
Xiang Ma1, Te Liu2
1 Shanghai Starriver Bilingual School
2 Shanghai Geriatric Institute of Chinese Medicine, Shanghai University of Traditional Chinese Medicine.
Abstract
Alzheimer’s Disease (AD) is a neurodegenerative disorder associated with ageing, cognitive decline, and behavioral impairment. However, the specific relationship between Klotho and AD is not well understood. Klotho is a gene mainly expressed in the kidneys and in the choroid plexus, associated with the suppression of senescence. The human KL gene encodes the α-Klotho protein, which is a multi-functional protein modulating phosphate salts, calcium, and vitamin D metabolism. We hypothesized that Klotho deficiency is related to early aging and cognitive impairment in AD transgenic mice. We established an early-onset triple-transgenic mouse model (APPKI/PS1KI/Klotho-/+) using 12 male mice in total, including wild-type (WT), APPKI/PS1KI, and APPKI/PS1KI/Klotho-/+ groups (n=4 per group). Behavioral assessments, including nest-building and novel object recognition (NOR) tests, suggested earlier cognitive and behavioral impairment in APPKI/PS1KI/Klotho-/+ mice than control groups. We also generated APPKI/PS1KI/Klotho-/+ murine brain organoids (mBOs). About 80 brain organoids are produced from one individual. Pathological staining analyses, including hematoxylin and eosin staining, Nissl staining, and senescence-associated β-galactosidase staining, demonstrated that APPKI/PS1KI/Klotho-/+ mBOs show pathological features of AD, including cell death, axonal atrophy, neuronal degeneration, and increased expression of senescence-associated β-galactosidase. In addition, 4D label-free quantitative proteomics analysis revealed differential expression of over 400 proteins between APPKI/PS1KI/Klotho-/+, APPKI/PS1KI, and WT mBOs. Bioinformatics predictive analysis indicated that most of these proteins were associated with the “Peroxisome” and “Nucleocytoplasmic transport” pathways. These findings suggest that Klotho deficiency is associated with rapid progression of cognitive impairment and early senescence in brain organoids derived from AD transgenic mice.
Keywords: Klotho, Alzheimer’s, organoids, brain, proteomics
Introduction
Alzheimer’s Disease (AD) is a type of dementia closely related to age. It is characterized by progressive loss of cognitive function (thinking, memory, and reasoning) and behavioral abilities, ultimately leading to death1. Mood disorders and hallucinations are also commonly observed in AD patients1. AD mainly affects the central nervous system in its onset. However, as the disease progresses, essentially all organ systems in the human body, such as the digestive system (for instance, via dysphagia) and the immune system (via neuroinflammation and abnormal microglial activity), would be affected2,3.
Recent studies estimate that about 32 million people suffer from overt clinical AD worldwide4. As a neurodegenerative disorder, AD primarily affects the elderly population aged 65 and above5. Early-onset AD accounts for approximately 5-6% of all cases, and the incidence rate is consistently reported to be higher in women than in men6,7. Projections indicate that “the number of US adults who will develop dementia each year was projected to increase from approximately 514,000 in 2020 to approximately 1 million in 2060,” and, since AD is the most common form of dementia, its incidence is expected to increase accordingly8. This trend underscores the urgent need for further research that aims to identify more effective ways to prevent and treat AD.
The pathogenesis of AD is highly complex and influenced by multiple factors. Some studies have pointed to genetic factors such as the amyloid precursor protein (APP) gene and the presenilin genes (PS-1, PS-2) on chromosomes 21, 14, and 1, respectively9. Two main mechanisms have been proposed to explain AD incidence: hyperphosphorylation of the tau protein and hyperaccumulation of beta-amyloid.
Tau is a major microtubule-associated protein (MAP) found mainly in the neuronal axons. Under normal physiological conditions, it is highly soluble, interacts with tubulin, and facilitates tubulin assembly into microtubules10. It has a role in resisting apoptosis: cells overexpressing tau are more resistant to external toxins such as H2O2, staurosporine, and camptothecin11. Phosphorylation of tau neutralizes the apoptosis-inducing effects of GSK-3β and stabilizes β-catenin, thereby ensuring cell survival even in environmental stress11. However, this anti-apoptotic effect, while neuroprotective under acute stress, may, in chronic contexts, allow damaged neurons to evade repair, leading to persistent dysfunction and neurodegeneration in the long term. Tau phosphorylation, a hallmark of AD, can lead to instability of tau-tubulin bonds, dissociation of tau from microtubules, and impaired microtubule polymerization12. In AD, pathological tau proteins are approximately three to four times more phosphorylated than transiently phosphorylated tau proteins in normal cells, may sequester normal tau into behaving in the same abnormal way as them, and self-assemble into toxic paired helical filaments (PHF) and straight filaments (SF), which in turn become neurofibrillary tangles that disrupt the integrity of the neuronal skeleton12. This, combined with the apoptosis-preventing effects of tau overphosphorylation, may result in the chronic death of neurons and eventual brain atrophy.
Βeta-amyloid proteins are derived from amyloid precursor proteins (APP), which have three types of processing secretase enzymes: ⍺, β, and ɣ13. Under normal circumstances, in an amyloidogenic pathway, APP is hydrolyzed by β secretase and then by ɣ secretase, releasing Aβ fragments, most of which (80-90%) have 40 amino acid residues in length (Aβ40) and less of which have 42 amino acid residues in length (Aβ 42)13,14. Aβ42 is more pathogenic and hydrophobic than Aβ4014. In the normal human brain, Aβ repairs the blood-brain barrier, supports brain recovery from injury, and modulates synaptic activity15. However, in AD, abnormal cleavage by secretases in the amyloidogenic pathway results in a high Aβ42/Aβ40 ratio14. Given Aβ42’s hydrophobic properties, its increased concentration may form neurotoxic extracellular plaques, leading to neuroinflammation, apoptosis, and eventual AD incidence14. Also, not only do Presenilins PS-1 and PS-2 promote aging, but they also serve as catalytic subunits of ɣ secretase, meaning that genetic mutations in them can lead to abnormalities in ɣ secretase cleavage of APP, which significantly heightens the Aβ42/40 ratio and promotes Aβ42 deposition16.
Nevertheless, we must not overstate the relationship between Aβ and AD. Research has pointed out that clinical trials with anti-amyloid drugs for AD treatment have repeatedly failed, and has proposed that faulty and reductive biological assumptions in the complex relationship between Aβ and AD could be one of the potential causes of this phenomenon17,18. Some studies even went to the extent of suggesting that amyloid is “neither necessary nor sufficient for AD-like brain atrophy”19. Collectively, these findings suggest that though Aβ seems to remain associated with AD, the specific causal relationship between them is under increasing scrutiny. Therefore, an investigation of other contributing factors, such as Klotho, is necessary for a more holistic understanding of AD pathology and incidence.
Given that AD is strongly associated with ageing, factors that regulate ageing-related processes may have important relevance to AD onset and progression. Klotho (KL) was initially identified as an anti-aging gene found on human chromosome 13. In humans, the Klotho protein has three forms: ⍺-Klotho, β-Klotho, and ɣ-Klotho20. Three forms of ⍺-Klotho products have been identified: secreted ⍺-Klotho, truncated soluble ⍺-Klotho, and full-length transmembrane ⍺-Klotho20. The Klotho protein is multifunctional, regulating metabolism (of phosphate, calcium, and vitamin D), reducing oxidative stress, and enhancing cognitive function20. It is predominantly found in the distal convoluted tubule of the kidney and the choroid plexus of the brain20.
There are mainly two mechanisms by which Klotho functions as an anti-aging gene. Studies report that it reduces phosphorylation of forkhead box proteins (FOXO), thereby allowing them to enter the nucleus20. The FOXO then activates a pathway that reduces reactive oxygen species (ROS) production and ROS-related oxidative stress20. Klotho is also an essential cofactor in FGF23 signaling pathways20. FGF23 regulates kidney phosphate excretion and systemic vitamin D activity, demonstrating that Klotho is also significant in these processes20.
In mice, knocking out the KL gene accelerates aging and shortens lifespan21. More specifically, Klotho-deficient (KL-/-) mice exhibit growth retardation, osteoporosis, ectopic calcification of soft tissues, premature tissue aging, and death at 9 weeks of age22. The skeletal muscle of these mice may also undergo severe atrophy22. In humans, the point mutation of the KL gene is associated with hypertension and kidney disease23. Conversely, significantly improved working and spatial memory, and better general cognition are observed after injecting KL protein at concentrations of 10 μg kg-1, with positive effects as early as 4 hours after administration24. Similarly, overexpression of KL products can prolong mouse lifespan21.
