
Abstract
Prion diseases are a class of neurodegenerative disorders that are caused by the accumulation of misfolded prion proteins in the brain. This narrative review assesses the recent advancements in both diagnostics and treatment strategies as pathological understanding continues to evolve in the field of prion disease research. Articles used for the purpose of this review were found through PubMed and Google Scholar and filtered to search for those published within the past decade to ensure relevance. Focusing on developing methodologies such as the study of differentially expressed genes (DEGs), morphological progressions, and novel, innovative remedies demonstrates key potential to improve the quality of life of those suffering from prion disease. Over the past decade, the research of DEGs in prion disease has made significant strides, going from detecting a large range of common DEGs between patients to pinpointing a small set of around 24 genes that can be linked to shifts in astrocyte and microglial cell expressions. Furthermore, novel methodologies, including the use of electron microscopy and 3-D voxel-based neuroimaging, for the mapping of morphological developments demonstrated significant potential to enable more precise and timely patient diagnoses. Recent advancements in research have also brought widely popular therapeutic techniques to the field of prion disease, including active immunization therapies, antisense oligonucleotides (ASOs), and stem cell therapies. However, these therapeutic strategies remain solely experimental at this point in time. Additionally, challenges remain associated with complex pathology, lack of data due to rarity of the disease, and major ethical concerns. Further research should aim to narrow down the list of DEGs to pinpoint those that are related to changes in glial cell activations, continue the study of innovative technologies, and understand deeper aspects of prion disease pathology to identify new therapeutic models and reduce the consequences of risky techniques. In overcoming these hurdles, the unraveling of key prion disease pathology yields the potential to improve quality of life and clinical outcomes for individuals with prion disease.
Keywords: Prion Disease, prion disease pathology, epigenetics, gene expression, differentially expressed genes, morphology, diagnostics, neurodegeneration
Introduction
Prion diseases are a type of neurodegenerative disease caused by the aggregation of abnormally shaped proteins, also known as prions (proteinaceous infectious particles)1. When the normal form of the prion protein, PrPC, is misfolded it becomes the infectious form known as PrPSc (the “Sc” stands for scrapie). Then, one of these misfolded prion proteins can produce a chain reaction. One PrPSc protein collides with another, thus misfolding a second prion protein. Next, the two misfolded proteins produce two more, and this cycle continues on and on until a colony of misfolded proteins has appeared1. This group of diseases comes in various forms, the main three subclasses being sporadic (spontaneous), genetic (familial), and acquired (infectious/transmitted). Sporadic Creutzfeldt-Jakob disease (sCJD) is one type of the sporadic prion diseases; it alone accounts for around 85% of all prion disease cases. Additionally, 10-15% of all cases are inherited through the genetic form of transmission; some types include genetic CJD (gCJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI). Lastly, around 1% of all cases are acquired through infectious particles, a comparatively rare form of transmission2. The wide variety of prion diseases gives rise to troublesome heterogeneity, which can alter a patient’s clinical course, prognosis, and their response to diagnostics tools based on the type of prion disease they have.
The symptomatic progression of prion disease can be visualized in three stages. The first stage encompasses basic abnormalities such as memory loss, fatigue, visual disturbances, etc. The second stage is where acute neurological symptoms begin to appear, including cognitive decline, motor impairment, language impairment, and sensory disturbances such as hallucinations. In the final stage, the severity steepens much faster. Immense challenges such as loss of consciousness, limb immobility, loss of reflexes, and overall weakness tend to emerge and make it difficult for the patient to go about normal life4. Eventually, complications such as pneumonia, heart disease, or respiratory failure are what inevitably lead to the patient’s death. This whole process, starting from the emergence of mild symptoms to inevitable death, also tends to unfold along different timelines. For some, this process of deterioration may last years. But for others, this process may unfold along the course of a couple quick months, underscoring the importance of prompt and accurate diagnosis.
One of the common methods used for prion disease diagnosis is cerebrospinal fluid (CSF) biomarker testing, which can contain pertinent proteins, such as 14-3-3 proteins and Total Tau proteins, that give insight to a patient’s neurological conditions and clinical concerns. Rapid neuronal degeneration can sometimes lead to leaking of 14-3-3 proteins into the CSF. In fact, in the late 1900s, various studies found a strong relationship between CJD patients and 14-3-3 protein aggregation in CSF4. As such, spinal taps used to extract trace amounts of the CSF can be very useful in detecting prion disease. Total Tau proteins are another similar CSF protein indicator that are connected to acute neurodegeneration but compared to the 14-3-3 protein biomarker they are less effective in accurate diagnosis because of overlap with cases of Alzheimer’s disease, another well-known neurodegenerative disorder4.
Recently, Magnetic Resonance Imaging (MRI) has also been a useful tool in early detection of prion disease. On its own, it may not have a high efficacy, but when paired with other methods of detection such as CSF biomarker testing, it becomes very pertinent. Recently, MRI scans have been used to detect abnormal signals of Diffusion-Weighted Imaging, which is the tracking of random water molecule movement in brain tissue across various regions of the brain. These regions include the frontal lobe, parietal lobe, occipital lobe, temporal cortex, caudate nucleus, putamen, or thalamus. If one or more of these regions exhibit serious change in signal intensities, then it can be used as a key criterion for detection of prion disease4.
