
Arjun S. Ulag1, Azhar Khandekar2
1 Lakeside School, Seattle, WA, USA
2 Fred Hutchinson Cancer Center, Seattle, WA, USA
Abstract
Tumor cells often contain small, round circular pieces of DNA floating outside their normal chromosomes, called extrachromosomal DNA (ecDNA). They influence how the cancer cells grow rapidly, evolve and resist treatment. ecDNA marks aggressive tumors and is the target for an emerging class of therapies, which makes the patient’s ecDNA status clinically actionable. However, identifying ecDNA today requires whole-genome sequencing (WGS), which is expensive, time consuming, and hence impractical for broad clinical use. We asked whether ecDNA could instead be detected from whole-exome sequencing (WES). This is the protein-coding region and roughly 1-2% of the human genome. The biggest advantage of WES is that it’s inexpensive and already routine in clinical labs. We hypothesized that features derived from mutational signals contained in WES are adequate to accurately determine ecDNA status. To test this, we used publicly available pan-cancer datasets, built machine learning models on a set of 227 WES-observable genomic features, and tested their ability to detect ecDNA status. Models were trained on WGS-labeled samples and tested using matched samples with WES data and known ecDNA status. The XGBoost model performed best with over 92% accuracy on the WES test set (n=56) and supporting our hypothesis. A WES-based ecDNA detection method could cost much less and put ecDNA detection within reach of everyday clinical practice for targeted therapies. However, the limited sample size and reliance on computationally inferred ecDNA labels make our study a proof-of-concept. Further work is required with larger and independent datasets to validate and refine it for clinical applicability.
Keywords: extrachromosomal DNA; whole-exome sequencing; machine learning; cancer genomics; bioinformatics
Introduction
Cancer is a disease caused by uncontrolled cell growth and this typically happens when genetic and epigenetic changes build up in the body over a long period1,2. At the genetic level, cancer is driven primarily by changes in two specific types of genes: oncogenes and tumor suppressor genes3. Oncogenes are responsible for signaling cells to grow, but when they become amplified, as in create too many copies of themselves, they drive rapid cell growth and proliferation beyond normal limits.
Oncogenes demonstrate a variety of alterations that result in their constitutional activation, such as point mutation, rearrangement, or amplification at the genomic level. Extrachromosomal DNA (ecDNA) is one specific mechanism very closely tied to gene amplification4,5. Unlike chromosomal DNA, ecDNA refers to circular DNA independent of any chromosome within the cell nucleus4. ecDNA-related research has received a lot of attention lately because of its vital role in driving tumor aggression and treatment resistance6.
ecDNA enables the cancer genes it carries to rapidly make copies of themselves, a process known as gene amplification. Gene copying is comparatively constrained in a normal linear chromosome that follows Mendelian inheritance. ecDNA doesn’t follow traditional rules of genetics and this allows the oncogenes it carries to reach high copy numbers and uncontrolled growth7 ,8,9,10. Its circular shape compounds the problem. The ecDNA replicates itself on its own and enables the oncogenes it carries to multiply at dangerously high levels that a normal chromosome could never do, all due to its circular shape lacking the normal regulatory constraints that keep a linear chromosome in check9,11.
ecDNA is rare in normal cells but typical in cancer cells4,7. If therapies targeted this unusually clean therapeutic target, it could not only shut down key cancer pathways, but do so without touching the normal, healthy tissue12,13. Due to this potential, several clinical trials are already underway testing this treatment strategy in selected cancer types13.
Although the precise mechanisms of ecDNA formation are varied and are largely unknown, several mutational processes and broader patterns of genome instability have been associated with ecDNA in specific cancer types14,15,16,17,18,19,20, including single-base substitution signatures such as APOBEC and copy number signatures CN21,22,23. These signatures consist of features including, but not limited to, i) mutations and their trinucleotide context ii) the size and microhomology length of insertions and deletions and iii) the size, heterozygosity state and total copy number of copy number alterations21,24.
Currently, various techniques are available for ecDNA detection, all differing in their sensitivity, costs, throughput, and detection reliability. Fluorescence in situ hybridization (FISH) is the gold standard method for ecDNA detection, but this method is low-throughput, expensive, and requires prior knowledge of which oncogenes are amplified on ecDNA. Recently, computational tools to detect ecDNA from whole-genome sequencing data have also been developed, which facilitate the unbiased detection of ecDNA25. Detecting ecDNA through assays other than whole genome sequencing (WGS), such as whole-exome sequencing (WES), is challenging because only WGS provides the ability to detect SVs and infer circularity from sequencing reads26,25. This represents a significant barrier for developing assays that can be utilized in a clinical setting, since implementing WGS in the clinic is currently not feasible due to high associated costs27. While existing computational tools can detect ecDNA from WGS by leveraging structural variant information and circular DNA reconstruction, these approaches are not applicable to WES data, which lacks genome-wide coverage and structural resolution. Despite the fact WES is widely used in clinical settings for various purposes, there is currently no established way of telling ecDNA presence using it. That leaves a major gap in an accessible method for detecting a highly clinically actionable therapeutic target. Our study is designed to address that very gap – as in, investigate whether mutation-derived features observable in WES contain sufficient signal to predict ecDNA status using machine learning.