However, research has also complicated this relationship between aging and Klotho. A study on a population-based sample of human adults age 55-87 years found that the KL-VS haplotype of the KL gene, which increases ⍺-Klotho levels, does not improve cognition25. This inconsistency may be due to the limitations of observational designs on humans (rather than an experimental design on other model organisms), which may not fully capture the consequences of Klotho deficiency on AD pathology. Variations in traits among samples from a population may add to the possibilities of research producing divergent results for an observational study. The fact that the relationship between Klotho and aging is not fully understood highlights the necessity of controlled experimental studies focused on the mechanisms of Klotho, such as this study.
Because AD is closely linked to ageing, the known anti-ageing effects of Klotho suggest that it may be relevant to AD. However, its relationship to AD has not been thoroughly studied. While studies have noted associations between reduced Klotho levels and cognitive decline, they remain mostly correlational and mechanistically shallow. As summarized above, the current literature has mostly focused on Aβ accumulation, tau hyperphosphorylation, and genetic mutations in APP or presenilins in AD pathology. No study to date has systematically investigated the impact of Klotho gene knockout on AD incidence and progression using a combined transgenic model, and the proteomic consequences of Klotho deficiency have never been characterized using brain organoids. This gap is a critical blind spot in terms of how we understand the intersection between aging-related molecular dysfunction and AD pathology.
Therefore, to address this research gap, we hypothesized that Klotho deficiency is strongly related to early aging and cognitive impairment in AD transgenic mice. To test this hypothesis, we constructed a triple transgenic mouse model of AD combined with aging (APPKI/PS1KI/Klotho-/+) and prepared brain organoids (mBOs) to examine the effects of Klotho deficiency on AD pathology from both in vivo and in vitro perspectives. The findings of this study may help explore how Klotho is one of the factors of AD incidence and potentially inspire the development of new medications based on Klotho for AD.
Materials and methods:
Construction of Transgenic Mice
The APPswe/PS1∆E9 (APPKI/PS1KI, APP+/PS1+) heterozygous double-transgenic mice used in this study all express a human/mouse chimeric amyloid precursor protein and a human mutant presenilin-1. They were purchased from Shanghai Model Organisms Center, Inc. (Shanghai, China). The C57BL/6J-Klem1Adiuj/J transgenic mice with α-Klotho heterozygous deficiency (Klotho-/+, Kl-/+), the traits of which are previously described26, were also purchased from Shanghai Model Organisms Center, Inc. (Shanghai, China). APPKI/PS1KI mice were intercrossed with Klotho-/+ transgenic mice and placed in separate cages. We selected 4 offspring with the genotype APPKI/PS1KI/Klotho-/+ (APP+/PS1+/Kl–) for our study. 12 mice in total were used in this research (4 Wild-type (WT) C57BL/6 mice; 4 APPKI/PS1KI heterozygous double transgenic mice; 4 APPKI/PS1KI/Klotho-/+ heterozygous tri-transgenic mice). All mice were 6-month-old males weighing 25±3 g. The aforementioned mice were housed in an identical environment (22±3°C, 60% relative humidity, 12-hour light/ 12-hour dark cycle), and ad libitum food and water were provided. All procedures were performed in accordance with the Guide for the Care and Use of Medical Laboratory Animals and the guidelines of the Shanghai University of Traditional Chinese Medicine (Shanghai, China) Laboratory Animal Care and Use Committee27. Animal suffering and discomfort were minimized.
Murine brain organoids (mBOs) established
Using protocols from previous research and after collecting mouse brain tissue from all three genotypes (WT, APPKI/PS1KI, APPKI/PS1KI/Kl-/+), we then added 5 mL of ice-pre-cooled Advanced DMEM/F12 cell culture medium to the brain tissues and homogenized them28,29. The cell suspensions were passed through a 200-mesh cell sieve, and the filtered cell suspension was collected. The filtered cell suspension was centrifuged to obtain a cell pellet, which was resuspended in 0.5 mL of ice-pre-cooled full-cell culture medium. 0.5 mL of ice-pre-cooled Matrigel was then added. The pellet was triturated to obtain a uniform mixture. A controlled rate of about 0.1 mL per drop was used to add the mixture dropwise to an ultra-low attachment cell culture dish. Then, the suspension was incubated in a cell culture incubator at 37°C with 5% CO2 for 15 minutes. 4 mL of pre-warmed full-cell-culture medium was next added, and the suspension was incubated in a cell culture incubator at 37°C with 5% CO2 again. The culture was maintained for about 10 days. Formation of clonal spheres of cells was subsequently observed. The full cell culture medium included the following: Neurobasal (75 mL), FBS (15 mL), Penicillin-streptomycin (1 mL), L-glutamine (1 mL), B27 supplement (2 mL), N2 supplement (1 mL), Activin A 10 ng/mL, bFGF 10 ng/mL, EGF 10 ng/mL, VEGF 10 ng/mL + Ascorbic acid 50 ng/mL, recombinant insulin like growth factor (R3-IGF-1; 10 ng/mL), hydrocortisone (10 ng/mL), and heparin (10 ng/mL). About 80 mBOs are established for each individual mouse.
Western blotting
12% denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins from the whole brain tissue of the three genotypes. The proteins were then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking and washing, the membranes were incubated with primary antibodies at 37°C for 45 minutes. The membranes were washed thoroughly and then incubated with the appropriate secondary antibodies at 37°C for 45 minutes. The membranes were washed four times with Tris-buffered saline-Tween 20 (TBST) at room temperature for 14 minutes each. The membrane was exposed on ECL-enhanced chemiluminescence (ECL kit, Pierce Biotechnology, Rockford, IL, USA) to visualize the immunoreactive proteins.
Hematoxylin and eosin staining (H&E)
4% paraformaldehyde was used to fix the organoids of the three genotypes. The fixed organoids were then dehydrated using an ethanol gradient, cleared using xylene, and embedded in paraffin. Thin slices of the organoids were cut at 4 μm thickness on a paraffin sectioning machine, and the slices were mounted on slides. Hematoxylin solution was added to the slides to stain them for 5 minutes at room temperature. Then ethanol fractionated with 1% hydrochloric acid was used to incubate the slides for 30 seconds. Light ammonia was added to the slides to restore the blue color for 1 minute. The slides were then rinsed with distilled water for 5 minutes. Subsequently, eosin staining solution was added to the slides at room temperature. The slides were incubated for 2 minutes. Next, the slides were rinsed with distilled water for 2 minutes. Following that, ethanol gradient decolorization was performed on the slides. Finally, neutral gum was used to seal the slides.
Nissl staining
Nissl staining was carried out according to the manufacturer’s instructions for a Nissl Staining Kit (Beyotime Biotechnology, Zhejiang, China). Mouse brain tissue embedded in paraffin was sectioned, deparaffinized, and rehydrated. Nissl staining solution was used to stain sections for 10 minutes. The sections were washed twice with distilled water for 10 minutes each, then dehydrated in 95% ethanol for 5 seconds. Xylene was used to clear the tissue before neutral resin was used for mounting. A light microscope was used to observe the pathological morphology of the sections.
Senescence-associated beta-galactosidase (SA-β-Gal) staining
The experiment was conducted following the manual of a SA-β-Gal Staining Kit (Beyotime Biotechnology, Hangzhou, China). Intact organoid cells cultured in a 24-well plate were used. The culture medium was discarded, and the cells were washed once with PBS (Gibco). One milliliter of staining fixative solution was added to the cells, which were fixed for 15 minutes at room temperature. After the fixative solution was discarded, the cells were washed 3 times with PBS, each for 3 minutes. One milliliter of staining working solution was added to each well, and the wells were incubated at 37°C overnight. Staining results were observed under a microscope on the following day.
Nest-building
According to previously established protocols, the mice were individually housed in a cage at 1900 hours, with one cotton fiber pad (5 cm × 5 cm; Ancare, San Jose, CA, USA) as nesting material30. Pictures of the nests were taken by a digital camera the next morning. The presence and quality of the nests were scored at a five-point scale from 1 to 5 as follows: 1=nestlet not noticeably touched, 2=nestlet partially torn up, 3=nestlet mostly shredded but often with no identifiable nest site, 4=an identifiable but flat nest, and 5=a near-perfect nest.
