
Abstract
Background: Viral mutation and immune evasion are major barriers in long-term vaccine efficacy and vaccine development. RNA viruses generally mutate at a higher rate and infect acutely, while DNA viruses possess highly complex immune evasion mechanisms to persist in the bodies of their hosts.
Objective: To examine how the mechanisms underlying mutation rates and immune evasion in RNA and DNA viruses influence vaccine development and efficacy, and to evaluate effective vaccine strategies.
Methods: A systematic literature search was conducted, combined with data compiling and analyses on RNA and DNA viruses, influenza, SARS-CoV-2, HSV, and the efficacy of different vaccine types from databases such as PubMed, ScienceDirect, and Nexstrain.
Results: The mutative nature of SARS-CoV-2 caused host immunity to wane within 6 months after vaccination. Moreover, there was a strong correlation between S1 mutation amount in SARS-CoV-2 and immune escape (r=0.91). mRNA vaccines achieved vaccine efficacy rates of around 95%, but declined against the highly mutative Omicron lineage, while protection against severe disease remained stable due to the usage of conserved epitopes for broad spectrum immunity. Although much slower at mutating, DNA viruses display immune evasion mechanisms that complicate vaccine design as well.
Conclusion: Viral evolution reflects both immune selective pressures and mutation rate. In the future, effective vaccine strategies must hybridize immunodominant but mutable antigens with conserved epitopes, use real-time phylogeny monitoring, and adopt booster schedules to combat waning immunity and antigenic drift.
Keywords: RNA virus, DNA virus, vaccine development, SARS-CoV-2, HSV, influenza, mutation rates, vaccine efficacy.
Introduction
While viral mutation is cited as the primary obstacle to vaccine development, immune evasion mechanisms also play a significant role. The mutation rates of viruses are heavily affected by numerous factors such as replication method, immune response, environment, viral polymerases, and host factors. These factors and immune evasion mechanisms create challenges in vaccine development and prevent effective treatment. This review will focus on the mechanisms of RNA virus mutation rates and the complex immune evasion of DNA viruses, their effects on treatment development, and compare real-world vaccine development challenges between RNA and DNA viruses. This will be achieved through using public databases on RNA and DNA viruses, viral mutation rates, host immune evasion mechanisms, and the vaccine development challenges that follow.
Methods
A systematic literature search was conducted in accordance with the PRISMA 2020 guidelines. The following electronic databases were searched: PubMed, ScienceDirect, Nature, ASM, Nextstrain, and GISAID (January 2003 to August 2025).
Inclusion Criteria: To be included, a manuscript had to address either RNA or DNA viruses, the mechanisms behind them, or vaccine development challenges faced because of the mechanisms behind RNA and DNA viruses. All papers had to be either randomized controlled trials, clinical trials, and written after 2010 to ensure more recent data collection, unless older papers were used to study the timeline of vaccine development and technological advancements in vaccine research. All papers were English only.
Exclusion Criteria: Articles were excluded if they were not peer reviewed. Papers that contained unique variables in their data when compared to other compiled papers were also excluded due to lack of comparatability. From downloaded datasets, listwise deletion was applied.
Mechanisms of RNA Virus Mutations
Viral mutations arise through a series of aforementioned factors, as well as recombination and reassortment that create markedly different mutation rates across RNA and DNA viruses, and single-stranded and double-stranded viruses. When comparing ssRNA viruses to dsRNA viruses, dsRNA viruses have slightly lower mutation rates than the minimum mutation rate of both virus types. However, ssRNA viruses have a much higher maximum mutation rate. Chemical damage has shown a potential correlation with increased mutation rates through experiments on the effects of ethanol on HCV1. +ssRNA viruses can almost immediately begin replicating after entering a host cell2. This increases the chemical damage that +ssRNA viruses receive, which could in turn increase mutation rates3. On the other hand, -ssRNA viruses must use RdRp within the virion to start translation2,4. This could explain why +ssRNA viruses have slightly higher mutation rates compared to -ssRNA viruses5. However, studies on the effects of chemical damage on ssRNA virus mutation rates are currently limited to HCV, thus it cannot be determined with complete assurance whether chemical damage and mutation rate are truly correlated or not6.
Meanwhile, experimentation on bacteriophages could help explain the difference in mutation rates experienced by dsDNA and ssDNA viruses. In DNA bacteriophages, access to post-replicative repair has shown mutation rate differences in single and double-stranded viruses3. Some ssDNA viruses such as bacteriophage ϕX174 cannot perform mismatch repair, meaning they have mutation rates in the upper echelons of mutation rates in DNA viruses3. The connection between post-replicative repair and mutation rate in eukaryotic viruses, however, is not established. Therefore, an explanation is still needed on the difference in mutation rates between ssDNA and dsDNA viruses.
RdRp is also a key factor in the difference in mutation rates between DNA and RNA viruses. In RNA viruses, some RdRp lack proofreading and often make mistakes during transcription, introducing mutations and mismatched bases. On the contrary, some DdDp have proofreading mechanisms, with several DNA viruses having mechanisms for repairing mismatched bases. This leads to higher mutation rates in RNA viruses than DNA viruses7. Influence from host cells can also strongly affect mutation rates in viruses. For instance, APOBEC–a family of deaminases that defend against retroviruses–are bound to by the viral protein Vif, encouraging proteasomal degradation of APOBECs3. This can cause mutations in viruses by encouraging DNA base editing by APOBECs and has been shown to increase mutation rates in HIV-1 by more than forty times3.
| Virus Type | Approximate range of mutation rate as substitutions per nucleotide per cell infection (s/n/c) |
| ssRNA (+) virus | 10⁻³–10⁻⁶ |
| ssRNA (-) virus | 10⁻⁵–10⁻⁶ |
| dsRNA virus | 10⁻⁶ |
| ssDNA virus | 10⁻⁶ |
| dsDNA virus | 10⁻⁷–10⁻⁸ |
| Retrovirus | 10⁻⁴–10⁻⁵ |
Table 1 shows that RNA viruses substitute anywhere from 1-100,000 times faster than DNA viruses, as RNA virus mutation rates range from 10⁻³ to 10⁻⁶ s/n/c, while DNA viruses range from 10⁻⁶ to 10⁻⁸ s/n/c. However, there is only one data point for both ssDNA and dsDNA virus mutation rates. The cellular environment in which viruses reside during the cell infection cycle has been shown to impact viral mutation rates. In retroviruses, imbalances in dNTP pools can affect mutation rates. This, along with host factors, can also greatly affect the mutation rate of viruses.
Consequences of High Mutational Drift
Viruses with high mutational drift constantly pose problems for vaccine development and treatment efficacy. Fitness landscapes shape evolution into an adaptive process that has multiple peaks and valleys. Peaks represent mutations that benefit organisms while valleys represent mutations that are detrimental to organisms. Therefore, viruses that are less fit will have a limited population, or gradually die out8. Moreover, quasispecies theory explains that rapidly replicating entities, such as viruses, contain a dynamic assortment of closely related genetic variants that form “mutant swarms” that compete with each other. This allows for the rapid adaptation of entities to changing environments, such as the development of resistance to vaccines and treatments9. Because selective pressures such as drugs, vaccines, and competing viruses push back on the populations of certain viral strains, viral strains further mutate and adapt to compete and evade other viral strains and immune responses, respectively. These evolutionary frameworks help explain the core causes behind the constant mutation and evolution of viruses.
One such case is antigenic drift and shift in influenza. Antigenic drift means that vaccines must be updated to maintain efficacy, while antigenic shift means that vaccine development needs to provide partial immunity to animal viruses. Vaccine development for influenza manipulates viruses to reassort and create new genetic variants. This allows vaccines to use genetically manipulated viruses for antigens, where the main virus contains the target strains while the second virus is used for additional safety measures. As viruses become more understood, manipulating viral polymerase or using treatments that increase mutation rates can decrease virus infectivity and a virus’s ability to replicate10.