The most relevant tool today for early diagnosis is Real-Time Quaking-Induced Conversion. This process of detecting prion disease involves shaking and breaking misfolded aggregates to track reproduction of the misfolded proteins; this is performed by comparing them against a recombinant normal protein via fluorescent labeling. First, a sample, such as CSF fluid, brain tissue, or blood, is extracted from the patient, sometimes through surgical methods. Recombinant PrPC, produced in bacterial or mammalian expression systems using a cloned PRNP gene, is then added to the reaction mixture. Then, the sample is shaken, also known as “quaking”, which breaks apart from newly misfolded proteins. Finally, the growth rate of the aggregates is tracked through the fluorescent dye Thioflavin T. A positive result of prion disease is correlated with a sigmoidal curve of fluorescence intensity over time. In essence, this method relies on the repeating patterns of quaking and then incubation, which allows for the breakage of aggregates and then the rapid growth following it5,6.
Despite these increasingly sensitive methods of detecting prion disease, which have allowed for increases in accurate and effective diagnoses, challenges continue to persist up to date with early diagnosis. This persistent lack of decisive tools is the reason why it can be challenging to make advancements in the field of prion disease.
One of the reasons why we lack effective diagnostic tools at this moment is because we cannot fully understand the pathways and mechanisms by which Prion proteins are misfolded and how its effector proteins are getting dysregulated. PrPSc aggregation has been connected to specific pathological changes, including reactive astro- and micro-gliosis7. Other pathologic changes within cells have been noted, such as vacuolation and progressive synaptic loss7. However, it is unclear how exactly PrPSc aggregation is linked to these changes and how it may be directly causing them. Gaining insight as to how and why specific pathological changes related to morphology and transcription occur could prove beneficial for early diagnosis and drug development efforts.
To understand how transcriptomic and morphological tools may advance prion diagnostics, it is useful to examine how similar approaches have transformed Alzheimer’s and Parkinson’s disease research.
For example, in a study conducted by Jin et al., the analysis of four different GEO datasets of Alzheimer’s patients led to a conclusive discovery that three specific differentially expressed genes could be tied to progression of the disease8. These genes included TXNIP, EGR1, and IGFBP5. The combination of multiple machine-learning processors found that each of these three genes were directly linked to Clinical Dementia Rating (CDR) scores and Braak staging, scoring systems that describe the progression and severity of a patient’s Alzheimer’s disease. Based on their findings, Jin et al. believe that the signature genes that they found can prove useful in the detection of Alzheimer’s disease and for future research in that field8. These findings do more than demonstrate advances in Alzheimer’s diagnostics, they provide a cohesive blueprint for prion disease research. Just as AD researchers narrowed broad transcriptomic information down to a few predictive genes, prion disease research must move toward identifying a similarly small, insightful gene pattern to strengthen diagnosis.
In a similar study, Li et al. analyzed various differentially expressed genes (DEGs) between cases of patients with Parkinson’s disease (PD) and healthy controls9. Based on the DEGs that were identified to be shared, two specific genes were found to be correlated to worsening motor progression in PD patients. These two genes were the LILRB3 gene, which is part of a family of receptors found mostly on immune cells, and the LRRN3 gene, which is linked to cell-to-cell signaling especially in the nervous system. Like Jin et al., Li et al propose the use of these two newly identified genes as a biomarker for effective Parkinson’s disease diagnosis9. The PD research also demonstrates that transcriptional biomarkers can correlate with clinical progression. Applying this strategy to prion disease may allow researchers to link DEGs not only to disease presence, but also to patient decline, which remains a major unmet diagnostic need in the field of prions.
My main focus is to enable prompt and accurate diagnosis so that patients have better disease prognosis. Therefore the central question in this review article is to better understand prion pathology and identify how this can enable researchers to improve upon existing diagnostic tools. For this it is important to understand the landscape of diagnostic tools. With these goals in mind, this review strives for a deeper understanding of all the interacting partners of prion disease pathogenesis and prion proteins to design better diagnostic tools. Once the specific disease pathways can be traced back to certain genes, an additional layer of diagnostics will be added to improve the overall efficiency, accuracy, and feasibility of current tools.
Methods
Articles used in this work were found on the PubMed and Google Scholar databases, and all articles used for the research in this article were peer-reviewed. Given the narrative nature of this review, literature published in English and accessible in full text was considered. Additionally, studies using both human models and animal models were considered. The search strings that were used for this research consisted of an arrangement of various keywords related to the hypothesis as well as Boolean operators. Keywords included “prion disease”, “gene expression”, “RNAseq”, “early diagnosis”, “differentially expressed genes”, etc. One example search string that was used was “Prion disease AND RNAseq.” The purpose of such keywords in the search strings used was to ensure that all articles found were in alignment with the scope of this article. Additionally, among the experimental studies found, both in vivo and in vitro studies were considered and used to gather information. Inclusion criteria also consisted of publication date: only articles published within the past 10 years were used to gather information. This stipulation was necessary to ensure that the topics reviewed in this article are the most recent advances within the field. Initial screenings of titles and abstracts were performed to determine relevance to prion disease diagnostics, transcriptomics, morphological research, or therapeutic advancements. Formal quality checklists such as PRISMA risk-of-bias assessments were not applied due to the narrative nature of this review.
