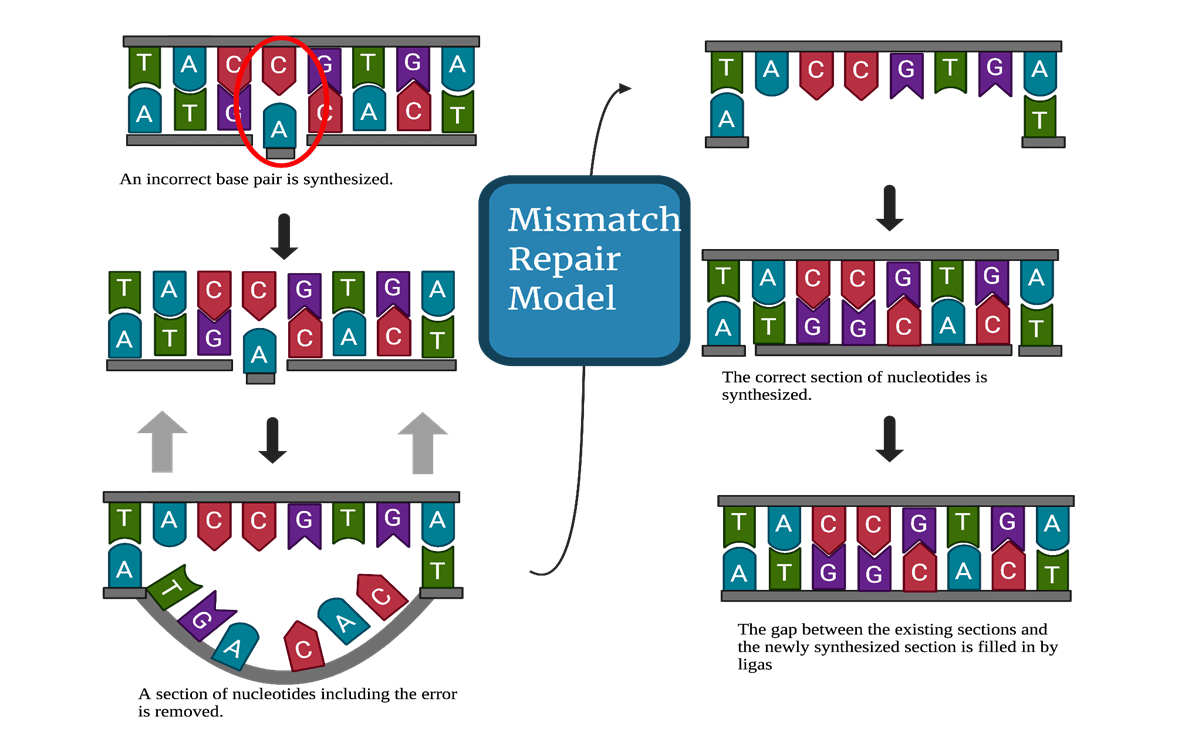
Finally, mismatch repair responds to an error occurring during the DNA replication process when two non-complementary bases are paired together. The repair mechanism is enacted through the signaling of DNA polymerase10. Figure 3 illustrates this process, with the synthesis of an incorrect pair being followed by the excision of a section of nucleotides with that includes the incorrect pair. The section that was excised is then replaced with the correct complementary base pairs, and the gaps between the newly synthesized portion and the existing strand are filled by ligase.
The previous mechanisms all occur before replication or during the replication process however, if that these mechanisms fail, there are additional barriers to prevent or remove mutations. These are post-replication repair pathways, including homologous recombination (HR) and non-homologous end joining (NHEJ).
Post-replicative DNA repair pathways
Post-replicative DNA repair pathways occur after replication and typically address damage incurred during the DNA replication process. This review further details three of these pathways: homologous recombination, non-homologous recombination, and transcription-coupled repair.
