
Abstract
Lung cancer is the deadliest type of cancer in both the United States and worldwide, accounting for roughly 1 in 5 cancer deaths each year. This high mortality rate is largely because it is often diagnosed at late stages, when treatment is less effective. About 85% of all lung cancer cases are classified as non-small cell lung cancer (NSCLC). Recently, immunotherapy has become an important treatment approach for NSCLC, aiming to boost the body’s own immune system to fight cancer. One immunotherapeutic strategy, chimeric antigen receptor (CAR) T-cell therapy, is FDA-approved for several blood cancers because it demonstrated strong efficacy. Despite its success in hematologic malignancies, CAR T-cell therapy has not yet been approved for the treatment of solid tumors. This project proposes a conceptual design for a bispecific TanCAR T-cell for NSCLC. The DepMap and GEPIA databases were used to analyze gene expression in NSCLC cell lines compared to healthy lung models to identify target genes highly expressed in cancer cells with minimal expression in normal tissue. From this analysis, MUC16 and LYPD3 were identified as promising target genes encoding surface proteins significantly overexpressed on NSCLC cells. Additionally, the project considers potential safety concerns such as cytokine release syndrome (CRS), using parameter correlation to identify which parameters of the CAR are most responsible for toxicity. Overall, this research proposes a data-driven design for CAR T-cell therapy in NSCLC, using a bispecific approach to represent a more specific and safer treatment strategy.
Keywords: NSCLC; immunotherapeutic; bispecific; CAR; CRS; immunological; in silico
Abbreviations
| Abbreviation | Definition |
| NSCLC | Non-Small Cell Lung Cancer |
| CAR | Chimeric Antigen Receptor |
| TanCAR | Tandem Chimeric Antigen Receptor |
| CRS | Cytokine Release Syndrome |
| scFv | Single-Chain Variable Fragment |
| CAF | Cancer-Associated Fibroblast |
| LUAD | Lung Adenocarcinoma |
| GEPIA | Gene Expression Profiling Interactive Analysis |
| TPM | Transcripts Per Million |
| MUC16 | Mucin 16 |
| LYPD3 | Ly6/PLAUR Domain Containing 3 |
| CD28 | Cluster of Differentiation 28 |
| CD3ζ | Cluster of Differentiation 3 Zeta Chain |
| 4-1BB | CD137 (4-1BB Costimulatory Receptor) |
| GTEx | Genotype-Tissue Expression Project |
| TCGA | The Cancer Genome Atlas |
Introduction
Impacts of Cancer
Cancer is the second leading cause of global mortality, following heart disease, with nearly 20 million new cases diagnosed annually1. Cancer is a group of diseases caused by the uncontrolled growth and spread of abnormal cells that lose control over their growth and no longer follow the usual cycle of division and death2. These changes are caused by genetic mutations that cause cells to develop a common set of traits known as the hallmarks of cancer. Some of these hallmarks include sustaining uncontrolled cell growth, evading signals that stop growth, resisting programmed cell death, becoming capable of unlimited division, expanding their blood supply (angiogenesis), and invading nearby tissues or spreading to other parts of the body through the blood stream or lymphatic system (metastasis)3,4
When cancer is localized, it can often be cured through targeted treatments like surgery or radiation, but metastasis makes the tumor cells much harder to eliminate permanently (Figure 1)5.
Current Cancer Treatments
There are four main types of cancer treatment that are often used in combination depending on tumor size, type, location, stage, as well as the patient’s medical history, mental health and other factors6. Surgery, chemotherapy, radiation, and immunotherapy aim to destroy cancer cells, slow their growth, and, or prevent them from spreading, with the overall goal of improving the patient’s quality of life. Surgery is a local treatment that treats only the part of your body with the cancer, by manually removing the tumor and surrounding tissues7. Surgery can be used to physically remove solid tumors that are generally contained in one location. Chemotherapy, administered intravenously or orally, uses large and small molecules that induce DNA damage, disrupt cell division, or interfere with typical cellular processes to stop further growth and spread8. Radiation therapy uses focused high energy rays to kill cancer cells and shrink tumors in specific areas while limiting damage to surrounding healthy tissue9. Immunotherapy is an advanced treatment option which enhances the immune system’s ability to recognize and attack cancer cells10. It works by stimulating the immune system to more effectively fight off diseases or by adding lab-made substances that act like components of the immune system. Types of immunotherapy drugs used in cancer treatment include checkpoint inhibitors, adoptive cell therapy (T-cell transfer), monoclonal antibodies, cancer vaccines, and immune system modulators. As the newest sector in cancer treatments, immunotherapy has significantly changed survival rates for certain cancers including melanoma, NSCLC, certain kidney cancers, and blood cancers like leukemia and lymphoma11.
Immune Response
Generally, the immune system recognizes and destroys cancer cells through a process known as the cancer immunity cycle. This process is self sustaining, because the death of tumor cells in one cycle releases more antigens to restart the immune response12. The cancer immunity cycle consists of 7 steps that recur (Figure 2). The cycle begins when dying cancer cells (due to apoptosis or other factors) release tumor specific proteins known as antigens or neoantigens, into the surrounding microenvironment13. Neoantigens are unique proteins produced by cancer cells due to mutations that can trigger an immune response14. Second, antigen presenting cells (APCs), such as dendritic cells (DCs), capture these antigens and travel to the lymph nodes or other lymph organs, where they process and display antigen fragments on the major histocompatibility complex (MHC) via MHC class I and II molecules, which are then recognized by CD8+ T cells, or CD4+ T cells, respectively15. After APCs present tumor antigens in the lymph nodes, T cells recognize these antigens and become primed to activate and target any cells showing those same biomarkers or antigens. Once activated, the effector T cells, short lived T lymphocytes, exit the lymph nodes and travel through the bloodstream toward the tumor, guided by chemical signals that help direct them to the tumor. This movement through the bloodstream is known as T-cell trafficking, and it allows activated T cells to circulate throughout the body in search of cancer cells that express the same antigens they were primed to recognize16. When they reach the tumor site, they perform T-cell infiltration, squeezing through blood vessel walls to enter the tumor microenvironment to be in direct contact with cancer cells.
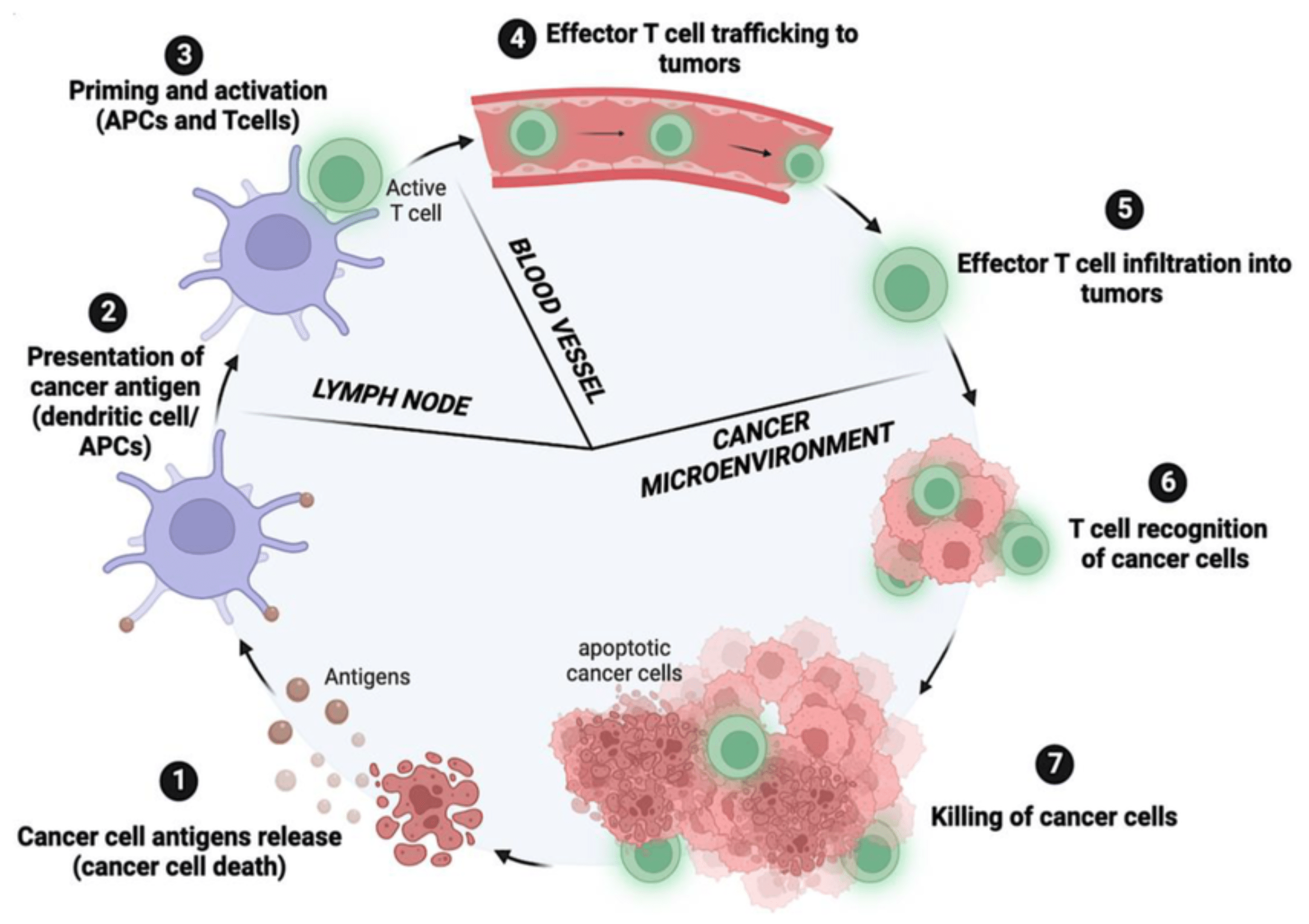
Inside the tumor, the T cells begin T-cell recognition, binding to neoantigens presented on cancer cells through MHC molecules to ensure they target the correct cells. Lastly, they initiate T-cell killing, releasing cytotoxic molecules (ie: perforin and granzymes) that induce tumor cell death17. The dying cancer cells then release additional neoantigens, restarting the cancer immunity cycle and enabling the immune system to continue its response.