Results
Disease Progression and Candidate Biomarkers
Morphological Changes
One very important area of prion disease studies relates to the actual morphology of the brain. This refers to the observable physical changes in the structure of neurons and brain cells that directly occur because of clinical prion disease progression. Studying these changes has proven to be valuable as it can help relate structural changes to clinical symptoms and stages of disease progression.
One such study does exactly that and investigates neuroanatomical markers, linking them to disease development. This study, conducted in 2016 by Vita et al., uses longitudinal high-resolution MRI, which involves a very strong magnetic resonance that is repeated rather than just scanned once, and voxel-based morphometry, a neuroimaging technique that creates a computer-based model of 3D pixels to map the brain, to track gray matter volume changes over time in human models (gray matter is the type of tissue that shows up on MRI scans and is composed of neurons and glial cells). Their findings demonstrated progressive shrinkage of the gray matter tissue over the course of disease progression in specific regions, namely the thalamus, basal ganglia, and cortical areas10. This study conducted by Vita et al. demonstrated a useful method of utilizing a combination of high-power MRI and 3D voxel-based neuroimaging to quantify brain tissue death, which was used in future studies as a building block10. That being said, although these voxel-based morphometry findings provide valuable insight into disease progression, they primarily serve as a research tool rather than a routine diagnostic tool in current clinical practice due to cost, accessibility, and limitations in flexibility.

Another study that highlights the use of researching prion disease morphology was conducted by Garcés et al. In their study, which was published in 2021, they ran an experiment comparing human patients with various strains of prion disease to human subjects that did not possess any form of neurodegeneration. To visualize morphological changes, Garcés et al. used various forms of electron microscopy (an immensely powerful form of microscopy that can magnify objects over 50 million times) and immunofluorescence techniques. Across all forms of CJD among test subjects, they found that there were shared glial cell abnormalities12. In addition, they found that microglial activation, a hallmark symptom in patients with neurodegeneration, was correlated with astrogliosis, the abnormal increase in astroglia cells12. Furthermore, this gliosis and glial activation occurred across all strains of CJD that were analyzed.
Another important result yielded from the study conducted by Garcés et al. was the correlation between gliosis, glial activation, and spongiosis. Spongiosis is a characteristic feature of prion diseases that occurs when bubbles of space form in the brain leading to a spongy appearance of brain tissue. Since the 1970s, the activation and abnormal growth of various glial cells in patients with prion disease have been viewed as a direct result of PrPSc accumulation. However, Garcés et al. presented the possibility that glial cell changes might be a distinct part of the disease process itself12. This idea stems from the fact that Garcés et al. compared glial cell changes directly to spongiosis rather than PrPSc aggregation. By conducting a systematic, quantitative, and focused analysis of glial cell abnormalities across various CJD subtypes, they reframed from the role of glia as central parts of prion disease pathology rather than just reactive bystanders12. This study highlights the diagnostic value of studying neuronal morphology across various disease stages. Such preclinical insights have the potential to help us understand disease mechanisms better. With this in mind, it is important to recognize that electron microscopy, although useful for research purposes, is not a feasible tool for routine clinical diagnosis because of its highly specialized nature and specific sampling needs.
Overall, these studies demonstrate correlation between glial activation, tissue degeneration, and disease pathology rather than establishing direct causality. Further, these studies may be influenced by sampling and selection bias. Most analyses with these techniques require relatively small cohorts and frequently rely on patients already identified with advanced disease, increasing the potential for such biases. Despite these limitations, these tools demonstrate the capability to further understanding of prion disease pathology for ongoing research in the field.
Genetic Changes
Due to recent advancements, studies revolving around prion disease have shifted from focus on classic diagnostics to a focus on modern transcriptomics, the study of changes in gene expressions, through RNA levels, rather than changes in the genetic code itself. However, the search into transcriptomic changes in prion disease models is not a strictly novel advance either. In their landmark publication in 2009, Hwang et al. investigated global gene expression changes in eight distinct mice models with eight different variations of prion disease13. This study allowed them to focus on specific host genetic impacts and the effects of prion strain on each mouse over the course of disease progression. From these eight models, they identified a core set of 333 shared differentially expressed genes (DEGs) which they then tied back to changes noticed throughout the experiment, including accumulation of PrPSc, microglial/astrocyte activation, synaptic loss, and ultimately neuronal death13.
Many articles written after the article by Hwang et al. have run experiments that aim to achieve a similar goal. For example, Cheng et al. conducted a study using the public dataset GSE160208, which consists of the expressional data of around 800 neuroinflammation-linked genes in the brain tissues of 27 sCJD patients and 20 normal individuals, to explore and identify connected genes and pathological mechanisms14. This study, which was published in 2023, looked specifically at around 800 genes that are linked to inflammation and identified which genes were associated with disease status as symptoms progressed. After using multiple tools to analyze these genes and correlate them to the disease status, Cheng et al. found 88 candidate genes that overlapped between the various testing methods14.