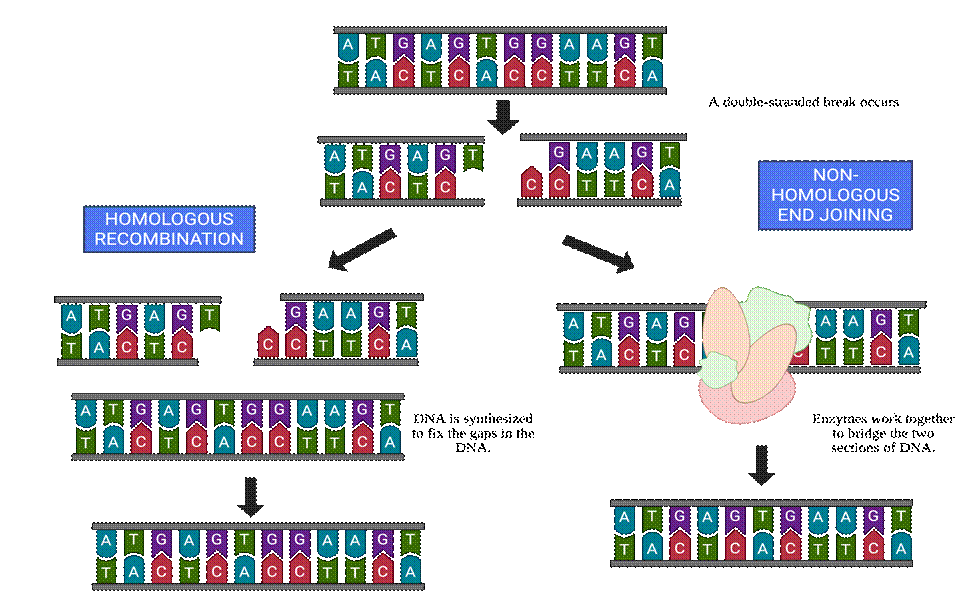
Homologous recombination repair relies on the principle that one of the two strands of the DNA double helix is undamaged10. During the DNA replication process, when DNA polymerase encounters the mutation, it is unable to copy it into the daughter strand, and replication is therefore halted10. Then, on the other side of the blockage, an Okazaki fragment is synthesized allowing DNA polymerase to continue replication10. This leaves a gap in the daughter strand. At this point, the undamaged parent strand is used as a template, and the gap is filled by extracting the same section from the parent strand10. Due to its complementary nature with the other parental strand, this new section is able to fill the gap left in the daughter strand caused by the mutation and the gap left on the undamaged parent strand is synthesized based on the already formed daughter strand with the help of DNA polymerase and the enzyme ligase10. This intricate process is demonstrated by the left path of Figure 4. These are DNA repair pathways most commonly used to control the daily damage DNA faces.
However, when a double stranded break occurs in the DNA strand, the DNA repair process of non-homologous end joining comes into play. If the DNA strands are broken into two segments, the process of non-homologous end joining includes the capture of these two segments, the molecular bridging between the two strands, and the re-ligation of the two into one strand, as seen in the right path of Figure 412. Unlike homologous recombination, non-homologous end joining does not involve the synthesis of new DNA in the site of error, but instead only joins the two fragmented ends together to be ligated12. This may result in the loss of certain nucleotides, as there is no synthesizing of the nucleotides damaged or lost in the original break; thus, the repaired DNA is not identical to the original strand of DNA, leading to a potential loss of genomic integrity12. On the other hand, the risk associated with homologous recombination is very small because of its template-based process. The only risk lies with accurate synthesis of the damaged strand, but when done correctly, the final strand should be identical to the original pre-damaged strand.
Transcription-coupled DNA repair
Transcription is the process in which mRNA is produced from a gene. The double-stranded DNA is separated, and RNA polymerase uses one strand as a template to craft a strand of mRNA. Transcription-coupled repair is a subset of nucleotide excision repair and is activated when RNA polymerase is halted in its elongation by lesions located on the DNA strands13
DNA repair in proliferating vs post-mitotic cells
Pre-replication and replication-coupled repair mechanisms (such as base-excision repair, nucleotide-excision repair, and mismatch repair) aim to preemptively catch lesions or errors in the DNA strand before the daughter strand has finished synthesizing. On the other hand, post-replication repair mechanisms (such as homologous recombination and non-homologous end joining) are essential to address lesions that persisted after initial repair mechanisms failed or lesions that occur after the replication process.
Specifically, neurons are post-mitotic cells which rely on the post-replicative pathways and faults in these pathways can lead to numerous neurological diseases. For example, R-loop associated DNA damage, typically repaired by non-homologous recombination, is correlated with Spinal Muscle Atrophy14.
Therefore, as showcased thus far, there are numerous DNA repair pathways, fine-tuned to catch and repair DNA errors associated with various cellular processes such as cell division, as well as environmental mutagenic agents, such as exposure to UV radiation. Despite their efficiency, DNA repair pathways are not completely accurate and can be altered so that they do not follow their intended purpose.
To demonstrate the role of DNA repair pathways in diseases affecting proliferating vs post-mitotic cells, this paper will be focusing on xeroderma pigmentosum (XP), affecting proliferating skin cells, and Alzheimer’s disease (AD), affecting post-mitotic neurons. The causes of common symptoms associated with each disease will be examined to identify which DNA repair pathway may be involved, and possible faults within the pathway will be discussed.
Xeroderma Pigmentosum
Xeroderma pigmentosum (XP) is a rare skin disease which causes an extreme sensitivity to sunlight or UV radiation15. XP affects cells that interact with UV radiation such as skin cells. Skin cells are mitotic cells that proliferate throughout their lifespan and as mentioned mitotic cells are susceptible to tumorigenic transformation. Thus, people with this condition have a higher risk of skin cancers and their life expectancy is much shorter due to the appearance of malignant skin tumors. Pyrimidine dimers that are caused by UV radiation are bulky DNA lesions that must be excised to prevent further issues such as skin cancers. Typically, the nucleotide-excision repair (NER) pathway excises these dimers; however, multiple mutations can occur throughout the process, which can cause the process to become faulty and error prone16
The most common subtype of this condition is caused by a mutation in the XPC gene, codes for an endonuclease on chromosome 3p2516. The mutated endonuclease is unable to sense UV damage the wildtype protein does, and without the sensor endonuclease alerting the body to damage, the DNA repair pathways are not activated to begin nucleotide excision and the damage persists16. Thus, with no repair pathways being activated, the UV damage builds to a dangerous level and the patient gains a heightened sensitivity to UV radiation and a much higher risk of malignant skin tumors.