Tumor Immune Evasion
Although the cancer immunity cycle is designed to continuously detect and eliminate abnormal cells, it is not flawless or universally effective. Many tumors develop ways to interrupt or weaken one or more steps of the cancer immunity cycle, allowing them to survive despite an active immune response18. Tumor immune evasion is the process by which cancer cells avoid detection and destruction by the immune system. Tumor cells evade the immune system through a variety of complex strategies, including altering their own properties and actively suppressing the immune response within the tumor microenvironment (TME)19. Additionally, several cells, including myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), Tumor-derived exosome (TEX), and Tumor-Associated Macrophage (TAM) demonstrate immunosuppressive qualities20.
Possible mechanisms of immune evasion include T cell dysfunction, the development of an immunosuppressive TME, and tumor antigen escape. In the TME the CD8+ cells that originally migrated to the tumor site during the cancer immunity cycle face exhaustion and dysfunction due to constant antigen exposure and T Cell Receptor signaling21. As a result, the CD8+ T cells stop producing essential cytokines (soluble signaling proteins) like IL-2, TNF- ɑ, and IFN-γ, signals crucial for activating, guiding, and amplifying the immune response and directly killing target cells. The surface of exhausted T cells also display co-inhibitory receptors (e.g., PD-1, CTLA-4, TIM-3, and LAG-3)22. When these receptors bind to their respective ligands on other cells (like cancer cells or antigen-presenting cells), they deliver strong inhibitory signals, effectively overriding any surrounding positive activation signals and shutting down the T cell’s activity.
Antigen escape is another key mechanism that allows a pathogen or tumor cell to evade the immune system by changing or hiding the antigens that immune cells recognize, either through antigen loss (downregulation), where the cell stops or reduces expression of the target entirely, or through antigen hiding (masking), where the antigen becomes covered or altered so the therapy can no longer target it.23,24. Even small changes in the antigen’s shape can be enough to prevent immune cells or therapeutic drugs from binding effectively. Antigen escape is a leading cause of reduced efficacy and relapse in CAR-T cell therapy, impacting 30%–70% of patients with hematologic malignancies who later relapse due to loss or alteration of the target antigen25. Increasing antigen specificity is also critical for safety, as excessive or off-target T-cell activation is a major contributor to CRS. Hence, a dual target approach would require tumor cells to alter two different antigens simultaneously, which is less likely to occur, and therefore decreases the likelihood of relapse.
T Cell Activation and Signaling
T Cell Activation and signaling are key processes in the cancer immunity cycle that are responsible for T cells identifying and responding to foreign antigens (Figure 2). When a DC presents these tumor antigens in the lymph node, T cells begin an activation process that determines whether they will launch an immune response26. The first signal (Signal 1) is activated when the T-cell receptor (TCR) binds to the antigen MHC complex displayed by the DC. The TCR is a specific receptor made of two protein chains (α and β) that form a binding site able to recognize only one particular antigen27. As a result, TCRs are highly specific for a single antigen. However, Signal 1 alone is not sufficient to fully activate the T cell. The T cell must also receive a second signal (Signal 2) also known as a costimulatory signal, before initiating an effector response. Signal 2 is mediated by costimulatory receptors on the T cell, such as CD28, which binds to cognate molecules like CD80 or CD86 expressed on the surface of a DC28. Without the costimulatory signal, the cell would reach a state of anergy or an unresponsive state29. The combination of Signal 1 and Signal 2 stabilizes the interaction between the T cell and dendritic cell, forming what is known as an immunological synapse. This synapse allows the T cell to receive an organized set of activation signals rather than briefly or accidentally30. This organized signaling through the immunological synapse is how T cells become fully activated, and when it is disrupted, as it often occurs in cancer immune evasion, the immune system’s ability to attack tumor cells is weakened.
Background of CAR T- cell Therapy
CAR T-cell therapy is an advanced and personalized form of immunotherapy. The concept of CAR‑T cells dates back over 30 years, first proposed in the late 1980s and refined through the 1990s and 2000s. In the early 2010s, this idea reached the clinic: the first adult to receive CAR‑T therapy was William Paul Ludwig, treated in 2010 for refractory chronic lymphocytic leukemia (CLL) at University of Pennsylvania. Shortly afterward, in 2012, 7 year old Emily Whitehead became the first pediatric patient treated with CAR‑T therapy, a major milestone in the treatment of pediatric leukemia. Their complete remissions demonstrated for the first time that genetically modified T cells had the potential to eradicate cancer in humans.
Currently, FDA-approved CAR T-cell products include Kymriah (for B-cell acute lymphoblastic leukemia and certain B-cell lymphomas), Yescarta (for large B-cell lymphoma and follicular lymphoma), Tecartus (for mantle cell lymphoma and relapsed/refractory B-cell ALL), Breyanzi (for various B-cell lymphomas), and Abecma and Carvykti (for relapsed or refractory multiple myeloma)31. CAR T-cell therapy is currently most effective in treating blood cancers because the cancer cells and the T cells are both in the bloodstream, making them easier to find and target. Treating solid tumors is more difficult because the tumors are in a localized area and have developed ways to create a hostile environment that prevents immune cells from attacking them32.
CAR Construct Design
The elements of a typical bispecific CAR include two recognition modules known as scFvs, a spacer domain, a transmembrane domain, an intracellular signaling domain, and one or more co-stimulatory domains33. An scFv is the small antigen binding region originally derived from an antibody, and in a CAR it allows the engineered T cell to specifically bind to the tumor associated proteins. In a tandem CAR (TanCAR) design, these two antigen-binding scFvs are linked together in series within a single CAR molecule by a flexible hinge, also called a spacer domain, allowing independent movement and binding to their respective antigen. A transmembrane domain anchors the engineered CAR to the T cell. CARs also include an intracellular signaling domain, which is a crucial component that translates external actions like the binding of a target antigen into an internal cellular response, such as proliferation, cytokine release, or cell killing. CARs also contain costimulatory domains within the intracellular region, which are essential for sustaining T-cell activity after antigen recognition34. While the CD3ζ chain provides the primary activation signal, costimulatory domains such as CD28 or 4-1BB provide a secondary signal that enhances T-cell survival and long-term persistence in the TME. Together, these signaling modules ensure that once the CAR binds to its target, the engineered T cell can fully activate, expand, and carry out an effective cytotoxic response35.
Administration and Delivery
Once the CAR T cells are fully engineered, they are administered to the patient through a multi-step process. First, T cells are collected from the patient’s blood via leukapheresis, separating them from other blood components before returning the remaining blood36. The collected T cells are then sent to a lab to be genetically modified to express chimeric antigen receptors (CARs), enabling them to recognize and attack specific cancer antigens37. Next, the engineered cells are expanded in the lab to create millions of cancer-fighting T cells. Before infusion, the patient receives conditioning chemotherapy to reduce existing immune cells and prepare the body for the new CAR T cells38. The modified CAR T cells are then introduced intravenously, typically in a hospital, followed by close monitoring for any adverse effects39. Following infusion, CAR T-cell activation can also lead to significant immune-mediated toxicities, most notably CRS
Safety and Toxicity Considerations – CRS
One of the most significant safety concerns in CAR T cell therapy is CRS. CRS occurs when activated CAR T cells release an excessive quantity of inflammatory cytokines into the bloodstream. These pro-inflammatory cytokines, such as IL-6 and IFN-γ, help immune cells communicate, but in excess they can result in inflammation throughout the body40. CRS can lead to symptoms like fever, fatigue, low blood pressure, and, in severe cases, problems with other organs.
CRS severity is evaluated using standardized grading frameworks, most commonly the ASTCT (American Society for Transplantation and Cellular Therapy) consensus grading system. This system classifies CRS from Grade 1 to Grade 4, based on symptoms such as fever, hypotension, hypoxia, and the need for medical intervention. Grade 1 CRS is generally mild and includes symptoms such as fever and fatigue, while Grades 2–4 represent increasingly severe and potentially life-threatening conditions, which often require hospitalization, oxygen supplementation, vasopressors, or intensive care unit (ICU) support41. The grading criteria are standardized for all clinical trials and FDA-approved CAR T-cell therapies to keep patient safety and clinical management consistent.
CAR-T therapies include black box warnings, which are the FDA’s strongest alerts for serious and potentially life threatening side effects. CRS remains the most prominent risk, experienced by a significant number of patients across approved CAR-T therapies: About 77% with KYMRIAH (ALL), 90% with YESCARTA (NHL), 91% with TECARTUS (MCL), 56% with BREYANZI (NHL), and 84% with CARVYKTI (multiple myeloma)42,43,44,45,46. Amongst them the prescription warnings emphasize that a substantial fraction of patients experienced moderate to severe CRS (Grades ≥2).
Proposal of Bispecific CAR T Therapy
CAR T-cells represent an effective strategy for the treatment of cancers. However, some major problems in CAR T-cell design include ineffectiveness in solid tumors, lack of specificity (where CAR T-cells may attack healthy cells; on-target, off-tumor effects; miss tumor cells due to antigen escape), and dangerous side effects like CRS. Hence, there is a need to design new CAR T-cells for solid tumors, while overcoming single antigen limitations and improving safety by requiring two target antigens. We hypothesized that integrating tumor-specific, bispecific targeting with correlation-based analysis of CAR design parameters associated with CRS can improve therapeutic specificity while informing safer CAR T-cell configurations. The goal of this study was to propose a novel bispecific TanCAR T-cell designed to recognize two proteins simultaneously while reducing adverse CRS effects. Therefore, the proposed bispecific design prioritizes antigen specificity as a strategy to improve efficacy while minimizing excessive immune activation associated with CRS.
