
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder with no cure, and JNK3, a kinase expressed in the brain, has emerged as a promising therapeutic target due to its role in neuronal death. This research uses computational approaches to identify small-molecule inhibitors of JNK3 that may interrupt disease progression and offer new therapeutic possibilities. Analysis of the JNK3 binding site, followed by virtual screening and docking, highlighted several compounds with strong predicted interactions at key residues. Subsequent ADME and toxicity assessments further narrowed the set to molecules with favorable drug-like characteristics and no major predicted safety concerns. While these results are predictive rather than experimental, they reveal promising chemical scaffolds that merit future biological validation and are promising starting points for future JNK3-targeted Alzheimer’s drug development.
Keywords: Drug discovery, Alzheimer’s disease, virtual screening, pharmacophore
Introduction
Alzheimer’s disease is a progressive and currently incurable neurodegenerative disorder characterized by memory loss, cognitive decline, and behavioral changes, and it remains one of the most significant global health challenges1‘2. Alzheimer’s disease is characterized by the accumulation of amyloid-beta (Aβ) plaques, tau neurofibrillary tangles, and chronic neuroinflammation3. Aβ plaques are formed when fragments of a protein called amyloid precursor protein (APP) are cut by enzymes like α-, β-, and γ-secretase4. At high concentrations, Aβ disrupts brain function by triggering inflammation and causing toxic depolarization of neurons5. Similarly, Tau protein, which normally helps stabilize the structure of neurons, becomes hyperphosphorylated in AD, forming neurofibrillary tangles that block cell transport and trigger cell death6‘7.
Current treatments, such as acetylcholinesterase inhibitors like Donepezil, temporarily improve symptoms by increasing acetylcholine availability, but do not alter the underlying disease processes8‘9‘10.
As neurodegenerational progresses, these symptomatic treatments lose effectiveness, underscoring the need for therapies that intervene directly in the molecular pathways driving AD pathology11.
One promising area of research focuses on enzymes called mitogen-activated protein kinases (MAPKs), especially a type called c-Jun N-terminal kinase 3 (JNK3)12‘13‘14. Elevated JNK3 levels have been found in the cerebrospinal fluid (CSF) of Alzheimer’s patients, correlating with cognitive decline and co-localizing with Aβ plaques, while JNK1 and JNK2 levels remain unchanged15‘16. In AD, JNK3 is highly active in regions with Aβ plaques and Tau tangles, and promotes Tau phosphorylation, neuroinflammation, mitochondrial dysfunction, and cell death–all key features of Alzheimer’s progression. JNK3 has been reported to phosphorylate APP at Thr668, and its levels are elevated in the cerebrospinal fluid of Alzheimer’s patients, where it is associated with Aβ plaques17. JNK3 has emerged as a highly attractive target for therapeutic intervention and inhibiting it could potentially slow or halt disease progression18.
Given its strong link to disease progression and pathology, scientists have been working on two main types of JNK3 inhibitors: reversible and irreversible19. Among the reversible inhibitors, SP600125 was found to competitively bind to the ATP-binding site of JNK3, thereby preventing activation of the JNK3 signaling pathway and reducing neuronal apoptosis20. Similarly, AS602801 has demonstrated the ability to block JNK3 phosphorylation and reduce the expression of caspase-3, a key protein involved in cell death pathways21. In addition to these reversible inhibitors, irreversible compounds like JNK-IN-8 bind permanently and have shown strong effects in reducing inflammation by calming overactive immune cells in the brain22. Beyond kinase inhibition, another approach targets the interaction between JNK3 and scaffold proteins such as JIP1, β-arrestin2, and PSD95, which organize JNK3 signaling at postsynaptic sites23. Peptide inhibitors like D-JNKI1 disrupt these protein-protein interactions and have been shown to prevent APP and Tau phosphorylation while reducing neurodegeneration in preclinical studies24. These protein-interaction inhibitors offer greater specificity than ATP-competitive molecules, helping minimize off-target effects arising from kinase homology within the JNK family25.
Methodology
Analysis of binding sites in JNK3
ProteinPlus
A structure-based computational approach was used to identify potential ligand-binding sites on JNK3. The crystal structure of JNK3 (PDB ID: 7KSJ) was selected for analysis because it represents a high-resolution co-crystal structure bound to a potent and selective small-molecule inhibitor. Binding site prediction was performed using the DoGSiteScorer module implemented in ProteinPlus (https://proteins.plus/), a web-based platform for protein structure analysis. Three-dimensional visualizations were analyzed to assess pocket accessibility and suitability for small-molecule binding.
FT site
After locating the binding sites, it was necessary to use FT site (https://ftside.bu.edu/) to evaluate the energetic favorability of the identified binding pockets and determine whether or not the interaction was favorable. The same JNK3 structure was submitted, and the tool identified residues forming energetically favorable binding regions through fragment-based mapping.