There are numerous other mutations in other genes that interrupt other parts of the repair pathway. For example, one rare subtype of xeroderma pigmentosum is caused by a mutation in the XPB gene which codes for excision repair cross-complementing 3 (ERCC3);16. This mutation disrupts the 9-subunit protein complex that ERCC3 is part of, leading to disruption in the formation of an open complex needed for DNA repair16
About 20% of patients with XP do not have mutations in the nucleotide excision repair pathway and are classified as XP variants (XPV)17. Mutated XPV produces a defective DNA polymerase n which typically synthesizes DNA to replace damaged sites (a post-replication pathway)18. Without complications, once sensed, the UV-damaged bases are ‘cut-out’ or excised from the larger double-stranded DNA using nucleotide excision and released in the form of single-stranded oligonucleotides19. This release of dimer-affected sections leaves a gap in the DNA which is filled with the synthesis of new DNA20. Patients with XP most commonly have mutations in one aspect of this pathway21.
Alzheimer’s disease
Evidence suggests that Alzheimer’s disease (AD), the most prevalent neurodegenerative disorder, is correlated with defects in the ability to repair oxidative damage22. The symptoms of Alzheimer’s typically present itself with amnestic cognitive impairment, but some patients experience non-amnestic cognitive impairment23. Other symptoms include lessened mental agility, visual impairment, and visuospatial awareness23.
In terms of neuropathology, there are multiple hallmarks of Alzheimer’s disease including positive lesions (typically a symptom that involves growth), such as the gain of tau-containing neurofibrillary tangles, Aβ-containing plaques, and negative lesions (typically a symptom that is the absence of something essential), such as the loss of neuronal network integrity24. A large sign of AD is deposits of Aβ aggregates in the central nervous system, and the disruption of Aβ homeostasis is one of the earliest stages of AD development. Genetic risk factors predisposing to AD include mutations in the APOE gene (especially carrying the APOE4 allele), as well as mutations in genes such as APP, PSEN1, and PSEN225. Evidence suggests that the APOE4 allele enhances Aβ pathology because carriers of this gene tend to have more aggregate deposits in their central nervous system.
There is evidence to suggest that defective DNA repair can contribute to neurological dysfunction, including in AD. For example, under normal conditions, oxygen byproducts naturally released during various metabolic processes in the cell accumulate in the body over time, and oxidative damage typically causes single-stranded breaks, which are fixed through base-excision26. Usually, brain cells have distinct repair pathways to regulate this accumulation. However, if DNA repair mechanisms such as base-excision repair are ineffective, defective DNA repair can contribute to neurological dysfunction.There is evidence to suggest that defective DNA repair can contribute to neurological dysfunction, including in AD. For example, under normal conditions, oxygen byproducts naturally released during various metabolic processes in the cell accumulate in the body over time, and oxidative damage typically causes single-stranded breaks, which are fixed through base-excision27. Usually, brain cells have distinct repair pathways to regulate this accumulation. However, if DNA repair mechanisms such as base-excision repair are ineffective, defective DNA repair can contribute to neurological dysfunction.
Other damage occurring through stimulation and other physiological tasks can cause double-stranded breaks which are usually fixed through non-homologous recombination. Thus, an error in non-homologous recombination could be a cause for the accumulation of double-stranded breaks in the brain. Since AD primarily affects cognitive functioning, the cells most intrinsically involved are neurons which are post-mitotic cells.
The goal of this review is to contrast two diseases, each concerning separate types of cells (proliferating and post-mitotic), to explore the different essential ways repair pathways work in the human body and to correlate errors in these essential pathways with debilitating conditions.
Methods
This paper is structured as a literature review and accordingly compiles information from primary and secondary research papers. The literature discussed in this review was collected from databases including PubMed and Google Scholar, identified using keywords such as “DNA repair mechanisms”,“DNA repair in Xeroderma Pigmentosum”, “DNA repair in Alzheimer’s disease”, “DNA repair failure”, “DNA repair in post-mitotic cells”, “DNA repair in proliferating cells”. Major focus is placed on describing and evaluating the results of 4 independent studies relating to DNA repair in Xeroderma Pigmentosum, and 4 independent studies relating to DNA repair in Alzheimer’s disease. The major inclusion criterion was that the studies are experimental and primarily rely on quantitative rather than qualitative methodology. The sources were evaluated by means of the impact factor of the Journal in which they are published and the citation index of the research paper and the authors, prioritising papers with a citation index > 15. Additional relevant literature was found in the reference list of recently published literature reviews. The date published was also considered to use up to date information in the course of this review, prioritising the consideration of papers published within the last 15 years.
Results
DNA Repair in Xeroderma Pigmentosum
In XP, patients present symptoms of sun sensitivity and increased likelihood of skin tumors, indicating an issue with the body’s response to UV radiation. This also suggests that patients suffer from inefficiency in the DNA repair pathway that responds to UV radiation, and multiple studies have been conducted to further categorize this molecular pathology.
