
Abstract
Cystic Fibrosis (CF) is an inherited autosomal recessive disease that affects  160,000 people worldwide. The mutation in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene impairs the proper folding of the CFTR protein, thereby inhibiting chloride and water transport in and out of pulmonary epithelial cells, which results in the production of thick, sticky mucus in the lungs and other organs. This mucus is unable to be cleared by the cilia, leading to chronic bacterial infections and potentially fatal breathing issues. Based on the mutation type, there are five classes of cystic fibrosis. Classes I, II, and III are generally more severe than classes IV and V; therefore, mutations associated with severe forms of the disease are reviewed here. The latest work suggests the use of a gene editing approach to address the mutation. Derivatives of the well-established CRISPR-Cas9 gene editing system, such as base and prime editing, have been used only in laboratory settings thus far to correct mutations by introducing an appropriate nucleotide. Seven CF mutations, all leading to a severe form of the disease, that were subjected to base and/or prime editing, are reviewed. The outcome of these procedures, their effectiveness, and sustainability as a long-term treatment as a standard clinical procedure are considered for this review. This technique, as promising as it sounds, demands a deeper study, overcoming the hurdles of funding, ethical issues, and bystander gene damage.
160,000 people worldwide. The mutation in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene impairs the proper folding of the CFTR protein, thereby inhibiting chloride and water transport in and out of pulmonary epithelial cells, which results in the production of thick, sticky mucus in the lungs and other organs. This mucus is unable to be cleared by the cilia, leading to chronic bacterial infections and potentially fatal breathing issues. Based on the mutation type, there are five classes of cystic fibrosis. Classes I, II, and III are generally more severe than classes IV and V; therefore, mutations associated with severe forms of the disease are reviewed here. The latest work suggests the use of a gene editing approach to address the mutation. Derivatives of the well-established CRISPR-Cas9 gene editing system, such as base and prime editing, have been used only in laboratory settings thus far to correct mutations by introducing an appropriate nucleotide. Seven CF mutations, all leading to a severe form of the disease, that were subjected to base and/or prime editing, are reviewed. The outcome of these procedures, their effectiveness, and sustainability as a long-term treatment as a standard clinical procedure are considered for this review. This technique, as promising as it sounds, demands a deeper study, overcoming the hurdles of funding, ethical issues, and bystander gene damage.
Keywords: Cystic Fibrosis, Prime Editing, Base Editing, CFTR Modulators, F508del
Introduction
Cystic Fibrosis is an inherited disease that affects about ∼160,000 people worldwide across 94 countries, but only 105,000 are diagnosed. This disease primarily affects Caucasians of European descent, with Ireland claiming the highest incidence rate of 2.64/10,000 inhabitants. Current estimates project that the number of CF patients living in North America and Europe is around 84,600, representing 80% of globally diagnosed cases. Other countries like Australia, Brazil, Canada, France, Germany, Italy, and Russia have between 2,500-10,000 patients, while both the UK and US have upwards of 10,000 CF patients each. All of these nations have a large percentage of White or Caucasian populations1. There are five different classes of CF. Classes I, II, and III cause more severe CFTR protein dysfunction as each has a closed ion channel with little to no CFTR protein made; Classes IV and V are milder versions of the disease with a low ion flow and protein production. CF is an autosomal recessive disease that can be caused by over 1,000 genetic mutations, but the most common mutation is the deletion of phenylalanine at position 508 in the CFTR protein (F508del) which affects nearly 89% of all CF patients who have at least one copy. The mutation impairs proper folding of the CFTR protein and thus inhibits chloride and water transport in and out of pulmonary epithelial, or respiratory, cells, causing thick mucus to build up in the lungs and other organs2. This mucus is unable to be cleared by the cilia, leading to chronic bacterial infections and potentially fatal breathing issues. Thus, CF is a chronic disease that persists for the patient’s entire life and often leads to an extremely premature death3. In the US, the median life expectancy for people with CF is 46 years old, while in countries with fewer treatment options, like Poland, it is under 25 years old. The worldwide average lifespan has been steadily increasing since the 1950s, when most CF patients died by age 5, due to medical advancements1.