Prankweb
The third computational experiment focused on validating whether at least one JNK3 binding site possessed both favorable geometric and energetic properties required for effective ligand binding. Binding site prediction was performed using PrankWeb (https://prankweb.cz/), which applies machine learning models trained on known protein–ligand complexes to identify and rank potential binding pockets. The JNK3 structure was analyzed to generate ranked binding sites along with residue-level probability scores and three-dimensional visualizations. These outputs were used to identify high-confidence binding pockets suitable for subsequent virtual screening and docking studies.
Virtual screening to discover small-molecule binders for JNK3
Pharmit
The virtual screening of JNK3 was conducted using Pharmit (https://pharmit.cs.pitt.edu/). Pharmacophore models were generated based on key interaction features derived from the predicted binding pocket, including hydrogen bond donors, hydrogen bond acceptors, hydrophobic features, and aromatic features, which represent common interactions required for effective kinase inhibition. Binding site water molecules were ignored during pharmacophore modeling. Multiple pharmacophore maps were constructed to account for potential variability in ligand–protein interactions. Virtual screening was conducted against the MolPort chemical library, which contains commercially available, drug-like compounds.
Molecular docking to evaluate binding affinity of selected compounds to JNK3
SwissDock
Following pharmacophore-based virtual screening, the top 15 compounds were selected for molecular docking analysis. This subset size was chosen to allow detailed evaluation within the time constraints of the study, as exhaustive docking of the full compound library was not feasible. Molecular docking was performed using the SwissDock web server (https://www.swissdock.ch/) to predict ligand–protein binding modes and relative binding affinity toward JNK3. Ligand structures were provided in Simplified Molecular Input Line Entry System (SMILES) format, obtained from the MolPort chemical library (https://www.molport.com/shop/index). The crystal structure of JNK3 was used as the target protein with the co-crystallized ligand removed and water molecules ignored. All protein chains were retained, and protonation states were automatically assigned by the server. Docking was conducted using a blind docking approach in which the entire protein surface was considered as the search space to avoid bias toward a predefined binding site. For each ligand, SwissDock generated multiple binding poses clustered based on structural similarity, and binding affinity estimates were reported using both the SwissParam scoring function and the AC score.
Determining drug suitability
SwissADME
To determine whether the top five docked compounds possessed favorable characteristics for use in the human body, their drug-likeness profiles were analyzed using SwissADME (https://www.swissadme.ch/). For each compound, the corresponding SMILES code was retrieved and inputted into SwissADME. The platform predicted key physicochemical and pharmacokinetic parameters relevant to oral drug development and were used to assess the suitability of each compound for further development as a potential therapeutic agent.
Toxicity assessment of selected compounds
ProTox
Following the identification of compounds that complied with Lipinski’s rule of five, toxicity risk was evaluated using ProTox 3.0 (https://tox.charite.de/protox3/index.php?site=home#). Ligand structures were provided in SMILES format. ProTox was used to generate in silico toxicity predictions, which were used to assess the relative safety of the selected compounds and prioritize candidates for further analysis.
Results
Detection of JNK3 Binding Sites
In the first digital experiment, the goal was to confirm the presence of potential binding sites on the JNK3 protein. The tool successfully identified 12 distinct binding pockets, each evaluated based on volume (Å3), surface area (Å2), and drug score. These parameters help assess how suitable a pocket may be for binding.
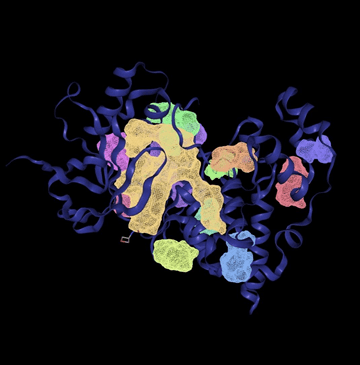
The visual 3D rendering of the protein structure in figure 1 shows all predicted binding sites in color. Larger pockets like P_0 (yellow) appear prominently, confirming their accessible positions on the protein surface.