Evidence Based on Human Cell Experiments
J.E. Cleaver conducted a study testing the efficiency of DNA repair in different diseases including Xeroderma Pigmentosum with controlled skin culture. The cultures were incubated in a dark room due to XP’s known sun sensitivity, and then were carefully exposed to UV radiation.
5-bromodeoxyuridine (³H-BUdr) was used to confirm weather normal DNA replication was occurring or DNA repair was occurring, after damage (caused by irradiation). ³H-BUdr substitutes for thymine during DNA replication, and is labelled with tritium, so it will increase the density of the DNA when incorporated. With ordinary DNA replication hybrid density DNA with H-BU is created, with a higher density than the DNA without 3H-BUdr substituting for thymine. However, if DNA repair occurs and H-BUdr is synthesized in ‘patches’, the density will stay relatively the same because the additions having such a low overall density. At the conclusion of the experiment, it was observed that in the control cultures, there was a large amount of tritium in the normal density regions (which is the region of peak absorbance of radiation). However, in XP, there are reduced amounts of tritium and no noticeable peaks in the normal density area. This difference hints at XP’s issue with replication-coupled repair as H-BUdr is not being synthesized in the patches. If it were being synthesized correctly as a form of repair then there would be evidence of tritium in the normal density area. It suggests that replication-coupled repair is either absent in XP or occurs at a much lower level than usual.
Another study, by Tsujimoto et al., aims to illustrate that there are faults in nucleotide-excision repair in XP. Tsujimoto et al. conducted a study on the use of antioxidant treatments on Xeroderma Pigmentosum cells. The first step was to study the difference in reaction to UV-B radiation. A dose of 800 J/m2 UV was used because this small dose would not greatly affect cell viability, with a survival rate of 90% in XP cells and 95% in normal cells. Then, the levels of 8-OHdG, which is a biomarker for UV-induced inflammation or oxidative damage, in both cell groups were measured at various points: 1.5, 3, 6, 9, and 24 h after irradiation. Normal cells had a lower level immediately after exposure than XP cells and, after 24 hours, had returned to their baseline level. However, in XP cells, 8-OHdG levels nearly doubled after exposure and remained at higher than baseline levels even after 24 hours had passed. This indicates the lack of nucleotide-excision repair to manage the UV-induced inflammation measured by 8-OHdG levels. The normal cells had processes that were actively and efficiently monitoring the inflammation, and this is reflected in the return to baseline levels within 24 hours; however, the XP cell did not return to baseline levels within this period, reflecting an issue in the repair pathway.
Evidence Based on Mice Experiments
In another study exploring DNA repair in XP, Miyauchi-Hashimoto et al. studied UV damage in XP and controls. Miyauchi-Hashimoto et al. conducted a study using mice that had XPA and XPC gene deficiencies, generated through genotyping. Ultimately, three groups of mice were used: wild type mice that had none of the XP genes, heterozygous mice, and homozygous mice with XPA gene-deficiency. After exposure to a dose of 250 mJ/cm2 of UVB radiation, the ears of the XPA gene-deficient mice became swollen while the other two groups experienced little swelling. A lower dose of 100 mJ/cm2 produced very similar results. A higher dose of 500 mJ/cm2 produced swelling in all three groups; however, the homozygous mice with XPA gene-deficiency experienced the swelling at a much greater level. The experiment continued with PUVA testing, which is a type of ultraviolet radiation testing. With the application of 8-MOI’ and a dose of 7.5J1cm2 of UVA two hours later, all three groups showed swelling, but again, the homozygous XPA gene-deficient mice experienced the swelling on a larger scale. This experiment showcased the sensitivity of XPA gene-deficient cells to radiation of UVB and PUVA. It also indicates a significant difference from the heterozygous mice and the homozygous wild-type mouse, as they experienced different results when exposed to the same scenarios and doses as the XPA gene-deficient mice.

This experiment aimed to illustrate the difference in sensitivity to UV radiation in various XP genes. Rob J. W. Berg et al. conducted an experiment involving various groups of mice with different XP gene statuses: gene-deficiency, heterozygous, and wild-type. The mice were exposed to UV radiation for up to 500 days using a Kromayer heat lamp. XPA gene-deficient mice were exposed for 1, 2, and 3 seconds and the other two groups were exposed for 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, and 30 seconds. The maximum effect of a single exposure to the lamp was observed after 48 hours in all three genotypes. In the first group (XPA gene-deficient), only the exposures of 2 and 3 seconds had an effect. For the other two groups, only the exposures of 16 seconds and longer had an effect. Thus, the ratio of sensitivity to UVB radiation effects with XPA gene-deficient cells versus the other cells is between 1:7 and 1:16. The wild-type and heterozygous mice developed very few early-stage tumors, which were disregarded in the experiment’s quantitative analysis. The XPA gene-deficient group, on the other hand, developed multiple skin tumors over the course of the experiment, demonstrating their increased sensitivity.
The aforementioned studies support the idea that DNA repair pathways failing to counteract UV-induced damage leads to sun sensitivity and an increased risk of malignant tumours, culminating in the disease of Xeroderma Pigmentosum.
DNA repair in Alzheimer’s Disease
In AD, patients face increasing cognitive decline due to double-stranded breaks accumulating in the brain. Treated through homologous recombination and nonhomologous end joining, double-stranded breaks in the brain impede many mental processes including the storage and retrieval of memory. The following studies analyze the correlations between these repair pathways and AD.
