
Background: Hereditary breast cancer constitutes 5-10% of all breast cancer cases. However, BRCA1/2 mutations only account for 20-25% of hereditary cases, leaving over 70% without known causes. This review collates existing evidence on mitochondrial DNA (mtDNA) variants and epigenetic changes in non-BRCA hereditary breast cancer to evaluate their importance in assessing risk, disease progression, and prognosis. The findings highlight key gaps in identifying genetic risk factors beyond mutations in nuclear genes.
Methods: This study is based on relevant literature focusing on human studies of hereditary breast cancers, especially non-BRCA cases. The findings have been interpreted from data related to mtDNA variants, epigenetic changes, and their links to cancer risk, progression, and outcomes.
Results: This review highlights the role of several mtDNA variants in the displacement loop (D-loop) region and in protein-coding genes in disrupting oxidative phosphorylation resulting in the production of reactive oxygen species. Paradoxically, higher burden of somatic mtDNA exhibits a direct correlation with better survival outcomes. Epigenetic changes including modifications in DNA methylation, histones, and microRNA associated with mitochondrial dysfunction have also been linked to these variants.
Conclusions: mtDNA variants and epigenetic changes are important contributors to non-BRCA hereditary breast cancer. They have the potential to provide treatment targets and better risk assessment methods. These findings warrant the integration of mitochondrial genomics into hereditary breast cancer research, marking a shift from focusing solely on nuclear genetic factors.
Keywords: mitochondrial DNA variants, non-BRCA hereditary breast cancer, epigenetic modifications, genetic predisposition, risk assessment, precision medicine, biomarkers, mitochondrial dysfunction
Introduction
Breast cancer is one of the most commonly diagnosed malignancies among women worldwide. Over 2.3 million new cases were reported globally in 2022 alone, which is approximately 11.7% of all cancer diagnoses1,2. Significant molecular and clinical heterogeneity are hallmarks of the disease and hereditary factors account for one-fourth of all breast cancer cases3,4. Although the genetic architecture that dictates susceptibility to breast cancer has been extensively studied, the complex interplay between inherited genetic variants, epigenetic modifications, and mitochondrial dysfunction continues to be challenging for conventional therapeutic approaches, carving opportunities for precision medicine approaches.
Background and Context
Hereditary breast cancers account for 5-10% of the diagnosed cases and are often attributed to germline mutations in genes involved in breast cancer predisposition4,5. The identification and characterization of high-penetrance susceptibility genes such as BRCA1 and BRCA2 has positively impacted our understanding of hereditary breast cancer5,6. The pivotal role of these genes in the functioning of the homologous recombination DNA repair pathway significantly increases the risk posed by pathogenic variants7,8. Despite their clinical significance, BRCA1 and BRCA2 mutations account for only a fraction of the hereditary breast cancer cases.
Moderate-penetrance genes, ATM, CHEK2, PALB2, RAD51C, and RAD51D, are associated with a 2-4-fold increase in the risk of developing breast cancer7,8. Although rare, some high-penetrance genes, TP53 (Li-Fraumeni syndrome), PTEN (Cowden syndrome), CDH1, and STK11 (Peutz-Jeghers syndrome), contribute to specific hereditary cancers7,9. Multi-gene panel testing has facilitated the identification of pathogenic variants across multiple susceptibility genes. High-risk populations have detection rates of 11.9-20%10,11.
The classification of breast cancer into subtypes based on molecular distinctions, luminal A and B, HER2-enriched, and triple-negative, underscores important genotype-phenotype correlations that have important implications for prognosis and treatment selection. For example, BRCA1-associated tumours predominantly exhibit triple-negative characteristics with high-grade morphology and are often diagnosed at a younger age, while BRCA2-associated cancers exhibit luminal features more frequently12.
Problem Statement and Rationale
Despite considerable progress in characterizing hereditary breast cancer, several critical gaps persist in the current perception of genetic predisposition mechanisms. While BRCA1 and BRCA2 mutations account for only 20-25% of breast cancer cases, more than 70% of genetically predisposed breast cancer cases are attributed to idiopathic causes6. These cases of non-BRCA hereditary breast cancer exhibit hereditary characteristics, but lack identifiable pathogenic variants in the known susceptibility genes. Intensive surveillance protocols, risk assessment models, and targeted therapeutic strategies are designed for BRCA1/2 carriers. This limits accessibility and impact for non-BRCA hereditary breast cancers. Current polygenic risk scores that are informative at the population level account for a modest proportion of the remaining hereditary risk only, adding further insult to injury7.
Recent research indicates that mitochondrial DNA (mtDNA) variants and epigenetic modifications account for the overlooked contributors of susceptibility to hereditary breast cancer. Evidence implicates mtDNA mutations in 60-73.7% of breast tumours suggesting biological and clinical significance of these alterations, rather than being merely bystander events1,2,3,4. Epigenetic modifications such as DNA methylation, histone modifications, and non-coding RNA regulation may complement or interact with genetic susceptibility factors, potentially influencing gene expression profiles and tumour behaviour13,14,15.
Significance and Purpose
Population-specific mtDNA variants such as the A8860G variant in Kurdish populations and distinct mutational patterns in African American versus European American women have been associated with the risk of developing breast cancer16,17. Existing risk-assessment models limit risk stratification in high-risk populations that test negative for pathogenic variants of known susceptibility genes. The inclusion of mtDNA variants and epigenetic biomarkers into existing risk assessment models would overcome these limitations. Additionally, the development of mtDNA-based liquid biomarkers that detect mtDNA mutations carrying tumour signatures in circulating extracellular vesicles with high specificity would promote non-invasive screening and recurrence monitoring, thereby proving beneficial to high-risk populations that are excluded from intensive surveillance, based on current guidelines17.
Elucidation of the role of mtDNA variants and epigenetic modifications in non-BRCA hereditary breast cancer has the potential to advance precision medicine approaches. Mitochondrial dysfunction and epigenetic dysregulation represent potential therapeutic targets. The development of therapeutic agents that target mtDNA mutation-associated oxidative phosphorylation defects is minimally toxic. Furthermore, the reversibility of epigenetic modifications makes them attractive targets for therapeutic intervention18.
Objectives
Our review aims to synthesize existing evidence that implicates mtDNA variants and epigenetic modifications in non-BRCA hereditary breast cancers and assess the clinical significance of mtDNA variants and epigenetic biomarkers in risk assessment, early detection, and prognosis. The study identifies research gaps, evaluates the potential of mitochondrial dysfunction and epigenetic dysregulation as therapeutic targets, investigates population-specific variations in mtDNA variants and epigenetic patterns, and provides recommendations for future research.
Scope and Limitations
Our review collates findings that highlight the role of mtDNA variants (point mutations, deletions, insertions, and copy number variations) and epigenetic modifications (DNA methylation, histone modifications, and non-coding RNA regulation) in predisposition risk, development, and progression of non-BRCA hereditary breast cancer. Although some relevant findings related to sporadic breast cancers have been discussed, non-hereditary or BRCA-associated breast cancers are not within the scope of this review. There is a dearth of studies that specifically investigate mtDNA variants and epigenetic modifications in well-defined non-BRCA hereditary breast cancer cohorts. This limits the generalizability of the findings. Furthermore, the cited studies refer to heterogeneous populations, provide inconsistent definitions of hereditary breast cancer, and may be subject to publication bias toward positive associations.
Theoretical Framework
Mitochondrial dysfunction resulting from mtDNA mutations affects the efficiency of oxidative phosphorylation, production of reactive oxygen species (ROS), and cellular metabolism. Furthermore, characteristics of mtDNA such as high rate of mutation, limited repair mechanisms, and maternal inheritance patterns increase vulnerability to inherited cancers.
Epigenetic inheritance is characterized by DNA methylation patterns and histone modifications caused by environmental influences getting transmitted across generations13,19.
Our study is grounded in a framework that acknowledges the crucial role of mtDNA variants and epigenetic modifications as contributing factors in non-BRCA hereditary breast cancers and highlights the complex genetic and epigenetic landscape of cancer susceptibility, tumour behaviour, and treatment response, rather than attributing the risks to single, high-penetrance causative factors. This framework draws from findings that indicate significant interactions between specific mtDNA polymorphisms suggesting that multiple mtDNA variants can interact to modify breast cancer risk.
Methodology Overview
We used a systematic approach to identify, evaluate, and synthesize current evidence regarding mtDNA variants and epigenetic modifications in non-BRCA hereditary breast cancers from existing literature. We followed predetermined inclusion criteria to ensure relevance in study selection.
Our review aims to collate relevant findings from existing studies to investigate and highlight the crucial role of mtDNA variants and epigenetic modifications in predisposition, progression, and prognosis of non-BRCA hereditary breast cancers. The findings from our review will contribute to more comprehensive approaches for risk assessment, early detection, and therapeutic intervention in the non-BRCA subset of hereditary breast cancers.