With the 88 candidate genes that were found, Cheng et al. cross referenced their results with yet another dataset. This time they used the public dataset GSE124571, which consists of ten samples of SCJD and ten samples of control brain tissue, and found that the 88 candidate genes demonstrated the same consistent gene up/down regulation in relation to disease status14. Compared to earlier works, such as the one by Hwang et al., Cheng et al. were able to take it one step further by analyzing real patient datasets to find distinct patterns. In doing so, their work advanced the field of diagnostics by connecting neuroinflammatory signals to a set of differentially expressed genes, thus adding a layer to diagnostic biomarkers for prion disease. Despite this, the available human datasets for prion disease research remain relatively small and reflect highly selected patient populations. The lack of data raises concerns for the statistical power of such a study and the potential for selection biases to skew results.
In a similar and interesting study conducted by Slota et al., mice infected with the Rocky Mountain Laboratory (RML) strain of prion disease were inspected using RNA-sequencing7. RNA sequencing is a powerful technique that sequences RNA molecules to reveal a full set of RNA transcripts in a sample. This tool can be used to detect whether specific genes are upregulated or downregulated. Using RNA-seq, Slota et al. investigated transcriptional changes in two main regions of the brain: the CA1 Hippocampal region and thalamus of the infected mice.
The experiment conducted by Slota et al. revealed two main themes. Firstly, they found a lack of transcriptional changes during the preclinical phase of disease for many mice7. Instead, the transcripts mostly changed when clinical disease marked by symptomatic progression occurred. The second finding was that neurons in different regions respond uniquely to prion pathology. Through their study, they found that 142 microglial transcripts were commonly upregulated in both CA1 and Thalamus of the mice7. In contrast, out of around 80 astrocyte-related genes that were altered in CA1 and 298 in the Thalamus, only 24 overlapped7. This demonstrated a level of regional specificity that was found across the transcriptional changes in the mice.
Through the studies discussed and the vast number of unexplored studies, it becomes clear that research focused on differentially expressed genes as well as transcriptional changes can prove useful insight. With that being said, there are some limitations to be addressed with these studies. For example, the studies by Hwang et al. and Slota et al. both used mouse-based models and transcriptional responses in mouse brains are known to not always perfectly mirror human biology. Differences in immune cell activation, disease rate and spread, and neuronal protection may limit translation of these findings to clinical, human-based trials.
Another important consideration is the variation between studies regarding sequencing platforms, analytic techniques, and tissue processing, which introduces heterogeneity and increases susceptibility to batch effects. These technical and biological implications make comparisons between studies challenging and may portray transcriptional signatures as more robust than they may truly be. Moreover, going from transcriptomic signatures to clinical biomarkers is a big leap. DEGS do not automatically equate to reliable diagnostic biomarkers. Instead, biomarkers must demonstrate reproducibility across separate groups, effectiveness through different disease stages, and detectability in common samples such as CSF or blood.
With these limitations in mind, the transcriptional findings of these studies should be viewed as hypothesis-generating rather than definitive diagnostic tools. Additional studies with multiple independent cohorts, along with careful attention to progressive and regional variation in gene expression, will be needed before DEGs can be defined as a tool for prion disease diagnostics.
Diagnostic Tools
14-3-3 Proteins
The use of 14-3-3 proteins as a biomarker for diagnosing prion disease has evolved over the past couple decades. Initially, when it was incorporated into diagnostic criteria in the mid 1990s, it was used in parallel to other common markers such as electroencephalogram (EEG) test results and clinical features. Around this time, 14-3-3 proteins were primarily detected via Western Blotting, a process in which a sample of proteins is put through gel electrophoresis and separated by size. However, for a long time this procedure was viewed as being limited by variability and labor-intensive techniques4.
In 2011, researchers developed the 14-3-3y Enzyme-Linked Immunosorbent Assay (ELISA) assay to overcome previous shortcomings. In this test, the walls of a microplate are coated with specific antibodies that bind to the 14-3-3y isoform of the 14-3-3 protein. When a sample of CSF is added to the microplate, the 14-3-3 proteins bind to the antibodies. After, specific enzymes that are also added to the mix attach to the antibody-protein linkage and this enzyme colors the proteins a specific color, which is then analyzed using a spectrometer to quantify the data. This relatively new model for detecting and quantifying 14-3-3 protein count is standardized and comparatively easy to utilize in a lab setting, making it very useful and widely adopted4. It is very important to recognize the fact that 14-3-3 testing has long been recognized as sensitive but less specific, in part because elevated 14-3-3 can be common in other rapidly progressive neurodegenerative or inflammatory processes, which can lead to variability among results based on cohorts used in studies.