We hypothesized that accurate ecDNA detection is possible from WES. In other words, we are able to predict with accuracy whether a cancer sample harbors ecDNA by training a machine learning model on features constructed from mutational signals contained in WES. To test this hypothesis, we constructed a model using mutational features restricted to those observable in WES, while leveraging WGS-based datasets for feature annotation28 and model training. Our results suggest that ecDNA status can be detected from WES with high accuracy, providing a first big step towards a practical and lower-cost alternative for detecting ecDNA in every day clinical setting. These findings provide an initial proof of concept for exploring ecDNA detection using mutation-derived features from WES, a widely used clinical assay.
Methods
Data Sources and Preprocessing
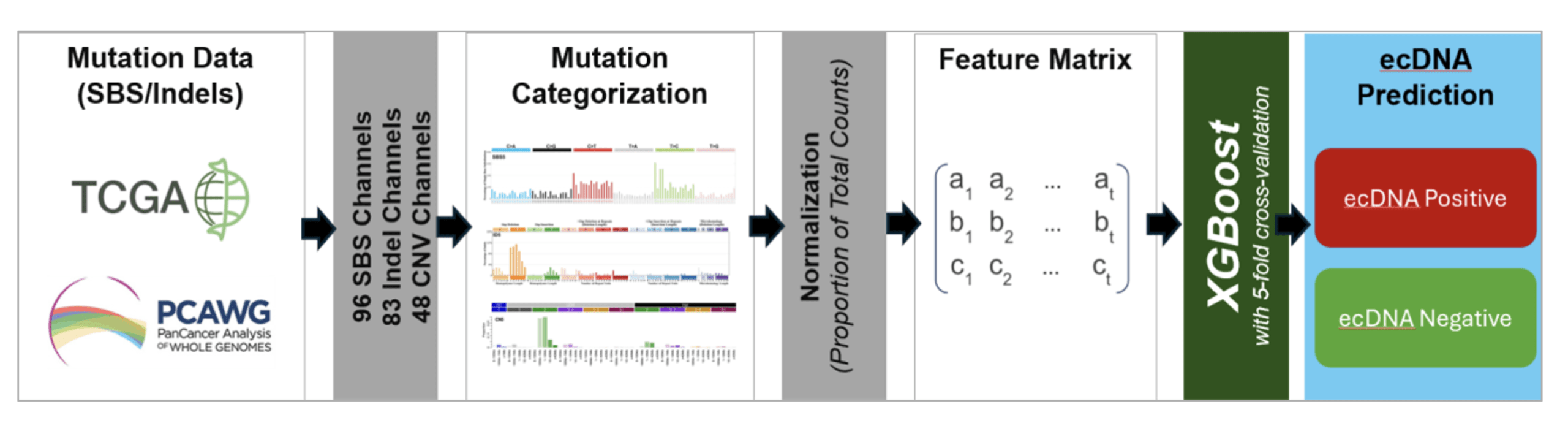
We obtained single-base substitution, insertion and deletion, and copy number mutation data from The Cancer Genome Atlas (TCGA) and the Pan-Cancer Analysis of Whole Genomes (PCAWG) pan-cancer databases29,30. All genomic data analyzed were publicly available and de-identified. Human subjects were not directly involved.
We categorized these mutations into 96 substitution, 83 indel, and 48 copy number mutational contexts according to the previously developed schema for classifying mutations for mutational signature analysis21. We converted the raw count matrices to proportions for each mutation type, and then all matrices were combined, resulting in a feature matrix consisting of a total of 227 features.
Copy number features were derived from WGS-based allele-specific copy number profiles generated using Allele-Specific Copy Number Analysis of Tumors (ASCAT)31 as part of the PCAWG consensus calls. These copy number profiles were intersected with exonic regions defined by Genome Annotation for the Encyclopedia of DNA Elements (GENCODE)32, and the resulting exonic-restricted segments were categorized using SigProfilerMatrixGenerator’s CN48 schema21,33. The same exonic restriction was applied to SBS96 and ID83 mutation features. Thus, all feature matrices were derived in the same way, as in from the WES-accessible genomic regions. ASCAT internally handles tumor purity and ploidy estimation34,31. This design isolates the question of whether ecDNA-discriminating signal is present within WES-accessible genomic territory, independent of additional noise introduced by native WES-based calling.