Methods
Search Strategy
We identified studies between 1995 and 2025 that investigate mtDNA variants and epigenetic modifications in non-BRCA hereditary breast cancers across 12 databases: PubMed MEDLINE, Embase, Web of Science Core Collection, and Scopus, as primary sources; Cochrane Library CENTRAL, CINAHL, ClinicalTrials.gov, and GeneReviews as speciality databases; and genetic registries including MITOMAP, ClinVar, gnomAD, TCGA/GDC, and GWAS catalog. . The Boolean search strings used comprised of multiple combinations of Medical Subject Headings (MeSH) terms and keywords (Table 1).
| Search Topic | Sub-topic | Boolean Search String |
| Mitochondrial DNA Variants & Hereditary Breast Cancer | Core mtDNA germline variants | (“mitochondrial DNA” OR “mtDNA” OR “mitochondrial genome”) AND (“breast cancer” OR “hereditary breast cancer” OR “HBOC” OR “non-BRCA”) AND (“variant” OR “mutation” OR “polymorphism” OR “haplogroup” OR “heteroplasmy” OR “homoplasmy”) NOT (“review” OR “editorial” OR “commentary”) |
| D-loop & control region specific | (“displacement loop” OR “D-loop” OR “control region” OR “m.16189” OR “m.16519” OR “m.310” OR “m.315”) AND (“breast cancer” OR “hereditary breast cancer”) AND (“germline” OR “hereditary”) | |
| OXPHOS gene variants | (“Complex I” OR “Complex III” OR “Complex IV” OR “ND1” OR “ND2” OR “ND5” OR “ND6” OR “CYTB” OR “ATP8” OR “ATP6” OR “COX1” OR “COX2” OR “COX3”) AND (“mitochondrial” OR “mtDNA”) AND (“breast cancer” OR “hereditary breast cancer”) AND (“mutation” OR “variant” OR “pathogenic”) | |
| Somatic mtDNA burden in tumors (hereditary context) | (“somatic” OR “tumor mtDNA” OR “tumor burden”) AND (“mitochondrial DNA” OR “mtDNA”) AND (“breast cancer”) AND (“hereditary” OR “BRCA-negative” OR “non-BRCA”) AND (“mutation frequency” OR “heteroplasmy” OR “homoplasmy” OR “copy number”) | |
| Population-specific mtDNA variants | (“mtDNA” OR “mitochondrial DNA”) AND (“breast cancer”) AND (“ancestry” OR “ethnic” OR “population” OR “African” OR “European” OR “Asian” OR “Hispanic” OR “haplogroup” OR “South Asian” OR “West African” OR “East Asian”) AND (“variant” OR “polymorphism” OR “association”) | |
| Epigenetic Modifications & mtDNA Dysfunction | DNA methylation & mtDNA interaction | (“DNA methylation” OR “methylome” OR “promoter methylation” OR “DNMT”) AND (“mitochondrial DNA” OR “mtDNA” OR “mitochondrial dysfunction” OR “OXPHOS”) AND (“breast cancer” OR “hereditary breast cancer”) AND (“epigenetic” OR “hereditary”) |
| Histone modifications & mitochondrial dysfunction | (“histone modification” OR “histone acetylation” OR “H3K4” OR “H3K9” OR “H3K27” OR “H4 acetylation” OR “chromatin remodeling”) AND (“mitochondrial” OR “mitochondrial dysfunction” OR “retrograde signaling” OR “mtDNA”) AND (“breast cancer” OR “cancer cells”) | |
| microRNA & mtDNA-related metabolic reprogramming | (“microRNA” OR “miRNA” OR “miR-” OR “ncRNA” OR “small RNA”) AND (“mitochondrial” OR “mtDNA” OR “OXPHOS” OR “metabolic reprogramming” OR “Warburg”) AND (“breast cancer” OR “cancer”) AND (“expression” OR “dysregulation” OR “biomarker”) | |
| Nuclear-mitochondrial crosstalk | (“retrograde signaling” OR “mitochondrial stress” OR “ATFS-1” OR “JNK signaling” OR “ROS” OR “reactive oxygen species”) AND (“mtDNA” OR “mitochondrial dysfunction”) AND (“gene expression” OR “epigenetic” OR “histone” OR “methylation”) AND (“cancer” OR “breast”) | |
| Epigenetic Biomarkers in Non-BRCA Hereditary BC | Epigenetic signatures in hereditary BC | (“hereditary breast cancer” OR “HBOC” OR “BRCA-negative” OR “non-BRCA hereditary”) AND (“epigenetic” OR “methylation” OR “histone” OR “microRNA” OR “chromatin”) AND (“biomarker” OR “signature” OR “classifier” OR “profiling”) AND (“risk assessment” OR “prognosis” OR “progression”) |
| ctDNA & mtDNA liquid biopsy in hereditary BC | (“circulating” OR “extracellular vesicles” OR “exosome” OR “cell-free”) AND (“mtDNA” OR “mitochondrial DNA” OR “tumor DNA”) AND (“breast cancer” OR “hereditary breast cancer”) AND (“detection” OR “biomarker” OR “monitoring” OR “screening”) | |
| Clinical Translation & Risk Models | Risk prediction models for non-BRCA hereditary BC | (“risk assessment” OR “risk prediction” OR “risk model” OR “risk score”) AND (“hereditary breast cancer” OR “BRCA-negative”) AND (“genetic variant” OR “polygenic” OR “mtDNA” OR “epigenetic”) AND (“validation” OR “calibration” OR “performance”) |
| Therapeutic targets in mtDNA-associated BC | (“mtDNA” OR “mitochondrial”) AND (“breast cancer” OR “cancer”) AND (“therapeutic” OR “drug target” OR “treatment” OR “intervention” OR “antioxidant” OR “metabolism”) AND (“OXPHOS” OR “ROS” OR “apoptosis” OR “chemoresistance”) |
Inclusion Criteria
Hereditary breast cancer was defined and studies were screened based on the following National Comprehensive Cancer Network (NCCN Version 3.2025) genetic/familial high-risk assessment criteria: (a) ≥2 first- or second-degree relatives affected by breast cancer; (b) diagnosis before or at 45 years of age; (c) diagnosis of triple-negative breast cancer at ≤60 years; (d) male breast cancer in the family; (e) both breast and ovarian cancer; (f) autosomal dominant or maternal inheritance; or (g) negative BRCA 1/2 status in ≥1 affected relative. Unselected population cohorts or sporadic breast cancer cases without family history were excluded.
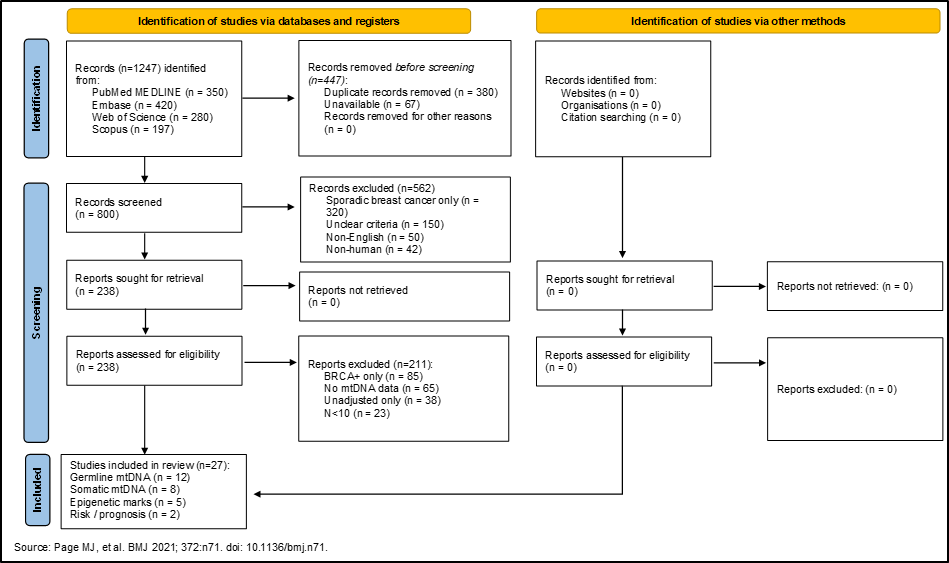
Peer-reviewed articles published in English between 1995 and 2025 that were available as full-text publications were included. Only human studies that investigated germline or somatic mtDNA variants (including point mutations, deletions, insertions, or copy number variations) or epigenetic modifications (including DNA methylation, histone modifications, or non-coding RNA alterations) associated with mtDNA dysfunction in relation to breast cancer risk, progression, or prognosis were identified and included. Studies that explicitly stated sequencing method, haplogroup assignment, nuclear-mitochondrial DNA segment (NUMT) filtering, heteroplasmy quantification, and HGVS nomenclature were prioritized. Although studies lacking these details were included, they were flagged for risk of bias assessment. We have depicted the identification and screening process of the literature in a PRISMA flow diagram (Figure 1).
Data Extraction and Synthesis
We recorded study characteristics (first author, publication year, country of origin, study design) and population details including sample size (N cases/ controls), ancestry/ethnicity classification, pedigree criteria for hereditary breast cancer definition, and BRCA testing status for all the selected references. While key findings related to the types of mtDNA variants studied (germline vs. somatic, specific genomic regions analysed, and detection methods used) were extracted from studies investigating the role of mtDNA in breast cancers, epigenetic studies were scanned for specific modifications (DNA methylation patterns, histone modifications, microRNA expression) that were associated with non-BRCA hereditary breast cancers. Risk of manifestation of non-BRCA hereditary breast cancer, tumour characteristics, treatment response, and prognostic outcomes were also documented.