MRI
The use of MRI as a marker has also been a technique that has developed during recent times and allowed for more accurate and timely diagnosis. In the late 1990s, the use of diffusion-weighted imaging (DWI) for prion disease patients revealed a characteristic signal change specific to those with forms of CJD4. Soon after, this method was improved and was made more accurate and was found to be useful in early clinical stages of prion disease4. Additionally, recently updated criteria have shifted from requiring multiple signals from different areas of the brain, to only needing to detect abnormalities in 1 of 7 brain regions4. This development has transformed the use of MRI for prion disease diagnosis, resulting in an increase in sensitivity from 83% to 90-95%4. It is still important to consider that MRI performance varies because sensitivity is influenced by disease progression and brain region specifics. It is possible that earlier disease or atypical presentations may lead to false negatives. In combination with other diagnostic tools, though, MRI has still proved useful as an evolving technique that yields great promise.
RT-QuIC Assay
The most recent tool that has transformed the norms for prion disease diagnosis has been the RT-QuIC assay. RT-QuIC is based around seeded conversion where small amounts of patient PrPSc become templates (“seeds”) to create misfolding of recombinant PrPC substrates under repeated shaking and incubation cycles6. As these misfolded aggregates grow in number, their formation is monitored using Thioflavin T, a fluorescent dye that binds β-sheet–rich amyloid structures; increasing fluorescence intensity over time, as a result, demonstrates progressive amyloid accumulation and serves as real-time detection of prion aggregation6. First-generation RT-QuIC assays significantly improved prion detection, but second-generation protocols, which involved improved substrates and optimized conditions, demonstrated further effectiveness, with shorter run times, greater sensitivity, and enhanced detection of a broader spectrum of prion phenotypes6. This second-generation is what the high performance values now reported in literature can be attributed to.
Various tests have demonstrated this technique to be highly useful with an estimated sensitivity between 80-100% and a specificity that is very close to 100%4. In fact, Europe adopted the RT-QuIC assay into their diagnostic criteria for prion disease in 201715. Although RT-QuIC is frequently reported to have very high specificity and excellent sensitivity, these values fluctuate across studies due to differences in sampling, RT-QuIC protocols, selection methods, and whether sporadic, genetic, or atypical prion phenotypes are used. With this in mind, RT-QuIC assays still can be valuable when used in conjunction with other detection tools. Overall, the RT-QuIC assay remains one of the most reliable and precise assays for detecting prion diseases to date.
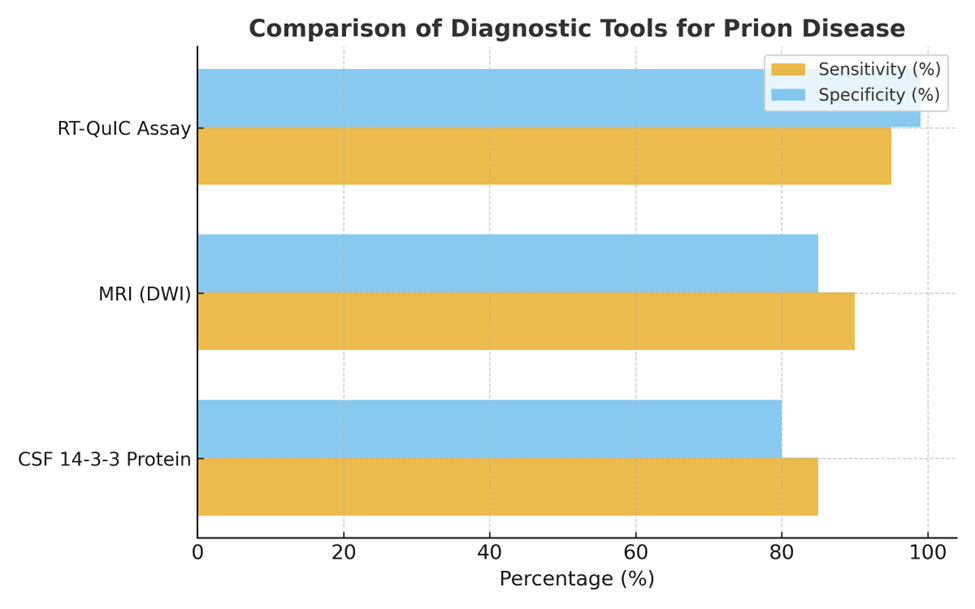
This schematic qualitatively illustrates relative diagnostic strengths based on consensus trends in recent literature rather than exact pooled statistical metrics Summary compiled and synthesized by the authors based on the cited literature. Created by the authors.
Interpreting Diagnostic Tests in Clinical Context
While sensitivity and specificity values are helpful for understanding relative diagnostic uses, they should not be interpreted as absolute measures. Performance varies across studies due to differences in patient demographics, disease progression, clinical protocols, and diagnostic criteria. Furthermore, atypical presentations and overlapping clinical disease can lead to false positives and false negatives. Thus, these values are best interpreted as indirect tools that add to, rather than replace, the current clinical diagnostic framework.