Methods
This in silico modeling was designed as a theoretical exploration rather than a predictive or experimentally validated model. As there is very limited publicly available data on cytokine release in bispecific TanCAR T-cell systems, simulated data was generated to examine how key design factors, such as antigen density and signaling strength, might influence cytokine release.
As a result, the outputs from this model, including correlations and feature importance, should not be interpreted as real biological findings. Instead, they reflect the assumptions built into the simulation and are meant to highlight possible trends. The purpose of this section is to explore relationships and help guide future experimental work, rather than to make quantitative claims.
DepMap
We used the DepMap (Cancer Dependency Map) database to identify genes that could be potential biomarkers for a dual-target CAR T cell therapy for NSCLC47. DepMap is a public data set of genetic and molecular information from both cancer and healthy cell lines, which allows for an effective comparison of gene expressions between cancerous and noncancerous cells. By targeting two different surface markers instead of one, the therapy becomes harder for the tumor to evade, since the tumor would need to alter both antigens at the same time to avoid detection, which is significantly less likely. This dual target strategy therefore adds additional reliability and decreases the chances of the tumor escaping the immune response.
GEPIA
In addition to DepMap, the GEPIA (Gene Expression Profiling Interactive Analysis) database was utilized to analyze gene expression in patient-derived NSCLC samples, and serve as validation for the DepMap results48. GEPIA combines RNA-sequencing data from TCGA and GTEx, to compare gene expression between tumor and normal tissue across large patient cohorts. By examining patient samples, it was confirmed that potential target genes are consistently overexpressed in NSCLC tumors relative to healthy lung tissue. GEPIA’s survival analysis feature was also used to assess whether expression levels of these genes correspond to overall patient’s survival rates48.
Computational Modeling and Parameter Correlation
Computational modeling was performed using Google Colab, an online, cloud based platform that allows researchers to use Python code for data analysis and modeling without specialized software or hardware. Google Colab is commonly used in scientific research because it supports reproducible analyses and provides an accessible environment for computational experiments. The code used for the computational analyses and figure generation is publicly available in a Digital Repository. Notably, the data used in this study were simulated and represent modeled relationships, not experimentally measured biological data.
Using Google Colab, an in silico approach was applied to assess how variations in CAR T-cell design related parameters could influence immune activation and cytokine release. The term in silico refers to experiments conducted through computer simulations rather than in laboratory (in vitro) or animal or human (in vivo) systems. In silico methods are particularly important in immunotherapy research because they allow early stage exploration of safety and design trade offs without biological risks.
Simulated datasets were generated to represent biologically reasonable ranges of CAR T-cell activation and cytokine signaling, informed by trends reported in prior CAR T-cell literature. Parameter values within the computational framework were selected based on biological characteristics that commonly differ across CAR generations and costimulatory domain designs reported in established CAR T-cell literature. These included differences in activation strength, cytokine production, activation kinetics, and T-cell persistence associated with different signaling domains. For example, CD28-associated signaling was modeled with stronger early activation behavior due to its known association with rapid T-cell activation and increased cytokine production, while 4-1BB-associated signaling is more commonly linked to slower activation and improved long-term persistence. Antigen density parameters were informed by relative expression trends observed in DepMap and GEPIA analyses, while hinge-related variables were included based on prior evidence that receptor spacing and flexibility can influence CAR signaling behavior. These variables were implemented as normalized, dimensionless parameters intended to model relative signaling trends within a conceptual in silico framework rather than experimentally measured biological constants. Key immune activation parameters were systematically set to observe their effects on predicted cytokine release levels.
Overall, the computational, in silico method helped study relationships between CAR T-cell design parameters and safety-related outcomes, specifically CRS.
Results
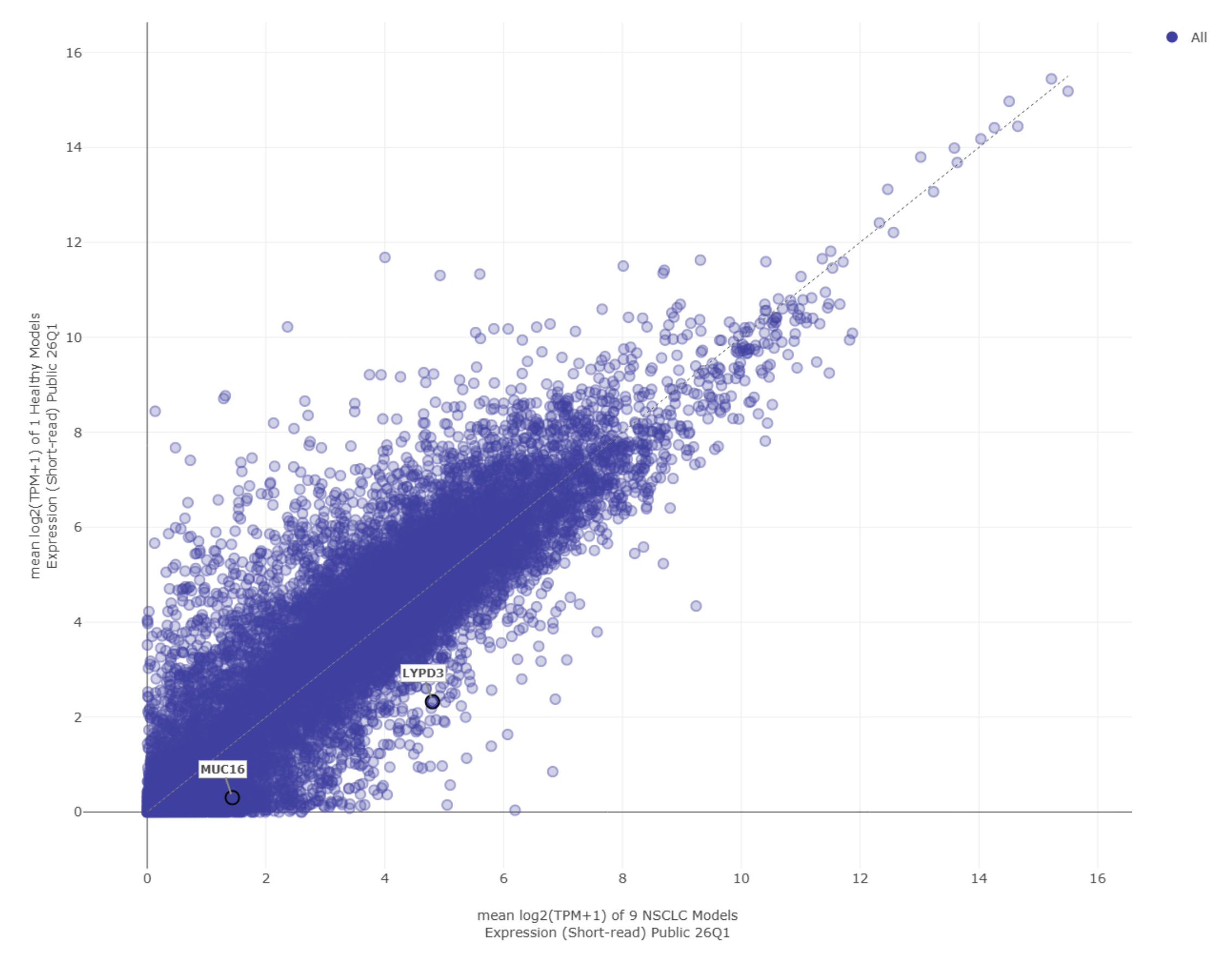
Using the Data Explorer tool in DepMap, we compared gene expression in NSCLC models and in immortalized lung cell models49. Each point on the scatter plot represents a gene, showing its level of expression in both cell types (Figure 3).
By analyzing the scatter plot, we identified genes that were expressed at higher levels in cancer cells than in immortalized lung cells. These genes have a high potential as CAR T-cell targets because they would allow the antigen recognition domain to distinguish between the cell surface proteins of healthy and cancerous tissue. From this analysis, two genes expressed in Chromosome 19, MUC16 and LYPD3, showed higher expression, like many others, in NSCLC cells than in immortalized “healthy” lung cells.
MUC16 is a gene encoding a large surface glycoprotein, Mucin 16, which helps cancer cells avoid detection by the immune system and promotes tumor growth. It has been highly expressed in ovarian, pancreatic, breast, and lung cancers50. LYPD3 is another gene encoding the cell surface protein, Ly6/PLAUR domain containing 3, which helps cancer cells stick to and migrate through tissues, contributing to tumor spread51. Because both MUC16 and LYPD3 are located on the cell surface, they are accessible to immune cells and therapeutic cells and therefore suitable for CAR T-cell targeting.