Findings with effect sizes, including odds ratios [OR], hazard ratios [HR], relative risks [RR] with 95% confidence intervals (CIs), were considered and multivariate-adjusted estimates with covariates were prioritized for survival analyses. We documented the presence or absence of ancestry adjustment (principal component analysis, matched controls) and flagged unadjusted findings or single-decimal precision reporting as quality concerns. Finally, we grouped the selected studies into those that investigated mitochondrial DNA variants in hereditary breast cancer, epigenetic modifications and their role in cancer predisposition, and the intersection between mitochondrial dysfunction and epigenetic regulation.
Risk of bias
We used the Newcastle-Ottawa Scale (NOS) to assess selection bias (population representativeness, control adequacy, participation rates), exposure (quality of mtDNA assay, NUMT filtering, heteroplasmy threshold specification), and outcome ascertainment (breast cancer diagnosis confirmation, hereditary criteria documentation, follow-up adequacy). Genotyping accuracy, population stratification controls, and replication status were assessed using Q-Genie. Finally, the grading of recommendations assessment, development, and evaluation (GRADE) tool helped us evaluate the risk of bias (NOS score), consistency across populations, relevance of outcome), precision, and publication bias. Table 2 shows the evidence tier for the studies based on their GRADE level.
| Evidence Tier | No. of Studies | % of Total | Newcastle-Ottawa Score (NOS) | Q-Genie Score (0–8) | Selection Quality | Genotyping Quality | Ancestry Adjustment | GRADE Certainty | Interpretation |
| Tier 1 | 8 | 29.63% | 7–9/9 (Low risk) | 7–8/8 (High) | Case-control matched; consecutive recruitment; clear hereditary criteria (a–g) | MAF reported; HWE tested; CR ≥95%; depth ≥1,000× | PC analysis OR matched controls OR stratified by ancestry (≥2 ancestries) | HIGH | Strong evidence; multivariate-adjusted; replicable across populations; ancestry-aware; suitable for clinical interpretation |
| Tier 2 (Germline) | 4 | 14.82% | 5–6/9 (Moderate risk) | 4–6/8 (Moderate) | Single cohort; some selection bias; hereditary criteria documented but limited | Partial QC (1–2 metrics); depth 500–1,000×; some genotypes unvalidated | Age-matched only; OR single ancestry; no PC adjustment | MODERATE | Moderate strength; design limitations (small N, limited adjustment); ancestry concerns; further replication needed |
| Tier 2 (Somatic) | 3 | 11.11% | 6–9/9 (Low-moderate risk) | 6–8/8 (High) | Tumour cohorts; well-documented; prospective | High sequencing depth; NUMT filtering documented | Mixed ancestry; somatic ≠ inherited risk | MODERATE | Moderate strength for somatic biology; NOT transferable to germline hereditary risk; mechanistic insight only |
| Tier 2 (Epigenetic) | 2 | 7.41% | 4–7/9 (Moderate-high risk) | 2–5/8 (Low-moderate) | Cell lines/cybrids; limited family-based cohorts; phenotype unclear | Epigenetic assay methods variable; validation limited | No ancestry stratification; mostly European cell lines | LOW | Low strength; mostly mechanistic/indirect; cell-line derived; very limited human hereditary BC validation |
| Tier 3 | 6 | 22.22% | <5/9 (High risk) | <4/8 (Low) | Sporadic/unselected BC; hereditary criteria absent or unclear | QC metrics absent; no depth/CR reported; validation lacking | Single ancestry; no adjustment; population stratification bias likely | VERY LOW | Very low strength; sporadic populations; small N; unadjusted; high publication/ selection bias; NOT hereditary BC; excluded from meta-analysis |
| Reviews | 4 | 14.81% | N/A (not empirical studies) | N/A | N/A | N/A | N/A | LOW | Useful for context/mechanism; NOT primary evidence; included for background |
Results and Discussion
Germline and Somatic mtDNA Variants in Non-BRCA Hereditary Breast Cancer
Common mtDNA Variants
Studies have identified a diverse spectrum of mtDNA variants associated with non-BRCA hereditary breast cancer1,2. These variants exhibit enrichment in specific genomic regions. The displacement loop (D-loop) and protein-coding genes essential for oxidative phosphorylation are the two primary regions that exhibit variant clusters.
D-loop region mutations are the most extensively studied variants1. Key polymorphisms including m.16519T>C, m.16189T>C, m.16290C>T, and various deletions and duplications at positions 310 and 315 have been identified in this region. The D-loop variants, m.310del (rs869289246), m.315dup (rs369786048), and m.16519T>C (rs3937033), have been linked with predisposition to breast cancer in multiple populations1,2. The m.16189T>C mutation and several D310 mutations in the D-loop region have also been associated with breast cancer susceptibility1,2. Focused-population studies demonstrate distinct patterns of the frequency at which variants occur and associations with haplogroups2,4,5.
Table 3 summarises the germline mtDNA variants associated with risk of non-BRCA hereditary breast cancer discussed in tier 1 and 2 studies based on specific variants and haplogroups across diverse ancestries.
| Variant/ Haplogroup | Study Design | Study population; No of cases; Ancestry | Study relevance | OR/RR [95% CI] | Adjusted for | Evidence Tier |
| m.16189T>C (D-loop); Multi Haplogroup | Case-control | Multi-ethnic; N=388; European American, African American, Malay6 | Criteria b, c (early-onset <45 yrs; triple-negative breast cancer (TNBC) cases included) | 2.1–2.8 | Age, ancestry | Tier 1 |
| m.310del (D-loop); Haplogroup H | Case-control (hereditary breast cancer cohort) | Italian; 194 cases/ 194 controls;Italian1 | Criteria a, b, g (≥ 2 affected relatives; early-onset; BRCA-negative confirmed) | 3.2 [1.8–5.6] | Age, BRCA status | Tier 1 |
| Haplogroup M | Case-control | Pakistani; 71 cases/60 controls; South Asian4 | Criteria a, b (familial clustering; early-onset cases) | 2.03 [0.66–6.29] | Age | Tier 2 |
| Haplogroup U | Case-control | Criteria a, b (familial clustering, early-onset cases) | 0.13 [3.02–20.54] |
OR/RR: odds ratio or relative risk with 95% confidence intervals; adjusted for: indicates covariates controlled in multivariate analyses; evidence tier 1: multivariate-adjusted, multiple populations, low risk of bias (NOS 7–9); tier 2: single cohort, moderate risk of bias (NOS 5–6); haplogroup multi: indicates mixed haplogroup background in multi-ethnic studies
Somatic mtDNA mutations and tumour behaviour
Multiple interconnected pathways that collectively compromise cellular homeostasis and promote tumorigenesis impact the role of mtDNA variants in breast cancer development. The primary mechanism involves disruption of oxidative phosphorylation (OXPHOS), resulting in a cascade of cellular changes that facilitate cancer development and progression20.
Somatic mtDNA mutations, found in 73.7% of breast cancer tumours, usually cluster in the promoter and replication regions and in genes coding for complex I of the electron transport chain (ETC)6. This clustering pattern indicates positive selection for mutations that affect critical mitochondrial processes. Accumulation of mtDNA mutations progressively destabilizes the OXPHOS system allowing cancer cells to adapt to new microenvironments and acquire metastatic potential. Some mutations are capable of totally inhibiting the functioning of the OXPHOS system and promote tumour development21,22,23
Irreversible damage to OXPHOS shifts the cellular energy production pathways toward aerobic glycolysis3, representing an early event in breast tumorigenesis. There is a compensatory increase in glycolytic ATP production that causes alterations in the ETC. While mutations in the coding mtDNA support tumour cell proliferation by altering protein function and OXPHOS efficiency, mutations in the non-coding regions affect replication, transcription, and structural organization of mtDNA21,24.