Therapeutic Strategies
Active Immunization Treatments
One recently studied area regarding potential treatment strategies for prion disease has been active immunization therapies – which can be challenging to develop as PrPC and PrPSc have different 3D structures but the same amino acid sequences, preventing normal immune responses based on peptide sequencing from playing a key role16. Multiple tests have been conducted to induce immunity within prion disease patients. For example, the introduction of modified prion peptides has been considered a stimulus for immune response. One method includes introducing truncated peptides that are known to produce increased immune response compared to normal peptide sequences. This technique aims to reduce self-tolerance to prions and help the immune system recognize these prions as foreign bodies. Another method includes introducing crosslinked or dimerized peptides that are abnormal and cause irregular 3D folding patterns. Once again, this technique aims to help the immune system recognize irregularly folded prion proteins so that immune response can play a larger role in therapy.
The primary goal of these techniques is to effectively trick the immune system into producing specific antibodies for irregular prions that have been introduced. In a study by Ishibashi et al., this treatment plan was able to prolong incubation periods in subject mice effectively demonstrating that altered prion peptides may one day be thoroughly utilized to slow prion disease17. At the same time, these strategies do showcase potential risks that need to be further investigated. Because the immune system is being trained to recognize and target a self-made protein, it is possible that the antibody produced may be more harmful to brain matter rather than helpful. This thought process is supported by tests run on Alzheimer’s patients where active immunization led to devastating toxicity in the brain16.
Despite encouraging immune responses in experimental models, active immunization strategies face further challenges. Dosing requirements are still unknown, administration would likely need to be repetitive, and targeting of the host’s proteins creates autoimmune toxicity risks. Other neurodegenerative vaccination attempts, including Alzheimer’s immunotherapy trials, demonstrated severe meningoencephalitis in some patients, which demonstrates the potential harm of such a therapy16. This method still remains in the preclinical and exploratory stage as there are no approved therapies and no clinical efficacy has been demonstrated in prion disease patients.
Antisense Oligonucleotide Therapy
A look into another recently developing strategy for treatment takes us to researcher Sonia Vallabh. Her most recent development has been the use of antisense oligonucleotides (ASOs) to treat the aggregation of misfolded prions18. These molecules are short strands of DNA that are made to directly bind to the mRNA of the Major Prion Protein gene. In binding to these mRNA sequences, a DNA-RNA hybrid chain is formed, which the body recognizes as an irregularity. As such, the enzyme RNase H is recruited and destroys the DNA-RNA hybrid, thus resulting in decreased protein synthesis through translation.
This technique aims to reduce the initial amount of PrPC proteins in the brain as for PrPSc to even begin aggregating. There must be a foundation of normal prion proteins to act as the starting point. As such, this method yields the potential to drastically slow down prion disease progression16. The main issue that comes along with this strategy is the unknown risks and uncertainties that have not been explored yet. The role of prion proteins in the human body is still largely unknown; so, while mice with depleted PrPC levels have shown normal growth and development, there is still no telling how the depletion of PrPC due to ASOs will impact humans. Though this concern is the main issue, there are other worries that limit this treatment strategy in current times, including the invasiveness of the technique and the ethical concerns of testing using human subjects16. Encouragingly, recent FDA approvals of ASO therapy for multiple rare, incurable diseases demonstrate the growing potential of this technique. For example, the use of ASO therapy for Duchenne Muscular Dystrophy, another rare, fatal genetic disease that has no current cure, has been largely successful in instances of its use. These advancements underscore the great potential of ASO therapy as a future method for combating prion disease19.
ASO therapy is a very scientifically advanced experimental strategy; however, significant issues arise as clinical trials attempt to progress. ASOs require intrathecal or intracerebroventricular administration, which requires repetitive, invasive dosing. Long-term suppression of PRNP in humans has unknown biological consequences. Although mice can tolerate it, we cannot assume the same for humans. While ASOs have achieved FDA approval in other neurological disorders (e.g., spinal muscular atrophy and other genetic neuromuscular diseases), ASO therapy for prion diseases continues to stay in preclinical development and is clearly limited by ethical concerns.
Stem Cell Therapy
On a different note, some recent studies have taken a dive into the world of stem cell therapy as a treatment for prion disease. Stem cells are a distinct class of cells that serve two main purposes: to develop into other forms of cells and to act as a repair system for the body. With these functions in mind, stem cells as a treatment for prion disease aim to repair and protect neuronal connections rather than aiming to reduce PrPSc aggregation. These recent studies have demonstrated the potential efficacy of stem cells in neurodegenerative diseases. The most promising types of stem cells include embryonic stem cells (ESCs), mesenchymal stem cells (MSCs), induced pluripotent stem cells (iPSCs), and neural stem cells (NSCs). A study in 2011 by Relaño-Ginés et al. revealed that intracerebral transplantation of fetal neural stem cells in mice subjects prolonged disease incubation and survivability20. At this stage, however, the risks are simply too large for stem cell therapy to be put in practice. The main concern surrounds the fact that risks include severe infections, immune complications, cancer development, and even death. While these risks seem too large to overcome, recent studies have taken on the challenge of testing stem cell therapy. In fact, there are a few scenarios in which the FDA allows for the use of stem cell therapy for humans. This method yields capacity for success in its ability to repair damaged brain tissue and restore neuronal connections.