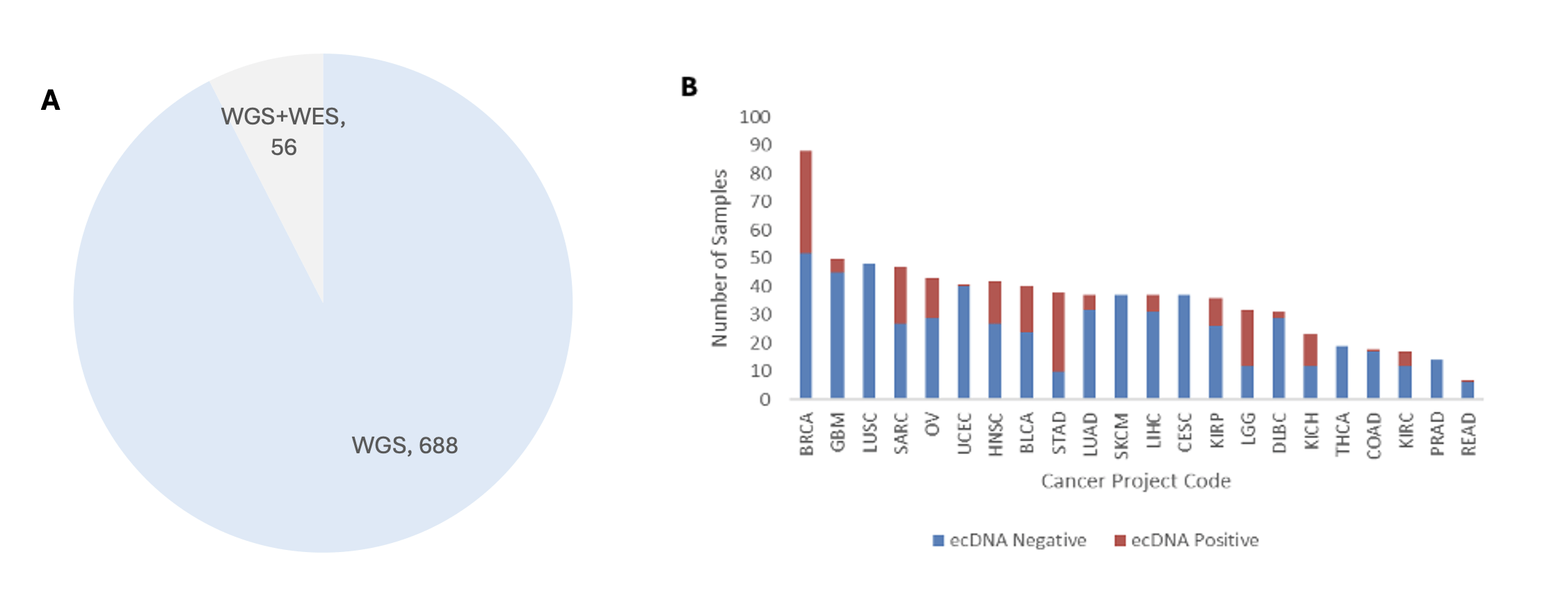
We derived the ecDNA status of each sample from previously published computational predictions from AmpliconArchitect software25. We used a total of 688 samples with only WGS data for training, and 56 samples with both whole-exome sequencing and whole-genome sequencing were used as the external test set. Although mutation features were derived from WGS-based datasets, the feature space was restricted to signals that are observable in WES data, enabling evaluation of model performance on WES samples.
Model Training
We selected XGBoost (Extreme Gradient Boosting) and a feed-forward neural network as the primary models. XGBoost is a gradient-boosting algorithm that combines decision trees to make accurate predictions. These models were selected due to their ability to handle complex patterns within the data, their resistance to noise within the data, and their resistance to overfitting when there is limited data.
For hyperparameter tuning, we tested a 3×3 grid across learning rate, n estimators, and maximum depth and optimized the grid based on our 10-fold cross-validation results from model selection. Because we are doing a binary classification task to identify ecDNA, we set the objective function to binary logistic loss. To keep the model from overfitting and ensure it generalizes well to new data, we also implemented early stopping. Since this project is an exploratory proof-of-concept, we used a smaller grid search to balance model optimization with reasonable training times.
We constructed a Multi-Layer Perceptron model to analyze the tabular data. MLP is an artificial neural network that uses overlapping dense layers to make predictions. We introduced non-linearity into the model using the ReLU (Rectified Linear Unit) activation function.
The network consisted of multiple hidden layers, with the first of the hidden layers containing 256 neurons and then decreased gradually to 16 neurons in the final layer to distill the information from the inputs. We applied L2 regularization across all layers with a penalty factor of 0.01 to avoid overfitting, and sigmoid activation for binary classification was used in the output layer. The model’s loss function was binary cross-entropy with Adam optimization to train it with a learning rate of 0.001.
We used 5-fold cross-validation for model training and performance assessments. Our goal was to make sure that the model didn’t overfit and worked well on new patient information. For each fold, we assessed the model using measures of accuracy, precision, recall, and F1-score. Both XGBoost and the neural network performance was improved by tweaking the hyperparameters.
Feature Engineering
We trained the model on frequencies of single-base substitutions (SBS), insertions/deletions (Indels), and copy number alterations (CNAs). We didn’t use raw counts and instead normalized the frequencies using proportions. The following formula was used to compute the proportions.
(1) 
where  is a mutation type (SBS, Indels, or CNAs).
is a mutation type (SBS, Indels, or CNAs).
These normalized proportions were then used as input data to the model. Raw mutation counts in a sample are not suitable by themselves for direct comparison across samples. They are influenced by many factors, including the sequencing depth and total mutation burden. And these factors have nothing to do with ecDNA. Feeding raw counts as input data to the model would make it key on the total number of mutations rather than the mutational patterns we are looking to learn from. We reduced this risk by converting raw mutation counts to their respective proportions. Now, we can credibly compare mutation patterns across datasets21,24. Further, only mutation signals measurable within WES data were used in feature construction, thereby ensuring compatibility with exome-based inputs. The computational workflow for feature construction and ecDNA prediction is shown in Figure 2.
Results
The dataset used in this study spans various cancer types, and these types are unevenly represented. Not just the sample count, but the ecDNA by cancer type is also varied. We keep this variation in mind during model interpretation and performance assessment. The pan-cancer dataset summary is available in Figure 1.
WES is a widely available clinical assay, however ecDNA detection currently depends on the expensive WGS. Therefore, we tested whether features derived from mutational signals available in WES can accurately predict ecDNA status. We trained the models on WGS-labeled samples and then tested them on an independent WES dataset. Our aim was to take a first big step towards making ecDNA detection widely accessible.