Discussion
Influenza A and B are the two most common types of influenza that can infect humans. Variants of Influenza A and B descend from two subtypes, A(H1N1) and A(H3N2), for Influenza A, and two unique lineages, B/Victoria and B/Yamagata, for Influenza B11. Influenza vaccines are most commonly inactivated, with an efficacy of 50%12. In influenza vaccines, the main virus is used to create immunity while the secondary virus is used for added safety, using the HA antigen to signal to the immune system what antibodies to produce. However, the HA antigen is constantly drifting, rendering previous vaccines ineffective as mutations alter the HA protein, which can fundamentally change the antigenicity of the virus, preventing antibody recognition. Therefore, to keep up with a virus that can mutate within half a year, vaccine development must predict the HA or NA antigens that are globally present after antigenic drift has occurred. This is currently possible, although difficult, as most viral mutations are deleterious, thus limiting the diversity of influenza variants. In fact, during the 2012-2013 season, a singular mutation K166Q in the HA head of Influenza A(H1N1) had masked an epitope that had previously been recognized by human monoclonal antibodies, preventing recognition by the latter13. These changes to the topology of the HA protein prevent the binding of antibodies to the protein, even if the protein remains functional. Middle-aged adults that had previously been exposed to the virus and had high neutralization antibody titers against that epitope before the 2012 season suddenly had low neutralization antibody titers during the 2012-2014 seasons due to the K166Q substitution13. The lack of efficacy has driven vaccine development for influenza but is held back by poor measurable signs of immunization. At this moment, the only acceptable correlate of protection is hemagglutination inhibition (HI) serum antibody titers of 1:32 to 1:40, with WHO stating that an HI test with an HAI titer of 40 or higher is considered an acceptable correlate of protection12. However, attenuated influenza vaccines have shown efficacy while being below the benchmark.
Due to the lack of efficacy and struggles in seasonal vaccines, universal vaccine development has been tested. One method is developing broadly neutralizing antibodies to the HA stalk, which can identify these structures, allowing for the induction of broadly neutralizing antibodies. Another method utilizes the sequences of circulating strains and creates an artificial sequence that broadly represents the group of strains, which should minimize the difference in the vaccine antigens and the circulating strains. As for universal influenza B vaccines, one influenza B vaccine targets the influenza B HA cleavage site in the HA stalk. Influenza vaccine development has made significant progress in creating universal vaccines, but antigenic drift and shift remain obstacles to work around12. Experimentation has also been done where mutations are introduced to the main virus to yield faster seed viruses12.
SARS-CoV-2, on the other hand, rapidly evolved at a rate higher than influenza, with variants popping up globally. Many of the first variants had mutations in the RBD in the S protein, which increased the transmission rates of these variants by 40-70%14. The increased transmission rate of these variants allowed the virus to spread rapidly to other countries. Approved vaccines were mostly spike protein-based such as the Moderna and Pfizer-BioNTech vaccines. However, there were also adenovirus vector-based vaccines such as AstraZeneca and Johnson & Johnson. Several studies on SARS-CoV-2 showed a link between the virus’s genomic changes and immune reactivity in patients, suggesting that the evolution of SARS-CoV-2 could render vaccines obsolete after enough mutations14. SARS-CoV-2 also displays antigenic drift, decaying efficacy quickly15.
SARS-CoV-2 variants were categorized into seven main groups of variants. SARS-CoV-2 variants mostly mutate in the spike proteins, with mutations such as E484K providing immune escape through methods such as reducing antibody binding13. The first variant, the Alpha variant, was extremely infectious and accounted for 92% of cases in Europe and 59% of cases in the US states in 202114. Later variants continued to mutate rapidly, resulting in greater immune escape, infectivity, and lethality. The Delta variants carried on the trend of spike protein deletions, such as Y144, further increasing infectivity14. Omicron was a variant that contained more than 30 mutations, with these mutations being associated with increased transmissibility, better antibody escape, and viral binding strength14. Furthermore, Omicron had higher antibody escape than previous variants, perhaps due to its difference in structure from other variants16.
Many viruses, like SARS-CoV-2, also enable immune escape through inhibiting pathogen recognition, preventing memory T-cell responses from activating. For example, the P272L variant of SARS-CoV-2 developed immune escape from a T-cell epitope that was dominant in killing the Wuhan variant, showing complete escape against the T-cell responses17. Thus, vaccines should incorporate conserved epitopes for more durable immunity that can prevent the complete immune escape that was demonstrated by the P272L variant.
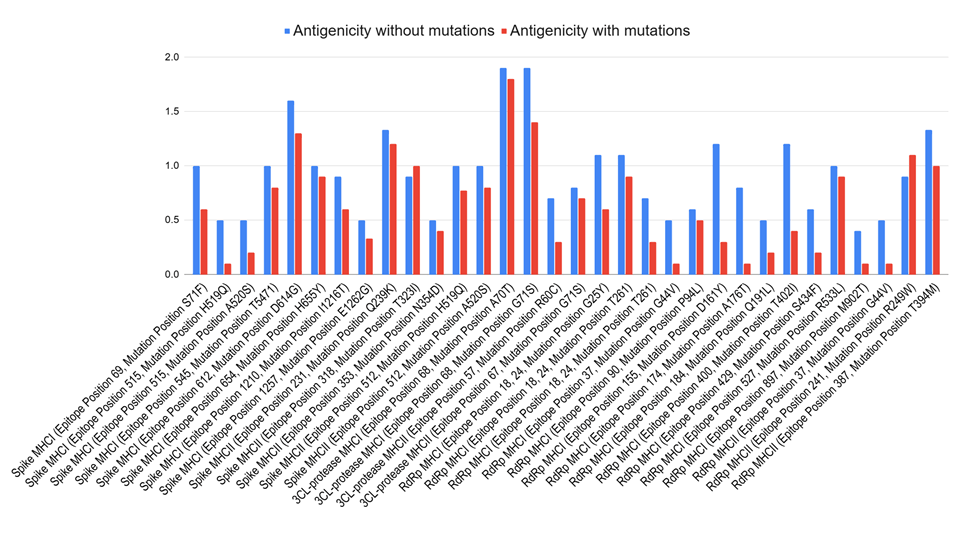
Mutations in the spike glycoprotein of SARS-CoV-2 either block the ACE2 binding site or compete with it, causing slight changes in amino acid chains that may alter the surface of viruses, preventing existing antibodies from being able to properly bind to viruses. Such structural changes cause a decrease in antigenicity. Figure 1 highlights this negative correlation between mutations and antigenicity, with almost every epitope tested showing a lowered antigenicity after mutating. A study on antibody evasion by SARS-CoV-2 Omicron subvariants found that mutations negatively impact ACE2 binding, causing complete or partial neutralizing antibody evasion. Furthermore, cryo-EM from a different study on the Omicron BA.2 spike revealed that the NTD loop, a hotspot for antigenic drift where many immunodominant antibodies in coronaviruses bind to, of the subvariant was structurally reorganized through the folding of the flexible N1 amino acid segment into the rigid β-strand18. The flexibility of loops such as the N1 loops often makes them the target of antibodies. Therefore, when altered into more rigid strands, antibodies could be prevented from binding to said surfaces, aiding in viral immune escape. On a larger scale, such structural changes to the NTD loop alter the 3D topology of the loop, potentially rendering viruses unrecognizable to antibodies even if the latter were to bind to ACE2. Such factors contributed to vaccine development challenges for SARS-CoV-2.