With this in mind, it is important to remember that stem cell therapies still face considerable obstacles. Successful treatment requires widespread delivery to affected brain regions, which continues to be difficult. Additionally, issues with dosing, survival of transplanted cells, immune rejection, and procedural risks persist as major unresolved safety concerns. While few rare clinical scenarios have allowed for the use of experimental stem cells, stem cell therapy for prion disease is still preclinical, and it has not demonstrated efficacy in any other human trials.
| Therapeutic Strategy | Pros | Cons |
| Active Immunization Therapy | • Stimulates immune system to recognize misfolded proteins • Shown to prolong incubation periods in mice | • Possible autoimmune toxicity • Risk of harmful inflammation or immune overactivation |
| Antisense Oligonucleotides (ASOs) | • Directly targets PRNP gene mRNA • Potential to slow disease onset | • Unknown long-term safety • Invasive delivery methods • Ethical concerns around early human trials |
| Stem Cell Therapy | • Aims to repair neuronal connections • Potential to support long-term brain recovery | • High risk of infection, immune reaction, and cancer development • Highly experimental now |
Each approach offers unique potential for slowing or preventing disease progression through different biological mechanisms. Active immunization therapy enhances immune recognition of misfolded prions but carries autoimmune risks. Antisense oligonucleotides (ASOs) reduce PRNP gene expression to prevent prion formation, though safety and ethical concerns remain. Stem cell therapy promotes neuronal repair and regeneration but is currently limited by high complication risks and experimental status. Summary compiled and synthesized by the authors based on the cited literature. Created by the authors.
Discussion
This review reflects on recent developments in the fields of diagnostics and treatment strategies within the realm of prion disease research. The findings presented in this article encompass a wide variety of concepts to pursue further research as well as techniques that deserve continued attention. However, despite advances in emerging diagnostic tools and therapeutic strategies, a fundamental challenge persists: our understanding of prion disease pathology remains incomplete.
Compared to older research, newer literature has explored novel ideas for diagnostic biomarkers and treatment plans. For example, the work done by Garcés et al. in connecting glial cell activation to spongiosis contributed to the novel idea that glia are key players in prion disease progression rather than just bystanders. Additionally, in studies focused on treatment, such as the research led by Sonia Vallabh, a significant limitation continues to be a lack of understanding of the role of prion proteins in the human body. By providing an overview of various areas of prion disease research and connecting a central issue between them all, this review contributes to current efforts in literature and highlights the potential behind shifting from a narrow emphasis on well-known information – such as PrPSc aggregation and its connection to clinical manifestations – towards a more comprehensive exploration of all interacting partners, no matter how subtle, that together shape prion disease pathology.
The objectives of this review were to consider recent advances in prion disease research and to identify gaps that were either unanswered or in the process of being studied. The information presented in this review delves into distinct areas of current research and outlines progress made in each area. Additionally, with the compilation of the information presented in this review, it is possible to present potential methods and suggestions for the advancement of diagnosis tools in the future.
With that in mind, future research should focus on overcoming limitations in all areas of prion disease research. For therapeutics, that involves gaining a wider understanding of the role of the PRNP gene in the human body and identifying solutions to mitigate consequences of risky techniques. For diagnostics and early detection of prion disease, studies should revolve around a combination of novel morphological analyses using innovative technology and the narrowing down of specific differentially expressed genes that are found to be tied to prion disease progression. If data of transcriptomics can be analyzed to zoom in on 2-3 specific genes, these different expressed genes could add a layer to current biomarker analysis as was done with other neurodegenerative diseases such as Alzheimer’s and Parkinson’s.
Some more concrete future directives could be multicenter RT-QuIC validation, longitudinal CSF reads, and harmonized prion tissue banks. Multicenter validation for RT-QuIC could go a long way in demonstrating the accuracy and efficacy of the technique. It could also help refine protocols to help standard future research. Additionally, longitudinal CSF, which is the progressive collection of a patient’s CSF over the course of prion disease development, could reveal early changes in patients prior to symptom onset and help track changes over the course of disease progression. Finally, harmonized prion tissue banks could help future analyses and improve the ability to cross-check between studies to compare results. These are all changes that would prove useful for future studies and aid in the improvement of diagnostic and therapeutic research.
Although prion diseases remain a largely mysterious and complicated topic, progress towards reaching definitive diagnosis methods and successful treatment plans has been gradually increasing in recent times. Continued research focused on better understanding of prion disease pathology and its implications for diagnostics and treatment solutions could contribute to a more refined perspective on prion disease and provide patients with the potential for an improved life.