Defective OXPHOS function increases the production of reactive oxygen species (ROS), resulting in genomic instability, cell transformation, and tumour progression. The increased oxidative stress causes accumulation of DNA damage and suppresses p53 function, activating intracellular signalling pathways. Mutations in mtDNA can also suppress apoptosis in cancer cells3. Therefore, the paradoxical association of higher number of mutations in mtDNA with better overall survival may be attributed to somatic tumour burden rather than inherited risks associated with mtDNA mutations6,7,8.
| Study design | Study population; No of cases; Ancestry | Study Relevance | Somatic Mutation Frequency | Heteroplasmy Status | Survival Association | Evidence Tier |
| Prospective cohort | 92 cases/ 92 controls; Mexican women (mainly TNBC)6 | Indirect: Somatic changes may interact with inherited mDNA background | 68/92 73.9% | 78.6% heteroplasmic; 21.4% homoplasmic | Better overall survival with higher burden | Tier 1 |
| Multi-cancer whole-genome sequencing | TCGA breast cancer subset; ~1000 cases; mixed ancestry9 | Mechanistic: Clonal evolution patterns relevant to inherited predisposition | 45.7%–73.7% | Clonal expansion documented | Clonal selection evidence | Tier 1 |
| Large cohort retrospective analysis | 1,300+ breast cancers (TCGA); ~80% European, 15% African10 | Indirect: Tumour mtDNA copy number as proxy for mitochondrial fitness | mtDNA copy number depletion common | Quantified across cancers | Lower copy number = worse survival | Tier 1 |
Inheritance Patterns and Haplogroups
The inheritance patterns of breast cancer-associated mtDNA variants exhibit unique characteristics that distinguish them from nuclear genetic factors. Mendelian inheritance does not explain the clustering patterns of non-BRCA hereditary breast cancers caused by mtDNA. Unlike nuclear DNA, mtDNA exhibits exclusive maternal inheritance1. This inheritance pattern accounts for instances of hereditary breast cancer that cannot be attributed to nuclear gene mutations.
Most breast tumours are homoplasmic, although many variants exist in the heteroplasmic state in normal tissues6,9. The dominance of homoplasmy is attributed to selective growth advantage offered by the tumour microenvironment to the mutated mtDNA and the over-replication of mutant mtDNA during tumorigenesis21. Tumour behaviour and treatment responses are heavily influenced by the transition from heteroplasmic to homoplasmic state25,26.
Significant ethnic and geographic variations have been observed in population-specific variants2,5. While haplogroups such as X and H are linked to BRCA1 and BRCA2 carriers, respectively27,28, haplogroup U protects against the risk of breast cancer2,29. mtDNA variation is highly structured in populations and susceptible to false-positive findings due to population stratification5,11,12. This warrants the need for replication studies to validate associations in independent populations30.
The population-specific breast cancer risk associations shown in Table 5 highlight mtDNA haplogroup variation and underscore the importance of ancestry-stratified analysis.
| Haplogroup4,5,12,31,32,33,24,34 | Primary Regions | Global Frequency (%)35 | Breast Cancer Risk Association |
| H | EUROPE (Central, Western, Northern) | ~40–50 | Neutral/Protective |
| L | AFRICA (Sub-Saharan) | ~35 (African pop.) | ↑ Risk |
| M | SOUTH ASIA (India, Pakistan, Bangladesh) | ~30–40 (SA only) | ↑ Risk (OR 2.03, p<0.001) |
| U | EUROPE (Eastern) + SOUTH ASIA | ~13.6–15% (Europeans); ~31.3% (Pakistani) | Protective (OR 0.13) |
| J/ T | MEDITERRANEAN | ~9–10% | ↑ Risk |
| V | NORTHERN EUROPE | ~4–5% | Neutral |
| W | WESTERN EUROPE | ~1–2% | Neutral |
| X | CENTRAL ASIA | ~2–3% | ↑ Risk |
| D5 | EAST ASIA (China) | Variable | ↑Risk (OR 2.789, p=0.007) |
| A/M7 | EAST ASIA (China, Vietnam, Mongolia) | ~10–15% | Protective (OR 0.195 multi-cancer) |
| B | EAST/SE ASIA (Japan, Thailand, Philippines) | ~10% | Unknown |
Epigenetic Modifications Associated with Mitochondrial Dysfunction
DNA Methylation Changes
Studies investigating the relationship between mtDNA variants and DNA methylation patterns suggest complex interactions between mitochondrial dysfunction and nuclear epigenetic regulation that may contribute to cancer development and progression. Current research on methylation patterns focuses on nuclear genes involved in mitochondrial biogenesis and function. Findings suggest that mtDNA mutations influence the methylation of nuclear genes encoding mitochondrial proteins36,17 resulting in mitochondrial dysfunction37,38,39. The involvement of retrograde signalling pathways that communicate mitochondrial dysfunction to the nucleus results in nuclear-mitochondrial crosstalk16,19, a critical mechanism by which mtDNA variants influence epigenetic regulation. Homeostasis of cellular energy also requires compensatory epigenetic changes that potentially contribute to cancer development38
DNA methylation patterns and histone modifications are affected by the increased ROS production associated with defective OXPHOS, creating a feedback loop between mitochondrial dysfunction and nuclear epigenetic dysregulation. This crosstalk accounts for the role of mtDNA variants in the development of cancer through mechanisms beyond direct effects on mitochondrial function. Evidence, albeit limited, indicates the influence of mitochondrial dysfunction on the expression of genes involved in cell cycle regulation, apoptosis, and DNA repair through epigenetic mechanisms. Epigenetic changes that alter the expression of tumour suppressors and oncogenes have been found to be involved in signalling pathways associated with OXPHOS defects40,17.
Histone Modifications
Although direct evidence that implicates specific histone modifications associated with mtDNA variants in breast cancer is limited17, potential mechanisms through which mitochondrial dysfunction influences chromatin structure and gene expression regulation have been suggested.
Chromatin changes affecting mitochondrial biogenesis represent an important mechanism by which cells respond to mtDNA-induced mitochondrial dysfunction16,17. The retrograde signaling pathways activated by mitochondrial dysfunction influence histone modifications at loci encoding mitochondrial proteins, potentially altering the expression of genes involved in mitochondrial biogenesis and function. Although these compensatory mechanisms initially protect against the consequences of mtDNA mutations, the resulting dysregulation of cellular homeostasis ultimately contributes to the development of cancer41.
The interaction of mtDNA with nuclear transcription factors is another potential mechanism by which mtDNA variants associate with epigenetic regulation. Mitochondrial dysfunction affects histone modifications, thus altering the expression of transcription factors that regulate both, mitochondrial and nuclear genes. The increased ROS production mediated by mtDNA mutations affects the function of the transcription factor and chromatin structure, contributing to the epigenetic dysregulation observed in cancer cells16,17. The effects of mtDNA variants on epigenetic regulation vary depending on cell types and tissues. However, tissue-specific patterns of histone modifications associated with mtDNA variants remain largely unexplored.
MicroRNA Regulation
mtDNA variants influence microRNAs (miRNAs) that target mitochondrial pathways42 by affecting mitochondrial function and cellular metabolism. Mitochondrial dysfunction associated with mtDNA mutations alters the expression of miRNAs that regulate genes involved in mitochondrial biogenesis, oxidative phosphorylation, and cellular metabolism. This dysregulation causes metabolic reprogramming in cancer cells and is a potential therapeutic target.
Dysregulation in mtDNA variant-associated breast cancers results from metabolic changes that influence miRNA expression patterns and contribute to cancer development and progression through effects on cell cycle regulation, apoptosis, and metastasis. The reversible nature of miRNA-mediated gene regulation makes this pathway an attractive target for therapeutic intervention, particularly in cancers associated with mitochondrial dysfunction42,20.
Mito-nuclear crosstalk and retrograde signaling
Germline mtDNA variants segregate in affected families, which partially elucidates the missing heritability among non-BRCA cases1,11. Somatic mtDNA mutations, on the other hand, occur during tumorigenesis and are often concentrated in Complex I genes6, suggestive of positive selection of mitochondrial alterations that favour the survival of cancer cells. Expansion of tumour clones results in a shift from heteroplasmy to homoplasmy6. The mitochondrial dysfunction resulting from accumulated mtDNA mutations and reduced mtDNA copy number causes compensatory reprogramming of gene expression by the cell nucleus in response to mitochondrial distress27. This retrograde signaling may potentially occur through the calcineurin-dependent pathways20 and alter nuclear pathways, which would in turn facilitate epigenetic modifications such as those that affect DNA methylation, histone modifications, and miRNA regulation. These epigenetic modifications could stabilize and amplify tumour-promoting effects resulting in EMT induction, expansion of tumour cells, aggressive tumour behaviour, and therapy resistance43,44.
Figure 2 is a schematic representation of mito‑nuclear crosstalk and retrograde signaling conceptualized based on indirect evidence derived from mechanistic studies. We emphasize that the proposed schematic is largely speculative due to a dearth of studies that provide direct evidence connecting specific mtDNA variants and epigenetic marks in hereditary breast cancer cohorts.