Due to the rapid development of new technology used in SARS-CoV-2 vaccines, extensive studies and trials were conducted to test the efficacy and safety of the vaccines. A study on an inactivated SARS-CoV-2 vaccine showed that the shorter the time gap between doses–less than three weeks–the more the immunogenicity decreased14. Other vaccines that used SARS-CoV-2 as a whole and not the variants showed similar results, but concerns regarding having to cover most immunological epitopes rose14. Improper inactivation processes could change the properties of these epitopes, causing non-neutralizing antibodies to be produced. This would allow SARS-CoV-2 to replicate more rapidly rather than stopping the virus from replicating.
Vaccine efficacy for SARS-CoV-2 is higher than influenza, with the Pfizer-BioNTech vaccine having an efficacy of 95% while the Moderna vaccine has an efficacy of 94.5%14. However, real world efficacy could be substantially lower given variable population response and diverse genes that give certain populations greater immunity, such as from HLA alleles. The mRNA vaccines are created by manipulating the spike protein mRNA to express genetic information for antibody production. The Pfizer-BioNTech vaccine decreased infection rates by 58% after 12-20 days and 72% after 45-59 days when compared to unvaccinated subjects14. Sinopharm, an inactivated vaccine, took over a month to develop antibody responses to SARS-CoV-2, while another inactivated vaccine, Covaxin, was able to retain substantial neutralizing antibodies for up to three months after the second dose16. However, these results are based on limited studies, as the durability of protection remains uncertain. Other vaccines had little information on them, but many vaccines that were not studied thoroughly or had little information on their efficacy were approved for emergency medical use14.
The Pfizer-BioNTech vaccine had such high short-term vaccine efficacy because early variants of SARS-CoV-2 had relatively few changes in the RBD and T-cell epitopes of the spike protein, meaning that early genomes contained highly conserved regions that allowed for high efficacy across variants. However, humoral and cellular immunity against SARS-CoV-2 variants waned after six months. There were discussions on further COVID vaccines that focused on more universal coverage through targeting conserved regions instead of spike proteins19. Despite this, multivalent vaccines that were developed and deployed further into the SARS-CoV-2 pandemic still primarily targeted the spike protein of SARS-CoV-2. Although developing a universal vaccine using conserved regions was never fully implemented for widespread public use for SARS-CoV-2, its potential is currently being studied for several viruses.
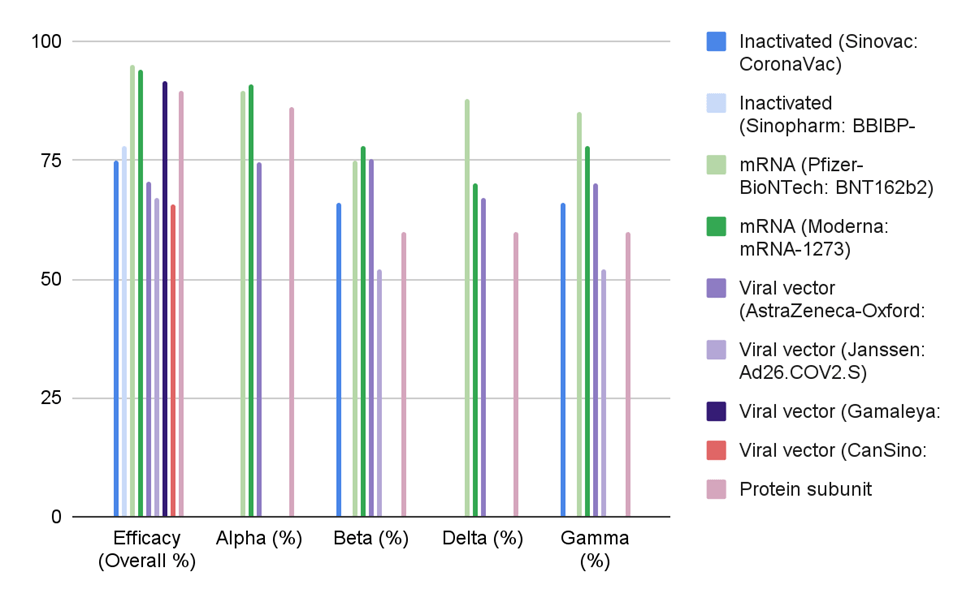
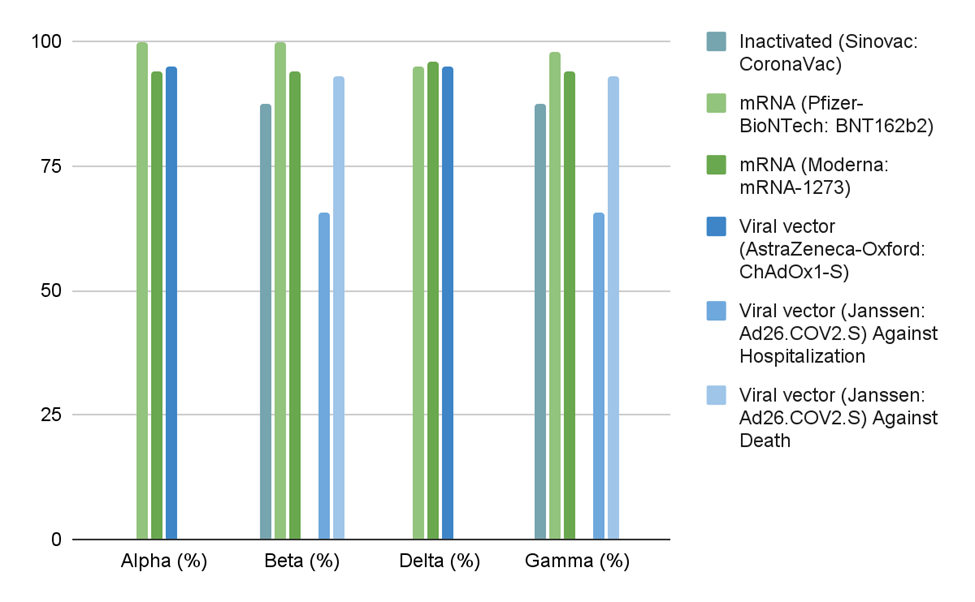
Figure 2 and Figure 3 both display vaccine efficacy but use different definitions: Figure 2 measures vaccine efficacy against infection, while Figure 3 measures vaccine efficacy against severe disease/death. In Figure 3, mRNA vaccines maintained high efficacy against severe disease/death across all four variants, and most vaccines performed well overall. However, Figure 2 shows reduced and varied protection against infection across variants for all platforms. Still, mRNA vaccines generally outperformed other vaccine types. Meanwhile, most inactivated and viral vector vaccines did not reach above 80% efficacy. The Novavax protein subunit vaccine did well against the Alpha variant, but struggled against Beta, Delta, and Gamma at 60% efficacy. Viral vector-based vaccines also showed low efficacy against infection across most SARS-CoV-2 variants tested. Still, vaccines in general reduced transmission risk by 88.5%, and vaccination, no matter what type, reduced transmission rates20,21. However, efficacy varied across variants and populations.