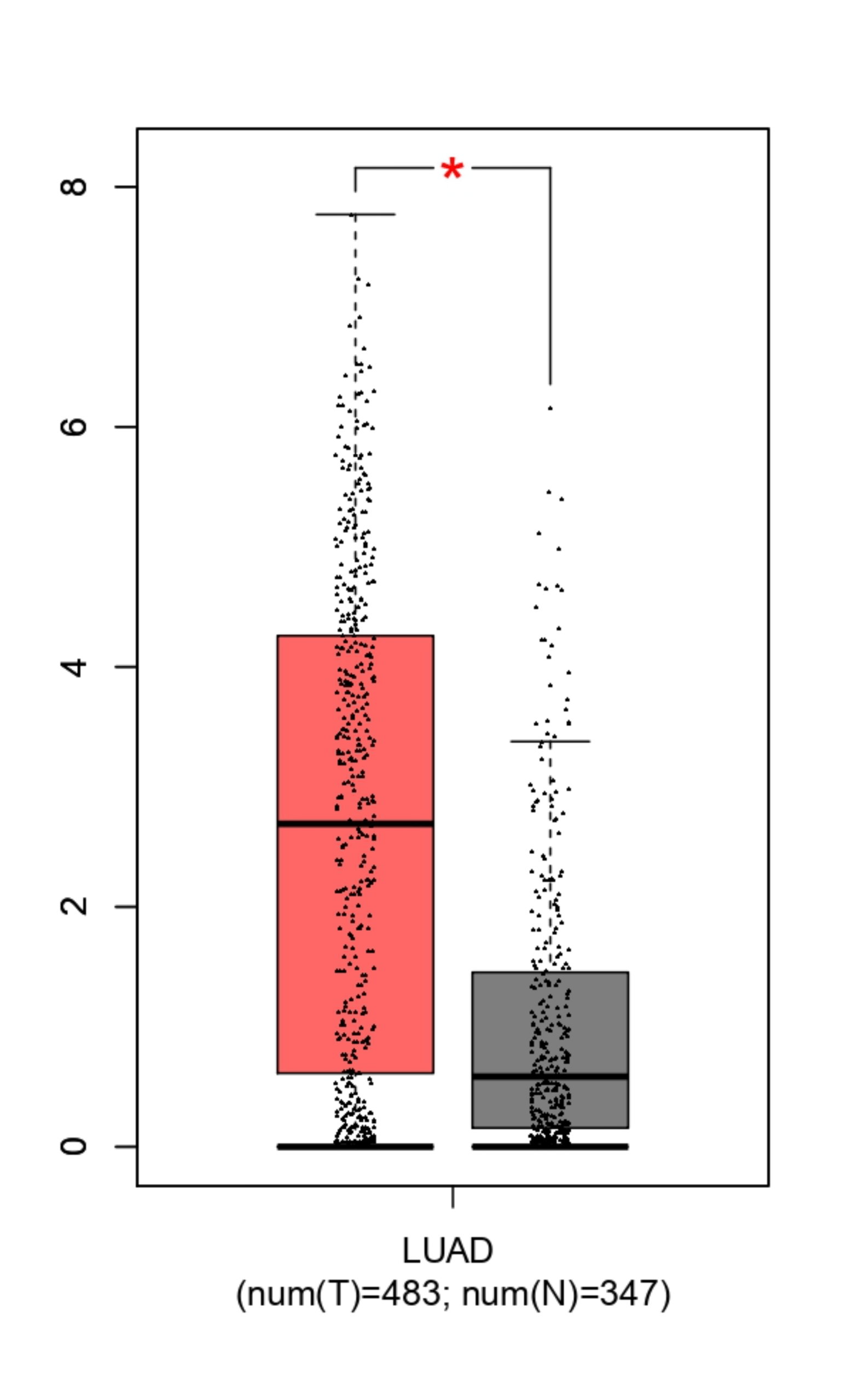
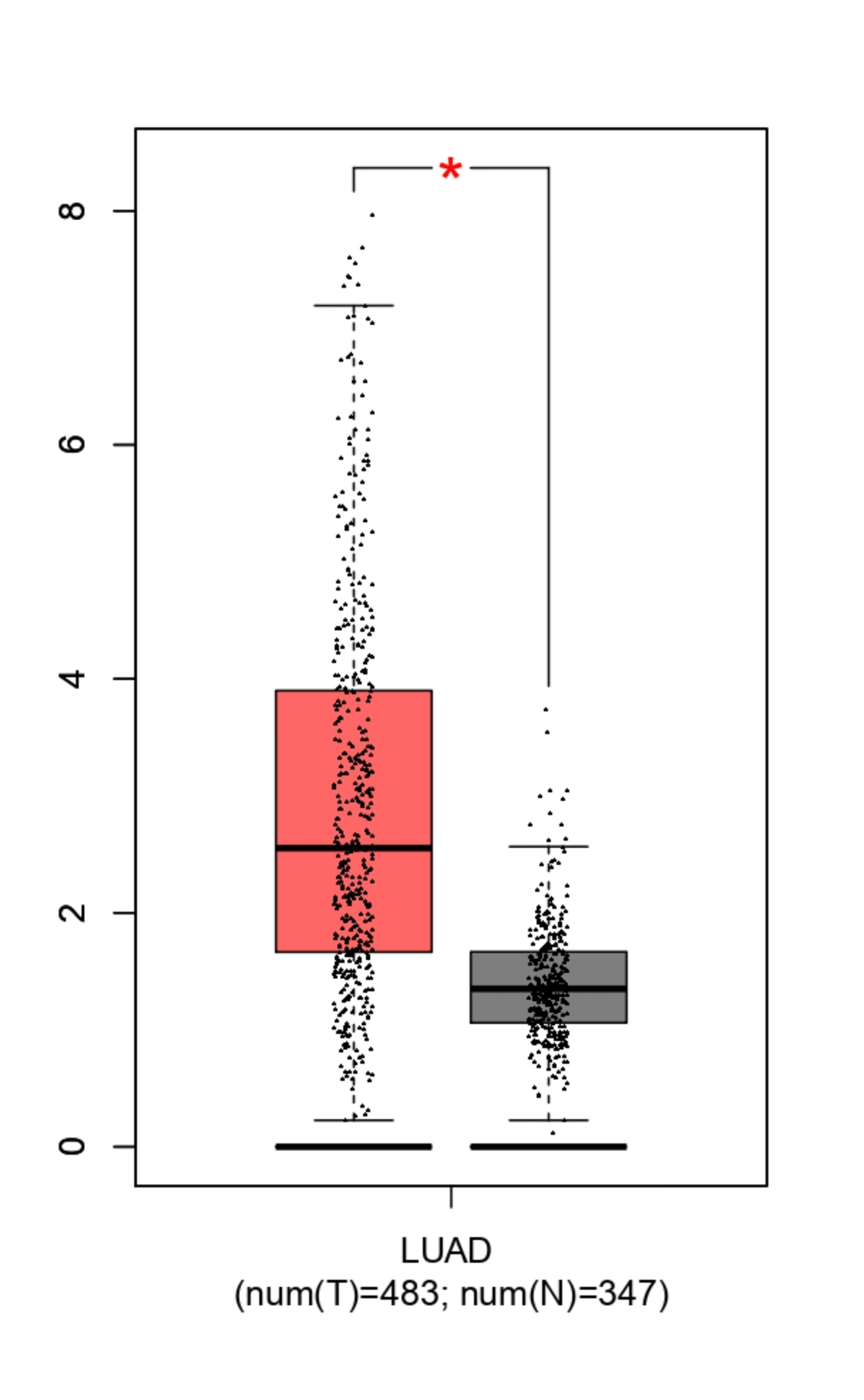
To validate the Depamp results, GEPIA was used to analyze LUAD (Lung Adenocarcinoma) samples. LUAD is the most common subtype of NSCLC, accounting for over 40% of cases, and is acceptable for NSCLC research because data derived from this major subtype directly contributes to overall statistics. The data showed that both MUC16 and LYPD3 are significantly overexpressed in tumor tissue compared to normal lung tissue. For LYPD3, tumor samples displayed a median log₂(TPM+1) expression of approximately 2.5–3, while normal lung tissue showed much lower expression, with a median near 0.5, showing significantly higher expression in cancer cells (Figure 4b). MUC16 displayed a similar pattern, with elevated expression levels and a wider distribution in tumor samples, whereas normal lung tissue remained clustered at low expression values (Figure 4a).
The statistical differences between tumor and normal groups (p < 0.05), determined using an unpaired Student’s t-test in GEPIA, along with the large sample sizes (483 tumor samples and 347 normal samples), indicate that the patterns are unlikely to result from random variation. A p-value represents the probability that the results occurred by chance, and a value below 0.05 is widely accepted as evidence that the result is statistically significant and reproducible. The consistently low expression in normal lung tissue suggests a reduced risk of targeting healthy cells. Together, these results validate the DepMap findings using patient-level data and support MUC16 and LYPD3 as tumor-associated antigens with sufficient differential expression to justify their use in a dual-target CAR T-cell strategy.
Survival analysis revealed that differences in survival between high and low expression groups were present but minimal, indicating that the importance of these genes lies in antigen specificity and tumor expression rather than in affecting patient survival. These results further support MUC16 and LYPD3 as suitable targets for a dual-target CAR T-cell approach based on both cell line and patient tumor expression. In the future, safety features like a “suicide switch” gene could be added to stop the CAR T cells if they cause excessive immune reactions, like CRS.
The in silico models by Google Colab incorporated several CAR T-cell design parameters as inputs, including signaling domain strength, antigen density, costimulatory domain strength, hinge length, and scFv affinity. The output variable was a CRS severity score, which represents the predicted magnitude of cytokine release for each respective parameter following CAR T-cell activation.