The following study aimed to establish a link between lipid metabolism and oxidative stress in Alzheimer’s and thus studied both connections with Alzheimer’s independently. Nie et al. collected plasma samples from three patient groups: those with Alzheimer’s, those with amnestic mild cognitive impairment (aMCI), and a control group. They used an analytical technique called LC-MS/MS to analyze the samples. They analyzed three different markers of oxidative stress: MDA (damage marker), SOD (antioxidant), and GSH-Px (antioxidant). They found that MDA levels were higher in the AD group and SOD levels were lower in the AD group, but GSH-Px levels were highest in the aMCI group and showed no significant difference from the control group. Overall, the study shows a correlation between Alzheimer’s and oxidative stress that is absent from the control group. Higher levels of one damage marker and lower levels of an antioxidant indicate an issue with regulation of oxidative damage in individuals with Alzheimer’s.
Starr Welty et al. investigated the accumulation of DNA double-stranded breaks (DSBs) in cells with AD compared to control cells. The molecular marker γ-H2AX was used to gauge accumulation of DSBs. Typically, in dividing cells, the activation of γ-H2AX is associated with a pause in cell cycle progression; however, in cells no longer able to undergo mitosis, levels of γ-H2AX continuously increase. Studies were done on tissue cells from postmortem human brain tissues with and without AD. A significantly larger number of neurons associated with γ-H2AX were found in the samples with AD than without. An ELISA test was used to quantify the observations of γ-H2AX found and it detailed that there were three times as many phosphorylated γ-H2AX in the hippocampus of the samples with AD than without. Thus, the study suggested significant accumulation of DNA double-stranded breaks in samples with AD. Therefore, there must also be an area in the normal repair pathways that prevents this accumulation in order to consolidate the stark difference between the normal group’s γ-H2AX levels and the AD group’s γ-H2AX levels. In addition, this study details an experiment conducted to record the levels of repair proteins typically involved in non-homologous end joining and homologous recombination, which are the two main repair pathways for DNA double-stranded breaks. They examined the mRNA expression level of these repair proteins and found reduced levels of repair proteins MRE11, RAD50, and BRCA1 in the hippocampus of the AD group compared to the non-AD group. This further indicates an error in the pathway with a lack of the proteins essential to the process. An accumulation of DNA DSB and reduced levels of certain essential repair proteins provide significant evidence of issues in the repair pathway of AD.
Another experiment aimed to correlate AD with excessive neuronal DSBs. Niraj M. Shanbhag et al. used postmortem brain tissues from patients with AD or mild cognitive impairment (MCI), patients with no mental impairments, and some control samples. Similar to the study conducted by Starr Welty et al. this study used γ-H2AX as a marker for DSBs. To ensure accuracy, they tried two different types of immunostaining to detect γ-H2AX: pan-nuclear and focal. Pan-nuclear immunostaining is a uniform staining of the entire nucleus while focal immunostaining highlights specific sites. Pan-nuclear immunostaining showed an increased number of γ-H2AX foci per neuron in both AD and MCI compared to samples without mental impairments. In addition, several AD cases and one MCI case showed a high number of γ-H2AX foci in proportion to NeuN-negative cells, which are markers for mature neurons. This shows that first, AD and MCI patients have more DSBs than those without mental impairments, providing significant evidence that accumulation of DSBs is associated with AD and MCI, and second, that there is an excessive amount of DSBs compared to normal neurons. In focal immunostaining, it was found that both AD and MCI had twice as many neurons with γ-H2AX foci in the frontal cortex compared to cognitively unimpaired controls and AD and MCI cases also had higher number of γ-H2AX foci per neuron for the other control group as well. In both immunostaining techniques, higher levels of γ-H2AX were found in the brains of those with mental impairments which further associates mental impairment with DSBs. This failure to address the accumulation of DSBs connects back to a failure of the repair pathway to function effectively in patients with AD and MCI.