Model Performance and Accuracy
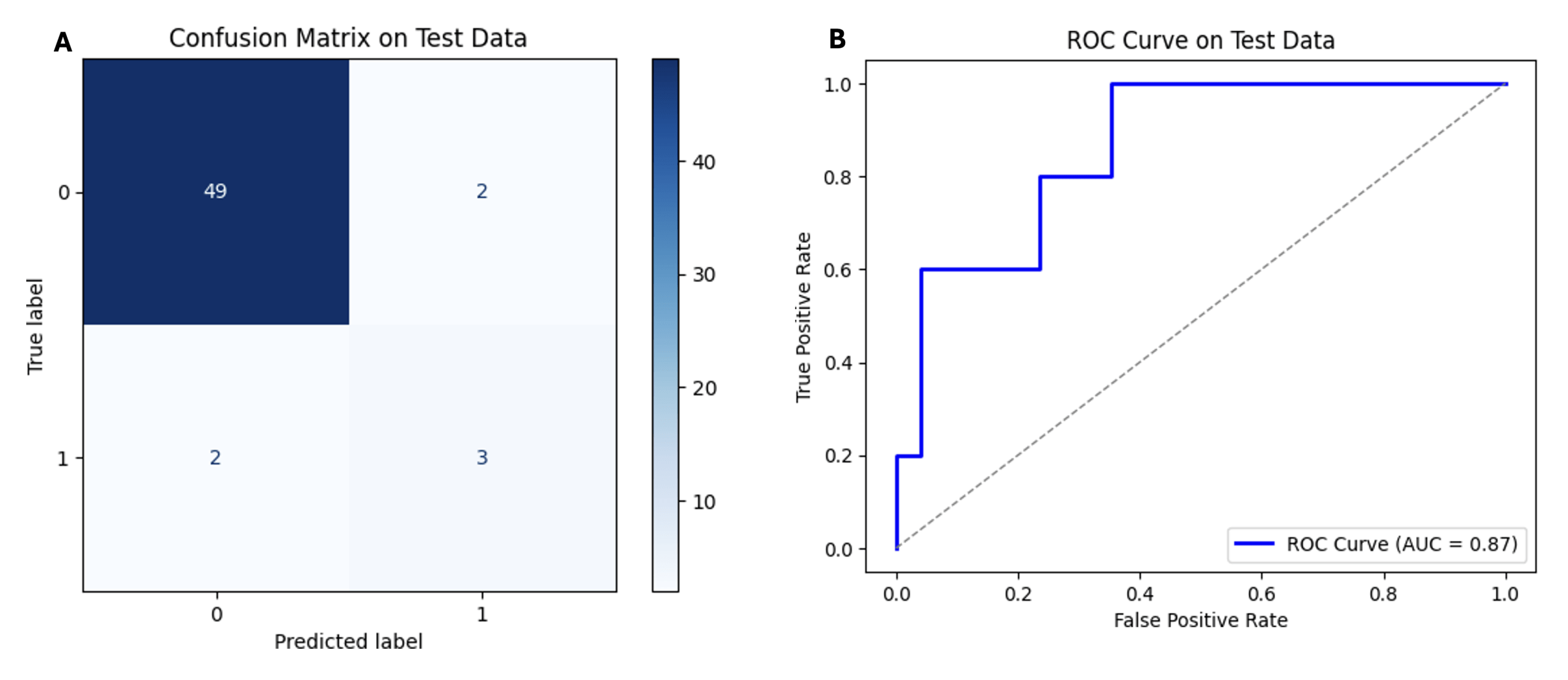
On the WES test set (n = 56), the XGBoost model achieved 92.86% accuracy and an area under the receiver operating characteristic curve (AUROC) of 0.87. Its confusion matrix shows 49 true negatives and 3 true positives, against 2 false positives and 2 false negatives. These measures show overall strong performance in ecDNA identification. Figure 3 shows the confusion matrix and ROC curve for the XGBoost model.
Comparatively, the MLP model achieved an accuracy of 87.50% on the WES test set. Its confusion matrix shows 49 true negatives and zero true positives, against 2 false positives and 5 false negatives. As the data shows, it failed to identify ecDNA-positive samples.
| Model | Accuracy | True Positives | True Negatives | False Positives | False Negatives |
| XGBoost | 92.86% | 3 | 49 | 2 | 2 |
| MLP | 87.50% | 0 | 49 | 2 | 5 |
Data summarized in the above table shows that both models perform well on ecDNA-negative samples, however XGBoost has better sensitivity in detecting ecDNA-positive cases.
There was a small number of ecDNA-positive samples (n = 5) in the test set. This test dataset imbalance limits the stability of the sensitivity estimates and so, further evaluation is required on larger and more balanced datasets.
Both models had errors with non-zero false positives and false negatives. Further model refinement can help reduce/eliminate these errors and achieve higher accuracy in ecDNA prediction using WES data.
Summarizing, the results show that WES data contain predictive signal associated with ecDNA. This is supported by XGBoost performance in ecDNA detection from analyzing features constructed based on mutation signals contained in WES data.
Discussion
Our study evaluated whether mutation signals contained in whole-exome sequencing (WES) could support accurate prediction of ecDNA status.
Current computational methods from whole-genome sequencing data identify both if a sample has ecDNA and the precise locations of the ecDNA25. However, in a clinical setting, just determining whether a patient has ecDNA or not can be valuable, given that current therapies in development target all ecDNA, irrespective of what oncogenes are on that ecDNA35. These findings suggest that ecDNA status may be inferred from WES-derived features, although further validation is required before considering clinical applications. Here, we show that ecDNA status can be predicted from WES-derived features, albeit from a very limited sample size. Expansion of the current feature set to include features beyond mutational contexts, such as quantifying the number of large shifts in copy number in adjacent segments, will improve the accuracy of detection.