| Name | Volume Å3 | Surface Area Å2 | Drug Score |
| P_0 | 1282.56 | 1509.52 | 0.81 |
| P_1 | 532.99 | 1200.52 | 0.84 |
| P_10 | 107.2 | 95.38 | 0.29 |
| P_11 | 105.34 | 271.56 | 0.25 |
| P_2 | 264.64 | 475.13 | 0.61 |
| P_3 | 247.42 | 320.31 | 0.6 |
| P_4 | 234.5 | 324.9 | 0.61 |
| P_5 | 180.99 | 394.74 | 0.31 |
| P_6 | 160.77 | 261.13 | 0.33 |
| P_7 | 144.13 | 312.79 | 0.31 |
| P_8 | 125.95 | 298.24 | 0.3 |
| P_9 | 112.7 | 300.34 | 0.37 |
A key concept in evaluating the size of the binding site is the Ångoström (Å), a unit of length commonly used to describe atomic and molecular dimensions. In this context, volume is expressed in cubic Ångoströms (Å3). A binding pocket’s volume is crucial–ideal volumes typically fall between 300 and 1200 Å3, which corresponds to the size range suitable for small molecule drugs. Pockets below 300 Å3 are generally too small to accommodate drug-like molecules, while pockets above 1200 Å3 may be too large. In the chart, only two pockets–P_0 and P_1– fall within or slightly above the optimal volume range. P_0 is 1282.56 Å3 with a drug score of 0.81 and P_1 is 532.99 Å3 with a drug score of 0.84. The remaining 10 pockets have volumes below 300 Å3, making them likely too small to serve as effective binding sites for drugs.
Following the identification of potential binding sites, the second experiment focuses on assessing the energetic favorability of small molecules binding at these sites. If the interaction at a given binding site is energetically unfavorable, small molecules would likely be “kicked out” or fail to bind effectively.
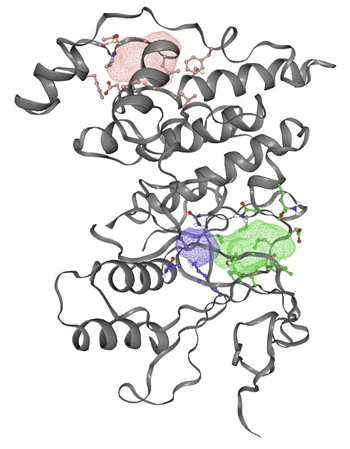
In figure 2, the gray ribbon represents the backbone of the JNK3 protein, while the colored mesh areas indicate the predicted binding sites based on energetic favorability. Any amino acid whose side chain is close enough (within 5 Å) to interact with the binding site is shown with detailed atoms (ball and stick).
The third experiment aimed to identify at least one binding site that met both key criteria: optimal size and favorable energetic interactions.
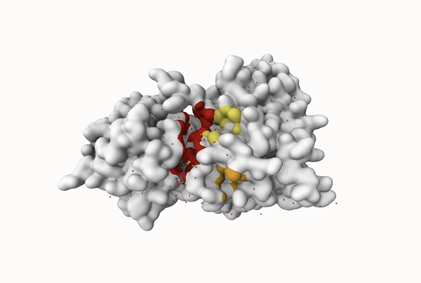
| Rank | Score | Probability | # of residues |
| 1 | 30.93 | 0.922 | 32 |
| 2 | 1.47 | 0.020 | 8 |
| 3 | 0.80 | 0.003 | 8 |
While only one such site would have been good, the experiment generated three predicted binding sites. In the chart above, the different binding sites were ranked. It also includes additional parameters such as probability, number of residues, and average conservation. The number of residues refers to the number of amino acids.
Binding site 1 stood out with a score of 30.93, a probability of 0.922, and 32 residues, reflecting strong binding site quality, as higher scores indicate better binding sites. In contrast, binding sites 2 and 3 had much lower scores (1.47 and 0.80, respectively), very low probabilities, and only 8 residues each.
Pharmacophore mapping and compound summary
Pharmacophore maps
A pharmacophore map represents the essential interactions a ligand must have to bind a target protein effectively. It consists of three elements: the number of interactions, the relative distances between them, and the type of interactions.
Drug compounds overlaid on pharmacophore maps
A total of 15 compounds were selected based on their low RMSD values across four different pharmacophore maps.