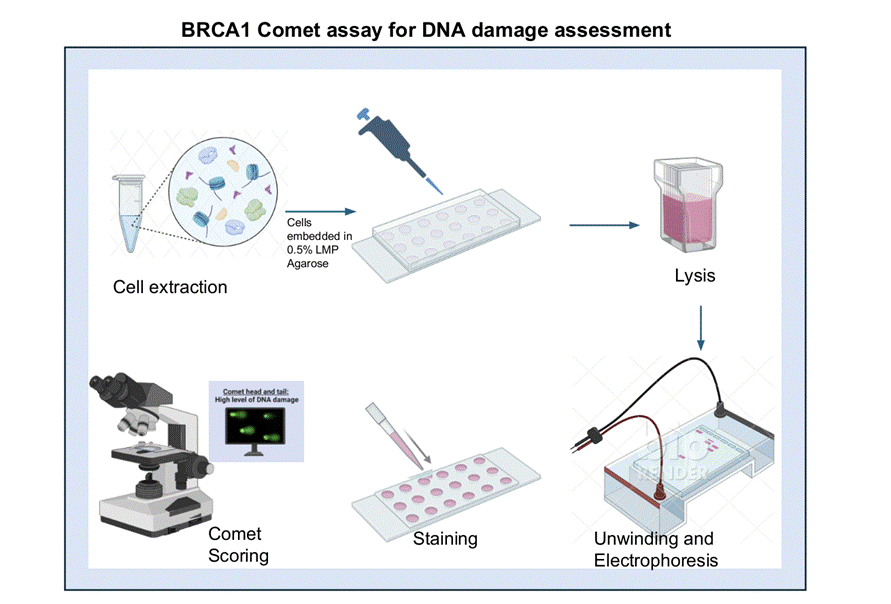
This next experiment also aims to correlate Alzheimer’s with repair pathways. Conducted by Elsa Suberbielle, the study focused on the reduction of BRCA1 in AD and correlating this reduction with homologous recombination. This is because BRCA1 directly binds to DNA double-stranded breaks to promote homologous recombination or repair. Thus, lower levels of BRCA1 may indicate that there is less homologous recombination activity as well. By linking BRCA1 reduction to AD, the study indicates that DNA repair is impeded in AD as well. To do this, the experiment analyzed post-mortem brain samples from people with no mental deficits and those with AD or (MCI). They found that in certain sections of the brain such as the CA1 and CA3 regions, the samples with AD or MCI had 65% lower amounts of BRCA1 and in the dentate gyrus it was 75% lower. In contrast to this, there were increased levels in histopathological lesions typically associated with AD. Overall, their BRCA1 levels in the brains of those with AD or MCI was 50-70% lower than those without any mental impairments. To correlate lowered levels of BRCA1 and damage to DNA integrity, they measured DNA fragmentation in a comet assay (a method to measure DNA breaks). The length of the tails was measured and the proportion of nuclei with these comet tails was calculated because it reflects the presence and range of DSBs. Lowered levels of BRCA1 were associated with a higher proportion of nuclei with tails, indicating that with the reduction of BRCA1 comes a loss of DNA integrity. Thus, the loss of BRCA1 is associated with AD, and due to BRCA1’s vital role in homologous recombination, this study provides evidence that links a critical failure in the homologous recombination pathway with AD.
The studies detailed above supported the idea that the failure to address the accumulation of DNA double-stranded breaks led to cognitive decline and disease-associated with mental impairments including Alzheimer’s Disease.
Discussion & Conclusion
XP is a disease associated with increased sensitivity to UV radiation and primarily affects proliferating cells in the skin. Through the studies shown above, the relationship between increased sensitivity to UV radiation was tested and correlated with DNA repair pathways. The experiments showcased this increased sensitivity in multiple ways such as increased swelling in XP cells in comparison to normal cells or a higher probability of tumor growth in XP cells. The main difference between the cells with the gene deficiency and the ones without is their reaction to UV radiation. Ordinarily, repair pathways are activated when in contact with UV radiation, specifically, the nucleotide excision repair pathway, but the cells with XP were not having the same success as the ones without, indicating a problem with the cells’ normal DNA repair response.
Multiple experiments were conducted on mice to investigate the effects of UV radiation on XP affected cells, and although this evidence does strongly support the hypothesis that issues in the repair mechanisms lead to conditions such as XP, it is not exact to how human cells may react. Although mice genes hold a high similarity to human genes, there is not complete accuracy in their cellular or genetic structure. There are many structural inconsistencies, complications due to size difference, and reactionary differences28. Additionally, laboratory mice are often inbred and thus tend to showcase a reduced genetic diversity to humans and are not exposed to a comparable environmental diversity to humans either. However, advancements in analytical techniques to delineate differences has essentially contributed to understanding murine and human biology28. Thus, discrepancies should be noted, but there is still validity to experiments involving murine samples. Conducting the same exact experiments where UV radiation is directly applied to humans may not be possible due to ethical concerns of subjecting patients to UV radiation when they have known sensitivity; however, experiments with modified conditions could be conducted in vitro with human tissues or Induced Pluripotent Stem Cells (IPSCs) collected from those affiliated with XP.
Now that the increased sensitivity and the cause and effect of the faulty repair pathways have been supported scientifically with experiments such as the ones summarized, a possible next step would be to conduct research on the repair pathways themselves and testing treatment methods that would prove effective on multiple variants of XP. Experiments should be conducted with independent variables that could affect each step of the repair pathway to locate the exact phase or transitional phase that is the cause of the error. The same experiments that are detailed above could be replicated, except the controls would be the untreated XP cells and the other cells would be treated with the proposed method.
AD severely impacts mental functioning and is primarily concentrated in the brain. The relationship between accumulated DSBs and this blockage of mental processes was supported in the studies discussed and thus correlates double-stranded breaks with the disease. A failure to carry out homologous recombination and nonhomologous end joining leads to the corrosion of DNA integrity in the brain.
It is possible that DNA damage may be simply a consequence of a disease or due to faulty repair mechanisms. An alternative explanation could be that repair mechanisms and DNA damage have a bidirectional relationship.
It’s important to acknowledge the complexity of AD. Although it is unlikely that errors in these mechanisms are the sole cause of AD, it is true and supported by the experiments that they are a significant factor and can worsen disease progression and pathology.