Currently, there is no cure because the symptoms that manifest from this genetic mutation occur before the patient is even born and can be prenatally diagnosed through ultrasounds showing abnormally large fetal intestines in the fetus3. In the US, all newborns are given a mandatory blood test as a preliminary disease screening. If symptoms of CF, such as salty sweat and thick mucus, or molecular signs are present, patients are diagnosed through a sweat chloride test as well as DNA testing that checks for one or more CF-related mutations. The most advanced clinical treatments for CF are CFTR Modulators, which are drugs that allow proper CFTR protein folding and elongate the time the chloride ion channel is left open3. By enabling more robust chloride transport, the pressure on the patient’s lungs is reduced, thinning the mucus and restoring normal breathing levels. The optimal standard modulator therapy for CF patients is the triple therapy called Trifakta, which consists of one potentiator Ivacaftor (VX-770) that helps the chloride channel stay open longer to most directly address Class III defects, and two correctors Elexacaftor (VX-445) and Tezacaftor (VX-661) to help misfolded CFTR protein reach the cell surface and treat Class II mutations. Yet, while CFTR modulators have the potential to extend a patient’s lifespan into their late 60s and 70s, they are not curative, require continuous administration, do not work on Class I mutations due to the lack of residual protein production, and cost between  310,000 annually for lifelong treatment1. This fee makes the treatment almost impossible to pay for without government or private insurer reimbursement. Thus, researchers have begun exploring alternative treatments that are longer-lasting and less burdensome on the patient.
310,000 annually for lifelong treatment1. This fee makes the treatment almost impossible to pay for without government or private insurer reimbursement. Thus, researchers have begun exploring alternative treatments that are longer-lasting and less burdensome on the patient.
A shift toward gene editing and gene therapy could result in a one-time treatment that permanently corrects the CFTR gene to express normal protein function and eliminate the subpar chloride transport causing life-threatening mucus secretions4. CRISPR-Cas9 was introduced as a revolutionary gene editing technique: it was the first to ensure specific gene targeting by using a guide RNA (gRNA) and Cas9 protein to make a double-stranded break (DSB) and repair DNA mutations5. Further gene editing techniques, though not clinically approved yet, have been derived from CRISPR-Cas9. One of these is prime editing (PE), in which a modified nickase Cas9 enzyme is coupled with a reverse transcriptase (RT) enzyme and an RNA molecule called prime editor gRNA (pegRNA) to help it reach the target site and make a single-strand DNA break. Then, RT uses the pegRNA’s template sequence to synthesize the new, edited DNA strand, modifying the genome and repairing gene expression for the desired protein. Its capabilities are broad, as it can go beyond single base-pair substitutions, and is perhaps safer than traditional CRISPR-Cas9 methods, since it does not use a DSB6. Another CRISPR-Cas9 modified approach is base editing (BE), which works by attaching an inactive Cas9 protein (dCas9) to a nucleotide, or base pair, modifying enzyme. Then, the gRNA forms a complex with dCas9 to guide it to the target site of the gene, where it attaches to either an adenine or cytidine deaminase enzyme to form a new, mutated structure known as an Adenine Base Editor (ABE) that changes an A-T base pair into a C-G nucleotide pair, or Cytosine Base Editor (CBE) to change C-G into A-T, helping to fix point mutations in genetic diseases without having to fully cut DNA strands and risk off-target effects (OTEs)7 While a gene editing therapy may be expensive, its position as a potential one-time intervention may make it more cost-effective and sustainable in the long run if and when it may surpass preclinical trials.
This paper will investigate the advantages and disadvantages of prime and base editing approaches in the treatment of CF. Special consideration will be given to the benefits and risks of each technology alongside additional factors like delivery cost, accessibility across the affected regions, and time needed to roll out these new treatments in order to propose an optimal path forward to the clinic.