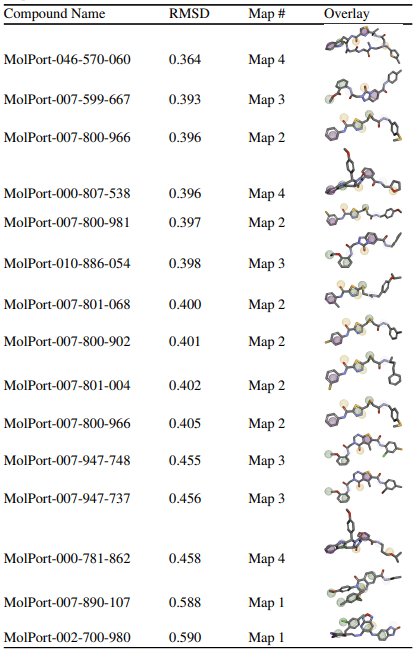
The goal of this virtual screening was to identify drug candidates that best fit the pharmacophore map of JNK3 by screening a large compound library. The quality of fit between each drug and the pharmacophore is quantitatively measured using the Root Mean Square Deviation (RMSD) score, which indicates how closely the chemical features of the compound align with the pharmacophore’s interaction points. RMSD values change from 0 to 1, where lower values represent better alignment and smaller deviations from the ideal pharmacophore geometry. Compounds were considered “top” candidates if they exhibited RMSD values below 0.6. Among the 15 top-ranked compounds identified, MolPort-046-570-060 demonstrated the strongest alignment with an RMSD of 0.364, followed closely by MolPort-007-599-667 (0.393) and MolPort-007-800-966 (0.396). The compounds were associated with pharmacophore maps 2, 3, and 4, suggesting that multiple interaction models of JNK3 may be suitable for ligand recognition. The majority of the selected compounds had RMSD values below 0.41, indicating a strong overall fit to their respective pharmacophore models. To further validate these pharmacophore-selected compounds and assess their binding feasibility within the JNK3 active site, molecular docking analyses were performed.
Docking site visualization and binding positions
Docking grid parameters and box size
Before initiating the molecular docking process, the docking grid was carefully defined. The grid box was centered at (19, 16, 25) with dimensions of 29 Å x 41 Å x 50 Å, ensuring full coverage of the protein. This is crucial because it allows the docking algorithm to explore all possible ligand binding conformations within the entire active site.
Docking results: binding poses of ligands

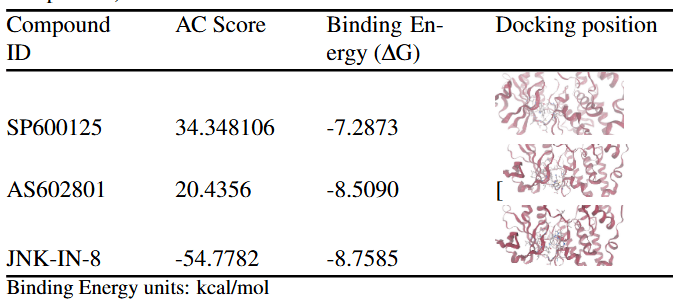
The Approximate Free Energy (AC) score provided by SwissDock is the primary metric used to evaluate and rank docking poses in this study. The AC score is a composite scoring function that estimates the favorability of ligand–protein interactions by combining several energetic contributions. Although reported in units of kcal/mol, the AC score represents a relative docking score rather than an experimentally derived thermodynamic binding free energy. In the context of SwissDock, more negative AC scores indicate more favorable predicted interactions between the ligand and the target protein. These values are best interpreted comparatively, allowing ligands docked to the same protein under identical conditions to be ranked based on their predicted binding favorability. For each compound, the reported results correspond to the most favorable docking pose from a single docking run, with no averaging across multiple independent runs. Across the 11 docked compounds, AC scores ranged from –22.76 to +39.99, indicating substantial variation in predicted binding favorability. The most favorable AC scores were observed for MolPort-007-890-107 (–22.76), MolPort-007-947-748 (–21.00), MolPort-007-801-004 (–17.70), MolPort-007-800-981 (–16.93), and MolPort-007-800-902 (–11.68). These compounds were therefore prioritized for further analysis, as their more negative AC scores suggest more stable predicted interactions with the JNK3 binding site. SwissParam scores were reported alongside AC scores as a supplementary metric but were not used for pose ranking, as they provide a rapid approximation rather than a rigorous ranking criterion. To provide a reference framework for interpreting docking results, three known JNK3 inhibitors (SP600125, AS602801, and JNK-IN-8) were redocked into the JNK3 structure using the same SwissDock protocol. Docking poses were ranked using the AC score, with SwissParam scores reported for comparison. JNK-IN-8 exhibited a markedly more favorable AC score (–54.78) than SP600125 (+34.35) and AS602801 (+20.44), consistent with its known high potency and irreversible binding mechanism. However, strong predicted binding alone does not guarantee in vivo effectiveness; therefore, ADME analysis was conducted to assess whether these compounds exhibit drug-like properties necessary for absorption.