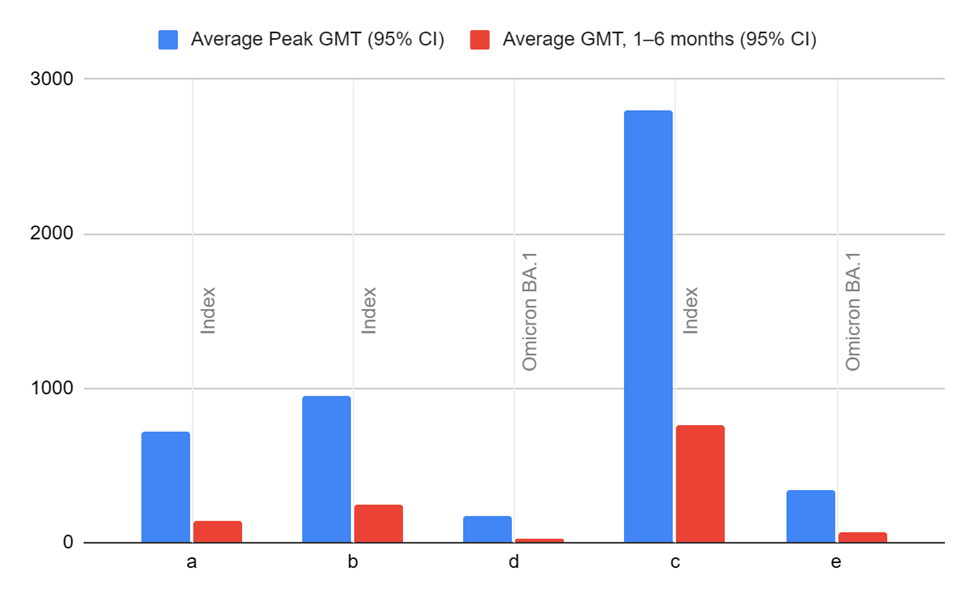
Vaccine performance cannot be concluded with full assurance due to many variables that affect both long-term immunity and short-term immunity. Waning immunity is a major factor that leads to substantial long-term declines in vaccine performance. Figure 4 demonstrates how waning immunity decreases GMTs, as all groups in the study showed substantially decreased GMTs 1-6 months after the initial or booster shot by anywhere from 2.4-9.0 folds against both Index and Omicron BA.1 variants. The GMTs were found to also decrease by 1.7-21.8 folds after those who were hybrid-immune were given a primary shot22. It is essential to know that the study that compiled and analyzed the waning immunity of SARS-CoV-2 vaccines admitted that many of the 26 studies used were either low in reliability or had no reliability due to lacking critical information.
Variable population responses can also create doubts about vaccine efficacy, as they form irregularities within the data. People who experienced prior infection from SARS-CoV-2, when vaccinated, were found to experience a potent immune response, with vaccination causing those with hybrid immunity to recall diverse and high quality memory B-cells. In fact, total B-cell count was found to increase by 5 to 10 fold in individuals with hybrid immunity when compared to those with natural immunity or vaccination23. Such differences in immune response can cause real-world populations to diverge from predicted outcomes.
Genetics also play a large part in immunity. HLA genes enhance the antibody response to initial SARS-CoV-2 vaccinations, with several HLA alleles independently influencing antibody response after SARS-CoV-2 vaccination24. Moreover, cumulative HLA variants have been found to have an effect on both the susceptibility and severity of breakthrough SARS-CoV-2, and are associated with anti-SARS-CoV-2 IgG levels, with this result being statistically significant genome-wide25. Specifically, the HLA-DQB1*06 allele family enhances antibody response, stimulates T-cells, and promotes spike-specific B-cell memory responses and neutralization antibody titers.
Variations in host immunity not only affect vaccine efficacy data, but also drive viral evolution. Under T-cell selective pressure, immune escape mutations developed in SARS-CoV-2, and antigenic drift and shift in influenza is partially caused by variable population antibody responses. Under these factors, uneven immune landscapes are created that viruses adapt to, fueling ongoing mutation and evolution.
Vaccine types address antigenic drift with varying levels of efficacy. Adenoviral vector-based vaccines showed decreased efficacy against emerging variants of SARS-CoV-2, with previous adenovirus immunity and antigenic drift contributing to lower immune responses to novel SARS-CoV-2 variants26. There were varying levels of decrease in neutralization titers for wild type SARS-CoV-2 and infectious variants that were subjected to the adenoviral vector-based vaccines. Much of the substantial decrease in neutralization antibody titers could be attributed to mutations within the RBD of the S protein in SARS-CoV-2 variants. Strains with RBD mutations exhibited reductions in neutralization titers from anywhere between 1.4-fold to 42.4-fold. Inactivated vaccines showed similar results, with antigenic drift potentially causing a decrease in efficacy. Protein subunit vaccines for influenza faced similar challenges with antigenic drift as well. Traditional protein subunit vaccines struggled to protect against antigenic drift due to the usage of surface proteins in vaccine development. However, recent advancements in vaccine technology such as the improvement in adjuvant technology shows promise in further improving the efficacy of protein subunit vaccines against influenza27.
Using conserved epitopes and regions in universal vaccines is integral to addressing antigenic drift within RNA viruses. Using multiple conserved epitopes in vaccine development prevents the vaccine from developing inefficacy over time due to mutations, allowing antibodies to recognize viruses that have largely mutated from the strains that the vaccines were developed with.
Such potential has already been noted in a multitude of vaccines. In protein subunit vaccine development for influenza, H1N1 HA-based vaccine design has shifted to targeting the stem of influenza viruses, masking or removing the HA head to expose the conserved regions for the creation of HA mini-stem vaccines. Such vaccines have been shown in preclinical studies to be able to protect against multiple different influenza subtypes through the induction of broadly neutralizing antibodies27. For adenoviral vector-based vaccines, the supplementation of the S protein with conserved T-cell epitopes led to strong humoral and cell-mediated immunity responses against the BA.2 strain of SARS-CoV-2. Furthermore, research into mRNA vaccine development using a conserved region of SARS-CoV-2, the N protein, showed incomplete but significant protection against infection28. Thus, multivalent vaccine development shows great promise in protecting against infection and antigenic drift. However, many conserved regions are hidden underneath the subunit proteins that are already being used in vaccine development. This could create weak immune responses when natural infection occurs. Overall, next-generation vaccines that incorporate conserved epitopes show great potential in resisting the effects of antigenic drift, although they do induce weaker immune responses than vaccines that use the main surface proteins of viruses.