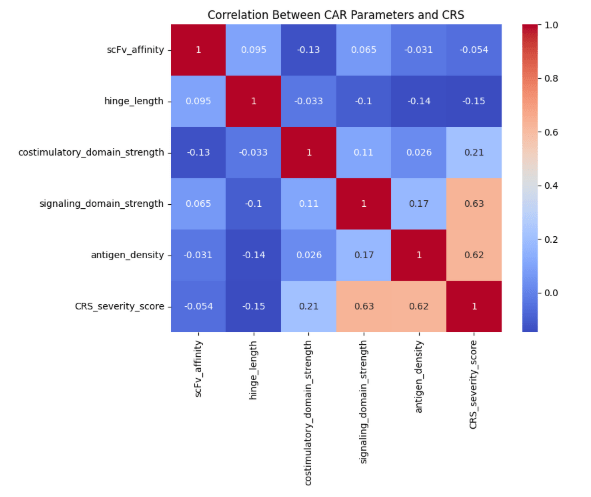
A correlation heatmap was generated to evaluate relationships between each CAR T-cell parameter and the CRS severity score. Because these data were acquired from a simulated dataset rather than real biological data, the observed correlations should be interpreted as preliminary findings that require further validation. This heatmap uses the Pearson correlation coefficients method (r), which ranges from −1 to +1. Values closer to +1 indicate a strong positive relationship, meaning that as one variable increases, the other also increases. Values near 0 indicate little to no relationship. Diagonal values of 1 were excluded from interpretation. The heatmap showed that signaling domain strength exhibited the strongest positive correlation with CRS severity (r ≈ 0.63). Antigen density also showed a strong positive correlation (r ≈ 0.62), indicating that a higher antigen availability on tumor cells is associated with increased, predicted cytokine release. Costimulatory domain strength had a weaker correlation (r ≈ 0.21), while hinge length (r ≈ −0.15) and scFv affinity (r ≈ −0.05) showed minimal association with CRS severity (Figure 5).
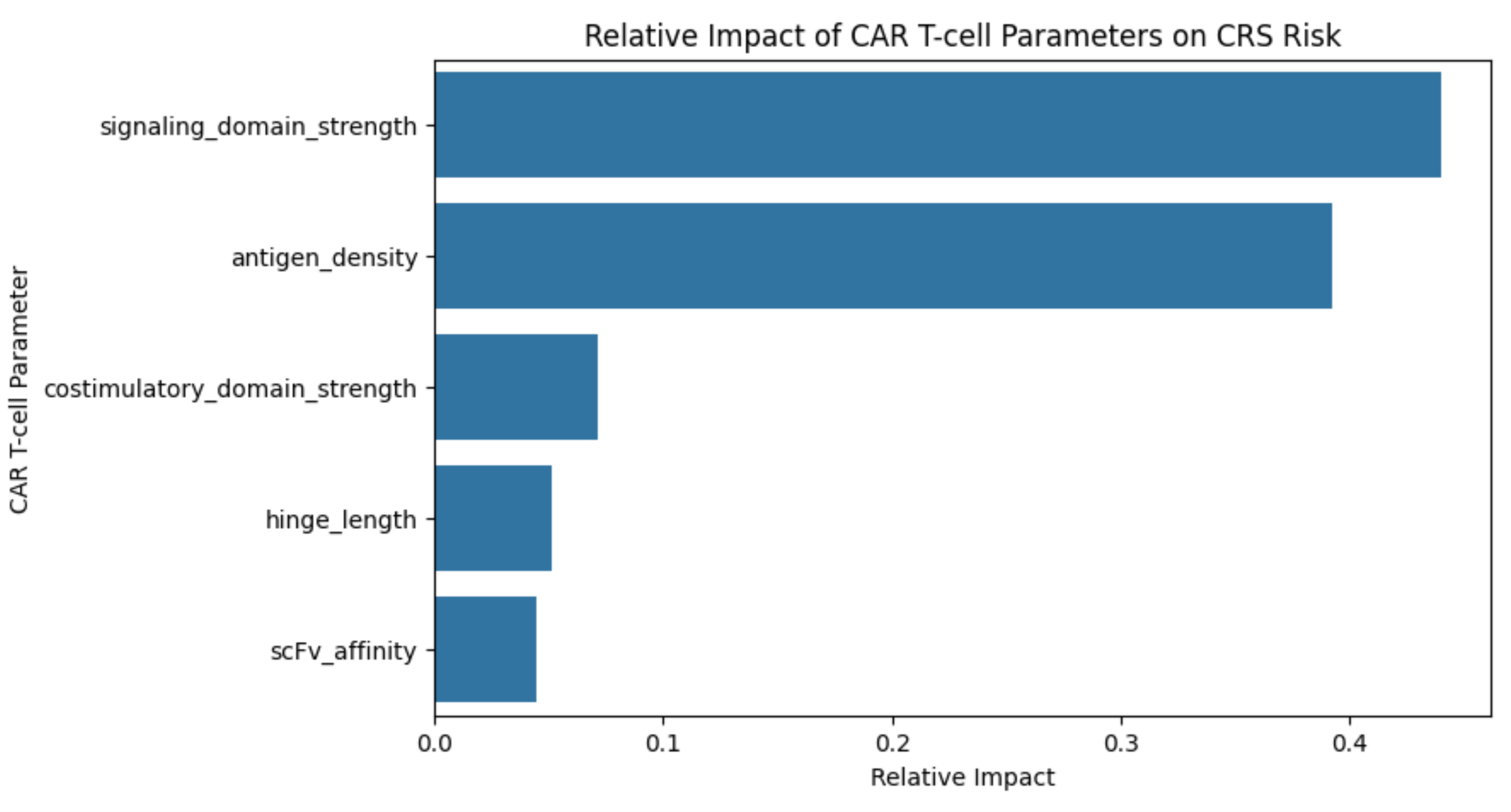
Figure 6a | Feature Importance plot: Visually represented as a horizontal bar graph comparing 5 individual CAR components with relative importance.
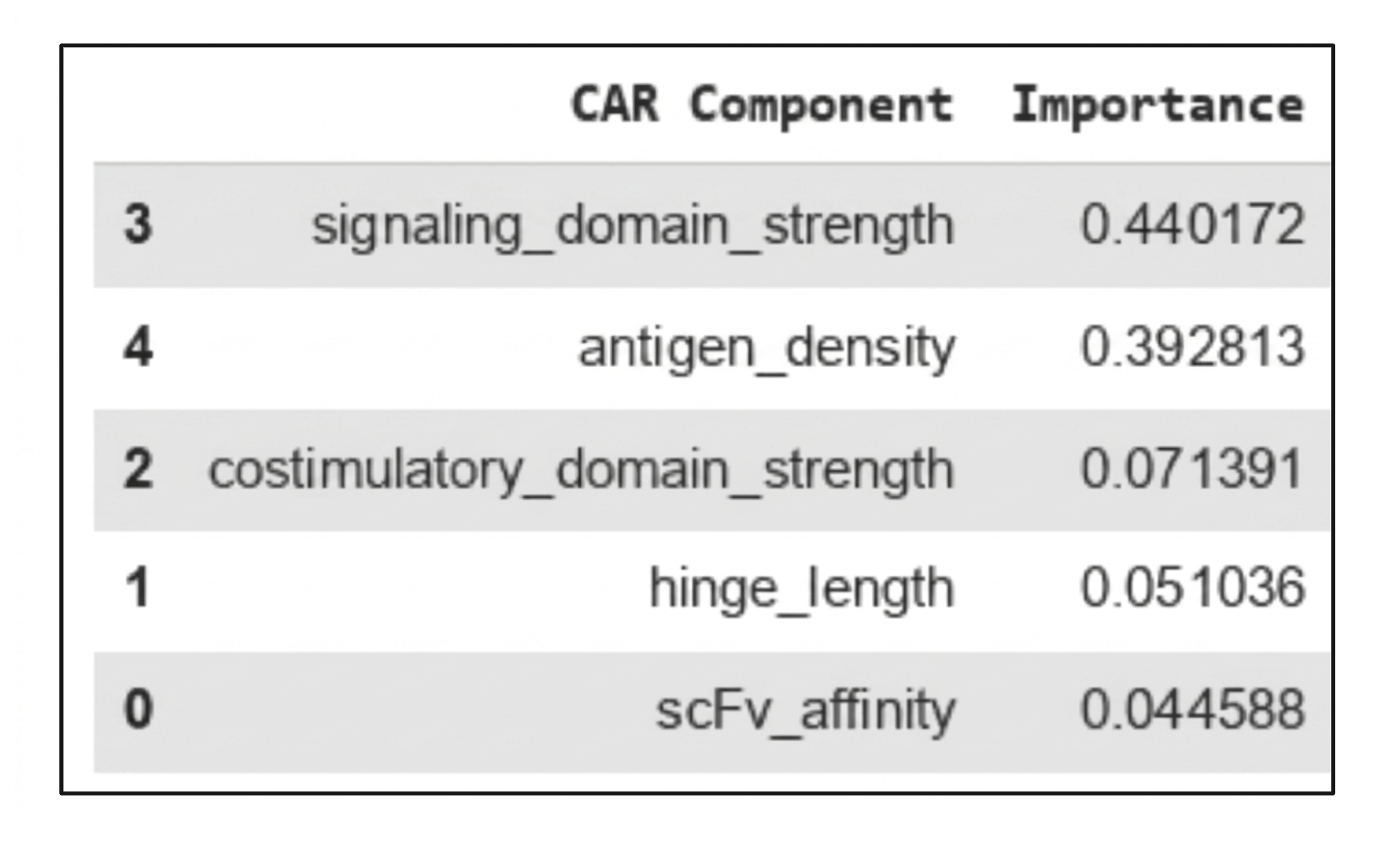
To further quantify the relative influence of each parameter, a random forest regression model was generated. This model evaluates how much each input contributes to predicting the output when all variables are considered together (Figure 6a). A feature importance analysis showed that the signaling domain strength accounted for approximately 44% of the model’s predictive contribution, making it the most influential parameter. Antigen density contributed approximately 39%, further supporting its role as a major driver of CRS risk. In comparison, costimulatory domain strength contributed about 7%, while hinge length (≈5%) and scFv affinity (≈4%) had relatively minor effects (Figure 6b). Because the feature importance analysis was based on a simulated dataset with assumed relationships, these importance values are meant to illustrate potential trends, not to serve as definitive predictive measures. We did not perform cross-validation or compute an R2, as the goal was to explore relationships that could inform future experimental design.