There were several limitations that may have affected our results. Firstly, the limited WES test dataset affected how well we could evaluate the model across each individual cancer type. Given the ecDNA prevalence was different across each individual cancer type, the model could have partially keyed-in on cancer-type-specific mutation patterns rather than ecDNA-associated signals themselves. Further, the training and test data were varied in both size and modality (WGS for training, WES for testing), thereby further impacting the model’s generalizability. While we restricted the feature set to signals observable in WES, the differences in their respective data sources and sequencing contexts can still pull feature distributions apart between the two sets. Given these limitations, establishing robust performance across settings requires deeper analyses with larger, more diverse WES cohorts stratified by cancer type.
Secondly, we used ecDNA labels from WGS-based computational tools. The model is therefore learning to approximate WGS-based inference from mutation-derived features rather than to detect ecDNA independently. This creates circular noise, meaning if the WGS-based computational tools make a mistake, our model will just repeat it. To prove our model really works, we need to test it against real-world lab experiments, such as fluorescence in situ hybridization (FISH), using independent WES cohorts.
Thirdly, we derived copy-number data from WGS-based ASCAT profiles restricted to exonic regions. We didn’t get this data from standard WES. We don’t know yet if the model would perform just as well if we used real-world exome software (like CNVkit or FACETS), which is a major next step for this research. Finally, we left out some genomic features that may be highly relevant for detecting ecDNA, such structural-variations. Adding these features in the future could make the model even more accurate.
These limitations notwithstanding, these results matter clinically. ecDNA is linked to aggressive tumors and is the target of a new class of treatment strategies, and its prediction is clinically actionable. Being able to predict a patient’s ecDNA status using WES, a widely available assay, could make screening more accessible in clinical settings12. However, our results only show correlations between mutation patterns and ecDNA status. Further analyses on larger and more diverse datasets is needed to establish causation.
There are few key next steps to build on this work. Firstly, and most importantly, we need a much larger dataset for model training and testing. Testing the model on much larger datasets and stratifying that data by cancer type would help us determine how well it performs across each individual cancer type. Secondly, it’s important to look inside the model through feature importance ranking or SHAP-based methods. This would help understand which mutation-derived features contribute most strongly to ecDNA detection, thereby elevating a predictive tool into a source of deep biological insight and moving from correlation to causation. Also expanding the genomic features, such as inclusion of structural variation signals, may help improve the model’s accuracy. We should also consider combining WES mutation data with other types of biological data to further improve the model’s accuracy and identify additional patterns are most closely associated with ecDNA.
Normalization to proportions improved comparability across samples, but it totally discarded absolute mutation burden, which may be biologically relevant. Taking both normalized and raw mutation counts into account may improve accuracy and interpretability of future models. Additionally, our hyperparameter search was deliberately narrow; expanding this using randomized or more extensive search strategies may help improve model performance.
Benchmarking against simpler baseline approaches, such as logistic regression or random classifiers, would show what the added model complexity actually gets for us. Finally, evaluating this approach on newly sequenced clinical samples will be important for understanding how it could be used in real-world settings.
In conclusion, our study provides an early demonstration that ecDNA status can be predicted from mutational signals observable in WES. However, given the limited sample size and reliance on computationally inferred labels, further validation on larger and independent datasets will be necessary to assess robustness and generalizability. With continued refinement, this approach may support future efforts to develop more accessible strategies for ecDNA characterization and improve understanding of ecDNA biology in cancer.