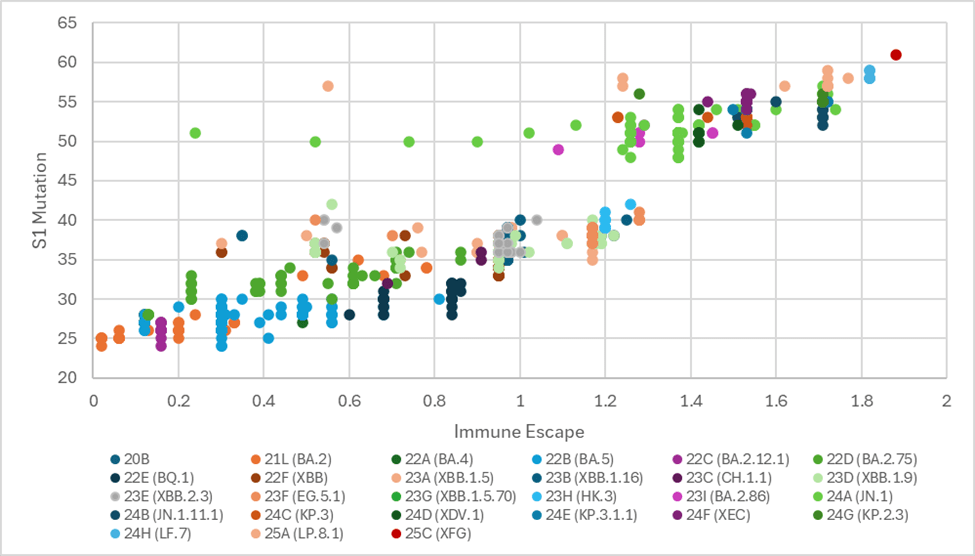
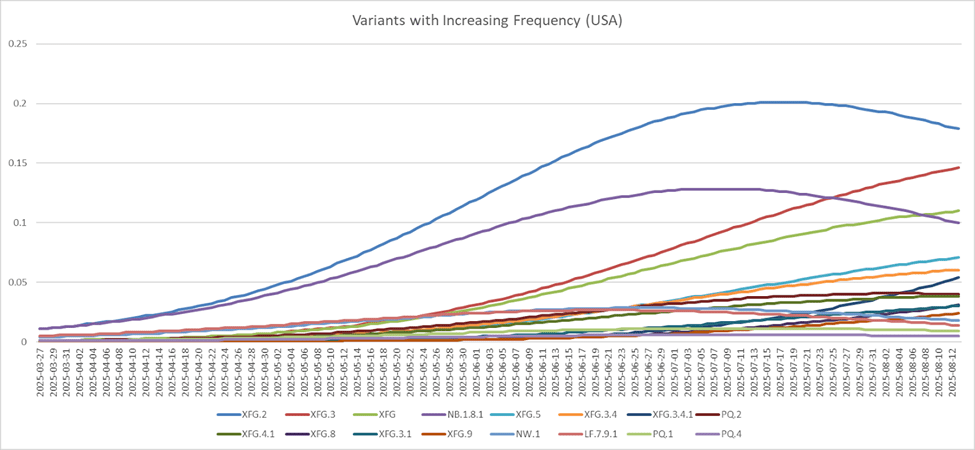
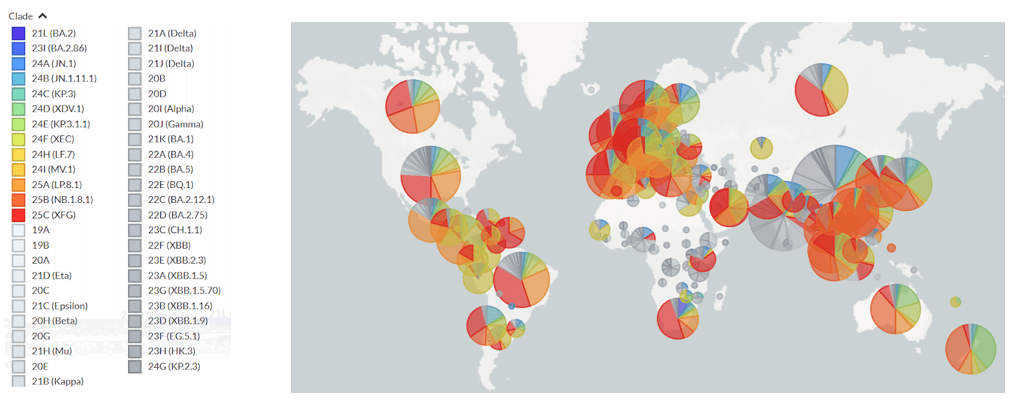
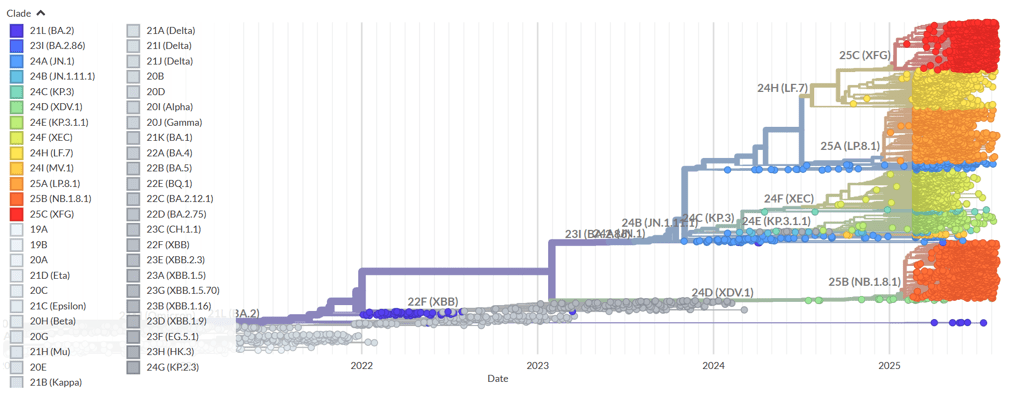
Interestingly, clade 25C(XFG) is prominent in Figure 5 and Figure 6, displaying the highest amount of S1 mutations and immune escape out of all of the clade memberships and lineages. In fact, out of all of the SARS-CoV-2 variants in the US, XFG variants are the most prominent variants that have increased in frequency in the United States, and have the highest lineage fitness, demonstrating how lineage fitness could be positively correlated with mutation frequency, variant frequency, and immune escape. Perhaps mutation frequency leads to increased lineage fitness as stronger variants of the lineage spread more frequently, resulting in a higher immune escape that along with the fitness of the variants, increases the frequency of the variants. Moreover, there could be a strong correlation between the fitness of SARS-CoV-2 variants, and the wealth and healthcare development of the country in which they are most prevalent. Figure 7 shows how globally, many of the strongest lineages of SARS-CoV-2 are most frequent in wealthy and healthcare-sufficient countries compared to their surrounding regions, such as the United States, Singapore, and Brazil. Interestingly, Figure 8 shows that two of the most common clades of SARS-CoV-2–XFG and LP.8.1–are descended from the same branch on the phylogeny tree: JN.1.11.1. The rise in frequency of XFG over the past six months indicates that a large part of the evolution of SARS-CoV-2 will occur through the XFG lineage. These analyses demonstrate how computational models and real-time tracking tools can predict mutation hotspots and likely escape variants, such as XFG. Therefore, it can potentially be concluded that mutation hotspots are likely to be highly developed countries around the globe, with most mutations of SARS-CoV-2 occurring within the S1 protein subunit of the virus.
In contrast, DNA viruses exhibit substantially lower mutation rates than RNA viruses, shifting the primary challenge to vaccine efficacy from mutations to immune evasion strategies. HSV-1 and HSV-2 are dsDNA viruses with no approved vaccines, although vaccines are being tested. Vaccine efforts at the moment have been focused on HSV-2. However, many HSV genes are involved in immune escape, meaning that vaccine development is incredibly difficult.
The genomes of HSVs are relatively stable compared to RNA viruses such as HIV-1 and HCV, although HSV-2 possesses an inherently higher mutation rate than HSV-129. Because HSVs are so stable, viral strains chosen for subunit vaccines or as parental viruses are unimportant.
HSV envelope glycoproteins gB, gD, and gH-gL bind viruses to host cells29. Potent-neutralizing antibodies are essential for gD to bind to receptors for adhesion and entry into cells. Therefore, the development of receptor-blocking compounds has been put into place. Moreover, HSV enters cells using proteins that also function in cellular communication and adhesion, making vaccination possible to elicit pathological effects. However, this did not occur in over 10,000 subjects tested29. Antibodies that delete gH have been proven to be very potent, as deletion of gH seems to cause HSV strains that have been tested as vaccines to be discontinuously replicated29. Without the gH, viruses cannot enter the next generation of cells. Because HSV infects through person-to-person contact, sterilization at the site of contact has been tested as a method of vaccination. However, only trials on mice have succeeded in sterilizing infections.
Despite lower mutation rates than RNA viruses, DNA viruses like HSV-1 possess sophisticated immune evasion mechanisms that can prevent critical antiviral responses, such as RNA and protein manipulation, latency, and viral mimicry. Such responses can lead to significant vaccine development and efficacy challenges.
One host shutoff approach used by viruses is the promotion of global RNA decay. Several herpesviruses have been thoroughly studied on their modulation of mRNA stability, as these herpesviruses are able to degrade mRNA through viral ribonuclease induction30. HSV-1 has a specific protein, UL41, that works as a virion host shutoff and an mRNA-specific degrader. UL41, upon cell entry, rapidly degrades both host and viral mRNAs prior to de novo viral gene expression, then seizes the synthesis of cellular protein and disaggregates preexisting polyribosomes, preventing the expression of cellular immune factors and diverting host organelles for viral replication. A study on mice found that mice that were infected with nonfunctional virion host shutoff had elevated levels of host-activated innate immune responses. Furthermore, wild-type HSV-1 was found to be capable of reducing levels of viperin mRNA, which builds the antiviral protein viperin, while HSV-1 viruses without UL41 did not.