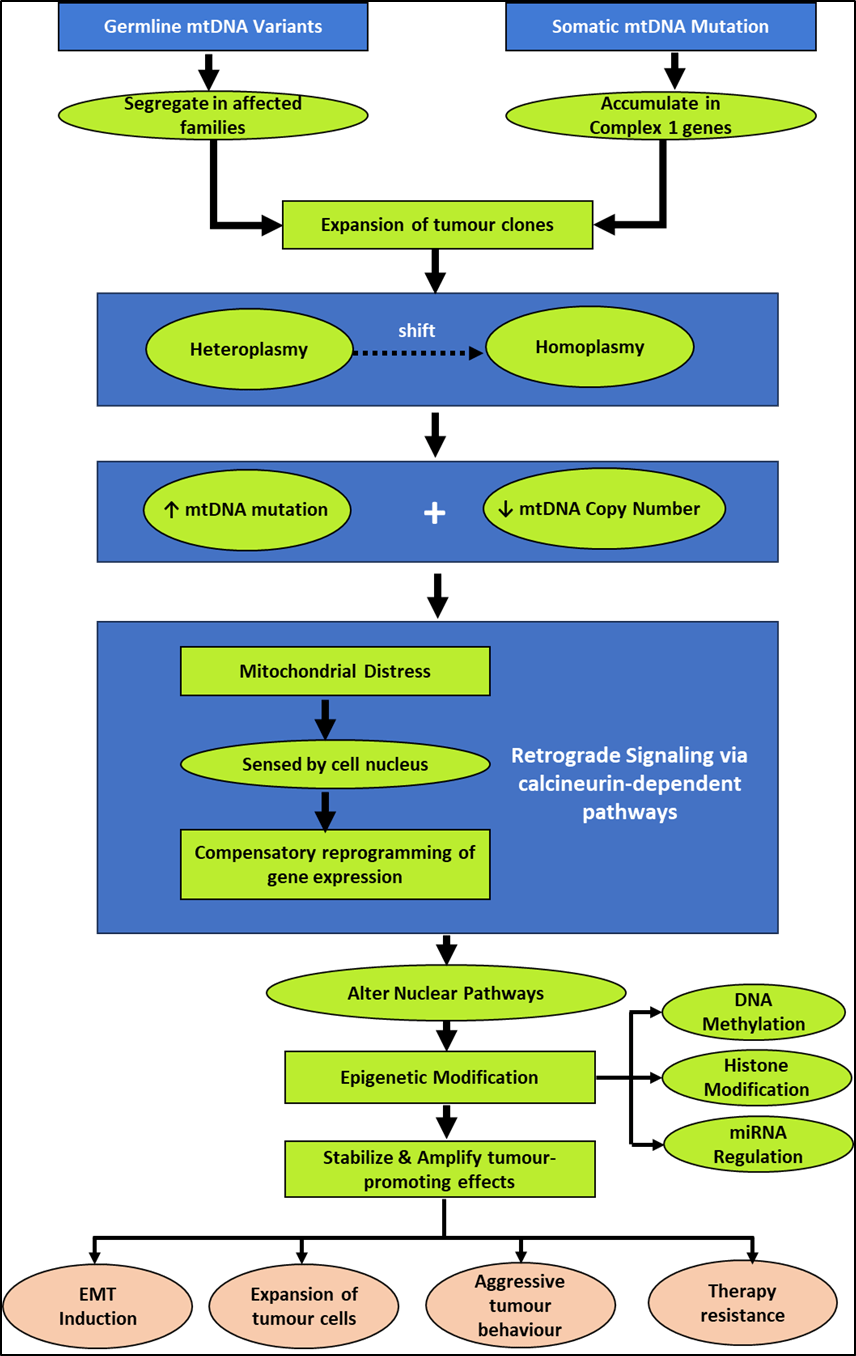
Table 6 represents a hypothetical matrix connecting mtDNA variant classes to proposed epigenetic effects, and observed phenotypes. In each context, the evidence strength and testable gaps have been highlighted.
| Variant Class | Proposed Epigenetic Effect | Observed Phenotype | Evidence Strength | Testable Gap |
| D-loop variants m.16189T>C, m.310del, m.315dup; germline1,2,11 | ↑DNA methylation at NRF1/TFAM promoters; miRNA dysregulation (miR-663 ↓) | ↓Mitochondrial biogenesis; altered metabolic fitness; predisposition to accumulation of somatic mutations | LOW | Cybrid models with specific D-loop variants (m.16189T>C vs. wild-type); allele-specific methylation mapping near NRF1 locus in patient fibroblasts |
| Complex I somatic mutations (ND1, ND2, ND5; tumour-acquired)6,9 | H3K4ac ↑ at OXPHOS genes; H3K27me3 ↑ at EMT-suppressor loci (E-cadherin); DNMT1/3A ↑ expression | EMT induction; cancer stem cell expansion; therapy resistance | MODERATE | Single-cell multi-omics (mtDNA genotype + methylome + transcriptome) in patient tumours; CRISPR-induced ND1/ND2 mutations in organoid models |
| mtDNA copy number reduction (copy number depletion; somatic)7,8,45 | Global DNAm remodeling; histone acetylation ↓ at metabolic genes; miRNA dysregulation (metabolic-associated miRNAs) | ↑Aerobic glycolysis; ↑Anti-apoptotic proteins (Bcl-2); ↑Therapy resistance/ paradoxical ROS-mediated chemosensitivity | MODERATE | Epigenome-wide association study (EWAS) correlating mtDNA copy number with DNAm patterns in TCGA tumours; mtDNA CN manipulation in patient-derived organoids |
| Haplogroup M (South Asian; germline)4,46 | Basal ↑DNMT expression; altered DNAm at mitochondrial biogenesis loci (PGC1α, SIRT3, NRF1); microRNA profile skew | Early age-of-onset breast cancer; ↑TNBC phenotype; potential ↑therapy sensitivity (ROS-mediated) | VERY LOW | Haplogroup-stratified EWAS in Pakistani/South Asian hereditary breast cancer families (n≥200 cases); baseline DNAm profiles (normal vs. tumour-adjacent) by haplogroup |
| Haplogroup U (European; germline background; protective)1,2,11 | Basal ↓DNMT expression OR optimal mitochondrial function → basal epigenetic stability | Lower BC risk; potentially delayed tumour progression; better survival outcomes | LOW | Haplogroup-specific epigenome profiling in normal breast tissue (haplogroup U vs. M vs. H carriers); baseline DNAm/histone landscape before tumour initiation |
| Heteroplasmy shift (heteroplasmic → homoplasmic; somatic clonal expansion)6,9 | Progressive DNAm changes at metabolic/survival genes during clonal expansion; histone remodeling tracking heteroplasmy levels | Clonal selection for intermediate heteroplasmy; EMT/CSC features correlate with heteroplasmy level; survival advantage47 | MODERATE | Longitudinal epigenome profiling (WGBS, ATAC-seq, single-cell) paired with mtDNA heteroplasmy quantification in patient tumours; isogenic cybrid lines with controlled VAF levels |
ATFS-1 – Activating Transcription Factor associated with Stress; CRISPR – Clustered Regularly Interspaced Short Palindromic Repeats; CSC – Cancer Stem Cell; DNAm – DNA Methylation; DNMT – DNA Methyltransferase; EMT Epithelial-Mesenchymal Transition; EWAS – Epigenome-Wide Association Study; H3K4ac – Histone H3 Lysine 4 Acetylation (active chromatin mark); H3K27me3 – Histone H3 Lysine 27 Trimethylation (repressive mark); ND1, ND2, ND5 – NADH Dehydrogenase Subunits 1, 2, 5 (Complex I genes); NRF1 – Nuclear Respiratory Factor 1; PGC1α – Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha; SIRT3 – Sirtuin 3 (NAD-dependent histone deacetylase); TCGA – The Cancer Genome Atlas; TFAM – Mitochondrial Transcription Factor A; VAF – Variant Allele Frequency; WGBS – Whole-Genome Bisulfite Sequencing
Clinical Implications and Translational Readiness
Currently, mtDNA variants and associated epigenetic markers are in the early stages of validation, making them exploratory biomarkers from a translational perspective. While assay variability, NUMT contamination, and inconsistent heteroplasmy thresholds limit analytical validity, clinical validity is unsubstantiated due to predominantly observational studies based on relatively small and ancestry-limited cohorts. Consequently, clinical implications of using mtDNA variants and epigenetic markers to design clinical risk models or surveillance protocols are presently hypothetical and subject to appropriate validation through future research.
Proposed framework for NUMT-aware assay pipeline validation
Presently, analytical approaches including high-depth next-generation sequencing (NGS) and heteroplasmy quantification methods for detection of mtDNA variants that provide reliable detection of homoplasmic and heteroplasmic variants above defined thresholds, about 5% variant allele frequency (VAF), in clinical settings are based on analytical validity (the laboratory assay’s ability to accurately identify mtDNA variants with appropriate sensitivity and specificity), clinical validity (the association between detected mtDNA variants and hereditary cancer risk), and clinical utility (whether detection of mtDNA variants improves patient management and health outcomes). However, substantial inter-laboratory variation in heteroplasmy threshold definition (ranging from 1% to 20%) and NUMT (nuclear mitochondrial DNA) handling protocols persists across platforms48,49,50,51.
In this review, we propose a conditional and exploratory framework comprising of three critical modifications to the existing assay pipeline. Mutation-specific heteroplasmy thresholds based on tissue-dependent biochemical penetrance can be implemented rather than universal cut-offs to account for VAF thresholds of pathogenic mtDNA variants below which phenotypic manifestation is unlikely, a heterogeneity that is poorly characterized in cancer predisposition52,53. Implementation of methylation-based separation methods would enable reliable distinction between mtDNA and NUMT contamination prior to variant calling. Although the significantly higher CpG methylation exhibited by NUMTs in comparison to mtDNA facilitates computational or biochemical filtering, routine diagnostic pipelines do not have standardized integrated protocols54,55,56. Finally, established haplogroup-stratified baseline methylation and mitochondrial function profiles would contextualize mtDNA variants within population-specific epigenetic backgrounds and ensure ancestry-calibrated processes52.