Novel Object Recognition (NOR) task
NOR testing was conducted for 4 days as described previously31,32,33. Each mouse received 1 day of testing per trial. Mice were given a 3-minute sample trial in which they were exposed to two identical objects and allowed to explore them freely. Sample trials were terminated when mice had accumulated 15 seconds of exploration time at each object or 3 minutes, whichever was shorter. Mice were then removed from the box and placed in a holding cage while the box was cleaned and configured for the test trial. Inter-trial interval was ∼2 minutes. In the test trial, a new copy of the object presented in the sample trial and a novel object were presented in the same environment for 3 minutes. Exploration of the objects was monitored and recorded via an overhead camera linked to a monitor and used for analysis. Object identity (novel and familiar) and presentation side were counterbalanced.
Recognition index (RI) and Discrimination index (DI) are calculated as follows:
![\[RI = \frac{Tn}{(Tn + T0)} \times 100%\]](https://nhsjs.com/wp-content/ql-cache/quicklatex.com-2426bd21f74441e12bdc61fe44797b8e_l3.png "Rendered by QuickLaTeX.com")
![\[DI = \frac{(Tn - T0)} {(Tn + T0)} \times 100%\]](https://nhsjs.com/wp-content/ql-cache/quicklatex.com-5b0c659d70d5085d98c3216d82ac675a_l3.png "Rendered by QuickLaTeX.com")
(Tn is the exploration time for the novel object, and T0 is the exploration time for the familiar object.)
An RI value greater than 50% or a DI value greater than 0% indicates a preference for the novel object, reflecting intact memory.
4D Label-free quantitative proteomics analysis
The 4D Label-free quantitative proteomics analysis was completed by Shanghai Personal Biotechnology Co., Ltd (Shanghai, China). 300 μL of 8M urea was added to the sample, and the protease inhibitor was added at a volume equal to 10% of the lysate volume. After centrifuging at 14,100 × g for 20 minutes, the supernatant was collected. The protein concentration was determined by the Bradford method, and the remainder was frozen at -80℃. A 100-µg aliquot of the extracted proteins from each sample was then reduced. 200 nM dithiothreitol (DTT) solution was added, and the proteins were incubated at 37 °C for 1 hour. The sample was diluted 4 times by adding 25 mM ammonium bicarbonate (ABC) buffer. Then trypsin (trypsin : protein =1:50) was added to the sample, which was incubated at 37℃ overnight. The next day, 50 μL of 0.1% FA was added to terminate the digestion. 100 μL 100% ACN was used to wash the C18 column, which was centrifuged at 1200 rpm for 3 minutes. The column was washed once with 100 μL of 0.1% FA and then centrifuged at 1200 rpm for 3 minutes. The collection tube was replaced, the sample was added, and the column was centrifuged at 1200 rpm for 3 minutes. The column was washed twice with 100 μL of 0.1% FA and then centrifuged at 1200 rpm for 3 minutes again. The column was washed once with 100 μL of pH 10 water. The collection tube was replaced, and the column was eluted with 70% ACN. The eluents of each sample were combined and lyophilized. The samples were stored at -80°C until loading. Nanoflow LC-MS/MS analysis of tryptic peptides was conducted on a quadrupole Orbitrap mass spectrometer (Q Exactive HF-X, Thermo Fisher Scientific, Bremen, Germany) coupled to an EASY nLC 1200 ultra-high-pressure system (Thermo Fisher Scientific) via a nano-electrospray ion source. 500 ng of peptides were loaded on a 25 cm column (150-μm inner diameter, packed using ReproSil-Pur C18-AQ 1.9- µm silica beads). Peptides were separated using a gradient from 8% to 12% B in 5 minutes, then 12% to 30% B in 33 minutes, and stepped up to 40% in 7 minutes, followed by a 15-minute wash at 95% B at 600 nl per minute, where solvent A was 0.1% formic acid in water and solvent B was 80% ACN and 0.1% formic acid in water. The total duration of the run was 60 minutes. The column temperature was maintained at 60 °C using an in-house-developed oven. The mass spectrometer was operated in “top-40” data-dependent mode, collecting MS spectra in the Orbitrap mass analyzer (120,000 resolution, 350–1500 m/z range) with an automatic gain control (AGC) target of 3E6 and a maximum ion injection time of 80 milliseconds. The most intense ions from the full scan were isolated with an isolation width of 1.6 m/z. Following higher-energy collisional dissociation (HCD) with a normalized collision energy (NCE) of 27, MS/MS spectra were collected in the Orbitrap (15,000 resolution) with an AGC target of 5E4 and a maximum ion injection time of 45 milliseconds. Precursor dynamic exclusion was enabled for 16 seconds.
All RAW files were analyzed using the Proteome Discoverer suite (version 2.4, Thermo Fisher Scientific). MS2 spectra were searched against the UP000000589 Mus musculus uniprot proteome database (54,742 target sequences downloaded on 2025-01-26). The Sequest HT search engine was used, and parameters were specified as follows: fully tryptic specificity, maximum of two missed cleavages, minimum peptide length of 6, fixed carbamidomethylation of cysteine residues (+57.02146Da), variable modifications for oxidation of methionine residues (+15.99492Da), precursor mass tolerance of 15 ppm and a fragment mass tolerance of 0.02Da for MS2 spectra collected in the Orbitrap. Percolator was used to filter peptide spectral matches and peptides to a false discovery rate (FDR) of less than 1%. After spectral assignment, peptides were assembled into proteins and were further filtered based on the combined probabilities of their constituent peptides to a final FDR of 1%. As default, the top matching protein or ‘master protein’ is the protein with the largest number of unique peptides and with the smallest value in the percent peptide coverage (that is, the longest protein). Only unique and razor (that is, parsimonious) peptides were considered for quantification.
Gene Ontology (GO) and InterPro (IPR) analysis were conducted using the interproscan-5 program against the non-redundant protein database, and the databases COG (Clusters of Orthologous Groups) and KEGG (Kyoto Encyclopedia of Genes and Genomes) were used to analyze the protein family and pathway. The probable interacting partners were predicted using the STRING-db server (http://string.embl.de/) based on the related species. STRING is a database of both known and predicted protein-protein interactions. The enrichment pipeline was used to perform the enrichment analysis of GO, and KEGG, respectively.
Statistical Analysis
Each experiment was performed at least three times and values were reported as mean ± standard error wherever it is applicable. Student’s t-test was used to evaluate differences (p < 0.05 indicated statistical significance). Statistical analyses were performed using GraphPad Prism v9.0.
Results
Klotho deficiency exacerbated cognitive impairment of APPKI/PS1KI mice:
APPKI/PS1KI mice and Klotho-/+ transgenic mice were intercrossed, resulting in the production of APPKI/PS1KI/Klotho-/+ heterozygous tri-transgenic mice (Figure 1A). Western blot results indicated that APP, PSEN1, and Aβ levels in the brain tissues of APPKI/PS1KI/Klotho-/+ mice were significantly higher than those in WT mice and were similar to those in the brain tissues of APPKI/PS1KI mice (Figure 1B). However, the expression level of Klotho protein in the brain tissue of APPKI/PS1KI/Klotho-/+ mice was significantly lower than that in WT mice and APPKI/PS1KI mice (Figure 1B). The Nest-Building experiment results showed that the nest-building score of APPKI/PS1KI/Klotho-/+ mice was significantly lower than that of WT mice, while no significant difference was observed between the nest-building scores of APPKI/PS1KI/Klotho-/+ mice and APPKI/PS1KI mice (Figure 1C). In addition, the experimental results of the Novel Object Recognition (NOR) task showed that the RI and DI scores of APPKI/PS1KI/Klotho-/+ mice were significantly lower than those of WT mice and APPKI/PS1KI mice (Figure 1D). These results suggest that Klotho deficiency could potentially exacerbate cognitive impairment in APPKI/PS1KI mice.