Acknowledgements
We thank The Cancer Genome Atlas (TCGA) Research Network and the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium for generating and making publicly available the genomic datasets used in this study.
Figures and Figure Titles/Captions
References
- E. N. Kontomanolis, A. Koutras, A. Syllaios, D. Schizas, A. Mastoraki, N. Garmpis, M. Diakosavvas, K. Angelou, G. Tsatsaris, A. Pagkalos, T. Ntounis, Z. Fasoulakis. Role of oncogenes and tumor-suppressor genes in carcinogenesis: a review. Anticancer Research. Vol. 40, pg. 6009–6015, 2020, https://doi.org/10.21873/anticanres.14622. [↩]
- R. L. Siegel, T. B. Kratzer, A. N. Giaquinto, H. Sung, A. Jemal. Cancer statistics, 2025. CA: A Cancer Journal for Clinicians. Vol. 75, pg. 10–45, 2025, https://doi.org/10.3322/caac.21871. [↩]
- E. N. Kontomanolis, A. Koutras, A. Syllaios, D. Schizas, A. Mastoraki, N. Garmpis, M. Diakosavvas, K. Angelou, G. Tsatsaris, A. Pagkalos, T. Ntounis, Z. Fasoulakis. Role of oncogenes and tumor-suppressor genes in carcinogenesis: a review. Anticancer Research. Vol. 40, pg. 6009–6015, 2020, https://doi.org/10.21873/anticanres.14622. [↩]
- Y. Dong, Q. He, X. Chen, F. Yang, L. He, Y. Zheng. Extrachromosomal DNA (ecDNA) in cancer: mechanisms, functions, and clinical implications. Frontiers in Oncology. Vol. 13, pg. 1194405, 2023, https://doi.org/10.3389/fonc.2023.1194405. [↩] [↩] [↩]
- R. G. W. Verhaak, V. Bafna, P. S. Mischel. Extrachromosomal oncogene amplification in tumor pathogenesis and evolution. Nature Reviews Cancer. Vol. 19, pg. 283–288, 2019, https://doi.org/10.1038/s41568-019-0128-6. [↩]
- M. Haughey, I. Noorani, C. Swanton, P. S. Mischel, B. Werner. Extrachromosomal DNA: shaping the evolutionary dynamics of cancer. Trends in Cancer. Vol. 11, pg. 901–916, 2025, https://doi.org/10.1016/j.trecan.2025.06.004. [↩]
- R. G. W. Verhaak, V. Bafna, P. S. Mischel. Extrachromosomal oncogene amplification in tumor pathogenesis and evolution. Nature Reviews Cancer. Vol. 19, pg. 283–288, 2019, https://doi.org/10.1038/s41568-019-0128-6. [↩] [↩]
- K. M. Turner, V. Deshpande, D. Beyter, T. Koga, J. Rusert, C. Lee, B. Li, K. Arden, B. Ren, D. A. Nathanson, H. I. Kornblum, M. D. Taylor, S. Kaushal, W. K. Cavenee, R. Wechsler-Reya, F. B. Furnari, S. R. Vandenberg, P. N. Rao, G. M. Wahl, V. Bafna, P. S. Mischel. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. Vol. 543, pg. 122–125, 2017, https://doi.org/10.1038/nature21356. [↩]
- H. Kim, N.-P. Nguyen, K. M. Turner, S. Wu, A. D. Gujar, J. Luebeck, J. Liu, V. Deshpande, U. Rajkumar, S. Namburi, S. B. Amin, E. Yi, F. Menghi, J. H. Schulte, A. G. Henssen, H. Y. Chang, C. R. Beck, P. S. Mischel, V. Bafna, R. G. W. Verhaak. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nature Genetics. Vol. 52, pg. 891–897, 2020, https://doi.org/10.1038/s41588-020-0678-2. [↩] [↩]
- K. Purshouse, J. Bartek, R. J. Gilbertson. Oncogene expression from extrachromosomal DNA is driven by copy number amplification and does not require spatial clustering in glioblastoma stem cells. eLife. Vol. 11, pg. e80207, 2022, https://doi.org/10.7554/eLife.80207. [↩]
- S. Wu, K. M. Turner, N.-P. D. Nguyen, R. Raviram, M. Erb, J. Santini, J. Luebeck, U. Rajkumar, J. Diao, B. Li, W. Zhang, C. James, R. G. W. Verhaak, V. Bafna, B. Ren, P. S. Mischel. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. Vol. 575, pg. 699–703, 2019, https://doi.org/10.1038/s41586-019-1763-5. [↩]
- H. Kim, N.-P. Nguyen, K. M. Turner, S. Wu, A. D. Gujar, J. Luebeck, J. Liu, V. Deshpande, U. Rajkumar, S. Namburi, S. B. Amin, E. Yi, F. Menghi, J. H. Schulte, A. G. Henssen, H. Y. Chang, C. R. Beck, P. S. Mischel, V. Bafna, R. G. W. Verhaak. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nature Genetics. Vol. 52, pg. 891–897, 2020, https://doi.org/10.1038/s41588-020-0678-2. [↩] [↩]
- J. Tang, N. E. Weiser, G. Wang, S. Chowdhry, E. J. Curtis, Y. Zhao, I. T.-L. Wong, G. K. Marinov, R. Li, P. Hanoian, E. Tse, S. G. Mojica, R. Hansen, J. Plum, A. Steffy, S. Milutinovic, S. T. Meyer, J. Luebeck, Y. Wang, S. Zhang, N. Altemose, C. Curtis, W. J. Greenleaf, V. Bafna, S. J. Benkovic, A. B. Pinkerton, S. Kasibhatla, C. A. Hassig, P. S. Mischel, H. Y. Chang. Enhancing transcription–replication conflict targets ecDNA-positive cancers. Nature. Vol. 635, pg. 210–218, 2024, https://doi.org/10.1038/s41586-024-07802-5. [↩] [↩]