With support for this relationship in the studies described above , the next step would be identifying even more factors involved in the repair pathway. BRCA1 is not the only factor that is essential to a healthy repair pathway. Only after identifying all such factors with testing can further steps be taken to limit the harm that faulty mechanisms can cause.
In one of the studies conducted they used both post-mortem brain samples from those with AD and other mild cognitive impairments. Since the focus was not solely on AD, data was collected through testing on both, and data was presented in a summary of both results together. Thus, the correlation discovered in the experiment is not as strong as it would have been had they only used samples from those with AD. There is no way of knowing if the data was misconstrued due to a basis on other deficits.
Based on the studies in the results section, evidence suggests that two different pathways are used in XP and AD pathology. In XP, the damage incurred is typically under the jurisdiction of pre-replicative pathways, but in AD, the damage incurred is typically under the jurisdiction of post-replicative pathways. Using these case studies, generalization is possible that diseases that affect proliferating cells, like XP, typically are exacerbated by the failure in pre-replicative pathways and that diseases that affect post-mitotic cells, like AD, are exacerbated by the failure in post-replicative pathways.
The above studies provide indications of a correlation between errors in nucleotide-excision repair and homologous recombination and pathology relating to Xeroderma Pigmentosum and Alzheimer’s disease respectively. Future work should aim to strengthen those correlations and investigate which molecular components of these intricate pathways tend to be at fault, contributing to disease. For example, future work could employ genomic editing using CRISPR-cas9-based tools to attempt to disrupt components of the DNA repair pathway that are indicated to be at fault in each disease respectively, and to study which perturbations recapitulate disease phenotypes. Similarly, CRISPR-cas9 can be used to correct any genetic mutations in the relevant DNA repair proteins in patient-derived iPSC lines, to attempt to alleviate disease symptoms, and strengthen the correlation between the DNA repair pathways and disease pathology/markers.
Correlations between these two genetic diseases and errors in repair pathways have been made. XP research has shown that with UV radiation, XP cells are unable to properly enact a nucleotide-excision repair pathway to prevent harmful damage. AD has shown that with the occurrence of DNA double-stranded breaks caused by many different issues, like oxidative damage, unlike ordinary cells, Alzheimer’s cells are not able to enact either a homologous recombination pathway or a non-homologous end joining pathway. Due to this inability to enact the correct pathway, the multiple intricacies of the disease appear. Thus, as multiple experiments have been conducted supporting this correlation, the next step would be experimenting with different enzymes or markers to discover how best to understand how these pathways function.
References
- Goldman, Aaron David, and Laura F. Landweber. “What Is a Genome?” PLoS Genetics, vol. 12, no. 7, Jul. 2016, p. e1006181, doi:10.1371/JOURNAL.PGEN.1006181. [↩]
- Song, Hao Yun, et al. “DNA Replication: Mechanisms and Therapeutic Interventions for Diseases.” MedComm, vol. 4, no. 1, Feb. 2023, p. e210, doi:10.1002/MCO2.210. [↩]
- Zhang, Zhe, et al. “Analyzing Effects of Naturally Occurring Missense Mutations.” Computational and Mathematical Methods in Medicine, vol. 2012, 2012, p. 805827, doi:10.1155/2012/805827. [↩]
- Zhang, Zhe, et al. “Analyzing Effects of Naturally Occurring Missense Mutations.” Computational and Mathematical Methods in Medicine, vol. 2012, 2012, p. 805827, doi:10.1155/2012/805827. [↩]
- Antonarakis, Stylianos E., et al. “Down Syndrome.” Nature Reviews. Disease Primers, vol. 6, no. 1, Jan. 2020, p. 9, doi:10.1038/S41572-019-0143-7. [↩]
- Alberts, Bruce, et al. An Overview of the Cell Cycle. 2002, https://www.ncbi.nlm.nih.gov/books/NBK26869/. [↩]
- Xia, Peng, et al. “Cell Cycle Proteins as Key Regulators of Postmitotic Cell Death.” The Yale Journal of Biology and Medicine, vol. 92, no. 4, Dec. 2019, p. 641, https://pmc.ncbi.nlm.nih.gov/articles/PMC6913832/. —. “Cell Cycle Proteins as Key Regulators of Postmitotic Cell Death.” The Yale Journal of Biology and Medicine, vol. 92, no. 4, Dec. 2019, p. 641, https://pmc.ncbi.nlm.nih.gov/articles/PMC6913832/. [↩]