Another method of RNA degradation is the usage of microRNAs to interfere with host translational processes. However, the roles of many miRNAs are still not yet known or understood, but it is currently known that viral miRNA target genes in immune and nonimmune cells, suggesting that miRNA is vital in subverting host antiviral immune responses.
DNA viruses exploit proteasome pathways to evade host immune responses to inhibit host defense factors, such as the encoding and recruitment of ligases that target the degradation of host proteins via the proteasome pathway. The viral E3 ubiquitin ligase performs such a task. One of the E3 ligase’s main targets is PML, which is a protein that acts as a central organizer or scaffolding for PML-NB. PML has been reported to silence a number of herpesviruses, due in part to the epigenetic silencing of viral genomes that PML-NBs are capable of30. Thus, when PML knockdown occurs, HSV-1 titers have been found to increase following infection in human fibroblasts. Several viral E3 ligases have been found to specifically target PML for proteasomal degradation, such as the ICP0 protein in HSV-1 that was found to directly target PML.
DNA viruses also degrade host Pattern Recognition Receptors (PRRs) to inhibit viral DNA recognition. The ICP0 protein in HSV-1 has also been found to target the viral DNA sensor DNA-PK. Inhibition of DNA-PK by ICP0 causes reductions in the half-life of DNA-PKcs. Moreover, ICP0 is essential and sufficient for the proteasomal degradation of DNA-PKcs, which encourages more efficient replication in DNA-PKcs depleted cells30.
In addition, DNA viruses hijack host defense proteins through functional inhibition and sequestration of the proteins. Immune system pathways that are activated by DNA virus infections intersect when transcription factors stimulate the process of gene transcription. Thus, many viruses have developed mechanisms to prevent the activation of proteins that cause the activation of gene expression. IRF-3 inhibits the gene expression of programmed cell death. The targeting of IRF-3 by viral proteins such as ICP0 could potentially establish and maintain persistent and latent infections, allowing DNA viruses to persist in the body of their host, and continuously replicate and infect their host. ICP0 binds to IRF-3, sequestering it between nuclear bodies, preventing the transcription factor association with host gene binding sites that is required for Type 1 IFN induction, which is important for host defense against viruses. Other viruses use other factors to inhibit IRF-3 and other proteins, displaying the diversity in complex immune evasion mechanisms that DNA viruses possess30.
DNA viruses also use viral mimicry to evade immune responses, such as cytokine mimicry. In herpesviruses, cytokine mimicry has been observed in the IL-10 cytokine, which is an anti-inflammatory cytokine that inhibits the production of pro-inflammatory cytokines. Viruses use these properties of IL-10 to mitigate host defenses through pro-inflammatory cytokine expression. cmvIL-10 is a gene that was most likely pirated from host genomes that binds to IL-10 receptors with high affinity. Despite only being 27% homogenous in sequence to human IL-10, cmvIL-10 is highly effective at promoting inflammatory cytokine expression. Dendritic cells (DC) are responsible for priming cellular immune responses. Plasmacytoid dendritic cells, which are major producers of type 1 interferons–which are antiviral proteins–during infection, had the expression of IFN⍺ in them severely reduced due to cmvIL-10, causing reductions in antiviral response.
In further detail, HSV can infect myeloid-lineage DC due to the HSV receptors that are expressed by the cells, which reduces the ability of DC to present antigens for immune responses. As mentioned before, HSV often induces IFNα, which increases T-cell survivability and supports Th1-like responses. IFNα is secreted by DC plasmacytoid in response to HSV, meaning that HSV must develop means to evade IFNα29. One way HSV evades IFN-α is by using the vhs protein. The vhs protein antagonizes IFNα, which produces the γ₁34.5 protein. This protein reduces the shutoff of host protein synthesis, which allows HSV to replicate more29. HSV can also infect T-cells, among several immunocompetent cells, which can influence the homeostasis of the CTL response to HSV31.
HSV inhibits antigens from causing antibody development in the HLA class I and class II pathways by preventing antigen processing and presenting. Inhibition of antigen-presenting is caused by interactions by ICP47 with the transporter responsible for antigen processing. The evasion of HLA class I-restricted responses is performed by the vhs gene29. In CD4 T-cells, not much is known about how HSV evades immune responses. However, it is known that ICP-22 is responsible for inhibiting antigen presentation, while HSV-infected B-cells are modified to present antigenic epitopes to CD4 T-cell clones inefficiently32.
Many vaccine formats have been explored in developing HSV vaccines. Peptide vaccines, which use a single epitope to elicit immune responses, have been proven to be protective on mice. However, this is not indicative of the efficacy on humans. Subunit vaccines present a more complex antigen to the immune system with recombinant protein formats being tested. They can only partially protect against HSV-2 seroconversion, with no protection against HSV-1 seroconversion. The GSK vaccine used a truncated form of the gD of HSV-2 and was estimated to have an efficacy of 75% for genital herpes29. In addition, truncated HSV glycoproteins had been used for immunotherapy in adults with symptomatic genital HSV-2. Immunotherapy using truncated HSV glycoproteins significantly reduced the number of genital herpes recurrences in a one-year period29. However, the development of the GSK genital herpes vaccine was discontinued due to dwindling funding. Killed-virus vaccines had been proven to protect animals29. However, because of a lack of data on efficacy and studies with a lack of placebo controls, there was no conclusive data on the efficacy of killed-virus vaccines29. Fractionated virus vaccines inactivate viruses in infected cells and partially purify subsets of viral proteins. A fractionated virus vaccine that was enhanced by HSV-2 enveloped glycoproteins appeared to be immunogenic and decreased the severity of recurrent HSV-2 disease. Further development saw an increasing trend in neutralizing and total antibodies, but no evidence supported that the vaccine had clinical benefits. Replication-deficient vaccines use the deletion of one or more genes required for viral replication to prevent viral infection or severity. They showed promising results in guinea pigs and entered clinical trials several years ago, but no final vaccine was developed. Attenuated vaccines stimulate a broad antibody response by presenting a mixture of epitopes from the entire HSV genome except from deleted genes. However, they could make vaccine strains either reactivate and recombine with wild viruses or be transmitted to immunocompromised patients29. These vaccines were all developed in the 2000s, and since then there have been innovations in vaccine technology. Moreover, all of the previously mentioned vaccines never succeeded. Although attenuated and subunit vaccines are still being researched and developed for HSV, nucleic acid vaccines are being developed for HSV. The main focus in the development of HSV nucleic acid vaccines has been DNA vaccines. One example of a DNA vaccine is COR-1, which codes for the HSV-2 envelope gD2 and a truncated gD2 fused with ubiquitin. This vaccine demonstrated humoral and immunal responses that protected against lethal viral challenge and reduced viral latency in murine models. Moreover, the vaccine has demonstrated, in clinical trials, safety and reduced viral shedding after administration into humans. It is important to note that no HSV vaccines have been released, as many are in clinical trials or were discontinued.
The nature of SARS-CoV-2 and influenza allows vaccines to be developed quickly due to the smaller genomes of RNA viruses. When comparing the 85 viral proteins that HSV contains to the 4->10 that influenza has and around 20 viral proteins that SARS-CoV-2 possesses, the genome size itself helps explain why HSV still does not have any approved vaccines33,34. The large genome and amount of immune-evading genes of HSV create major challenges to vaccine development. Moreover, dwindling funding for many HSV vaccines forced many vaccines that were in clinical trials or development to be cancelled. HSV vaccines could become approved shortly, but with many vaccines having gone through late-stage trials without being approved, it is unpredictable when vaccines for HSV will be available.