It is important to reiterate that clinical biopsy-based mtDNA variant detection cannot be recommended as a standard-of-care risk stratification tool until the assay pipeline is standardized across independent laboratories, replicability of effect sizes is comparable to established moderate-penetrance nuclear genes, the assay is validated in large, ancestry-diverse, family-based cohorts with long-term follow-up, and mtDNA variant detection is shown to improve clinical decision-making beyond existing nuclear gene testing.
Risk Assessment
Existing prediction models do not account for genetic predisposition of breast cancers that test negative for the known susceptibility genes57,58. mtDNA variants fit into this critical gap in risk assessment for breast cancer. The maternal inheritance pattern of mtDNA variants explains cancer clustering in families that do not exhibit traditional autosomal dominant inheritance patterns1,27, potentially resolving unexplained hereditary cases of breast cancer. The prediction models would also benefit significantly from the integration of population-specific and haplogroup-specific considerations and patterns.
Disease Progression
mtDNA variants influence breast cancer progression through complex mechanisms affecting tumour behaviour and treatment response. While higher frequency of somatic mtDNA mutations in tumours is typically associated with aggressive disease progressive and poor prognosis, higher frequency of mtDNA mutations in somatic tumours6,7,8 associate with better overall survival, independent of tumour stage and hormone receptor status. The high levels of oxidative stress resulting from the mtDNA mutations increase susceptibility to anticancer agents. mtDNA variants also impact the metastatic potential of breast cancers. While severe mutations increase tissue susceptibility, milder mutations enable adaptation to new microenvironments. mtDNA-associated tumours also gain survival advantages in diverse metastatic sites owing to metabolic reprogramming facilitating the shift to aerobic glycolysis3. Metabolic adaptations have the potential to enhance sensitivity to ROS-inducing chemotherapies, although the resulting apoptosis suppression and AKT pathway activation culminates in resistance to treatment.
Prognosis and Survival
Analysis of somatic mtDNA mutations in tumours has promising prognostic implications. Depletion of somatic mtDNA is a potential biomarker for early detection and prognosis59,60,61. The transition of heteroplasmic tumours to the homoplasmic state is a good indicators of treatment response patterns. The positive correlation between the frequency of somatic mtDNA mutations and survival, notwithstanding traditional prognostic factors, highlights the potential of mtDNA analysis as a prognostic beyond conventional markers6,7.
Current Challenges and Limitations
Studies investigating mtDNA variants in breast cancer face several significant challenges that limit the translation of research findings into clinical applications. The number of copies of mtDNA per cell is very high, which makes accurate quantification of heteroplasmy levels and detection of low-level variants challenging. Nuclear mtDNA sequences (NUMTs) interfere with sequencing and analysis of mtDNA, increasing the likelihood of false-positive results62. Furthermore, the circular structure of mtDNA and the presence of homopolymeric regions, particularly in the D-loop, poses additional sequencing challenges that require specialized analytical approaches. Separation of mitochondrial and nuclear DNA fractions is a crucial aspect of mtDNA analysis. The complexity of the process requires specialized sample processing skills and have the potential to introduce variability in the results.
The lack of standardized protocols for mtDNA extraction, sequencing, and analysis makes it difficult to compare results across studies and laboratories. Methodologies for quantification of heteroplasmy are inconsistent across studies, with different thresholds used to define heteroplasmy. Additionally, the definition of pathogenic and benign mtDNA variants lacks consensus. Functional validation of most reported variants is very limited. In comparison to nuclear DNA analysis, quality control standards for mtDNA sequencing and analysis remain underdeveloped.
Although we identified more than 100 studies as eligible for this review, only five studies focused on mtDNA germline variants and somatic mutations and reported findings based on ≥2 independent cohorts, included multivariate analysis, and exhibited ancestry-aware categorization1,4,6,9,11. While most of the remaining studies were based on either single large cohort or ≥2 smaller cohorts, included multivariate analysis, mechanistic studies, and population-specific analyses, some of the studies were predominantly descriptive, unadjusted, comprised of case reports or small case series, and did not have definite inclusion criteria. The heterogeneous nature of mtDNA variants warrants the need for large, well-characterized cohorts to identify clinically meaningful associations. Longitudinal studies with extended follow-up periods could potentially highlight prognostic implications of mtDNA variants and their interactions with treatment factors.
Most of the identified studies focus on particular populations and ethnic groups, with the exception of a few studies4,11, limiting diversity of ethnic groups in study populations. Non-European ancestries are mostly underrepresented, resulting in findings being derived exclusively from European cohorts. This is a critical limitation that affects the generalizability of research findings. mtDNA haplogroups and variants are population-specific and findings based on diverse study populations underscore the clinical implications of mtDNA variants across different ethnic groups. The lack of diversity in study populations often perpetuates health disparities and limits the clinical applications of mtDNA-based approaches in diverse patient populations. The scarcity of studies focused on non-BRCA hereditary breast cancer mtDNA that are based on larger, well-designed, ancestry-stratified cohorts warrants caution in clinical translation of the current research findings.
Implications and Significance
Our study contributes significantly to the current knowledge of non-BRCA hereditary breast cancer by collating relevant research findings and suggesting that mitochondrial DNA variants and epigenetic modifications are significant contributors to cancer risk, progression, and prognosis. We have highlighted that mtDNA variants have measurable effect sizes comparable to moderate-penetrance nuclear genes associated with hereditary breast cancer. This fills a critical gap in explaining breast cancer cases that test negative for BRCA1/2, but are genetically predisposed. Our findings underscore the shortcomings of conventional assumptions about the associations between mutational frequency and overall survival that could potentially inform therapeutic strategies. This review also emphasizes the need to focus on the maternal inheritance patterns of mtDNA variants as an alternative explanation when traditional autosomal dominant inheritance models fall short, thereby resolving diagnostic uncertainties and improving genetic counselling approaches. Furthermore, the collated insights on population-specific distributions of mtDNA variants discussed in our review highlight the importance of diverse study populations in reducing health disparities and ensuring equitable translation of precision medicine approaches across different ethnic groups.
Future Directions
There is a compelling need for functional studies using cellular and animal models for the advancement of mtDNA variant research. Study populations spanning multi-ethnic groups and large sample sizes are crucial for validating associations and identifying population-specific effects. Longitudinal cohort studies conducted pre-diagnosis play a pivotal role in revealing temporal relationships and early biomarkers. Systematic development of standardized, clinically-validated diagnostic assays with next-generation sequencing panels for comprehensive mtDNA analysis, supported by quality assurance programs and point-of-care testing capabilities are warranted for reliable clinical translation. Pharmacogenomic studies and metabolic profiling have the potential to guide therapy selection for personalized treatment approaches.
Risk prediction models would benefit from the incorporation of mtDNA variants. The development of therapeutic agents that target epigenetic changes and exploit the unique biology of mtDNA variants such as those that target oxidative phosphorylation defects, mitochondrial-targeted antioxidants, and metabolic inhibitors exploiting glycolytic vulnerabilities in combination with conventional chemotherapy, immunotherapy, and autophagy modulators could potentially reverse dysfunction-associated changes. Single-cell techniques could be explored to quantify heteroplasmy. Leveraging technological advances such as machine learning algorithms for variant interpretation, deep learning for pattern identification, and AI-driven drug discovery, supported by multi-omics integration platforms is important for managing data complexity to ensure comprehensive analysis of large-scale datasets.
Limitations
We acknowledge several important limitations of our study. Our ability to draw definitive conclusions and provide a meta-analysis of the findings was limited due to the heterogeneous nature of study populations, limited representation of diverse ethnic groups, relatively small sample sizes, and definitions of hereditary breast cancer across included studies. We recognize the potential for variability in results across studies due to the lack of standardized protocols for heteroplasmy quantification and variant interpretation. Most associations derived from our findings are correlative rather than causative owing to the limited functional validation of identified variants. Furthermore, our interpretations of the functional associations between mitochondrial dysfunction and nuclear gene regulation may reflect the scarcity of evidence regarding epigenetic modifications specifically associated with mtDNA variants.
Conclusion
This comprehensive review highlights the relevance of mtDNA variants and epigenetic modifications in predisposition to hereditary breast cancers that are not attributed to mutations in known nuclear DNA genes. The findings underscore the critical need to expand beyond nuclear-centric approaches in cancer genetics. It redefines our assumptions about correlations between mutational frequency and survival outcomes.
This study collates knowledge about existing therapeutic approaches and interprets the emerging evidence for epigenetic modifications associated with mitochondrial dysfunction to suggest novel therapeutic vulnerabilities that could be exploited for precision medicine approaches. This review emphasizes the importance of studies with large, diverse, and inclusive populations by providing evidence of the population-specific distributions of mtDNA variants.
The integration of mitochondrial genomics into hereditary breast cancer research represents a fundamental paradigm shift that recognizes the intricate interplay between nuclear and mitochondrial genes in genetic predisposition to cancer.