Together, these findings indicate that CRS risk in the model is primarily driven by CAR T-cell activation signaling and tumor antigen density, rather than by receptor structure or binding. This highlights the importance to carefully optimize signaling and costimulatory modules while designing CAR T-cells to balance efficacy with safety.
Design
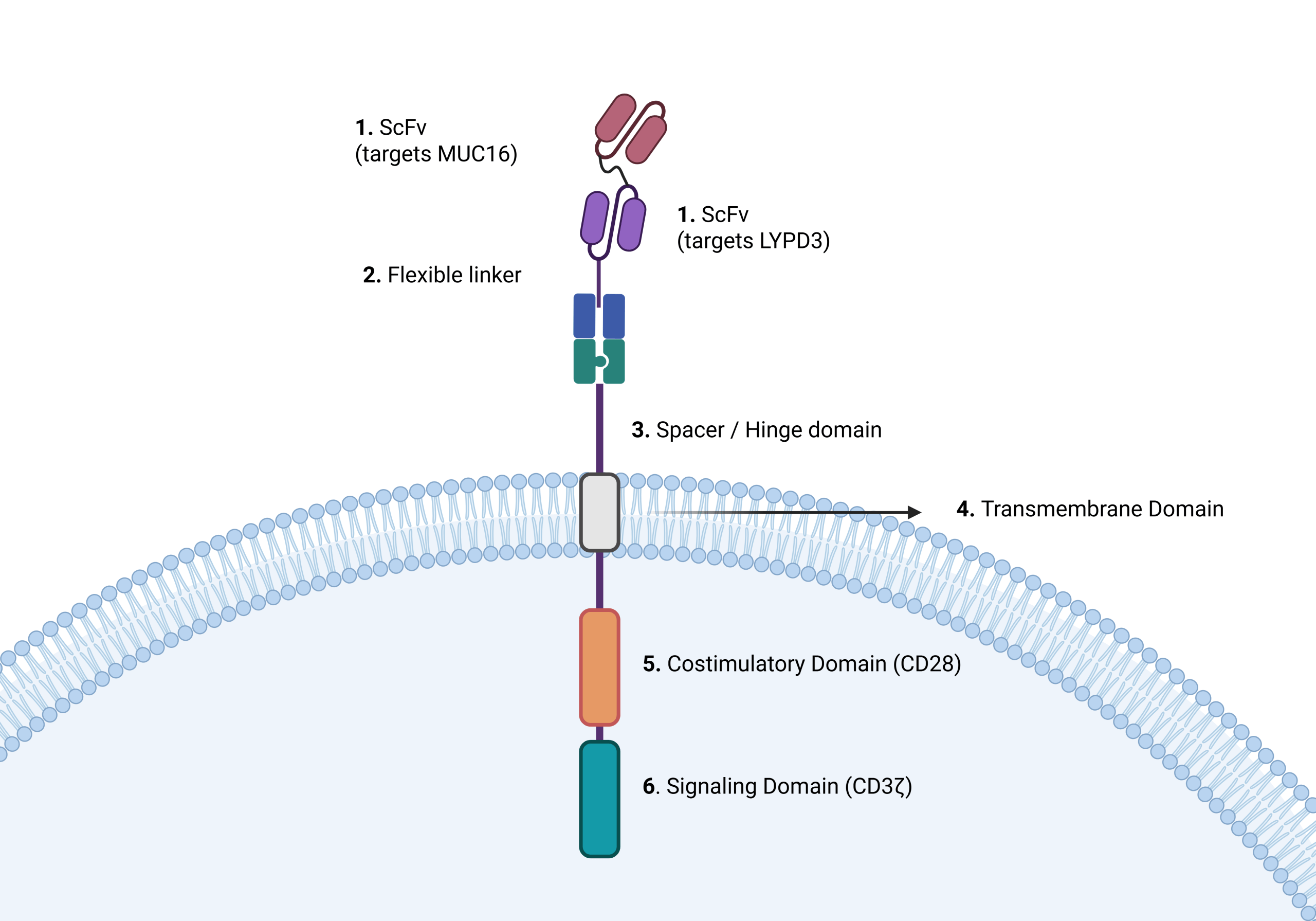
With all data in consideration, a design was proposed. The proposed TanCAR T-cell design is a bispecific, dual-antigen receptor engineered to recognize MUC16 and LYPD3 simultaneously on NSCLC tumor cells while minimizing CRS effects. The targeting domains used in the proposed TanCAR construct were based on previously studied antibody binders against MUC16 and LYPD3. The anti-MUC16 targeting module was derived from validated binders used in prior CAR-T and solid tumor targeting studies, particularly the 4H11-based MUC16 targeting approaches described in previous CAR-T research52 , while the anti-LYPD3 targeting module was based on previously characterized monoclonal antibody research involving LYPD3-targeted therapies in epithelial cancers53 . These scFv targeting domains were included as part of a theoretical TanCAR design framework and were not experimentally optimized or newly engineered in this study. These antigens were selected based on DepMap and GEPIA analyses demonstrating significantly higher expression in NSCLC tumors compared to normal lung tissue, with relatively low expression in healthy samples. Requiring simultaneous engagement of two tumor-associated antigens increases targeting specificity and reduces the likelihood of antigen escape, as tumor cells would need to downregulate or alter both antigens at once to evade immune recognition.
Structurally, the CAR resembles the second generation CAR, because it incorporates one costimulatory domain (CD28), one signaling domain (CD3ζ) as per the results in the parameter correlation, two single-chain variable fragments (scFvs) arranged in tandem (linked sequentially), each specific to one antigen (MUC16 and LYPD3). These scFvs are connected by a flexible peptide linker that allows independent binding to each antigen while maintaining stability. The two scFv domains were arranged in a tandem configuration and connected by a flexible glycine-serine (G4S) linker to allow independent antigen binding while reducing steric hindrance between the domains. The MUC16-targeting scFv was positioned closer to the N-terminal extracellular region of the construct, followed by the LYPD3-targeting scFv. This arrangement was chosen to support efficient access to both antigens on NSCLC tumor cells, including heterogeneous tumor populations where one or both antigens may be variably expressed. Overall, this configuration was designed to maintain binding accessibility and ensure that engagement of one scFv does not interfere with the function of the other. The scFv domains are followed by a hinge and transmembrane region, which connect to the intracellular signaling domains. Based on the computational CRS correlation analysis, the hinge length was chosen to be average rather than extended, as hinge length showed minimal correlation with CRS severity (r ≈ −0.15) and therefore does not need aggressive changes that could compromise functionality.
The intracellular signaling module was intentionally designed to prioritize safety while preserving cytotoxic function. The CAR includes a single CD3ζ signaling domain with a moderate-strength costimulatory domain, such as CD28 rather than 4-1BB. The CD28 costimulatory domain was selected to provide rapid T-cell activation and enhanced initial effector response, which is particularly relevant for heterogeneous NSCLC tumor environments requiring immediate cytotoxic activity. In contrast, 4-1BB-based signaling is associated with slower activation kinetics but improved T-cell persistence and reduced exhaustion. In this study, CD28 was prioritized to support stronger early anti-tumor responses, while CD3ζ was used as the primary signaling domain to initiate T-cell activation. This design choice is consistent to standard second-generation CAR architectures and was optimized with the computational modeling framework used to evaluate activation strength and CRS-associated signaling parameters. This choice is also supported by the in silico modeling results, which showed that the signaling domain strength was the strongest predictor of CRS risk, responsible for approximately 44% of CRS, while costimulatory domain strength contributed only ~7% (Figure 6b). Using a less aggressive costimulatory signal helps limit excessive cytokine release while maintaining T-cell persistence and metabolism in the solid TME54.
Additionally, the bispecific design helps reduce the effects of antigen density, which is known to increase the risk of CRS. (r ≈ 0.62; 39% feature importance). By requiring a dual target, tandem approach, the T-cell activation becomes more localized to tumor cells with high co-expression of MUC16 and LYPD3, rather than targeting high densities of a single antigen alone. This method helps reduce widespread T-cell activation and lowers the probability of CRS risk throughout the body.
Overall, this CAR T-cell design integrates tumor-specific antigen selection with CRS-informed receptor engineering, balancing efficacy and safety. The design directly reflects computational findings that excessive intracellular signaling and antigen density drive CRS, while structural features such as hinge length and scFv affinity play a smaller role. By combining bispecific targeting with controlled signaling strength, this CAR T-cell configuration aims to improve therapeutic precision in NSCLC while minimizing immune-mediated toxicity.
Discussion
In a lab setting, several key tests would be necessary to demonstrate the efficacy of our bispecific TanCAR T design. Pre-clinical studies would need to confirm that the TanCAR T cell can recognize tumor cells and induce tumor cell death in vivo and in vitro55. Subsequently, the persistence, trafficking, activation, and infiltration of T cells into solid tumor tissue could be tested using in vivo models of mice that spontaneously develop NSCLC-like disease, or are implanted with human NSCLC xenograft tissue56. Additional assays that measure cytokine release, off-target effects, and TME activity would help verify that the therapy is both potent and safe. Together, these evaluations serve as essential steps before any bispecific CAR T therapy can advance to clinical testing.
Following successful preclinical validation, an initial Phase I clinical trial would likely enroll patients with advanced or treatment-refractory NSCLC whose tumors express both MUC16 and LYPD3. The primary objectives would be to evaluate safety, tolerability, and the optimal dose of the engineered TanCAR T cells. Patients would be closely monitored for adverse events associated with CAR T-cell therapy, including CRS, immune effector cell-associated neurotoxicity syndrome (ICANS), and potential on-target, off-tumor toxicity. Additional monitoring of CAR T-cell persistence, expansion, and tumor response would help assess both safety and preliminary therapeutic efficacy.