However, HSV is not the only disease where vaccine development has been plagued by shaky investment and technological limitations. These issues are universal to vaccine development35. The development process is long and costly, with every stage in the process holding inherent risks that can create financial burdens on vaccine manufacturers. Inherent technological limitations can lead to ineffective vaccines, such as the failure to select a pathogen’s correct genetic sequence for antigen coding. Moreover, next-generation vaccines require special refrigeration containers, as improperly preserved mRNA vaccines can lead to reduced effectiveness. This restriction negatively affects the scale at which next-gen vaccines are deployed globally, and hinder worldwide vaccination attempts. In manufacturing, the inherent high cost and requirement for highly qualified individuals, as well as manufacturing limitations for certain vaccines also lead to discouragement in vaccine development. Moreover, vaccines benefit groups outside of the stakeholders, decreasing the return on investment and discouraging investors from investing in the vaccine market. This underinvestment reduces the incentive of vaccine manufacturers to develop new vaccines, which can cause the technology and market behind vaccine development to plateau. However, when it comes to extreme financial incentives, such as in the case of the SARS-CoV-2 pandemic, where governments will invest heavily to rapidly develop vaccines, vaccine development rapidly progresses and new technologies are quickly improved. These are rare cases, however, meaning that most vaccines take several years to develop instead of the one year it took for SARS-CoV-2 vaccines to be developed. Although technological advances have greatly progressed the efficiency and efficacy of vaccine development, such as the invention of the mRNA vaccine, these new vaccine types come with limitations that once again burden vaccine development. Hindrances such as these are real-world challenges that governments must address.
Conclusion
Viral mutations prevent vaccines from retaining efficacy and often cause vaccine development struggles through antibody escape mutations. However, advancements in vaccine technologies show great promise, such as the hybridization of immunodominant viral regions with conserved epitopes for broad spectrum protection. Multivalent vaccines, despite being weaker than monovalent vaccines, are more durable and do well when viral strains mutate, showing promise in dealing with antigenic drift. Pursuing vaccines that utilize the genomes of multiple strains through conserved epitopes are the way forward in dealing with RNA viruses. Yet still, despite advancements having been made in dissecting viral genomes and abilities to manipulate these genomes, much about viruses remains unknown. The complex immune evasion techniques of DNA viruses still provide great challenges in vaccine development for viruses such as HSV, and vaccine development is still largely limited by technological and financial boundaries. Furthermore, it is still relatively unknown why retroviruses perform reverse transcription and the processes behind the difference in mutation rate between single-stranded and double-stranded viruses. Viruses must be closely examined in the future to prevent and mitigate further pandemics like SARS-CoV-2.
References
- S. Seronello, J. Montanez, K. Presleigh,M. Barlow, S. B. Park, J. Choi. Ethanol and Reactive Species Increase Basal Sequence Heterogeneity of Hepatitis C Virus and Produce Variants with Reduced Susceptibility to Antivirals. PloS One. 6. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0027436 (2011). [↩]
- S. Rampersad, P. Tennant. Replication and expression strategies of viruses. Viruses. 55-82. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC7158166/#:~:text=Genomic %20dsRNA%20is%20transcribed%20into,evades%20the%20hosts’%20immune%20system (2018). [↩] [↩]
- R. Sanjuán, P. Domingo-Calap. Mechanisms of viral mutation. Cellular and Molecular Life Sciences: CMLS. 73, 4433–4448. https://pmc.ncbi.nlm.nih.gov/articles/PMC5075021/ (2016). [↩] [↩] [↩] [↩] [↩]
- D. Clark, N. Pazdernik. Viruses. Molecular Biology (Second Edition). 649-685. http://www.sciencedirect.com/science/article/abs/pii/B9780123785947000214 (2013). [↩]
- L. Bono, D. Siobain. Mechanisms of RNA virus evolution. Encyclopedia of Virology (Fourth Edition). 1, 62-70. ww.sciencedirect.com/science/article/abs/pii/B9780128145159000138 (2021). [↩]
- S. Seronello, J. Montanez, K. Presleigh,M. Barlow, S. B. Park, J. Choi. Ethanol and Reactive Species Increase Basal Sequence Heterogeneity of Hepatitis C Virus and Produce Variants with Reduced Susceptibility to Antivirals. PloS One. 6. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0027436 (2011). [↩]
- M. Pérez-Losada, M. Arenas, J. C. Galán, F. Palero, F. González-Candelas. Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infection, Genetics and Evolution: Journal of Molecular Epidemiology and Evolutionary Genetics in Infectious Diseases. 30,296-307. https://www.sciencedirect.com/science/article/pii/S156713481400478X (2015). [↩]
- P. Schuster. Quasispecies on Fitness Landscapes. Curr Top Microbiol Immunol. 392, 61-120. https://pubmed.ncbi.nlm.nih.gov/26597856/ (2016). [↩]
- J. Sardanyés, C. Perales, E. Domingo, S. F. Elena. Quasispecies theory and emerging viruses: challenges and applications. npj Viruses.19. https://www.nature.com/articles/s44298-024-00066-w#:~:text=Quasispecies%20theory%20revolutionized %20our%20understanding,drug%20resistance%2C%20and%20viral%20emergence. (2024). [↩]
- Center for Disease Control and Prevention. How flu viruses can change: ‘Drift’ and ‘Shift.’ Centers for Disease Control and Prevention. http://www.cdc.gov/flu/about/viruses/change.htm (2022). [↩]
- H. Kim, R. G. Webster, R. J. Webby. Influenza virus: dealing with a drifting and shifting pathogen. Viral Immunology. 31, 174-183. https://pubmed.ncbi.nlm.nih.gov/29373086/ (2018). [↩]
- H. Kim, R. G. Webster, R. J. Webby. Influenza virus: dealing with a drifting and shifting pathogen. Viral Immunology. 31, 174-183. https://pubmed.ncbi.nlm.nih.gov/29373086/ (2018). [↩] [↩] [↩] [↩]
- A. Yasuhara, S. Yamayoshi, P. Soni, T. Takenaga, C. Kawakami, E. Takashita, Y. Sakai-Tagawa, R. Uraki, M. Ito, K. Iwatsuki-Horimoto, T. Sasaki, K. Ikuta. S. Yamada, Y. Kawaoka. Diversity of antigenic mutants of influenza A(H1N1)pdm09 virus escaped from human monoclonal antibodies. Scientific Reports. 7. https://pmc. ncbi.nlm.nih.gov/articles/PMC5735164/#:~:text=These%20middle%2Daged%20adults%20suffered,of%20the%20wild%2Dtype%20virus (2017). [↩] [↩] [↩]