References
- S. Weigl, A. Paradiso, S. Tommasi. Mitochondria and Familial Predisposition to Breast Cancer. Vol. 14 Current Genomics, 2013. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Tommasi S, Favia P, Weigl S, Bianco A, Pilato B, Russo L, Paradiso A, Petruzzella V. Mitochondrial DNA variants and risk of familial breast cancer: an exploratory study. Vol. 5 Int J Oncol, 2014. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- N. Yadav, D. Chandra. Mitochondrial DNA mutations and breast tumorigenesis. Vol. 1836 Biochimica et Biophysica Acta – Reviews on Cancer, 2013. [↩] [↩] [↩] [↩] [↩]
- N. Khalid, M. U. Khan, R. Rehman, S. Kanwal, T. Zahid, M. U. Ghani, A. Iftikhar, Q. Ali, M. A. Javed. Unraveling the genetic connections for mitochondrial DNA control region and breast cancer susceptibility. Vol. 15 Scientific Reports, 2025. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- V. Riley, A. M. Erzurumluoglu, S. Rodriguez, C. Bonilla. Mitochondrial DNA haplogroups and breast cancer risk factors in the avon longitudinal study of parents and children (alspac). Vol. 9, Genes, 2018. [↩] [↩] [↩] [↩] [↩] [↩]
- C. J. Pérez-Amado, H. Tovar, L. Gómez-Romero, F. O. Beltrán-Anaya, V. Bautista-Piña, C. Dominguez-Reyes, F. Villegas-Carlos, A. Tenorio-Torres, L. A. Alfaro-Ruíz, A. Hidalgo-Miranda, S. Jiménez-Morales. Mitochondrial DNA mutation analysis in breast cancer: shifting from germline heteroplasmy toward homoplasmy in tumors. Vol. 10, Frontiers in Oncology, 2020. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- M. J. A. Weerts, A. M. Sieuwerts, M. Smid, M. P. Look, J. A. Foekens, S. Sleijfer, J. W. M. Martens. Mitochondrial DNA content in breast cancer: Impact on in vitro and in vivo phenotype and patient prognosis. Vol. 7 Oncotarget, 2016. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- S. Chen, Z. Li, Y. He, F. Zhang, H. Li, Y. Liao, Z. Wei, G. Wan, X. Xiang, M. Hu, K. Xia, X. Chen, J. Tang. Elevated mitochondrial DNA copy number in peripheral blood cells is associated with childhood autism. Vol. 15, BMC Psychiatry 2015. [↩] [↩] [↩] [↩] [↩]
- T. C. Larman, S. R. DePalma, A. G. Hadjipanayis, A. Protopopov, J. Zhang, S. B. Gabriel, L. Chin, C. E. Seidman, R. Kucherlapati, J. G. Seidman. Spectrum of somatic mitochondrial mutations in five cancers. Vol. 109, Proceedings of the National Academy of Sciences of the United States of America, 2012. [↩] [↩] [↩] [↩] [↩] [↩]
- E. Reznik, M. L. Miller, Y. Şenbabaoğlu, N. Riaz, J. Sarungbam, S. K. Tickoo, H. A. Al-Ahmadie, W. Lee, V. E. Seshan, A. A. Hakimi, C. Sander. Mitochondrial DNA copy number variation across human cancers. Vol. 5 eLife, 2016. [↩] [↩]
- D. Covarrubias, R. K. Bai, L. J. C. Wong, S. M. Leal. Mitochondrial DNA variant interactions modify breast cancer risk. Vol. 53, Journal of Human Genetics, 2008. [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Y. Li, E. E. Giorgi, K. B. Beckman, C. Caberto, R. Kazma, A. Lum-Jones, C. A. Haiman, L. Le Marchand, D. O. Stram, R. Saxena, I. Cheng. Association between mitochondrial genetic variation and breast cancer risk: the multiethnic cohort. Vol 14 PLoS ONE, 2019. [↩] [↩] [↩]
- D. Doupa, M. N. Badji, F. Mbaye, S. Ka, A. Dem, M. Kane, M. Sembène. Benign breast tumors among senegalese women: diversity and genetic evolution of d-loop. Vol. 02, OALib, 2015 [↩] [↩]
- S. L. Mitchell, R. Goodloe, K. Brown-Gentry, S. A. Pendergrass, D. G. Murdock, D. C. Crawford. Characterization of mitochondrial haplogroups in a large population-based sample from the United States. Vol. 133 Human Genetics, 2014. [↩]
- A. M. Dworkin, T. H. M. Huang, A. E. Toland. Epigenetic alterations in the breast: implications for breast cancer detection, prognosis and treatment. Vol. 19 Seminars in Cancer Biology,2009. [↩]
- D. Yang, J. Kim. Mitochondrial retrograde signalling and metabolic alterations in the tumour microenvironment. Vol. 8 Cells, 2019 [↩] [↩] [↩] [↩]
- Z. Kang, J. Wang, J. Liu, L. Du, X. Liu. Epigenetic modifications in breast cancer: from immune escape mechanisms to therapeutic target discovery. Vol. 16 Frontiers in Immunology, 2025. [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- D. Doupa, M. N. Badji, F. Mbaye, S. Ka, A. Dem, M. Kane, M. Sembène. Benign breast tumors among senegalese women: diversity and genetic evolution of d-loop. Vol. 02, OALib, 2015 [↩]
- M. Guha, S. Srinivasan, G. Ruthel, A. K. Kashina, R. P. Carstens, A. Mendoza, C. Khanna, T. Van Winkle, N. G. Avadhani. Mitochondrial retrograde signaling induces epithelial–mesenchymal transition and generates breast cancer stem cells. Vol. 33, Oncogene, 2014. [↩] [↩]
- T. Carden, B. Singh, V. Mooga, P. Bajpai, K. K. Singh. Epigenetic modification of mir-663 controls mitochondriatonucleus retrograde signaling and tumor progression. Vol. 292, Journal of Biological Chemistry, 2017. [↩] [↩] [↩]
- D. Chen, Z. Yan, Q. Yuan, F. Xie, Y. Liu, Z. Feng, Z. Wang, F. Zhou, J. Xing, Z. Zhang, F. Wang, X. Guo. Mitochondrial dna haplogroups and snps: risk factors in multiple cancers based on a cross-tumor analysis in chinese population. Vol. 33 Cancer Epidemiol Biomarkers Prev, 2024. [↩] [↩] [↩]
- L. Ma, Q. Fu, B. Xu, H. Zhou, J. Gao, X. Shao, J. Xiong, Q. Gu, S. Wen, F. Li, L. Shen, G. Chen, H. Fang, J. Lyu. Breast cancer-associated mitochondrial dna haplogroup promotes neoplastic growth via ros-mediated akt activation. Vol. 142 Int J Cancer, 2018. [↩]
- B. Humphries, Z. Wang, C. Yang. MicroRNA regulation of epigenetic modifiers in breast cancer. Vol. 11 Cancers, 2019. [↩]
- R. K. Bai, S. M. Leal, D. Covarrubias, A. Liu, L. J. C. Wong. Mitochondrial genetic background modifies breast cancer risk. Vol. 67 Cancer Research, 2007. [↩] [↩]
- B. Humphries, Z. Wang, C. Yang. MicroRNA regulation of epigenetic modifiers in breast cancer. Vol. 11 ,Cancers, 2019. [↩]
- R. S. Lee, K. Sad, D. V. Fawwal, J. M. Spangle. Emerging role of epigenetic modifiers in breast cancer pathogenesis and therapeutic response. Vol. 15, Cancers, 2023. [↩]
- L. Shen, J. Wei, T. Chen, J. He, J. Qu, X. He, L. Jiang, Y. Qu, H. Fang, G. Chen, J. Lu, Y. Bai. Evaluating mitochondrial DNA in patients with breast cancer and benign breast disease. Vol. 137, Journal of Cancer Research and Clinical Oncology, 2011. [↩] [↩] [↩]
- K. S. Vikramdeo, S. Anand, S. K. Sudan, P. Pramanik, S. Singh, A. K. Godwin, A. P. Singh, S. Dasgupta. Profiling mitochondrial dna mutations in tumors and circulating extracellular vesicles of triple-negative breast cancer patients for potential biomarker development. Vol. 5, FASEB BioAdvances, 2023. [↩]
- D. Covarrubias, R. K. Bai, L. J. C. Wong, S. M. Leal. Mitochondrial DNA variant interactions modify breast cancer risk. Vol. 53, Journal of Human Genetics, 2008. [↩]
- K. S. Vikramdeo, S. Anand, S. K. Sudan, P. Pramanik, S. Singh, A. K. Godwin, A. P. Singh, S. Dasgupta. Profiling mitochondrial dna mutations in tumors and circulating extracellular vesicles of triple-negative breast cancer patients for potential biomarker development. Vol. 5, FASEB BioAdvances, 2023. [↩]