PRISMA Flow
A search with the initial filtering for publication date (2015–2025) years and full, free text availability across the Google Scholar and the PubMed database yielded a total of 1,200 records. After removing 860 non-prime or non-base editing articles as well as 260 duplicates, only 80 unique studies remained for screening. After a title/abstract review, 55 records were excluded due to lack of original data. With 25 sought for retrieval, a full-text assessment excluded 7 more reports due to irrelevance, leaving 18 studies related to base and prime gene editing for CF to be analyzed in the paper. The remaining studies listed in the bibliography had a similar screening process and were scanned for key words such as “CFTR Modulator” and “in vivo delivery”
Prime editing to correct CF
Prime editing to correct F508del in CF
The versatility of prime editing and its ability to edit hundreds of nucleotides without a double-stranded break is being investigated as a potentially promising approach to treat CF. In a 2024 pre-clinical study by Alexander Sousa et al., researchers used a prime editor to target the F508del Class II mutation and prepare the repaired DNA for functional gene expression. Sousa and his team, with their correction strategy, achieved an average of 11%, 58%, and 25% F508del correction in human-derived kidney and epithelial cells of CF patients (Table 1)8. Furthermore, Sousa’s team tested various gRNAs and DNA sequencing methods to detect and minimize the OTEs (Fig. 1).
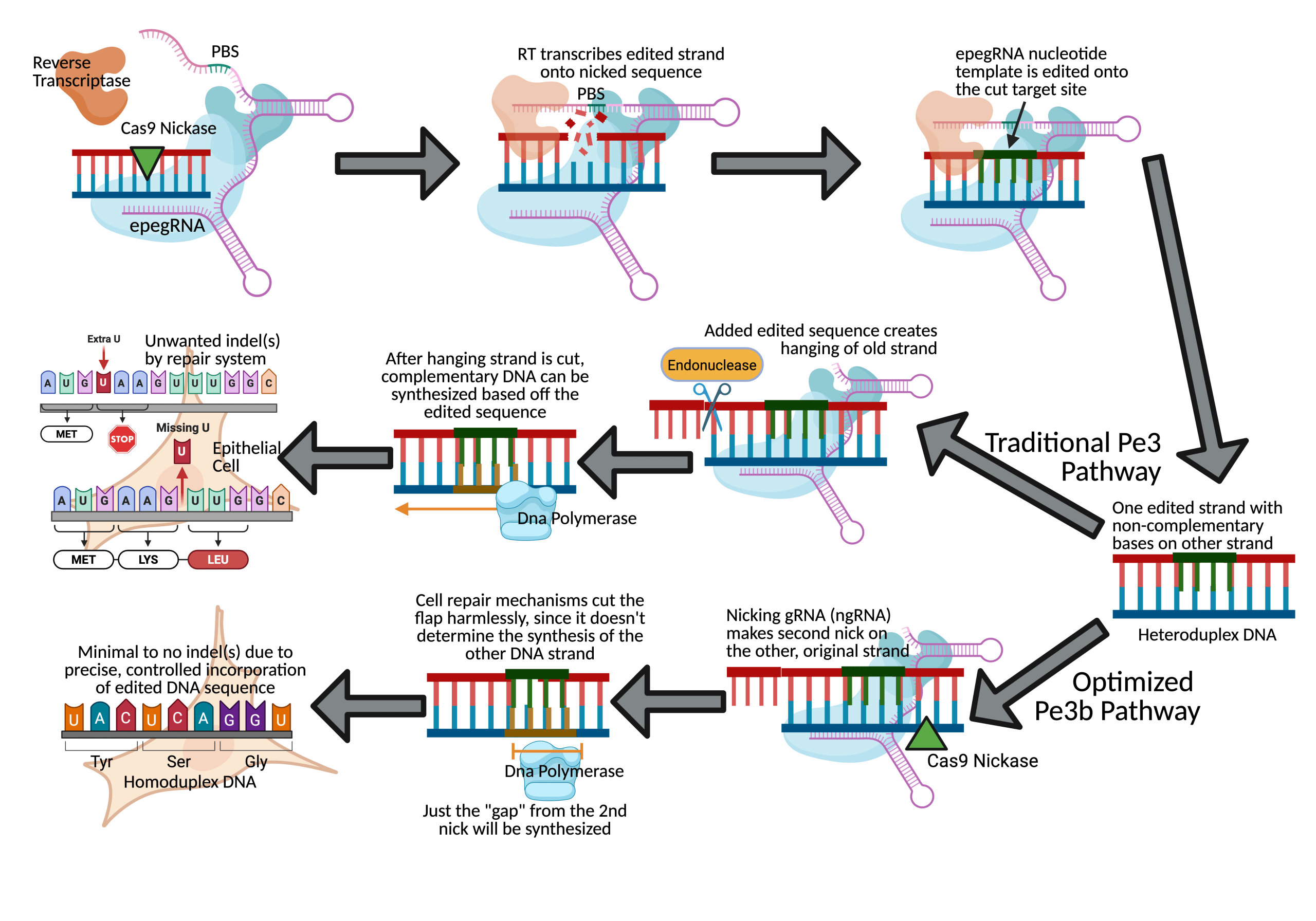
They used engineered pegRNAs (epegRNAs), nicking gRNAs (ngRNAs), which makes a second DNA nick to improve editing efficiency, and a double-stranded gRNA (dsgRNA) as their optimal gRNA (Fig. 1). Using these, they observed only  0.1% potential off-target editing at four distinct insertion/deletion (indel) sites of primary airway epithelial cells of CF patients across all gRNA conditions (Table 1)8. The researchers also accounted for scaffolding, an example of off-target editing at the target site that is more common with advanced PE methods. They observed that their epegRNA successfully avoided scaffold incorporation in an average of 99.2% of edited sequences across three edited donor cell lines (Table 1). Another problem that often occurs in PE is the amount of unedited target sequences due to the lack of PE recognition. Recognition of the Protospacer Adjacent Motif (PAM) and other protospacer regions by a prime editor is necessary to edit the gene. However, 3.6% of the epithelial cells did not show the CTT insertion to correct F508del even after the PAM and protospacer sequences were recognized, making these sequences “un-targetable”8. Still, the mean insertion frequency with PAM and protospacer incorporation was 23%, yielding a much higher success percentage than the unrecoverable cells8.