References
- J. Geschwind. Prion diseases. Continuum. Vol. 21, pg. 1612–1638, 2015, DOI: 10.1212/CON.0000000000000245. [↩] [↩]
- Q. Shi, C. Chen, W. Gao, J. Zhang, P. Li. The global epidemiology of prion diseases: A review. Prion. Vol. 16, pg. 15–28, 2022, DOI: 10.1080/19336896.2022.2036208. [↩]
- F. Eriso. Toxic prion: Translation of a genome’s gene with a toxic change. ResearchGate. 2022. https://www.researchgate.net/publication/366688090_Toxic_Prion_Translation_of_a_Genome%27s_gene_with_a_toxic_change [↩]
- M. Shimamura, K. Satoh. Recent advances in the diagnosis of human prion diseases. Viruses. Vol. 17, pg. 210, 2025, DOI: 10.3390/v17020210. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- P. Baranová, K. Fialová, A. Rehakova, L. Holada. Real-Time Quaking-Induced Conversion (RT-QuIC) assay in prion disease diagnosis. Microorganisms. Vol. 12, pg. 468, 2024, DOI: 10.3390/microorganisms12020468. [↩]
- R. Atarashi, K. Satoh, T. Sano, T. Fuse, M. Yamaguchi, K. Ishibashi, K. Yamanaka, M. Nakamura, M. Shinagawa, S. Kitamoto. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nature Methods. Vol. 8, pg. 1005–1010, 2011, DOI: 10.1038/nmeth.1701. [↩] [↩] [↩] [↩]
- E. Slota, A. Booth, D. Clark, L. Piccardo, B. Chesebro. Regional and temporal transcriptional changes in prion disease. Scientific Reports. Vol. 12, pg. 21544, 2022, DOI: 10.1038/s41598-022-21544-7. [↩] [↩] [↩] [↩] [↩] [↩]
- M. Jin, R. Li, Y. Chen, L. Zhang. Identification of key differentially expressed genes in Alzheimer’s disease through bioinformatic analysis. BMC Neurology. Vol. 23, pg. 94, 2023, DOI: 10.1186/s12883-023-03188-5. [↩] [↩]
- X. Li, J. Zhang, P. Wang, S. Huang. Integrated analysis of gene expression in Parkinson’s disease reveals novel biomarkers. Frontiers in Neuroscience. Vol. 17, pg. 1123456, 2023, DOI: 10.3389/fnins.2023.1123456. [↩] [↩]
- M. De Vita, L. Portelius, M. Mielke, A. Alcolea. Longitudinal voxel-based morphometry reveals progressive gray matter atrophy in prion disease. NeuroImage: Clinical. Vol. 13, pg. 408–417, 2017, DOI: 10.1016/j.nicl.2016.12.023. [↩] [↩]
- N. Desai, T. Purzycki. Early diagnostic measures to confirm the diagnosis of human prion diseases. Cureus. Vol. 15, pg. e39412, 2023, DOI: 10.7759/cureus.39412. [↩]
- R. Garcés, M. López-García, E. De Pablo, A. Nieto. Astrogliosis and microgliosis as central mechanisms in Creutzfeldt–Jakob disease. Acta Neuropathologica Communications. Vol. 9, pg. 44, 2021, DOI: 10.1186/s40478-021-01137-3. [↩] [↩] [↩] [↩]
- D. Hwang, I. Y. Lee, H. Yoo, N. Gehlenborg, J.-H. Cho, B. Petritis, D. Baxter, R. Pitstick, R. Young, D. Spicer, N. D. Price, J. G. Hohmann, S. J. DeArmond, G. A. Carlson, L. E. Hood. A systems approach to prion disease. Molecular Systems Biology. Vol. 5, pg. 252, 2009, DOI: 10.1038/msb.2009.10. [↩] [↩]
- Y. Cheng, T. Chen, J. Hu. Genetic analysis of potential biomarkers and therapeutic targets in neuroinflammation from sporadic Creutzfeldt–Jakob disease. Scientific Reports. Vol. 13, pg. 14122, 2023, DOI: 10.1038/s41598-023-41066-9. [↩] [↩] [↩]
- European Centre for Disease Prevention and Control. Variant Creutzfeldt–Jakob disease: EU case definition. https://www.ecdc.europa.eu/en/infectious-disease-topics/variant-creutzfeldt-jakob-disease/disease-information/eu-case-definition, 2025. [↩]
- J. Liu, M. Yang, H. Zhang. Emerging immunotherapeutic approaches in prion disease: Advances and challenges. International Journal of Molecular Sciences. Vol. 25, pg. 10283, 2024, DOI: 10.3390/ijms25102583. [↩] [↩] [↩] [↩] [↩]
- K. Ishibashi, T. Yamanaka, M. Nakamura. Active immunization with prion peptides prolongs incubation period in mice. Journal of Virology. Vol. 81, pg. 12864–12873, 2007, DOI: 10.1128/JVI.01234-07. [↩]
- S. Vallabh, E. Minikel, S. Schreiber. Targeting prion protein RNA with antisense oligonucleotides. Neuron. Vol. 108, pg. 218–229, 2020, DOI: 10.1016/j.neuron.2020.07.022. [↩]
- U.S. Food and Drug Administration. FDA approves first gene therapy treatment for certain patients with Duchenne muscular dystrophy. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-certain-patients-duchenne-muscular-dystrophy, 2023. [↩]
- A. Relaño-Ginés, S. Lehmann, T. Caillaud, V. Tilly, A. Grassi, A. Brillaud, C. Reine, V. Andreoletti, M. V. Didier, F. Andreoletti. Stem cell therapy extends incubation and survival time in prion-infected mice in a time window–dependent manner. Journal of Infectious Diseases. Vol. 204, pg. 1038–1045, 2011, DOI: 10.1093/infdis/jir520. [↩]



{kind=link}