- D. Chen, Z. Yan, Q. Yuan, F. Xie, Y. Liu, Z. Feng, Z. Wang, F. Zhou, J. Xing, Z. Zhang, F. Wang, X. Guo. Mitochondrial DNA haplogroups and snps: risk factors in multiple cancers based on a cross-tumor analysis in chinese population. Vol. 33Cancer Epidemiol Biomarkers Prev, 2024 [↩]
- L. Ma, Q. Fu, B. Xu, H. Zhou, J. Gao, X. Shao, J. Xiong, Q. Gu, S. Wen, F. Li, L. Shen, G. Chen, H. Fang, J. Lyu. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ros-mediated akt activation. Vol. 142 Int J Cancer, 2018 [↩]
- K. K. Singh, M. Kulawiec. Mitochondrial DNA polymorphism and risk of cancer. Vol. 471 Methods in Molecular Biology, 2009. [↩]
- D. Doupa, M. N. Badji, F. Mbaye, S. Ka, A. Dem, M. Kane, M. Sembène. Benign breast tumors among senegalese women: diversity and genetic evolution of d-loop. Vol. 02, OALib, 2015 [↩]
- S. L. Mitchell, R. Goodloe, K. Brown-Gentry, S. A. Pendergrass, D. G. Murdock, D. C. Crawford. Characterization of mitochondrial haplogroups in a large population-based sample from the United States. Vol. 133 Human Genetics, 2014 [↩]
- A. M. Dworkin, T. H. M. Huang, A. E. Toland. Epigenetic alterations in the breast: implications for breast cancer detection, prognosis and treatment. Vol. 19 Seminars in Cancer Biology, 2009. [↩]
- D. Doupa, M. N. Badji, F. Mbaye, S. Ka, A. Dem, M. Kane, M. Sembène. Benign breast tumors among senegalese women: diversity and genetic evolution of d-loop. Vol. 02, Open Access Library Journal, 2015. [↩]
- S. L. Mitchell, R. Goodloe, K. Brown-Gentry, S. A. Pendergrass, D. G. Murdock, D. C. Crawford. Characterization of mitochondrial haplogroups in a large population-based sample from the united states. Vol. 133 Human Genetics, 2014. [↩] [↩]
- A. M. Dworkin, T. H. M. Huang, A. E. Toland. Epigenetic alterations in the breast: implications for breast cancer detection, prognosis and treatment. Vol. 19, Seminars in Cancer Biology, 2009. [↩]
- A. M. Dworkin, T. H. M. Huang, A. E. Toland. Epigenetic alterations in the breast: implications for breast cancer detection, prognosis and treatment. Vol. 19 Seminars in Cancer Biology,2009. [↩]
- D. Doupa, M. N. Badji, F. Mbaye, S. Ka, A. Dem, M. Kane, M. Sembène. Benign breast tumors among senegalese women: diversity and genetic evolution of d-loop. Vol. 02, OALib, 2015. [↩]
- B. Humphries, Z. Wang, C. Yang. MicroRNA regulation of epigenetic modifiers in breast cancer. Vol. 11, Cancers, 2019. [↩] [↩]
- B. Humphries, Z. Wang, C. Yang. MicroRNA regulation of epigenetic modifiers in breast cancer. Vol. 11, Cancers, 2019. [↩]
- R. S. Lee, K. Sad, D. V. Fawwal, J. M. Spangle. Emerging role of epigenetic modifiers in breast cancer pathogenesis and therapeutic response. Vol. 15 Cancers, 2023. [↩]
- E. Reznik, M. L. Miller, Y. Şenbabaoğlu, N. Riaz, J. Sarungbam, S. K. Tickoo, H. A. Al-Ahmadie, W. Lee, V. E. Seshan, A. A. Hakimi, C. Sander. Mitochondrial DNA copy number variation across human cancers. Vol. 5 eLife, 2016. [↩]
- K. S. Vikramdeo, S. Anand, S. K. Sudan, P. Pramanik, S. Singh, A. K. Godwin, A. P. Singh, S. Dasgupta. Profiling mitochondrial DNA mutations in tumors and circulating extracellular vesicles of triple-negative breast cancer patients for potential biomarker development. Vol. 5, FASEB BioAdvances, 2023. [↩]
- C. J. Pérez-Amado, H. Tovar, L. Gómez-Romero, F. O. Beltrán-Anaya, V. Bautista-Piña, C. Dominguez-Reyes, F. Villegas-Carlos, A. Tenorio-Torres, L. A. Alfaro-Ruíz, A. Hidalgo-Miranda, S. Jiménez-Morales. Mitochondrial DNA mutation analysis in breast cancer: shifting from germline heteroplasmy toward homoplasmy in tumors. Vol. 10, Frontiers in Oncology, 2020. [↩]
- A. Abicht, F. Scharf, S. Kleinle, U. Schön, E. Holinski-Feder, R. Horvath, A. Benet-Pagès, I. Diebold. Mitochondrial and nuclear disease panel (mito-and-panel): combined sequencing of mitochondrial and nuclear DNA by a cost-effective and sensitive ngs-based method. Vol. 6 Molecular Genetics and Genomic Medicine, 2018. [↩]
- E. K. K. Ip, M. Troup, C. Xu, D. S. Winlaw, S. L. Dunwoodie, E. Giannoulatou. Benchmarking the effectiveness and accuracy of multiple mitochondrial DNA variant callers: practical implications for clinical application. Vol. 13 Frontiers in Genetics, 2022 [↩]
- W. Burke. Genetic tests: clinical validity and clinical utility. Current Protocols in Human Genetics, 2014. [↩]
- C. J. Mattocks, M. A. Morris, G. Matthijs, E. Swinnen, A. Corveleyn, E. Dequeker, C. R. Müller, V. Pratt, A. Wallace. A standardized framework for the validation and verification of clinical molecular genetic tests. Vol. 18, European Journal of Human Genetics, 2010. [↩]
- I. A. Sobenin, K. Y. Mitrofanov, A. V. Zhelankin, M. A. Sazonova, A. Y. Postnov, V. V. Revin, Y. V. Bobryshev, A. N. Orekhov. Quantitative assessment of heteroplasmy of mitochondrial genome: perspectives in diagnostics and methodological pitfalls. Vol. 2014 BioMed Research International, 2014. [↩] [↩]
- K. K. Smith, J. D. Moreira, C. R. Wilson, J. O. Padera, A. N. Lamason, L. Xue, D. M. Gopal, D. B. Flynn, J. L. Fetterman. A systematic review on the biochemical threshold of mitochondrial genetic variants. Vol. 34 Genome Research, 2024. [↩]
- Y. Zhong, M. Okuno, N. Tsutsumi, S. I. Arimura. Mitochondrial DNA and the largest nuclear-mitochondrial DNA in arabidopsis can be separated by their methylation levels. Vol. 197 Plant Physiology, 2025. [↩]
- S. N. Cox, A. S. Varvara, G. Pesole. MitSorter: a standalone tool for accurate discrimination of mtDNA and numt ont reads based on differential methylation. Vol. 5, Bioinformatics Advances, 2025. [↩]
- W. Wei, K. R. Schon, G. Elgar, A. Orioli, M. Tanguy, A. Giess, M. Tischkowitz, M. J. Caulfield, P. F. Chinnery. Nuclear-embedded mitochondrial DNA sequences in 66,083 human genomes. Vol. 611 Nature, 2022. [↩]
- J. Geary, L. H. Gerido, A. M. Gutierrez, K. J. Dale. Equity and inclusion in assessing hereditary cancer risk: insights from excluded communities, structured interviews, and population genetics. Vol. 32 Cancer Control, 2025. [↩]
- E. M. Swisher, H. M. Harris, S. Knerr, T. N. Theoryn, B. M. Norquist, J. Brant, B. H. Shirts, F. Beers, D. L. Cameron, E. J. Dusic, L. A. Riemann, B. Devine, M. L. Raff, R. Kadel, H. J. Cabral, C. Wang. Strategies to assess risk for hereditary cancer in primary care clinics: a cluster randomized clinical trial. Vol. 8 JAMA Network Open, 2025. [↩]
- E. Reznik, M. Miller, Y. Şenbabaoğlu, N. Riaz, J. Sarungbam, S. Tickoo, H. Al-Ahmadie, W. Lee, V. Seshan, A. Hakimi, C. Sander. Mitochondrial DNA copy number variation across human cancers. Vol. 5 Elife, 2016. [↩]
- N. Chen, S. Wen, X. Sun, Q. Fang, L. Huang, S. Liu, W. Li, M. Qiu. Elevated mitochondrial DNA copy number in peripheral blood and tissue predict the opposite outcome of cancer: a meta-analysis. Vol. 6 Scientific Reports, 2016. [↩]
- K. Manto, S. Ustun Yilmaz, Z. Pala Kara, H. Kara, F. Tokat, C. B. Akyerli, C. Uras, M. Muftuoglu, U. Özbek. Association of mitochondrial DNA copy number variations with triple-negative breast cancer: a potential biomarker study. Vol. 13 Diseases, 2025. [↩]
- F. Peng, S. Wang, Z. Feng, K. Zhou, H. Zhang, X. Guo, J. Xing, Y. Liu. Circulating cell-free mtDNA as a new biomarker for cancer detection and management. Vol. 21 Cancer Biology and Medicine, 2024. [↩]