Predicted pharmacokinetic properties
| Compound | Molecular weight (g/mol) | # H-bond acceptors | # H-bond donors | iLOGP | Solubility | GI absorption |
| MolPort-007-890-107 | 487.61 | 4 | 1 | 4.69 | poor | high |
| MolPort-007-947-748 | 500.93 | 6 | 2 | 3.58 | poor | low |
| MolPort-007-801-004 | 458.57 | 5 | 2 | 2.60 | poor | low |
| MolPort-007-800-981 | 446.52 | 6 | 2 | 3.20 | moderate | low |
| MolPort-007-800-902 | 430.52 | 5 | 2 | 3.11 | moderate | low |
| MolPort-007-800-966 | 444.59 | 4 | 2 | 3.29 | poor | low |
| MolPort-007-801-068 | 470.61 | 5 | 2 | 3.73 | poor | low |
| Compound | Molecular weight (g/mol) | # H-bond acceptors | # H-bond donors | iLOGP | Solubility | GI absorption |
| SP600125 | 220.23 | 2 | 1 | 1.49 | moderate | high |
| AS602801 | 457.55 | 7 | 0 | 3.88 | moderate | high |
| JNK-IN-8 | 507.59 | 6 | 3 | 4.09 | moderate | high |
Following molecular docking, the top seven compounds were further analyzed to assess their drug-likeness and pharmacokinetic potential using SwissADME. This evaluation was based on Lipinski’s Rule, a set of guidelines that help predict whether a compound is likely to be effective in the human body. According to the rule, a compound is considered suitable if it has a molecular weight under 500 g/mol, as smaller molecules can be quickly absorbed and metabolized. Also, it cannot have more than 10 hydrogen bond acceptors, no more than 5 hydrogen bond donors, and a calculated logP no greater than 5, which ensures a balance between aqueous solubility and lipid membrane permeability. Most of the seven compounds fully satisfied all four Lipinski criteria, suggesting favorable drug-like properties. MolPort-007-801-068, MolPort-007-800-966, MolPort-007-800-902, and MolPort-007-800-981 met all requirements, exhibiting optimal molecular weights, acceptable hydrogen bonding profiles, and iLogP values in the range of 3.11–3.73, indicating good membrane permeability without excessive lipophilicity. MolPort-007-947-748 slightly exceeded the molecular weight cutoff (500.93 g/mol), but only marginally, suggesting it may still retain favorable drug-like characteristics. MolPort-007-890-107 had an iLogP of 4.69, close to the upper limit of 5, indicating it strongly favors the lipid phase, which could affect solubility. Conversely, MolPort-007-801-004 had a slightly lower iLogP (2.60), suggesting it is less lipophilic and may have reduced membrane permeability compared to the other top compounds. SwissADME analysis was also performed on known JNK3 inhibitors (SP600125, AS602801, and JNK-IN-8) with all three showing high predicted gastrointestinal absorption. JNK-IN-8 slightly exceeded the 500 g/mol weight limit and had a relatively high iLogP (4.09), indicating greater lipophilicity, while SP600125 met all Lipinski criteria but had a very low iLogP (1.49), making it highly hydrophilic. In contrast, AS602801 satisfied all requirements and exhibited balanced properties. Given that many drug candidates fail during development due to unacceptable toxicity despite promising pharmacokinetics, toxicity prediction was conducted as a critical next step.
Toxicity profiles and FDA comparative analysis
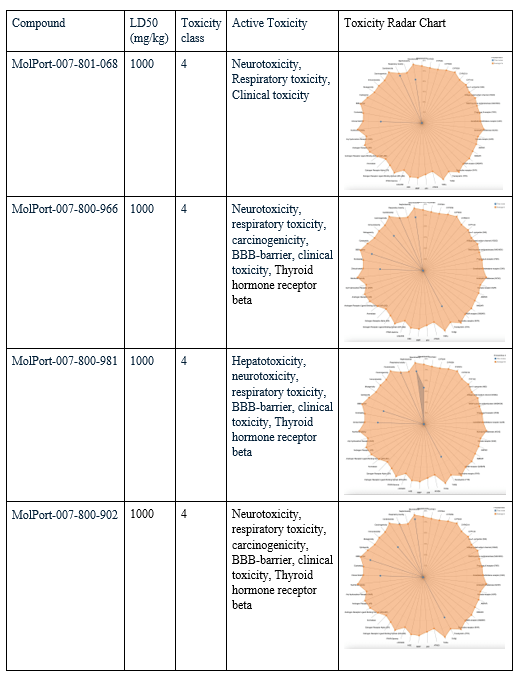
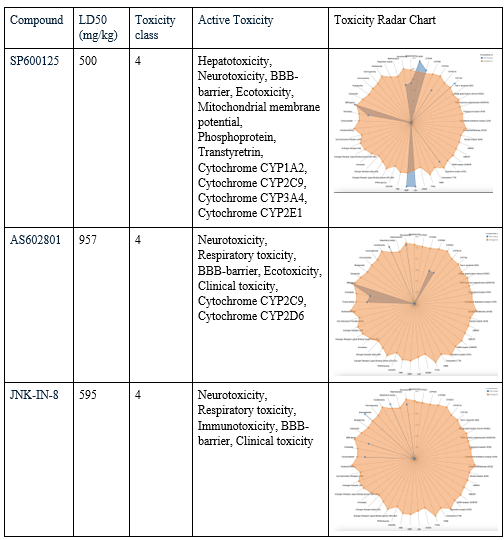
After choosing the compounds with the best ADME profile, their toxicity was evaluated using two key measures: LD50 and toxicity class. The LD50 (lethal dose 50%) represents the estimated amount of a substance required to cause death in 50% of a test population. The higher the value, the less toxic the compound is considered. Toxicity class ranks compounds from 1 (extremely toxic) to 6 (non-toxic), with class 3 or higher generally considered acceptable for drug development. All four selected compounds had an LD50 value of 1000 mg/kg, placing them in toxicity class 4, considered low in toxicity. To evaluate their toxicity in greater detail, radar charts were used to visualize each compound’s active toxicity endpoints. In these charts, the orange dot represents the average for active molecules, and predicted endpoints (blue) below this limit are considered less concerning in terms of