If this CAR T-cell were made in a lab, we could insert the bispecific CAR gene into T cells using gene delivery methods such as lentiviral transduction, which allows the new gene to become a permanent part of the engineered T cell’s DNA. Another method, electroporation, creates temporary pores in the cell’s membrane, allowing substances like DNA, RNA, proteins, and drugs to enter cells efficiently. This would allow the CAR T-cell to deliver the gene without using a virus, making it a safer but more short term method. Once engineered, the modified T cells could be tested in culture to see how effectively they recognize and kill cancer cells that express MUC16 and LYPD3.
Although the steps of the cancer immunity cycle show how T cells can recognize and destroy tumor cells, this process is not always efficient in real biological settings. The same limitations also apply to engineered T cells, including CAR T cells. While CAR T therapy has shown strong efficacy for the treatment of blood cancers, it has not been as successful for solid tumors. Numerous clinical trials have been conducted and many are ongoing, but as of late 2024, no CAR T-cell therapies have received general FDA approval for solid tumors57. Early trials often showed limited efficacy, with low overall response rates (around 9% in one meta-analysis)58. Responses, when they occurred, were often partial or not substantial, with most patients experiencing tumor relapse.
There are several potential explanations for the limited efficacy of CAR T cells in solid tumors. Unlike blood cancers where cancer cells circulate freely and are easily accessible to T cells in the bloodstream and lymphoid tissues, solid tumors are surrounded by an inhibitory microenvironment comprising healthy tissue, or stroma, abnormal vasculature, and low levels of chemokines needed for T-cell trafficking and infiltration59.The TME can therefore limit the access of CAR T cells to the tumor, and also suppress the cytotoxic activity of CAR T cells in the local environment. In addition, NSCLC tumors often produce immunosuppressive cytokines such as TGF-β and IL-10, which can reduce T-cell activation and promote T-cell exhaustion. Immune checkpoint pathways, including PD-1/PD-L1 signaling, may further impair CAR T-cell function after tumor infiltration. Together, these mechanisms create a highly suppressive microenvironment that represents a major obstacle to durable CAR T-cell responses in solid tumors.
In addition, many solid tumors are heterogeneous, meaning that they do not express one single, uniform antigen across all cells of the tumor60. Instead, antigen expression varies from cell to cell, which increases the chance of antigen escape. This means that a CAR targeting only one antigen may eliminate some tumor cells but ignore others, allowing the tumor to regrow. Antigen expression may also vary between major NSCLC subtypes, including lung adenocarcinoma and squamous cell carcinoma. Such differences could influence the effectiveness of antigen-targeted therapies, as the abundance and distribution of target antigens may not be uniform across patient populations. While this study evaluated MUC16 and LYPD3 expression in NSCLC broadly, future investigations should examine subtype-specific expression patterns to determine whether distinct therapeutic strategies or patient selection criteria are needed for adenocarcinoma and squamous cell carcinoma.
Bispecific CAR T cells could potentially address the limitations of CAR T cell therapy by allowing the CAR T cells to recognize more than one tumor antigen at once, thereby reducing the likelihood of antigen escape, increasing the specificity of antigen recognition, and potentially enhancing the cytotoxic activity of the CAR T cells. The selection of MUC16 and LYPD3 was further supported by analyses of tumor-to-normal expression ratios across multiple datasets, which suggested relatively tumor-restricted expression patterns and a potentially reduced risk of on-target, off-tumor toxicity. However, comprehensive preclinical safety studies would still be required to fully evaluate antigen expression in healthy tissues and assess potential toxicities prior to clinical application.
Other strategies for improving the efficacy and safety of CAR T cells are currently being studied. For instance, there is ongoing interest in designing CAR T-cells that can respond to features of the TME. The TME of solid tumors may contain several non-cancer cells, such as cancer-associated fibroblasts (CAFs), regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs), all of which can suppress T cells trafficking and activation at the tumor site.
Studies are ongoing to investigate the use of bispecific or combination approaches that help CAR T cells break down physical barriers created by CAFs, resist immunosuppressive signals from Tregs and MDSCs, or function better in the exhaustive conditions inside solid tumors61. Although these strategies are still being tested, they could represent novel approaches by which the efficacy of CAR T cell designs could be improved in solid tumors. Advancing CAR T-cell designs could one day transform how we fight even the most challenging cancers, including NSCLC.
Conclusion
This paper proposes a dual-target bispecific TanCAR T-cell therapy for NSCLC, using MUC16 and LYPD3 as tumor-specific targets to improve specificity while potentially reducing CRS. This approach is important because it addresses major challenges in immunotherapy in solid tumor treatment, including CRS, antigen escape, on-target, off-tumor effects, and an overall lack of specificity which has limited the success of current CAR T therapies. Moving forward, preclinical testing and further optimization of this design could lead to safer, effective immunotherapies and potentially expand CAR T-cell therapy to solid tumors like NSCLC.
Acknowledgments
I would like to thank my mentors and teachers who guided me through this research and provided valuable feedback. I am also grateful to the creators and entire team responsible for the DepMap and GEPIA databases, as well as the developers of BioRender and Google Colab, which made the data analysis and visualizations possible. Finally, I appreciate my family, whose encouragement and support helped me complete this project.
References
- S. Li, Y. He, J. Liu, et al. An umbrella review of socioeconomic status and cancer. Nat Commun. Vol. 15, pg. 9993, 2024, https://doi.org/10.1038/s41467-024-54444-2. [↩]
- National Cancer Institute. What is cancer?, https://www.cancer.gov/about-cancer/understanding/what-is-cancer, 2007. [↩]
- D. Hanahan, R. A. Weinberg. Hallmarks of cancer: the next generation. Cell. Vol. 144, pg. 646-674, 2011, https://doi.org/10.1016/j.cell.2011.02.013. [↩]
- K. B. Blackburn. Metastatic cancer: what happens when cancer spreads?, https://www.mdanderson.org/cancerwise/metastatic-cancer–what-happens-when-cancer-spreads.h00-159460845.html, 2025. [↩]
- C. N. Qian, Y. Mei, J. Zhang. Cancer metastasis: issues and challenges. Chin J Cancer. Vol. 36, pg. 38, 2017, https://doi.org/10.1186/s40880-017-0206-7. [↩]
- National Cancer Institute. Types of cancer treatment, https://www.cancer.gov/about-cancer/treatment/types, 2017. [↩]
- National Cancer Institute. Surgery to treat cancer, https://www.cancer.gov/about-cancer/treatment/types/surgery, 2024. [↩]
- National Cancer Institute. Chemotherapy to treat cancer, https://www.cancer.gov/about-cancer/treatment/types/chemotherapy, 2015. [↩]
- National Cancer Institute. Radiation therapy for cancer, https://www.cancer.gov/about-cancer/treatment/types/radiation-therapy, 2015. [↩]
- Cleveland Clinic. What is immunotherapy?, https://my.clevelandclinic.org/health/treatments/11582-immunotherapy, 2025. [↩]
- P. Garg, S. Pareek, P. Kulkarni, D. Horne, R. Salgia, S. S. Singhal. Next-generation immunotherapy: advancing clinical applications in cancer treatment. J Clin Med. Vol. 13, pg. 6537, 2024, https://doi.org/10.3390/jcm13216537. [↩]
- I. Mellman, D. S. Chen, T. Powles, S. J. Turley. The cancer-immunity cycle: indication, genotype, and immunotype. Immunity. Vol. 56, pg. 2188-2205, 2023, https://doi.org/10.1016/j.immuni.2023.09.011. [↩]
- S. K. Kim, S. W. Cho. The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Front Pharmacol. Vol. 13, pg. 868695, 2022, https://doi.org/10.3389/fphar.2022.868695. [↩]
- Signal Transduction and Targeted Therapy. Neoantigens: promising targets for cancer therapy, 2022, https://doi.org/10.1038/s41392-022-01270-x. [↩]
- C. Wang, Y. Shi, D. Zhang, et al. Generalization of neoantigen-based tumor vaccine by delivering peptide-mhc complex via oncolytic virus. EMBO Mol Med. Vol. 17, pg. 1118-1152, 2025, https://doi.org/10.1038/s44321-025-00225-3. [↩]
- D. J. Aires, M. Yoshida, S. K. Richardson, et al. T cell trafficking plays an essential role in tumor immunity. Lab Investig J Tech Methods Pathol. Vol. 99, pg. 85-92, 2019, https://doi.org/10.1038/s41374-018-0124-6. [↩]