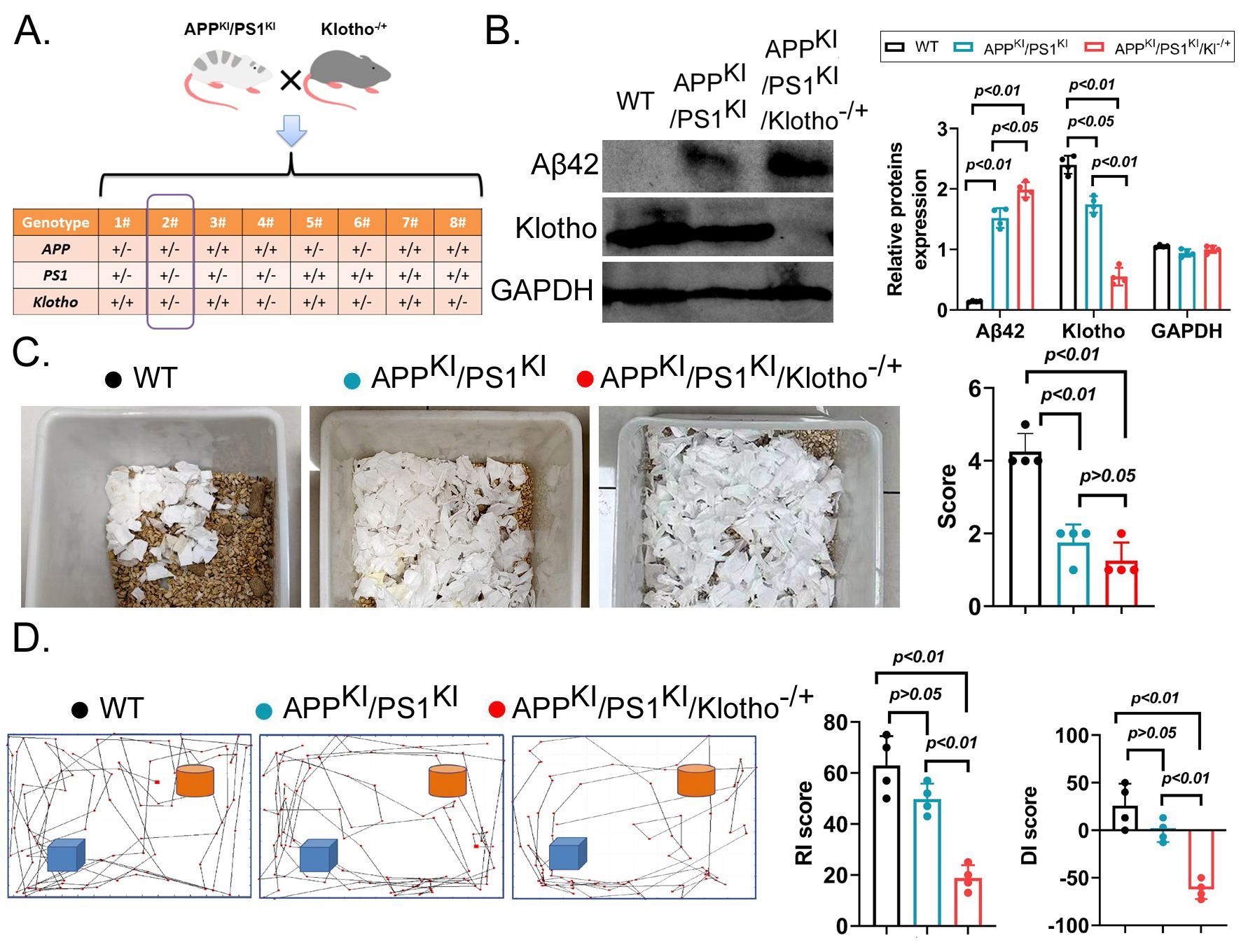
(A) Screening strategy for APPKI/PS1KI/Klotho-/+ triple transgenic mice.
(B) Western blot detection results (n=4 per group, p-values are displayed on the figure)
(C) The Nest-Building experiment results indicated that the Nest-Building score of APPKI/PS1KI/Klotho-/+ mice was significantly lower than that of WT mice (n=4 per group, p-values are displayed on the figure) and noticeably lower than that of APPKI/PS1KI mice, but the latter difference was not significant.
(D) The results of the NOR task experiment showed that the RI and DI scores of APPKI/PS1KI/Klotho-/+ mice were significantly lower than those of the control groups (n=4 per group, p-values are displayed on the figure).
Klotho deficiency induced neuronal damage and aging on APPKI/PS1KI/Klotho-/+ mBOs:
mBOs were generated from brain tissue of each group of mice and cultured in suspension. Under light microscopy, mBOs derived from WT mice and APPKI/PS1KI mice exhibited a clonal morphology with larger size and an apparently higher number (Figure 2A). The colony spheres of mBOs derived from APPKI/PS1KI/Klotho-/+ mice were noticeably smaller. Many floating cells were observed in the culture medium, suggesting increased cell death (Figure 2A). The results of H&E staining and Nissl staining showed that multiple neuron-like cells were visible in the mBO clonal spheres derived from WT mice and APPKI/PS1KI mice. These cells had large somata and prominent nuclei (Figure 2B, 2C). However, in the mBO clonal spheres derived from APPKI/PS1KI/Klotho-/+ mice, most neuronal cells exhibited atrophy and unclear nuclear morphology, showing marked cell deterioration and suggesting cell death (Figure 2B, 2C). Meanwhile, the SA-β-gal staining results also revealed the presence of multiple blue-green staining-positive cells in the colony spheres derived from mBOs of APPKI/PS1KI/Klotho-/+ mice, indicating significant senescence in these mBOs (Figure 2D).
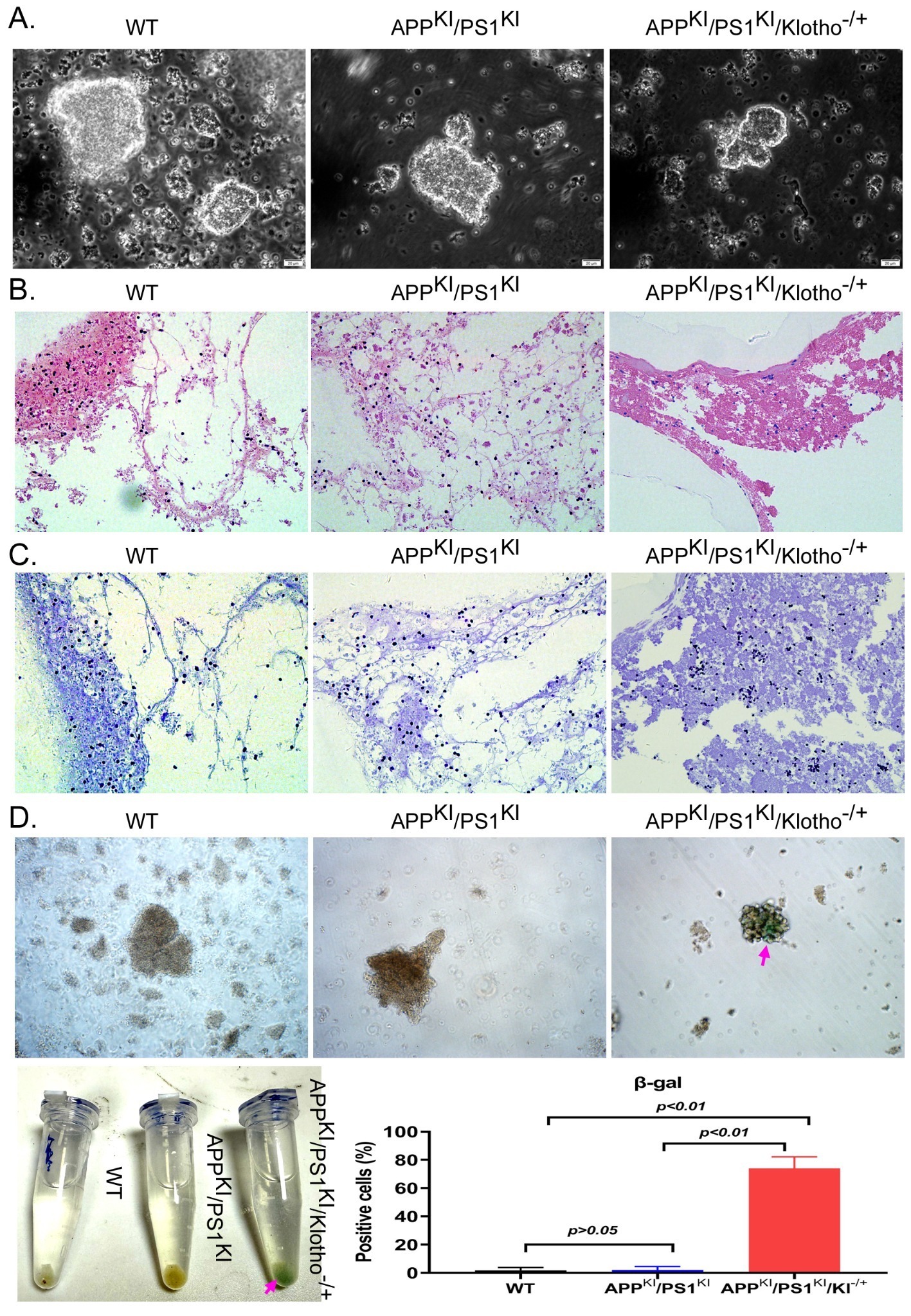
(A) Field of view under light microscope for mBOs. Magnification: 200x (n=4 per group, representative images shown).