toxicity. MolPort-007-801-068 showed three active toxicity endpoints, all of which were below the reference limit. MolPort-007-800-966 exhibited multiple predicted endpoints, with respiratory toxicity reaching the reference threshold while all other endpoints remained below it. MolPort-007-800-902 displayed six active toxicity endpoints, with respiratory toxicity slightly exceeding the reference limit and the remaining endpoints falling below it. MolPort-007-800-981 also showed six active toxicity endpoints; however, five were below the reference threshold and only one reached the average activity level. Although some compounds had one active toxicity endpoint at or slightly above the reference, all four compounds remain eligible for advancement, as drugs may still proceed if no more than one active toxicity endpoint exceeds the defined limit.
Known JNK3 inhibitors were also evaluated using ProTox to provide a reference for interpreting the toxicity profiles of the screened compounds. SP600125 was predicted to have 11 active toxicity endpoints. Of these, six endpoints exceeded the reference threshold indicated by the orange marker in the toxicity radar chart, suggesting a comparatively higher predicted toxicity burden. AS602801 showed seven active toxicity endpoints, with one endpoint exceeding and one marginally exceeding the reference threshold, while the remaining endpoints stayed below the limit. JNK-IN-8 displayed five active toxicity endpoints, two of which crossed the reference threshold. Overall, while all three reference inhibitors exhibited acceptable acute toxicity classifications, their toxicity radar profiles revealed multiple elevated endpoints.
Discussion
Several JNK3 inhibitors, including SP600125, AS602801, and JNK-IN-8, have been studied extensively but face limitations that reduce their clinical potential. Poor membrane permeability, indicated by very low iLogP values (e.g., SP600125, iLogP = 1.49), limits their ability to efficiently cross lipid membranes and reach intracellular targets, potentially reducing efficacy in vivo. Additionally, toxicity concerns associated with these inhibitors, including hepatotoxicity and neurotoxicity, may hinder their therapeutic development. In contrast, my computational analysis identified novel compounds, including MolPort-007-890-107, MolPort-007-947-748, MolPort-007-801-004, and MolPort-007-800-981, with stronger predicted binding to JNK3, reflected by their highly negative AC scores. Beyond binding affinity, these compounds also show favorable ADME profiles, with appropriate molecular weight, hydrogen bonding capacity, and lipophilicity, suggesting improved pharmacokinetic properties over the reference inhibitors. Toxicity predictions indicate minimal active endpoints exceeding reference limits for the newly identified compounds, highlighting a lower potential for side effects compared to the known inhibitors. SP600125, AS602801, and JNK-IN-8 showed multiple active toxicity endpoints above the reference threshold, with SP600125 exhibiting six such endpoints, suggesting a higher risk of adverse outcomes that could limit their in vivo applicability. Notably, compounds like MolPort-007-800-981 and MolPort-007-800-902 combine moderate solubility with balanced lipophilicity, which may further enhance in vivo absorption and distribution. These findings have meaningful implications for therapeutic development. Safer and more permeable JNK3 inhibitors could achieve effective target inhibition in the brain, offering potential new strategies for treating neurodegenerative diseases. Additionally, reduced predicted toxicity suggests these compounds may be suitable for chronic administration with lower risks of liver or neurological side effects. Collectively, the combination of predicted efficacy, favorable pharmacokinetics, and improved safety supports the potential of these compounds as candidates for further in vivo validation. While these predictions are encouraging, experimental studies are required to confirm binding affinity, pharmacokinetics, and safety, which will determine their true translational potential as therapeutics.