- J. C. A. Janeway, P. Travers, M. Walport, M. J. Shlomchik. Immunobiology: the immune system in health and disease. 5th Edition, 2001, https://www.ncbi.nlm.nih.gov/books/NBK27101/. [↩]
- M. Tufail, C. H. Jiang, L. Li. Immune evasion in cancer: mechanisms and cutting-edge therapeutic approaches. Signal Transduct Target Ther. Vol. 10, pg. 227, 2025,https://doi.org/10.1038/s41392-025-02280-1. [↩]
- G. W. Tormoen, M. R. Crittenden, M. J. Gough. Role of the immunosuppressive microenvironment in immunotherapy. Adv Radiat Oncol. Vol. 3, pg. 520-526, 2018, https://doi.org/10.1016/j.adro.2018.08.018. [↩]
- J. Yu, L. Fu, R. Wu, et al. Immunocytes in the tumor microenvironment: recent updates and interconnections. Front Immunol. Vol. 16, 2025, https://doi.org/10.3389/fimmu.2025.1517959. [↩]
- W. Jiang, Y. He, W. He, et al. Exhausted cd8+t cells in the tumor immune microenvironment: new pathways to therapy. Front Immunol. Vol. 11, pg. 622509, 2021, https://doi.org/10.3389/fimmu.2020.622509. [↩]
- D. S. Chen, I. Mellman. Oncology meets immunology: the cancer-immunity cycle. Immunity. Vol. 39, pg. 1-10, 2013, https://doi.org/10.1016/j.immuni.2013.07.012. [↩]
- H. Lin, X. Yang, S. Ye, L. Huang, W. Mu. Antigen escape in car-t cell therapy: mechanisms and overcoming strategies. Biomed Pharmacother. Vol. 178, pg. 117252, 2024, https://doi.org/10.1016/j.biopha.2024.117252 [↩]
- A. Kallingal, M. Olszewski, N. Maciejewska, W. Brankiewicz, M. Baginski. Cancer immune escape: the role of antigen presentation machinery. J Cancer Res Clin Oncol. Vol. 149, pg. 8131-8141, 2023, https://doi.org/10.1007/s00432-023-04737-8. [↩]
- A. Mishra, R. Maiti, P. Mohan, G. Gupta. Antigen loss following car-t cell therapy: mechanisms, implications, and potential solutions. Eur J Haematol. Vol. 112, pg. 211-222, 2024, https://doi.org/10.1111/ejh.14101. [↩]
- A. M. McDonnell, B. W. S. Robinson, A. J. Currie. Tumor antigen cross-presentation and the dendritic cell: where it all begins?. Clin Dev Immunol. Vol. 2010, pg. 539519, 2010, https://doi.org/10.1155/2010/539519. [↩]
- ScienceDirect Topics. T lymphocyte receptor – an overview, https://www.sciencedirect.com/topics/neuroscience/t-lymphocyte-receptor, 2025. [↩]
- J. H. Esensten, Y. A. Helou, G. Chopra, A. Weiss, J. A. Bluestone. Cd28 costimulation: from mechanism to therapy. Immunity. Vol. 44, pg. 973-988, 2016, https://doi.org/10.1016/j.immuni.2016.04.020. [↩]
- J. C. Ribot, A. deBarros, B. Silva-Santos. Searching for “signal 2”: costimulation requirements of γδ t cells. Cell Mol Life Sci CMLS. Vol. 68, pg. 2345-2355, 2011, Searching for “signal 2”: costimulation requirements of γδ t cells. Cell Mol Life Sci CMLS. Vol. 68, pg. 2345-2355, 2011, https://doi.org/10.1007/s00018-011-0698-2. [↩]
- Z. Chao, M. Mei, Y. Yang, et al. Immunological synapse: structures, molecular mechanisms and therapeutic implications in disease. Signal Transduct Target Ther. Vol. 10, pg. 254, 2025, https://doi.org/10.1038/s41392-025-02332-6. [↩]
- National Cancer Institute. CAR t-cell therapy approved for children, young adults with leukemia, https://www.cancer.gov/news-events/cancer-currents-blog/2017/tisagenlecleucel-fda-childhood-leukemia, 2017. [↩]
- Blood Cancer Journal. CAR-t cell therapy: current limitations and potential strategies, 2021, https://doi.org/10.1038/s41408-021-00459-7. [↩]
- J. Jayaraman, M. P. Mellody, A. J. Hou, et al. CAR-t design: elements and their synergistic function. EBioMedicine. Vol. 58, pg. 102931, 2020, https://doi.org/10.1016/j.ebiom.2020.102931. [↩]
- Z. L. Chang, Y. Y. Chen. Cars: synthetic immunoreceptors for cancer therapy and beyond. Trends Mol Med. Vol. 23, pg. 430-450, 2017, https://doi.org/10.1016/j.molmed.2017.03.002. [↩]
- E. Roselli, J. C. Boucher, G. Li, et al. 4-1bb and optimized cd28 co-stimulation enhances function of human mono-specific and bi-specific third-generation car t cells. J Immunother Cancer. Vol. 9, pg. e003354, 2021, https://doi.org/10.1136/jitc-2021-003354. [↩]
- CAR T-Cell Therapy Patient Education. Step 1: cell collection, http://cartpatient.mypreview.site/step-1-cell-collection.html, 2025. [↩]
- CAR T-Cell Therapy Patient Education. Step 2: car t-cell manufacturing, http://cartpatient.mypreview.site/step-2-car-t-cell-manufacturing.html, 2025. [↩]
- CAR T-Cell Therapy Patient Education. Step 4: preparative chemotherapy, http://cartpatient.mypreview.site/step-4-preparative-chemotherapy.html, 2025. [↩]
- CAR T-Cell Therapy Patient Education. Step 5: car t-cell infusion, http://cartpatient.mypreview.site/step-5-car-t-cell-infusion.html, 2025. [↩]
- A. A. Al-Qahtani, F. S. Alhamlan, A. A. Al-Qahtani. Pro-inflammatory and anti-inflammatory interleukins in infectious diseases: a comprehensive review. Trop Med Infect Dis. Vol. 9, pg. 13, 2024, https://doi.org/10.3390/tropicalmed9010013. [↩]
- D. W. Lee, B. D. Santomasso, F. L. Locke, et al. Astct consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. Vol. 25, pg. 625-638, 2019, https://doi.org/10.1016/j.bbmt.2018.12.758. [↩]
- Novartis. Kymriah® (tisagenlecleucel) | official patient website, https://us.kymriah.com/, 2026. [↩]
- Kite Pharma. Yescarta® (axicabtagene ciloleucel) patient & caregiver site | car t-cell therapy for certain types of non-hodgkin lymphoma in adults, https://www.yescarta.com/, 2024. [↩]
- Kite Pharma. Tecartus® (brexucabtagene autoleucel) patient & caregiver site | car t-cell therapy for mantle cell lymphoma, https://www.tecartus.com/, 2023. [↩]
- Bristol Myers Squibb. Home | breyanzi® (lisocabtagene maraleucel) car t cell therapy, https://www.breyanzi.com/home, 2026. [↩]
- Janssen Biotech. Official patient website | carvykti® (ciltacabtagene autoleucel), https://www.carvykti.com/, 2026. [↩]
- J. Mao, Z. Wang, J. Geng, et al. DEPMP: a deep learning-based framework for predicting peptide-mhc binding affinity. Brief Bioinform. Vol. 25, pg. bbad514, 2024, https://doi.org/10.1093/bib/bbad514. [↩]
- Z. Tang, B. Kang, C. Li, T. Chen, Z. Zhang. Gepia2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. Vol. 47, pg. W556–W560, 2019, https://doi.org/10.1093/nar/gkz430. [↩] [↩]
- J. Mao, Z. Wang, J. Geng, et al. DEPMP: a deep learning-based framework for predicting peptide-mhc binding affinity. Brief Bioinform. Vol. 25, pg. bbad514, 2024, https://doi.org/10.1093/bib/bbad514. [↩] [↩]
- Y. Zhang, L. L. Hong, Z. Q. Ling. Muc16: clinical targets with great potential. Clin Exp Med. Vol. 24, pg. 101, 2024, https://doi.org/10.1007/s10238-024-01365-5. [↩]
- T. Hu, Y. Zhang, T. Yang, Q. He, M. Zhao. Lypd3, a new biomarker and therapeutic target for acute myelogenous leukemia. Front Genet. Vol. 13, pg. 795820, 2022, https://doi.org/10.3389/fgene.2022.795820. [↩]
- H. K. Lin, D. A. Blake, T. Liu, R. Freeman, G. B. Lesinski, L. Yang, S. Rafiq. MUC16CD is a novel CAR T cell target antigen for the treatment of pancreatic cancer. Molecular Therapy – Oncology. Vol. 33, 2024, https://pubmed.ncbi.nlm.nih.gov/39346763/. [↩]
- T. Neumann, E. Hartung, J. Gellert, L. Weiß, M. Weiske, N. Kast, S. Gurka, S. Marinoff, A. Jäkel, A. Danielczyk, P. Kehler. Targeting a cancer-specific LYPD3 glycoform for tumor therapy. Frontiers in Drug Discovery. Vol. 3, 2023, https://doi.org/10.3389/fddsv.2023.1298916. [↩]
- M. M. Honikel, S. H. Olejniczak. Co-stimulatory receptor signaling in car-t cells. Biomolecules. Vol. 12, pg. 1303, 2022, https://doi.org/10.3390/biom12091303. [↩]
- X. Si, L. Xiao, C. E. Brown, D. Wang. Preclinical evaluation of car t cell function: in vitro and in vivo models. Int J Mol Sci. Vol. 23, pg. 3154, 2022, https://doi.org/10.3390/ijms23063154. [↩]
- J. Gehl. Electroporation: theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol Scand. Vol. 177, pg. 437-447, 2003, https://doi.org/10.1046/j.1365-201X.2003.01093.x. [↩]
- G. Escobar, T. R. Berger, M. V. Maus. Car-t cells in solid tumors: challenges and breakthroughs. Cell Rep Med. Vol. 6, 2025, https://doi.org/10.1016/j.xcrm.2025.102353. [↩]
- B. Hou, Y. Tang, W. Li, Q. Zeng, D. Chang. Efficiency of car-t therapy for treatment of solid tumor in clinical trials: a meta-analysis. Dis Markers. Vol. 2019, pg. 3425291, 2019, https://doi.org/10.1155/2019/3425291. [↩]
- Innovative Genomics Institute. Crispr clinical trials: a 2022 update, https://innovativegenomics.org/news/crispr-clinical-trials-2022/, 2022. [↩]
- S. J. Diaz-Cano. Tumor heterogeneity: mechanisms and bases for a reliable application of molecular marker design. Int J Mol Sci. Vol. 13, pg. 1951-2011, 2012, https://doi.org/10.3390/ijms13021951. [↩]
- L. A. Kankeu Fonkoua, O. Sirpilla, R. Sakemura, E. L. Siegler, S. S. Kenderian. Car t cell therapy and the tumor microenvironment: current challenges and opportunities. Mol Ther Oncolytics. Vol. 25, pg. 69-77, 2022, https://doi.org/10.1016/j.omto.2022.03.009. [↩]
and Physical and Mental State for Healthy Youth")


{kind=link}