(B) H&E staining detection results of mBOs (n=4 per group).
(C) Nissl staining detection results of mBOs (n=4 per group).
(D) SA-β-gal staining detection results of mBOs (n=4 per group, p-values are displayed on the figure).
Klotho deficiency significantly influenced the proteomics landscape of APPKI/PS1KI/Klotho-/+ mBOs:
The differences in protein expression among the three groups of mBOs were systematically analyzed using 4D Label-free quantitative proteomics (Figure 3A). The LC-MS/MS detection results indicated that the numbers of peptides with protein quantification values (Complete Identifications) across the three samples were almost consistent (Figure 3B), suggesting similar sample quality and depth of detection. The sample overlap analysis indicated that all three groups of mBOs contained their respective proteins, but only 2146 common peptides were identified (Figure 3C). By performing median normalization on the original detection data, we made peptide intensities comparable across samples, reducing systematic differences in the experiments (Figure 3D). Finally, Heatmap statistical results indicated that 2,253 unique peptide sequences were detected in mBOs derived from APPKI/PS1KI/Klotho-/+ mice, that 2,530 unique peptide sequences were detected in mBOs derived from WT mice, and that 2,541 unique peptide sequences were detected in mBOs derived from APPKI/PS1KI mice (Figure 3E). Meanwhile, the Pearson product-moment correlation coefficient indicated that the peptide sequences detected across the three groups of mBOs exhibited a strong linear positive correlation (≥0.98; Figure 3F). By setting certain thresholds (signed Fold Change > 1.2 for significant upregulation; signed Fold Change < -1.2 for significant downregulation), differences between the protein abundance levels of the three groups of mBOs were determined. Specifically, in the comparison between the APPKI/PS1KI group and the WT group, there were 236 proteins with significantly upregulated expression and 233 proteins with significantly downregulated expression. In the comparison between the APPKI/PS1KI/Klotho-/+ group and the WT group, there were 184 proteins with significantly upregulated expression and 235 proteins with significantly downregulated expression. In the comparison between the APPKI/PS1KI/Klotho-/+ group and the APPKI/PS1KI group, there were 200 proteins with significantly upregulated expression and 216 proteins with significantly downregulated expression (Figure 4A).
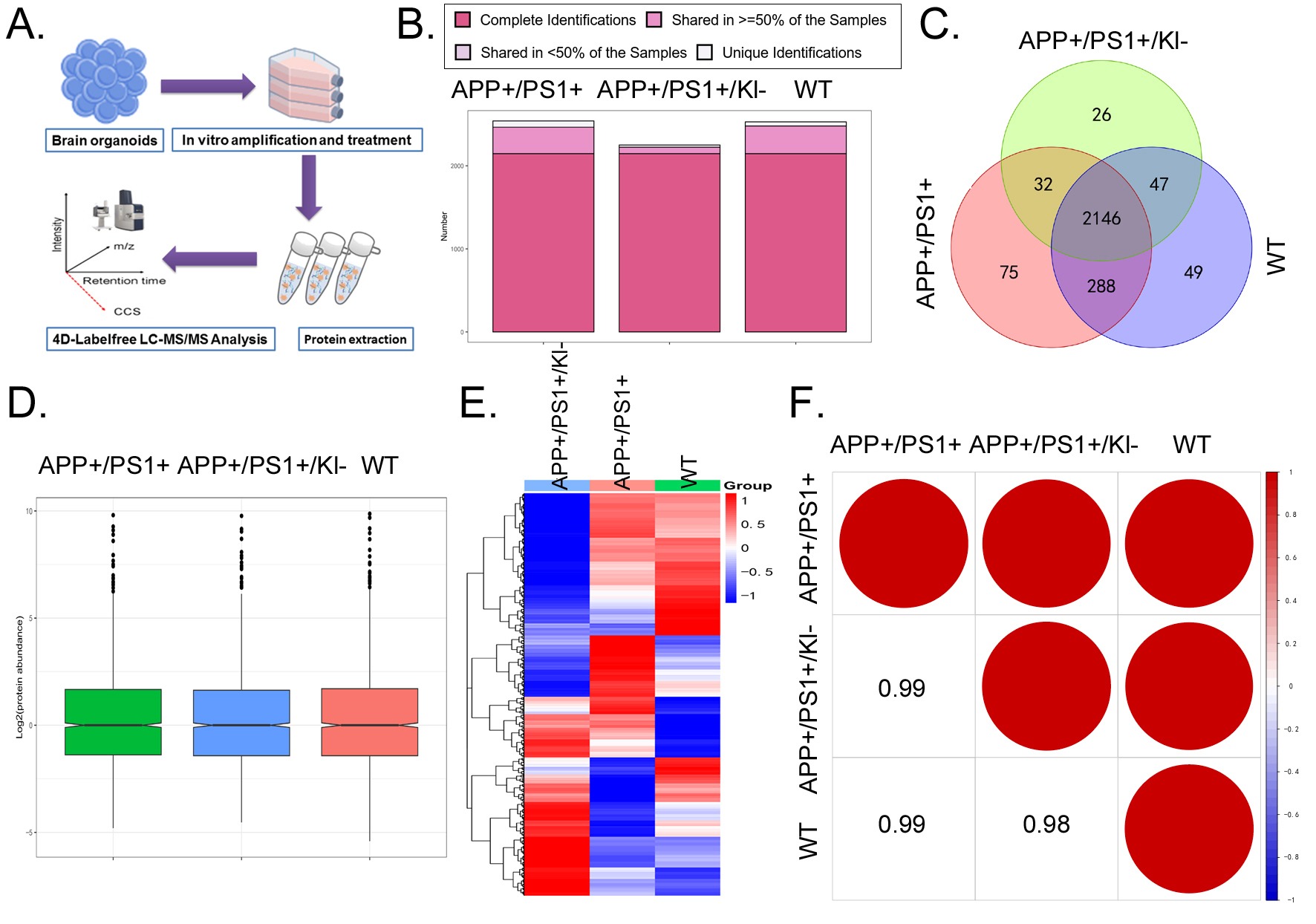
(A) High-throughput proteomics detection strategy.
(B) Statistical results of the number of peptide sequences with Complete Identifications in the sample.
(C) The number of peptide sequences shared by the three groups of mBOs is 2146.
(D) The result of median normalization processing on the original detection data.
(E) Heatmap representation of proteomics results.
(F) Statistical results of Pearson product-moment correlation coefficient.
Based on the Clusters of Orthologous Groups (COG) algorithm for homologous classification of the gene products, we predicted the functional categories of proteins with high abundance in the three groups of mBOs (Figure 4B). Among them, the top three categories were “Signal transduction mechanisms (T)”, “Posttranslational modification, protein turnover, chaperones (O)”, and “General function prediction only (R)”. We also used the Gene Ontology (GO) analysis algorithm to predict the functions, localizations, and activities of proteins with significant differences among the three groups. GO analysis results indicated that proteins with significant differences in expression between the APPKI/PS1KI and WT groups were mainly distributed in “Nucleocytoplasmic transport (Biological process)”, “Cell surface (Cellular component)”, and “Translational initiation factor activity (Molecular function)” (Figure 4C). The proteins with significant differences in expression between the APPKI/PS1KI/Klotho-/+ group and the WT group were mainly distributed in “Lipid metabolic process (Biological process)”, “P-body (Cellular component)”, and “Translational initiation factor activity (Molecular function)” (Figure 4D).