Limitations
While this study presents promising drug candidates for JNK3 inhibition, several limitations should be acknowledged. Only three high quality binding sites were identified, restricting the range of possible interactions with small-molecule inhibitors. Additionally, the limited number and narrow shape of these pockets may pose challenges in designing diverse and selective compounds. Additionally, in the virtual screening phase, only four pharmacophore maps were used, despite the availability of a wider variety that could have captured additional structural features. Furthermore, only the top 15 compounds were selected and screened from the MolPort database, despite the vast number of small molecules available. The limited scope may have excluded other strong candidates that fell just outside the top RMSD rankings. Another important limitation emerged during ADME analysis—almost all of the compounds showed low gastrointestinal (GI) absorption, which may impact their effectiveness as orally administered drugs.
Conclusion
Alzheimer’s disease is a devastating progressive brain condition characterized by ongoing brain cell damage, memory loss, and cognitive decline. JNK3 is a critical kinase predominantly expressed in the brain and plays a central role in the progression of Alzheimer’s disease by promoting neuronal death and inflammation. Its strong involvement in these damaging processes make JNK3 a highly important and promising target for developing new strategies. Unlike current treatments that only alleviates symptoms, targeting JNK3 offers the potential to directly slow or stop disease progression. By designing small molecules that selectively inhibit JNK3’s activity, researchers aim to intervene in these critical pathways.
To identify potential JNK3 inhibitors, five computational experiments were conducted using a range of specialized web-based tools. Binding site analysis (ProteinPlus, FT site, PrankWeb) revealed three high-quality pockets, with PrankWeb’s top site scoring 30.93. Virtual screening of MolPort compounds via Pharmit against 4 pharmacophore maps yielded 15 candidates with low RMSD values, including MolPort-046-570-060 (0.364), MolPort-007-599-667 (0.393), and MolPort-007-800-966 (0.396), showing excellent structural compatibility. Molecular docking with SwissDock found MolPort-007-890-107 had the strongest predicted binding (-22.76), followed by MolPort-007-947-748 (–20.997), MolPort-007-801-004 (–17.7039), and MolPort-007-800-981 (–16.9256), and these compounds were therefore prioritized for further analysis. To evaluate sustainability in the human body, 7 compounds were analyzed using SwissADME for drug-likeness. MolPort-007-801-068, MolPort-007-800-966, MolPort-007-800-902, and MolPort-007-800-981 all had optimal molecular weight (<500 g/mol), good hydrogen bond parameters, and logP values within the optimal range (3.11-3.73), indicating good permeability and balanced lipophilicity. Among these, only MolPort-007-800-981 and MolPort-007-800-902 exhibited moderate solubility, while the rest of the compounds showed low solubility, meaning they are more likely to dissolve adequately, improving absorption. Toxicity assessment using ProTox showed that all four compounds had predicted LD50 values of 1000 mg/kg, placing them in toxicity class 4, which corresponds to low acute toxicity. MolPort-007-801-068 displayed no active toxicity endpoints above reference limits, while MolPort-007-800-966 and MolPort-007-800-902 each showed a single endpoint at the reference threshold. MolPort-007-800-981 exhibited multiple predicted toxicity endpoints, with five remaining below the reference limit and one at the threshold. Overall, MolPort-007-800-981 emerged as the most promising candidate, combining a favorable AC score (–16.9256), an ideal molecular weight (446.52 g/mol), moderate solubility, a balanced iLogP value (3.20), and an acceptable predicted toxicity profile, making it a compelling lead for further investigation.
It is important to note that this study is limited by its reliance on computational predictions, including single docking runs, in silico ADME and toxicity assessments, and the absence of experimental confirmation. These limitations mean that actual biological activity and safety may differ from the predicted results, underscoring the need for subsequent laboratory validation.
Looking ahead, the next steps will focus on translating these computational findings into experimental validation. In the short term, the most promising drug candidate—particularly MolPort-007-801-068—will be obtained for in vitro testing to evaluate their actual binding affinity, selectively, and biological activity in controlled lab settings. In the long term, successful compounds will be advanced in vivo studies using animal models of Alzheimer’s disease to examine their pharmacokinetics, therapeutic potential, and safety in a more complex biological system. These future efforts will be critical in determining whether the identified inhibitors can serve as viable leads for clinical development.