- J. A. Malik, S. Ahmed, A. Mir, M. Shinde, O. Bender, F. Alshammari, M. Ansari, S. Anwar. The SARS-COV-2 mutations versus vaccine effectiveness: new opportunities to new challenges. Journal of Infection and Public Health. 15, 228-240. www.sciencedirect.com/science/article/pii/S1876034122000028 (2022). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Y. Finkel, O. Mizrahi, A. Nachshon, S. Weingarten-Gabbay, D. Morgenstern, Y. Yahalom-Ronen, H. Tamir, H. Achdout, D. Stein, O. Israeli, A. Beth-Din, S. Melamed, S. Weiss, T. Israely, N. Paran, M. Schwartz, N. Stern-Ginossar. The coding capacity of SARS-COV-2. Nature. 589, 125–130. www.nature.com/articles/s41586-020-2739-1#Sec1 (2020). [↩]
- J. A. Malik, S. Ahmed, A. Mir, M. Shinde, O. Bender, F. Alshammari, M. Ansari, S. Anwar. The SARS-COV-2 mutations versus vaccine effectiveness: new opportunities to new challenges. Journal of Infection and Public Health. 15, 228-240. www.sciencedirect.com/science/article/pii/S1876034122000028 (2022). [↩] [↩]
- G. Dolton, C. Rius, M. S. Hasan, A. Wall, B. Szomolay, E. Behiry, T. Whalley, J. Southgate, A. Fuller, The COVID-19 Genomics UK (COG-UK) consortium, T. Morin, K. Topley, L. R. Tan, P. Jr Goulder, O. B. Spiller, P. J. Rizkallah, L. C. Jones, T. R. Connor, A. K. Sewell. Emergence of immune escape at dominant SARS-CoV-2 killer T cell epitope. Cell. 185, 2936–2951. https://pmc.ncbi.nlm.nih.gov/articles/PMC9279490/#sec2 (2022). [↩]
- G. Cerutti, Y. Guo, L. L.H. L. Y. Liu, Z. Zhang, Y. Luo, Y. Huang, H. Wang, D. D. Ho, Z. Sheng, L. Shapiro. Cryo-EM structure of the SARS-CoV-2 Omicron spike. Cell Rep. 38.https://pmc.ncbi.nlm.nih.gov/articles/PMC8818377/ (2022). [↩]
- K. Y. Leong, S. K. Tham, C. L. Poh. Revolutionizing immunization: a comprehensive review of mRNA vaccine technology and applications. Virol J. 22. https://pmc.ncbi.nlm.nih.gov/articles/PMC11900334/#:~:text=To%20overcome% 20the%20lack%20of,based%20neutralization%20studies%20in%20mice (2022). [↩]
- J. R. Marcelin, A. Pettifor, H. Janes, E. R. Brown, J. G. Kublin, K. E. Stephenson. COVID-19 Vaccines and SARS-CoV-2 Transmission in the Era of New Variants: A Review and Perspective. Open Forum Infect Dis. 9. https://pmc.ncbi.nlm.nih.gov/articles/PMC8992234/#s2 (2022). [↩]
- A. Oordt-Speets, J. Spinardi, C. Mendoza, J. Yang, G. Morales, J. M. McLaughlin, M. H. Kyaw. Effectiveness of COVID-19 Vaccination on Transmission: A Systematic Review. COVID. 3, 1516-1527. https://www.mdpi.com/2673-8112/3/10/103 (2023). [↩]
- H. Jacobsen, I. Sitaras, M. Katzmarzyk, V. C. Jiménez, R. Naughton, M. M. Higdon, M. D. Knoll. Systematic review and meta-analysis of the factors affecting waning of post-vaccination neutralizing antibody responses against SARS-CoV-2. NPJ Vaccines. 8. https://pmc.ncbi.nlm.nih.gov/articles/PMC10589259/#Sec2 (2023). [↩]
- M. Brockman, F. Mwimanzi, H. R. Lapointe, Y. Sang, O. Agafitei, P. K. Cheung, S. Ennis, K. Ng, S. Basra, L. Y. Lim, F. Yaseen, L. Young, G. Umviligihozo, F. H. Omondi, R. Kalikawe, L. Burns, C. J. Brumme, V. Leung, J. S. G. Montaner, D. Holmes, M.L.DeMarco, J. Simons, R.Pantophlet, M. Niikura, M.G. Romney, Z. L. Brumme. Reduced Magnitude and Durability of Humoral Immune Responses to COVID-19 mRNA Vaccines Among Older Adults. J Infect Dis. 225, 1129–1140. https://pmc.ncbi.nlm.nih.gov/articles/PMC8689804/#s10 (2021). [↩]
- J. Xie, B. Mothe, M. A. Herraiz, C. Li, Y. Xu, A. M. Jödicke, Y. Gao, Y. Wang, S. Feng, J. Wei, Z. Chen, S. Hong, Y. Wu, B. Su, X. Zheng, C. Cohet, R. Ali, N. Wareham, D. P. Alhambra. Relationship between HLA genetic variations, COVID-19 vaccine antibody response, and risk of breakthrough outcomes. Nature Communications. 15. https://www.nature.com/articles/s41467-024-48339-5 (2024). [↩]
- M. Esposito, F. Minnai, M. Copetti, G. Miscio, R. Perna, A. Piepoli, G. D. Vincentis, M. Benvenuto, P. D’Addetta, S. Croci, M. Baldassarri, M. Bruttini, C. Fallerini, R. Brugnoni, P. Cavalcante, F. Baggi, E. M. G. Corsini, E. Ciusani, F. Andreetta, T. A. Dragani, M. Fratelli, M. Carella, R. E. Mantegazza, A. Renieri, F. Colombo. Human leukocyte antigen variants associate with BNT162b2 mRNA vaccine response. Communications Medicine. 4. https://www.nature.com/articles/s43856-024-00490-2 (2024). [↩]
- M. S. T. Murala, V. Gairola, E. E. Sayedahmed, S. K. Mittal. Next-Generation Adenoviral Vector-Based Vaccines for Severe Acute Respiratory Syndrome Coronavirus-2. Vaccines (Basel). 13. https://pmc.ncbi.nlm.nih.gov/articles/PMC12031563/ (2025). [↩]
- Y. Zhang, J. Gao, W. Xu, X. Huo, J. Wang, Y. Xu, W. Ding, Z. Guo, R. Liu. Advances in protein subunit vaccines against H1N1/09 influenza. Frontiers in Immunology. 15. https://pmc.ncbi.nlm.nih.gov/articles/PMC11621219/#s7 (2024). [↩] [↩]
- R. L. Hajnik, J. A. Plante, S. R. Bonam, G. H. Rafael, Y. Liang, N. C. Hazell, J. Walker, R. A. Reyna, D. H. Walker, M.-G. Alameh, D. Weissman, S. C. Weaver, K. S. Plante, H. Hu. Broad protection and respiratory immunity of dual mRNA vaccination against SARS-CoV-2 variants. npj Vaccines. 9. https://www.nature.com/articles/s41541-024-00957-2#:~:text=A%20broadly%20protective%20SARS%2DCoV,variants%20in%20animal%20models11. (2024). [↩]
- D. M. Koelle, L. Corey. Recent progress in herpes simplex virus immunobiology and vaccine research. Clinical Microbiology Reviews. 16, 96-113. www.ncbi.nlm.nih.gov/pmc/articles/PMC145296/ (2003). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- M. S. Crow, K. K. Lum, X. Sheng, B. Song, I. M. Cristea. Diverse mechanisms evolved by DNA viruses to inhibit early host defenses. Crit Rev Biochem Mol Biol. 51, 452–481. https://pmc.ncbi.nlm.nih.gov/articles/PMC5285405/#S2 (2016). [↩] [↩] [↩] [↩]
- D. M. Koelle, L. Corey. Recent progress in herpes simplex virus immunobiology and vaccine research. Clinical Microbiology Reviews. 16, 96-113. www.ncbi.nlm.nih.gov/pmc/articles/PMC145296/ (2003). [↩]
- D. M. Koelle, L. Corey. Recent progress in herpes simplex virus immunobiology and vaccine research. Clinical Microbiology Reviews. 16, 96-113. www.ncbi.nlm.nih.gov/pmc/articles/PMC145296/ (2003). [↩]
- Y. Finkel, O. Mizrahi, A. Nachshon, S. Weingarten-Gabbay, D. Morgenstern, Y. Yahalom-Ronen, H. Tamir, H. Achdout, D. Stein, O. Israeli, A. Beth-Din, S. Melamed, S. Weiss, T. Israely, N. Paran, M. Schwartz, N. Stern-Ginossar. The coding capacity of SARS-COV-2. Nature. 589, 125–130. www.nature.com/articles/s41586-020-2739-1#Sec1 (2020). [↩]
- Y. N. Gong, G. W. Chen, C. J. Chen, R. L. Kuo, S. R. Shih. Computational analysis and mapping of novel open reading frames in influenza A viruses. PloS One. 9. https://pmc.ncbi.nlm.nih.gov/articles/proteins (2014). [↩]
- Vaccine Development: Capabilities and Challenges for Addressing Infectious Diseases. GAO. 22. https://www.gao.gov/products/gao-22-104371 (2021). [↩]



{kind=link}