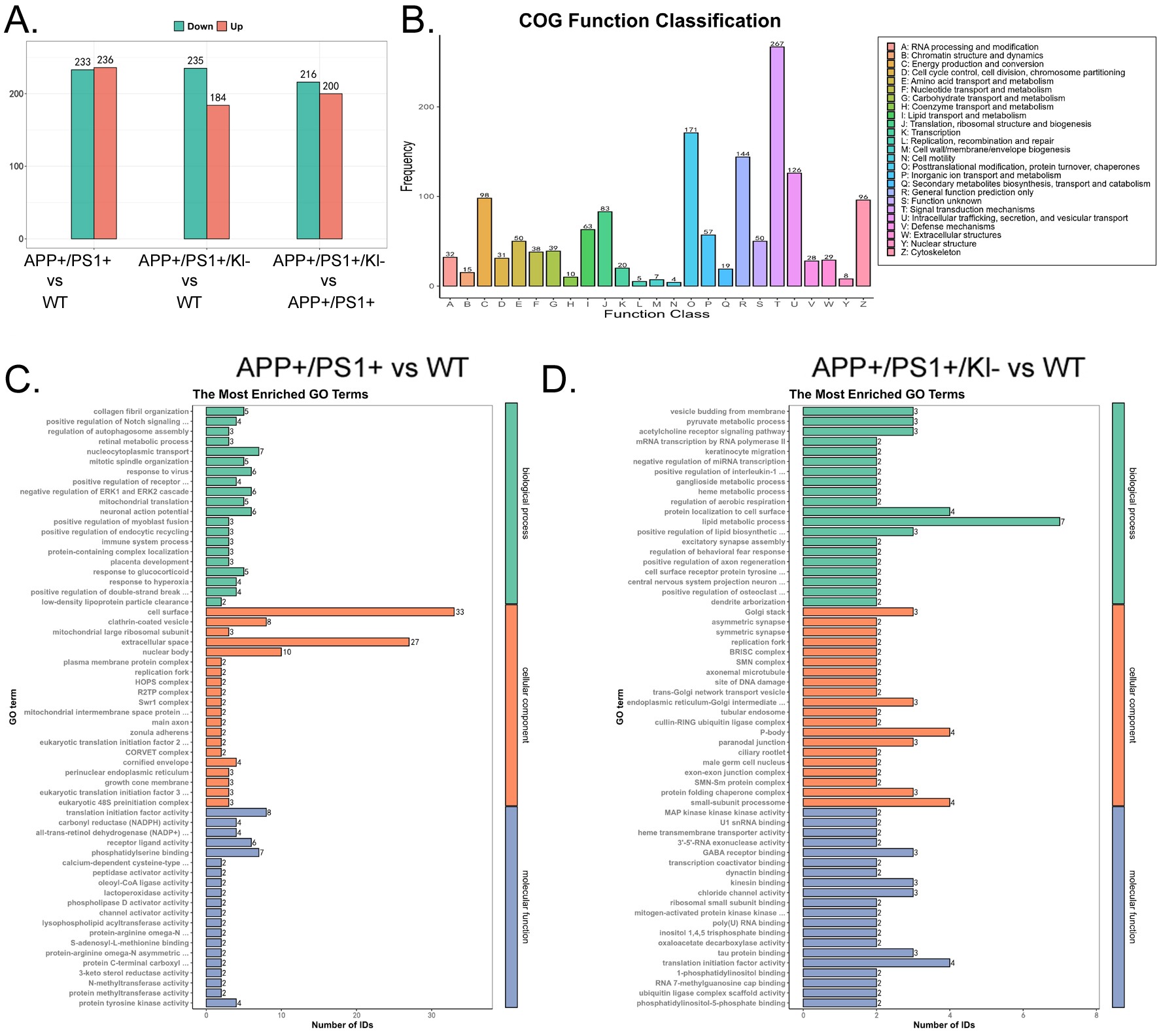
(A) Statistical results of the number of peptides with significantly upregulated and downregulated expression in the three groups of samples.
(B) COG functional enrichment results.
(C) GO functional prediction results of peptides with significant differential expression between APPKI/PS1KI group and WT group.
(D) GO functional prediction results of significantly differentially expressed peptides in APPKI/PS1KI/Klotho-/+ group compared to WT group.
Finally, we predicted signaling pathways underlying the differential protein expression profiles. The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that the proteins with significantly upregulated expression in the APPKI/PS1KI group compared to WT group were mostly enriched in signaling pathways such as “Peroxisome” and “Nucleocytoplasmic transport”, whereas the proteins with significantly downregulated expression were mostly enriched in signaling pathways such as “Nucleocytoplasmic transport” and “mRNA surveillance pathway” (Figure 5A). In the APPKI/PS1KI/Klotho-/+ group, the proteins with significantly upregulated expression were mostly enriched in signaling pathways such as “Peroxisome” and “Mitophagy – animal”, whereas the proteins with significantly downregulated expression were mostly enriched in signaling pathways such as “Nucleocytoplasmic transport” and “Ribosome” compared to the WT group (Figure 5B). Finally, we identified the intersection and found that the proteins with significantly upregulated expression across the three groups were mostly enriched in the “Peroxisome” signaling pathway, whereas those with significantly downregulated expression were mostly enriched in the “Nucleocytoplasmic transport” signaling pathway (Figure 5C, 5D).
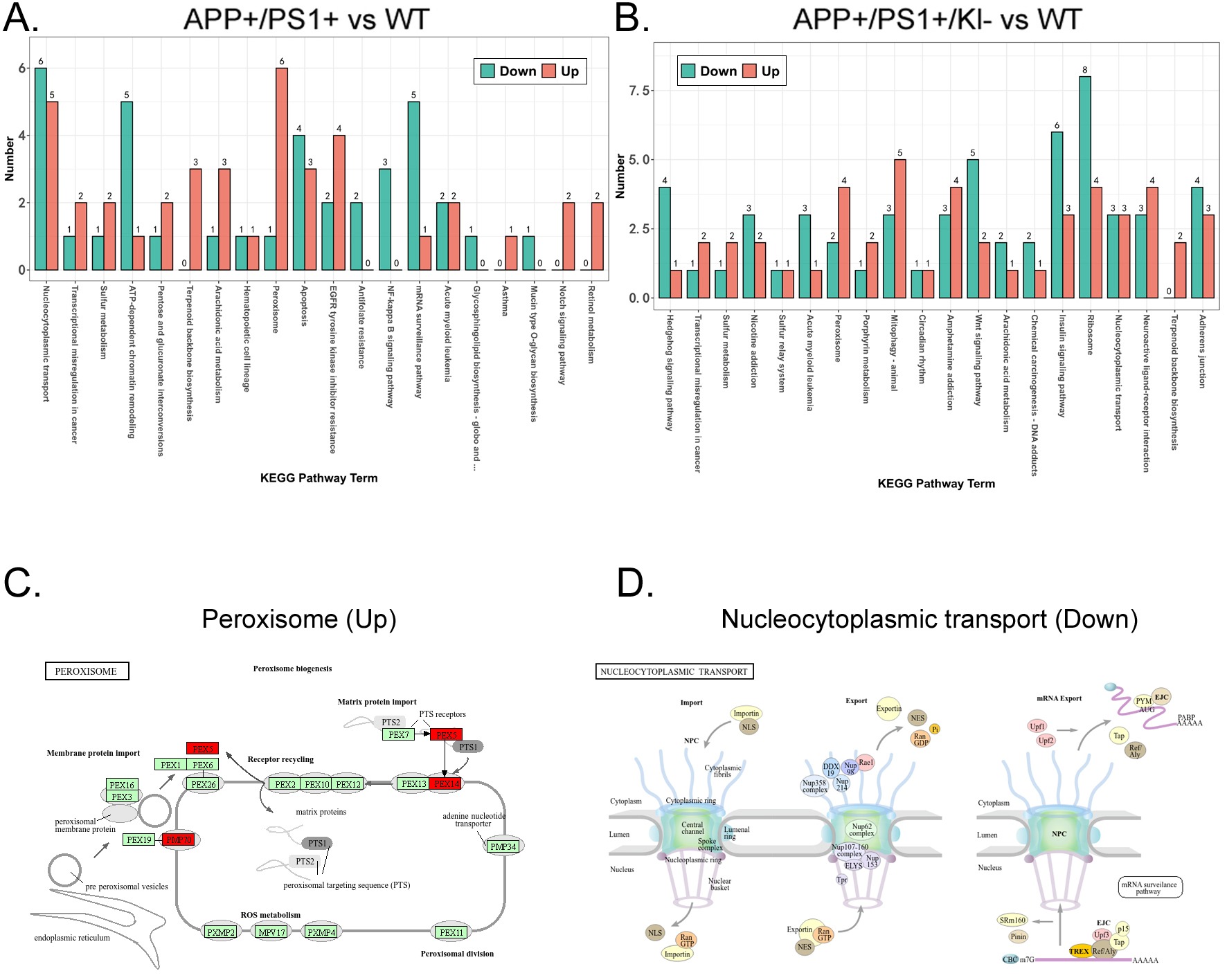
(A) KEGG prediction results of significantly differentially expressed peptides in APPKI/PS1KI group compared to WT group.
(B) KEGG prediction results of peptides with significant differential expression between APPKI/PS1KI/Klotho-/+ group and WT group.
(C) The proteomics signal transduction pathway involved in the most significantly upregulated peptide sequence common to all three samples.