- L. B. Alexandrov, J. Kim, N. J. Haradhvala, M. N. Huang, A. W. T. Ng, Y. Wu, A. Boot, K. R. Covington, D. A. Gordenin, E. N. Bergstrom, S. M. A. Islam, N. Lopez-Bigas, L. J. Klimczak, J. R. McPherson, S. Morganella, R. Sabarinathan, D. A. Wheeler, V. Mustonen, PCAWG Mutational Signatures Working Group, PCAWG Consortium, G. Getz, S. G. Rozen, M. R. Stratton. The repertoire of mutational signatures in human cancer. Nature. Vol. 578, pg. 94–101, 2020, https://doi.org/10.1038/s41586-020-1943-3. [↩]
- PCAWG Evolution and Heterogeneity Working Group, PCAWG Consortium. The evolutionary history of 2,658 cancers. Nature. Vol. 578, pg. 122–128, 2020, https://doi.org/10.1038/s41586-019-1907-7. [↩]
- C. Swanton, N. McGranahan, J. Starrett, C. C. Harris. Cancer evolution and heterogeneity. Nature. Vol. 581, pg. 66–77, 2020, https://doi.org/10.1038/s41586-020-2292-6. [↩]
- S. Nik-Zainal, L. B. Alexandrov, D. C. Wedge, P. Van Loo, C. D. Greenman, K. Raine, D. Jones, J. Hinton, J. Marshall, L. A. Stebbings, A. Menzies, S. Martin, K. Leung, L. Chen, C. Leroy, M. Ramakrishna, R. Rance, K. W. Lau, L. J. Mudie, I. Varela, D. J. McBride, G. R. Bignell, S. L. Cooke, A. Shlien, J. Gamble, I. Whitmore, M. Maddison, P. S. Tarpey, H. R. Davies, E. Papaemmanuil, P. J. Stephens, S. McLaren, A. P. Butler, J. W. Teague, G. Jönsson, J. E. Garber, D. Silver, P. Miron, A. Fatima, S. Boyault, A. Langerød, A. Tutt, J. W. M. Martens, S. A. J. R. Aparicio, Å. Borg, A. V. Salomon, G. Thomas, A.-L. Børresen-Dale, A. L. Richardson, M. S. Neuberger, P. A. Futreal, P. J. Campbell, M. R. Stratton. Mutational processes molding the genomes of 21 breast cancers. Cell. Vol. 149, pg. 979–993, 2012, https://doi.org/10.1016/j.cell.2012.04.024. [↩]
- P. J. Stephens, C. D. Greenman, B. Fu, F. Yang, G. R. Bignell, L. J. Mudie, E. D. Pleasance, K. W. Lau, D. Beare, L. A. Stebbings, S. McLaren, M.-L. Lin, D. J. McBride, I. Varela, S. Nik-Zainal, C. Leroy, M. Jia, A. Menzies, A. P. Butler, J. W. Teague, M. A. Quail, J. Burton, H. Swerdlow, N. P. Carter, L. A. Morsberger, C. Iacobuzio-Donahue, G. A. Follows, A. R. Green, A. M. Flanagan, M. R. Stratton, P. A. Futreal, P. J. Campbell. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. Vol. 144, pg. 27–40, 2011, https://doi.org/10.1016/j.cell.2010.11.055. [↩]
- S. C. Baca, D. Prandi, M. S. Lawrence, J. M. Mosquera, A. Romanel, Y. Drier, K. Park, N. Kitabayashi, T. Y. MacDonald, M. Ghandi, E. Van Allen, A. Kryukov, A. Sboner, J. P. Theurillat, H. Beltran, R. Beroukhim, T. R. Golub, M. A. Rubin, L. A. Garraway, M. Meyerson, G. Getz, O. Elemento, F. Demichelis. Punctuated evolution of prostate cancer genomes. Cell. Vol. 153, pg. 666–677, 2013, https://doi.org/10.1016/j.cell.2013.03.021. [↩]
- C. Bailey, O. Pich, K. Thol, T. B. K. Watkins, J. Luebeck, A. Rowan, G. Stavrou, N. E. Weiser, B. Dameracharla, R. Bentham, W.-T. Lu, J. Kittel, S. Y. C. Yang, B. E. Howitt, N. Sharma, M. Litovchenko, R. Salgado, K. L. Hung, A. J. Cornish, D. A. Moore, R. S. Houlston, V. Bafna, H. Y. Chang, S. Nik-Zainal, N. Kanu, N. McGranahan, Genomics England Research Consortium, A. M. Flanagan, P. S. Mischel, M. Jamal-Hanjani, C. Swanton. Origins and impact of extrachromosomal DNA. Nature. Vol. 635, pg. 193–209, 2024, https://doi.org/10.1038/s41586-024-08107-3. [↩]
- L. B. Alexandrov, J. Kim, N. J. Haradhvala, M. N. Huang, A. W. T. Ng, Y. Wu, A. Boot, K. R. Covington, D. A. Gordenin, E. N. Bergstrom, S. M. A. Islam, N. Lopez-Bigas, L. J. Klimczak, J. R. McPherson, S. Morganella, R. Sabarinathan, D. A. Wheeler, V. Mustonen, PCAWG Mutational Signatures Working Group, PCAWG Consortium, G. Getz, S. G. Rozen, M. R. Stratton. The repertoire of mutational signatures in human cancer. Nature. Vol. 578, pg. 94–101, 2020, https://doi.org/10.1038/s41586-020-1943-3. [↩] [↩] [↩] [↩] [↩]
- T. I. Zack, S. E. Schumacher, S. L. Carter, A. D. Cherniack, G. Saksena, B. Tabak, M. S. Lawrence, C.-Z. Zhang, J. Wala, C. H. Mermel, C. Sougnez, S. B. Gabriel, B. Hernandez, H. Shen, P. W. Laird, G. Getz, M. Meyerson, R. Beroukhim. Pan-cancer patterns of somatic copy number alteration. Nature Genetics. Vol. 45, pg. 1134–1140, 2013, https://doi.org/10.1038/ng.2760. [↩]
- C. H. Mermel, S. E. Schumacher, B. Hill, M. L. Meyerson, R. Beroukhim, G. Getz. GISTIC2.0 facilitates sensitive and confident localization of focal somatic copy-number alteration targets in human cancers. Genome Biology. Vol. 12, pg. R41, 2011, https://doi.org/10.1186/gb-2011-12-4-r41. [↩]