References
- C. A. Lane, R. Hardy, J. C. Schott. Alzheimer’s disease. European Journal of Neurology. Vol. 25, pg. 59–70, 2018. [↩]
- E. Passeri, K. Elkhoury, M. Morsink, et al. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int J Mol Sci. Vol. 23, pg. 13954, 2022. [↩]
- A. D. Snow, K. R. Jakubowski, R. C. Dickey, et al. In vitro comparison of major memory-support dietary supplements for their effectiveness in reduction/inhibition of beta-amyloid protein fibrils and tau protein tangles: key primary targets for memory loss. Scientific Reports. Vol. 11, pg. 3001, 2021. [↩]
- J. Y. Hur. γ-Secretase in Alzheimer’s disease. Experimental & molecular medicine. Vol. 54, pg. 433-446, 2022. [↩]
- P. Scheltens, B. De Strooper, et al. Alzheimer’s disease. Lancet. Vol. 397, pg. 1577–1590, 2021. [↩]
- J. R. Dickson, S. Schwab, M. P. Kuret, et al. Alzheimer disease-associated tau post-translational modification mimics impact tau propagation and uptake. Journal of Neuropathology and Experimental Neurology. Vol. 84, pg. 459–470, 2025. [↩]
- S. Kumari et al. Apoptosis in Alzheimer’s disease: insight into the signaling pathways and therapeutic avenues. Apoptosis. Vol. 28, pg. 943-957, 2023. [↩]
- J. S. Birks, et al. Donepezil for dementia due to Alzheimer’s disease. The Cochrane Database of Systematic Reviews. Vol. 6, CD001190, 2018. [↩]
- L. Jean, et al. In vivo localization of human acetylcholinesterase-derived species in a β-sheet conformation at the core of senile plaques in Alzheimer’s disease. The Journal of Biological Chemistry. Vol. 294, pg. 6253–6272, 2019. [↩]
- J. Kim, et al. Donepezil regulates LPS- and Aβ-stimulated neuroinflammation through MAPK/NLRP3 inflammasome/STAT3 signaling. International Journal of Molecular Sciences. Vol. 22, pg. 10637, 2021. [↩]
- Z. Breijyeh, R. Karaman. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules. Vol. 25, pg. 5789, 2020. [↩]
- C. A. Musi et al. JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases. Cells. Vol. 9, pg. 2190, 2020. [↩]
- L. Resnick, M. Fennell. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Discov Today. Vol. 9, pg. 932–939, 2004. [↩]
- Y. Zhao et al. Hypothesis: JNK signaling is a therapeutic target of neurodegenerative diseases. Alzheimers Dement. Vol. 18, pg. 152-158, 2022. [↩]
- Z. Li et al. Targeting protein kinases for the treatment of Alzheimer’s disease: Recent progress and future perspectives. Eur J Med Chem. Vol. 261, pg. 115817, 2023. [↩]
- S.P. Bhujbal, J.M. Hah. Advances in JNK inhibitor development: therapeutic prospects in neurodegenerative diseases and fibrosis. Arch Pharm Res. Vol. 48, pg. 858-886, 2025. [↩]
- N.B. Mañucat-Tan, K. Saadipour, et al. Cellular Trafficking of Amyloid Precursor Protein in Amyloidogenesis: Physiological and Pathological Significance. Mol Neurobiol. Vol. 56, pg. 812-830, 2019. [↩]
- P. Qin, Y. Ran, Y. Liu, C. Wei, X. Luan, H. Niu, et al. Recent advances of small molecule JNK3 inhibitors for Alzheimer’s disease. Bioorg Chem. Vol. 128, pg. 106090, 2022. [↩]
- G. Li et al. Recent Advances in c-Jun N-Terminal Kinase (JNK) Inhibitors. Curr Med Chem. Vol. 28, pg. 607-627, 2021. [↩]
- M. Shvedova, Y. Anfinogenova, E.N. Atochina-Vasserman, et al. c-Jun N-Terminal Kinases (JNKs) in Myocardial and Cerebral Ischemia/Reperfusion Injury. Front Pharmacol. Vol. 9, pg. 715, 2018. [↩]
- Y. Wu et al. JNK3 inhibitors as promising pharmaceuticals with neuroprotective properties. Cell adhesion & migration. Vol. 18, pg. 1-11, 2024. [↩]
- C. A. Musi et al. Colocalization and Interaction Study of Neuronal JNK3, JIP1, and β-Arrestin2 Together with PSD95. International journal of molecular sciences. Vol. 23, pg. 4113, 2022. [↩]
- R. Nakano, T. Nakayama, H. Sugiya. Biological Properties of JNK3 and Its Function in Neurons, Astrocytes, Pancreatic β-Cells and Cardiovascular Cells. Cells. Vol. 9, pg. 1802, 2020. [↩]
- A. A. Eshraghi, M. Aranke, R. Salvi, D. Ding, J. K. M. Coleman Jr., E. Ocak, R. Mittal, T. Meyer. Preclinical and clinical otoprotective applications of cell-penetrating peptide D-JNKI-1 (AM-111). Hearing Research. Vol. 368, pg. 86–91, 2018. [↩]
- M.T.H. Duong, J.H. Lee, H.C. Ahn. C-Jun N-terminal kinase inhibitors: Structural insight into kinase-inhibitor complexes. Comput Struct Biotechnol J. Vol. 18, pg. 1440–1457, 2020. [↩]



{kind=link}