(D) The proteomics signal transduction pathway involved in the most significantly down-regulated peptide sequence common to all three samples.
Discussion
Our findings suggest that Klotho deficiency is associated with accelerated aging in AD transgenic mice. This may exacerbate their cognitive dysfunction and promote the early emergence of AD symptoms.
Our study consisted of both in vitro and in vivo analyses of WT, APPKI/PS1KI, and APPKI/PS1KI/Klotho-/+ mice, using organoids derived from these mice. We discovered that Klotho deficiency could potentially exacerbate cognitive impairment in APPKI/PS1KI mice, as the worse performance in NOR tests by APPKI/PS1KI/Klotho-/+ mice compared to WT mice and APPKI/PS1KI mice suggests. In addition, Klotho deficiency induced neuronal damage and aging on APPKI/PS1KI/Klotho-/+ mBOs, demonstrated by the significantly different morphologies of APPKI/PS1KI/Klotho-/+ mBOs compared to WT and APPKI/PS1KI mBOs when they are assessed by Nissl staining, H&E staining, and SA-β-gal staining. The proteomics landscape of Klotho-deficient mice is also significantly different from that of WT and APPKI/PS1KI mBOs, as shown by analyses using 4D Label-free quantitative proteomics, COG, GO, and KEGG.
Previous research has already situated Klotho as an important anti-ageing factor, and our research generally aligns with this perspective20,22,23. Mechanistically, researchers have illustrated that Klotho plays a crucial role in cell-sustaining pathways through mechanisms such as reducing phosphorylation of FOXO proteins and acting as a cofactor in FGF23 signaling pathways20,23. Similarly, our research suggests the beneficial effects of Klotho, demonstrating that its deficiency is associated with earlier and more severe AD incidence and progression. However, our study places Klotho not only as a protein associated with aging but also as a modulator of the pathological onset and severity of AD. This extends prior research by pointing out that, in addition to resisting aging, Klotho may also modulate bodily responses to specific neurodegenerative diseases.
Our research results are, in general, quite consistent with the conclusion that Klotho deficiency accelerates aging in AD transgenic mice, which exacerbates their cognitive dysfunction and promotes the early emergence of AD symptoms. The only inconsistency may be that while APPKI/PS1KI/Klotho-/+ mice performed worse than APPKI/PS1KI mice in the nest-building tests, this difference was not statistically significant. This could be attributed to a floor effect. Here, the AD symptoms in APPKI/PS1KI mice might already have interfered with their ability to build nests, to the extent that further impairment (such as Klotho deficiency) becomes less noticeable and its impact becomes statistically insignificant in our study. In comparison, differences in NOR test performance, which aligns with the conclusion reached in this paper, could be potentially more sensitive to Klotho levels than the nest-building tests.
Limitations still exist in this research. First, the sample size is relatively small, with only 12 mice (4 from each of the three genotypes) used in this study. This may result in this study having relatively low statistical power and a higher risk of producing inaccurate results. Future studies should possibly use a larger sample size. Second, only one AD model (APPKI/PS1KI) is used. This means that research findings could be limited to this model rather than AD in general. Future studies should experiment with other standardized AD models such as TAPP, BRI-Aβ42A, and PDAPP34. Third, we did not establish specific mechanisms underlying the relationship between Klotho and AD. To our knowledge, this study is among the first to systematically analyze the effects of Klotho gene knockout on AD incidence and progression. Although this study provides convincing evidence that Klotho deficiency is closely associated with AD incidence and progression, it does not explain the specific pathways and mechanisms underlying this relationship. Even though this study involves proteomics analyses, it establishes only correlations, not causation. Future research could focus on the specific mechanisms and pathways as to how Klotho deficiency promotes AD incidence and worsens AD symptoms. Fourth, the extent to which mBOs are representative of in vivo pathology is worthy of consideration. While mBOs derived from the mice in this study demonstrated pathological features consistent with AD, they nonetheless represent a simplified system. They lack the full complexity of the in vivo brain environment. Factors such as the circulatory system and the presence of full, intact neural networks, which are lacking in mBOs, may influence the progression of AD-like symptoms. Nonetheless, the neuronal changes observed in the mBOs are still consistent with and supported by the behavioral studies and proteomics analysis results, suggesting that the insights we gained from mBOs may reflect real, existing effects of Klotho deficiency, instead of artifacts of the in vitro mBO system. That said, future studies could still employ more complex models incorporating more complex factors, such as the circulatory system and the immune system, to better imitate in vivo conditions.
Collectively, the research findings presented in this paper suggest that Klotho deficiency accelerates aging in AD transgenic mice, which exacerbates their cognitive dysfunction and promotes the early emergence of AD-like phenotypes. The conclusions of this paper may serve as a foundation for future research on new medications for AD. However, this paper should not be seen as a final resolution of AD. Further research, perhaps on the underlying mechanisms of the relationship between Klotho and AD, is still needed to address this most common neurodegenerative disease in the world.
References
- X.-L. Li, N. Hu, M.-S. Tan, J.-T. Yu, L. Tan. Behavioral and psychological symptoms in alzheimer’s disease. BioMed Research International. Vol. 2014, pg. 1–9, 2014 https://doi.org/10.1155/2014/927804 [↩] [↩]
- A. Mira, R. Gonçalves, I. T. Rodrigues. Dysphagia in alzheimer’s disease: a systematic review. Dementia & Neuropsychologia. Vol. 16, pg. 261–269, 2022 https://doi.org/10.1590/1980-5764-dn-2021-0073 [↩]
- M. T. Heneka, M. J. Carson, J. E. Khoury, G. E. Landreth, F. Brosseron, D. L. Feinstein, A. H. Jacobs, T. Wyss-Coray, J. Vitorica, R. M. Ransohoff, K. Herrup, S. A. Frautschy, B. Finsen, G. C. Brown, A. Verkhratsky, K. Yamanaka, J. Koistinaho, E. Latz, A. Halle, G. C. Petzold, T. Town, D. Morgan, M. L. Shinohara, V. H. Perry, C. Holmes, N. G. Bazan, D. J. Brooks, S. Hunot, B. Joseph, N. Deigendesch, O. Garaschuk, E. Boddeke, C. A. Dinarello, J. C. Breitner, G. M. Cole, D. T. Golenbock, M. P. Kummer. Neuroinflammation in alzheimer’s disease. The Lancet Neurology. Vol. 14, pg. 388–405, 2015 https://doi.org/10.1016/S1474-4422(15)70016-5 [↩]
- A. Gustavsson, N. Norton, T. Fast, L. Frölich, J. Georges, D. Holzapfel, T. Kirabali, P. Krolak‐Salmon, P. M. Rossini, M. T. Ferretti, L. Lanman, A. S. Chadha, W. M. Van Der Flier. Global estimates on the number of persons across the alzheimer’s disease continuum. Alzheimer’s & Dementia. Vol. 19, pg. 658–670, 2022 https://doi.org/10.1002/alz.12694 [↩]
- Y. Liu, Y. Tan, Z. Zhang, M. Yi, L. Zhu, W. Peng. The interaction between ageing and alzheimer’s disease: insights from the hallmarks of ageing. Translational Neurodegeneration. Vol. 13, pg. 7, 2024 https://doi.org/10.1186/s40035-024-00397-x [↩]