- T. I. Zack, S. E. Schumacher, S. L. Carter, A. D. Cherniack, G. Saksena, B. Tabak, M. S. Lawrence, C.-Z. Zhang, J. Wala, C. H. Mermel, C. Sougnez, S. B. Gabriel, B. Hernandez, H. Shen, P. W. Laird, G. Getz, M. Meyerson, R. Beroukhim. Pan-cancer patterns of somatic copy number alteration. Nature Genetics. Vol. 45, pg. 1134–1140, 2013, https://doi.org/10.1038/ng.2760. [↩] [↩]
- V. Deshpande, J. Luebeck, N.-P. D. Nguyen, M. Bakhtiari, K. M. Turner, R. Schwab, H. Carter, P. S. Mischel, V. Bafna. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nature Communications. Vol. 10, pg. 392, 2019, https://doi.org/10.1038/s41467-018-08200-y. [↩] [↩] [↩] [↩]
- H. Kim, S. Kim, T. Wade, E. Yeo, A. Lipsa, A. Golebiewska, K. C. Johnson, S. An, J. Ko, Y. Nam, H. Y. Lee, S. Kang, H. Chung, S. P. Niclou, H. E. Moon, S. H. Paek, V. Bafna, J. Luebeck, R. G. W. Verhaak. Mapping extrachromosomal DNA amplifications during cancer progression. Nature Genetics. Vol. 56, pg. 2447–2454, 2024, https://doi.org/10.1038/s41588-024-01949-7. [↩]
- M. Réda, C. Richard, A. Bertaut, J. Niogret, T. Collot, J.-D. Fumet, J. Blanc, C. Truntzer, I. Desmoulins, S. Ladoire, A. Hennequin, L. Favier, L. Bengrine, J. Vincent, A. Hervieu, J.-G. Dusserre, C. Lepage, P. Foucher, C. Borg, B. Chauffert, F. Ghiringhelli. Implementation and use of whole-exome sequencing for metastatic solid cancer. EBioMedicine. Vol. 51, pg. 102624, 2020, https://doi.org/10.1016/j.ebiom.2019.102624. [↩]
- H. Carter, K. Chen, M. Isik, S. Tyekucheva, V. E. Velculescu, K. W. Kinzler, B. Vogelstein, R. Karchin. Cancer-specific high-throughput annotation of somatic mutations. Cancer Research. Vol. 69, pg. 6660–6667, 2009, https://doi.org/10.1158/0008-5472.CAN-09-1133. [↩]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. Vol. 578, pg. 82–93, 2020, https://doi.org/10.1038/s41586-020-1969-6. [↩]
- Cancer Genome Atlas Research Network, J. N. Weinstein, E. A. Collisson, G. B. Mills, K. R. Mills Shaw, B. A. Ozenberger, K. Ellrott, I. Shmulevich, C. Sander, J. M. Stuart. The Cancer Genome Atlas Pan-Cancer analysis project. Nature Genetics. Vol. 45, pg. 1113–1120, 2013, https://doi.org/10.1038/ng.2764. [↩]
- P. Van Loo, S. H. Nordgard, O. C. Lingjærde, H. G. Russnes, I. H. Rye, W. Sun, V. J. Weigman, P. Marynen, A. Zetterberg, B. Naume, C. M. Perou, A.-L. Børresen-Dale, V. N. Kristensen. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences. Vol. 107, pg. 16910–16915, 2010, https://doi.org/10.1073/pnas.1009843107. [↩] [↩]
- A. Frankish, M. Diekhans, A.-M. Ferreira, R. Johnson, I. Jungreis, J. Loveland, J. M. Mudge, C. Sisu, J. Wright, J. Armstrong, I. Barnes, A. Berry, A. Bignell, C. Carbonell Sala, J. Chrast, F. Cunningham, T. Di Domenico, S. Donaldson, I. T. Fiddes, C. García Girón, J. M. Gonzalez, T. Grego, M. Hardy, T. Hourlier, T. Hunt, O. G. Izuogu, J. Lagarde, F. J. Martin, L. Martínez, S. Mohanan, P. Muir, F. C. P. Navarro, A. Parker, B. Pei, F. Pozo, M. Ruffier, B. M. Schmitt, E. Stapleton, M. M. Suner, I. Sycheva, B. Uszczynska-Ratajczak, J. Xu, A. Yates, D. Zerbino, Y. Zhang, J. Aken, J. S. Choudhary, M. Gerstein, R. Guigó, T. J. P. Hubbard, M. Kellis, B. Paten, M. L. Tress, P. Flicek. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Research. Vol. 47, pg. D766–D773, 2019, https://doi.org/10.1093/nar/gky955. [↩]
- S. M. A. Islam, R. Díaz-Gay, A. Wu, M. M. Barnes, J. V. Vangara, E. N. Bergstrom, Y. He, A. Vella, Y. Wang, J. Teague, L. B. Alexandrov. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genomics. Vol. 2, pg. 100179, 2022, https://doi.org/10.1016/j.xgen.2022.100179. [↩]
- S. L. Carter, A. Cibulskis, E. Helman, A. McKenna, H. Shen, T. Zack, P. W. Laird, R. C. Onofrio, W. Winckler, B. A. Weir, R. Beroukhim, D. Pellman, D. A. Levine, E. S. Lander, M. Meyerson, G. Getz. Absolute quantification of somatic DNA alterations in human cancer. Nature Biotechnology. Vol. 30, pg. 413–421, 2012, https://doi.org/10.1038/nbt.2203. [↩]
- J. Tang, N. E. Weiser, G. Wang, S. Chowdhry, E. J. Curtis, Y. Zhao, I. T.-L. Wong, G. K. Marinov, R. Li, P. Hanoian, E. Tse, S. G. Mojica, R. Hansen, J. Plum, A. Steffy, S. Milutinovic, S. T. Meyer, J. Luebeck, Y. Wang, S. Zhang, N. Altemose, C. Curtis, W. J. Greenleaf, V. Bafna, S. J. Benkovic, A. B. Pinkerton, S. Kasibhatla, C. A. Hassig, P. S. Mischel, H. Y. Chang. Enhancing transcription–replication conflict targets ecDNA-positive cancers. Nature. Vol. 635, pg. 210–218, 2024, https://doi.org/10.1038/s41586-024-07802-5. [↩]


and Physical and Mental State for Healthy Youth")
{kind=link}