- Campisi, Judith, and Huber R. Warner. “Aging in Mitotic and Post-Mitotic Cells.” Advances in Cell Aging and Gerontology, vol. 4, no. C, Jan. 2001, pp. 1–16, doi:10.1016/S1566-3124(01)04024-X. [↩] [↩] [↩]
- Aranda-Anzaldo, Armando. “The Post-Mitotic State in Neurons Correlates with a Stable Nuclear Higher-Order Structure.” Communicative & Integrative Biology, vol. 5, no. 2, Mar. 2012, p. 134, doi:10.4161/CIB.18761. [↩]
- DNA Repair – The Cell – NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK9900/. Accessed 3 Apr. 2025. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Gutierrez, Roberto, and Timothy R. O’Connor. “DNA Direct Reversal Repair and Alkylating Agent Drug Resistance.” Cancer Drug Resistance, vol. 4, no. 2, 2021, p. 414, doi:10.20517/CDR.2020.113. [↩] [↩]
- Weterings, Eric, and David J. Chen. “The Endless Tale of Non-Homologous End-Joining.” Cell Research 2008 18:1, vol. 18, no. 1, Jan. 2008, pp. 114–24, doi:10.1038/cr.2008.3. [↩] [↩] [↩]
- Spivak, Graciela. “Transcription-Coupled Repair: An Update.” Archives of Toxicology, vol. 90, no. 11, Nov. 2016, p. 2583, doi:10.1007/S00204-016-1820-X. [↩]
- Cuartas, Juliana, and Laxman Gangwani. “R-Loop Mediated DNA Damage and Impaired DNA Repair in Spinal Muscular Atrophy.” Frontiers in Cellular Neuroscience, vol. 16, Jun. 2022, p. 826608, doi:10.3389/FNCEL.2022.826608/FULL [↩]
- Leung, Alexander K. C., et al. “Xeroderma Pigmentosum: An Updated Review.” Drugs in Context, vol. 11, 2022, pp. 2022-2–5, doi:10.7573/DIC.2022-2-5. [↩]
- Lucero, Renee, and David Horowitz. “Xeroderma Pigmentosum.” StatPearls, Jul. 2023, https://www.ncbi.nlm.nih.gov/books/NBK551563/. [↩] [↩] [↩] [↩] [↩]
- Karass, Michael, et al. “Xeroderma Pigmentosa: Three New Cases with an in Depth Review of the Genetic and Clinical Characteristics of the Disease.” Fetal and Pediatric Pathology, vol. 34, no. 2, Apr. 2015, pp. 120–27, doi:10.3109/15513815.2014.982336,. [↩]
- Karass, Michael, et al. “Xeroderma Pigmentosa: Three New Cases with an in Depth Review of the Genetic and Clinical Characteristics of the Disease.” Fetal and Pediatric Pathology, vol. 34, no. 2, Apr. 2015, pp. 120–27, doi:10.3109/15513815.2014.982336,. [↩]
- Kemp, Michael G., and Aziz Sancar. “DNA Excision Repair: Where Do All the Dimers Go?” Cell Cycle, vol. 11, no. 16, Aug. 2012, p. 2997, doi:10.4161/CC.21126. —. “DNA Excision Repair: Where Do All the Dimers Go?” Cell Cycle, vol. 11, no. 16, Aug. 2012, p. 2997, doi:10.4161/CC.21126. [↩]
- Kemp, Michael G., and Aziz Sancar. “DNA Excision Repair: Where Do All the Dimers Go?” Cell Cycle, vol. 11, no. 16, Aug. 2012, p. 2997, doi:10.4161/CC.21126. —. “DNA Excision Repair: Where Do All the Dimers Go?” Cell Cycle, vol. 11, no. 16, Aug. 2012, p. 2997, doi:10.4161/CC.21126. [↩]
- Kemp, Michael G., and Aziz Sancar. “DNA Excision Repair: Where Do All the Dimers Go?” Cell Cycle, vol. 11, no. 16, Aug. 2012, p. 2997, doi:10.4161/CC.21126. —. “DNA Excision Repair: Where Do All the Dimers Go?” Cell Cycle, vol. 11, no. 16, Aug. 2012, p. 2997, doi:10.4161/CC.21126. [↩]
- Buccellato, Francesca Romana, et al. “Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery.” Antioxidants, vol. 10, no. 9, Sep. 2021, p. 1353, doi:10.3390/ANTIOX10091353. [↩]
- Knopman, David S., et al. “Alzheimer Disease.” Nature Reviews Disease Primers 2021 7:1, vol. 7, no. 1, May 2021, pp. 1–21, doi:10.1038/s41572-021-00269-y. [↩] [↩]
- Knopman, David S., et al. “Alzheimer Disease.” Nature Reviews Disease Primers 2021 7:1, vol. 7, no. 1, May 2021, pp. 1–21, doi:10.1038/s41572-021-00269-y. [↩]
- Tanzi, Rudolph E. “The Genetics of Alzheimer Disease.” Cold Spring Harbor Perspectives in Medicine, vol. 2, no. 10, 2012, p. a006296, doi:10.1101/CSHPERSPECT.A006296. [↩]
- Canogovi et al. zzLin; Xiaozeng, et al. “Contributions of DNA Damage to Alzheimer’s Disease.” International Journal of Molecular Sciences, vol. 21, no. 5, Mar. 2020, p. 1666, doi:10.3390/IJMS21051666. [↩]
- Lin, Xiaozeng, et al. “Contributions of DNA Damage to Alzheimer’s Disease.” International Journal of Molecular Sciences, vol. 21, no. 5, Mar. 2020, p. 1666, doi:10.3390/IJMS21051666. [↩]
- Rydell-Törmänen, Kristina, and Jill R. Johnson. “The Applicability of Mouse Models to the Study of Human Disease.” Mouse Cell Culture, vol. 1940, 2018, p. 3, doi:10.1007/978-1-4939-9086-3_1. [↩] [↩]