- C. R. Beam, C. Kaneshiro, J. Y. Jang, C. A. Reynolds, N. L. Pedersen, M. Gatz. Differences between women and men in incidence rates of dementia and alzheimer’s disease. Journal of Alzheimer’s Disease. Vol. 64, pg. 1077–1083, 2018 https://doi.org/10.3233/JAD-180141 [↩]
- M. F. Mendez. Early-onset alzheimer disease and its variants. Continuum. Vol. 25, pg. 34–51, 2019 https://doi.org/10.1212/CON.0000000000000687 [↩]
- M. Fang, J. Hu, J. Weiss, D. S. Knopman, M. Albert, B. G. Windham, K. A. Walker, A. R. Sharrett, R. F. Gottesman, P. L. Lutsey, T. Mosley, E. Selvin, J. Coresh. Lifetime risk and projected burden of dementia. Nature Medicine. Vol. 31, pg. 772–776, 2025 https://doi.org/10.1038/s41591-024-03340-9 [↩]
- R. N. Rosenberg, D. Lambracht-Washington, G. Yu, W. Xia. Genomics of alzheimer disease: a review. JAMA Neurology. Vol. 73, pg. 867, 2016 https://doi.org/10.1001/jamaneurol.2016.0301 [↩]
- R. Medeiros, D. Baglietto‐Vargas, F. M. LaFerla. The role of tau in alzheimer’s disease and related disorders. CNS Neuroscience & Therapeutics. Vol. 17, pg. 514–524, 2011 https://doi.org/10.1111/j.1755-5949.2010.00177.x [↩]
- H.-L. Li, H.-H. Wang, S.-J. Liu, Y.-Q. Deng, Y.-J. Zhang, Q. Tian, X.-C. Wang, X.-Q. Chen, Y. Yang, J.-Y. Zhang, Q. Wang, H. Xu, F.-F. Liao, J.-Z. Wang. Phosphorylation of tau antagonizes apoptosis by stabilizing β-catenin, a mechanism involved in alzheimer’s neurodegeneration. Proceedings of the National Academy of Sciences. Vol. 104, pg. 3591–3596, 2007 https://doi.org/10.1073/pnas.0609303104 [↩] [↩]
- X. Zhang, J. Wang, Z. Zhang, K. Ye. Tau in neurodegenerative diseases: molecular mechanisms, biomarkers, and therapeutic strategies. Translational Neurodegeneration. Vol. 13, pg. 40, 2024 https://doi.org/10.1186/s40035-024-00429-6 [↩] [↩]
- V. W. Chow, M. P. Mattson, P. C. Wong, M. Gleichmann. An overview of app processing enzymes and products. NeuroMolecular Medicine. Vol. 12, pg. 1–12, 2010 https://doi.org/10.1007/s12017-009-8104-z [↩] [↩]
- M. P. Murphy, H. LeVine. Alzheimer’s disease and the amyloid-β peptide. Journal of Alzheimer’s Disease. Vol. 19, pg. 311–323, 2010 https://doi.org/10.3233/JAD-2010-1221 [↩] [↩] [↩] [↩]
- S. A. Kent, T. L. Spires-Jones, C. S. Durrant. The physiological roles of tau and aβ: implications for alzheimer’s disease pathology and therapeutics. Acta Neuropathologica. Vol. 140, pg. 417–447, 2020 https://doi.org/10.1007/s00401-020-02196-w [↩]
- J.-Y. Hur. γ-secretase in alzheimer’s disease. Experimental & Molecular Medicine. Vol. 54, pg. 433–446, 2022 https://doi.org/10.1038/s12276-022-00754-8 [↩]
- P. Modrego, A. Lobo. A good marker does not mean a good target for clinical trials in alzheimer’s disease: the amyloid hypothesis questioned. Neurodegenerative Disease Management. Vol. 9, pg. 119–121, 2019 https://doi.org/10.2217/nmt-2019-0006 [↩]
- O. V. Forlenza, B. J. A. P. Barbosa. What are the reasons for the repeated failures of clinical trials with anti-amyloid drugs for ad treatment? Dementia & Neuropsychologia. Vol. 19, pg. e2025E001, 2025 https://doi.org/10.1590/1980-5764-dn-2025-e001 [↩]
- A. M. Fjell, K. B. Walhovd. Neuroimaging results impose new views on alzheimer’s disease—the role of amyloid revised. Molecular Neurobiology. Vol. 45, pg. 153–172, 2012 https://doi.org/10.1007/s12035-011-8228-7 [↩]
- G. D. Dalton, J. Xie, S.-W. An, C.-L. Huang. New insights into the mechanism of action of soluble klotho. Frontiers in Endocrinology. Vol. 8, pg. 323, 2017 https://doi.org/10.3389/fendo.2017.00323 [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- H. Kurosu, M. Yamamoto, J. D. Clark, J. V. Pastor, A. Nandi, P. Gurnani, O. P. McGuinness, H. Chikuda, M. Yamaguchi, H. Kawaguchi, I. Shimomura, Y. Takayama, J. Herz, C. R. Kahn, K. P. Rosenblatt, M. Kuro-o. Suppression of aging in mice by the hormone klotho. Science. Vol. 309, pg. 1829–1833, 2005 https://doi.org/10.1126/science.1112766 [↩] [↩]
- M. Kuro-o, Y. Matsumura, H. Aizawa, H. Kawaguchi, T. Suga, T. Utsugi, Y. Ohyama, M. Kurabayashi, T. Kaname, E. Kume, H. Iwasaki, A. Iida, T. Shiraki-Iida, S. Nishikawa, R. Nagai, Y. Nabeshima. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. Vol. 390, pg. 45–51, 1997 https://doi.org/10.1038/36285 [↩] [↩] [↩]
- Y. Xu, Z. Sun. Molecular basis of klotho: from gene to function in aging. Endocrine Reviews. Vol. 36, pg. 174–193, 2015 https://doi.org/10.1210/er.2013-1079 [↩] [↩] [↩]
- S. A. Castner, S. Gupta, D. Wang, A. J. Moreno, C. Park, C. Chen, Y. Poon, A. Groen, K. Greenberg, N. David, T. Boone, M. G. Baxter, G. V. Williams, D. B. Dubal. Longevity factor klotho enhances cognition in aged nonhuman primates. Nature Aging. Vol. 3, pg. 931–937, 2023 https://doi.org/10.1038/s43587-023-00441-x [↩]
- B. W. Müller, A. Hinney, N. Scherbaum, C. Weimar, C. Kleinschnitz, T. Peters, L. Hochfeld, S. Pechlivanis, A. Stang, M. Jokisch, B. Kowall. Klotho kl-vs haplotype does not improve cognition in a population-based sample of adults age 55–87 years. Scientific Reports. Vol. 11, pg. 13852, 2021 https://doi.org/10.1038/s41598-021-93211-x [↩]
- L. Lai, Y. Li, J. Liu, L. Luo, J. Tang, J. Xue, T. Liu. Bovine serum albumin aggravates macrophage m1 activation and kidney injury in heterozygous klotho-deficient mice via the gut microbiota-immune axis. International Journal of Biological Sciences. Vol. 17, pg. 742–755, 2021 https://doi.org/10.7150/ijbs.56424 [↩]
- Ministry of Health of the People’s Republic of China. Implementation Rules for the Administration of Medical Laboratory Animals. 1998 [↩]
- H. Chen, Y. Wen, Z. Yu, X. Du, W. Pan, T. Liu. Codonopsis pilosula polysaccharide alleviates rotenone-induced murine brain organoids death through downregulation of gene body dna methylation modification in the zic4/pgm5/camta1 axis. Biochemistry and Biophysics Reports. Vol. 37, pg. 101593, 2024 https://doi.org/10.1016/j.bbrep.2023.101593 [↩]
- Y. Huang, X. Liu, Y. Feng, X. Nie, Q. Liu, X. Du, Y. Wu, T. Liu, X. Zhu. Rotenone, an environmental toxin, causes abnormal methylation of the mouse brain organoid’s genome and ferroptosis. International Journal of Medical Sciences. Vol. 19, pg. 1184–1197, 2022 https://doi.org/10.7150/ijms.74569 [↩]
- Y. Lv, B. Meng, H. Dong, T. Jing, N. Wu, Y. Yang, L. Huang, R. E. Moses, B. W. O’Malley, B. Mei, X. Li. Upregulation of gsk3β contributes to brain disorders in elderly regγ-knockout mice. Neuropsychopharmacology. Vol. 41, pg. 1340–1349, 2016 https://doi.org/10.1038/npp.2015.285 [↩]
- A. P. Swiercz, M. C. Tsuda, H. A. Cameron. The curious interpretation of novel object recognition tests. Trends in Neurosciences. Vol. 48, pg. 250–256, 2025 https://doi.org/10.1016/j.tins.2025.02.003 [↩]
- J. A. Ainge, C. Heron-Maxwell, P. Theofilas, P. Wright, L. De Hoz, E. R. Wood. The role of the hippocampus in object recognition in rats: examination of the influence of task parameters and lesion size. Behavioural Brain Research. Vol. 167, pg. 183–195, 2006 https://doi.org/10.1016/j.bbr.2005.09.005 [↩]
- D. I. G. Wilson, R. F. Langston, M. I. Schlesiger, M. Wagner, S. Watanabe, J. A. Ainge. Lateral entorhinal cortex is critical for novel object‐context recognition. Hippocampus. Vol. 23, pg. 352–366, 2013 https://doi.org/10.1002/hipo.22095 [↩]
- A. M. Hall, E. D. Roberson. Mouse models of alzheimer’s disease. Brain Research Bulletin. Vol. 88, pg. 3–12, 2012 https://doi.org/10.1016/j.brainresbull.2011.11.017 [↩]



{kind=link}