0.1% potential off-target editing at four distinct insertion/deletion (indel) sites of primary airway epithelial cells of CF patients across all gRNA conditions (Table 1)8. The researchers also accounted for scaffolding, an example of off-target editing at the target site that is more common with advanced PE methods. They observed that their epegRNA successfully avoided scaffold incorporation in an average of 99.2% of edited sequences across three edited donor cell lines (Table 1). Another problem that often occurs in PE is the amount of unedited target sequences due to the lack of PE recognition. Recognition of the Protospacer Adjacent Motif (PAM) and other protospacer regions by a prime editor is necessary to edit the gene. However, 3.6% of the epithelial cells did not show the CTT insertion to correct F508del even after the PAM and protospacer sequences were recognized, making these sequences “un-targetable”8. Still, the mean insertion frequency with PAM and protospacer incorporation was 23%, yielding a much higher success percentage than the unrecoverable cells8.
These results were achieved by silent edits inserted to evade the cell’s mismatch repair system (MMR); these silent edits did not cause expression of other proteins and also prevented harmful post-editing Cas9 re-engagement8. With their optimization of the in-vitro correction strategy, Sousa et al. reported that their average of 25% F508del correction restored CFTR protein chloride channel function to more than 50% of wild-type (WT) airway epithelial cells: comparable to the success of traditional Cas9 nuclease Homology Directed Repair (HDR) method (Table 1)8. Nevertheless, while there was elimination of scaffolding and optimization of PE recognition, this success is still in early ex-vivo testing phases of laboratory models; in reality, both are significant technical barriers to make PE safe for clinical usage9.
Prime editing to correct N1303K and L227R in CF
In a separate pre-clinical study done in 2024, two additional Class II CF-causing missense mutations that are unresponsive to CFTR Modulator treatment, L227R and N1303K, were tested with PE for functional recovery by Mattjis Bulcaen and his team. L227R is an ultra-rare mutation–with only 29 recorded cases ever–in which a guanine base is changed to thymine at position 680, whereas N1303K is the fourth-most prevalent mutation in which cytosine is converted to guanine, changing amino acid (AA) 1303 from asparagine to lysine10. Though these are both singular nucleotide base changes, traditional BE cannot fix either of the two nucleotide changes due to its limited conversion capabilities7. Thus, PE3b ngRNAs were introduced to increase PE efficiency by encouraging the cellular repair system to adopt the edited strand over the original. By combining the pegRNA+13 C A, which changes cytosine to adenine 13 bases after the nick, with the new PE3b ngRNAs, an average of 25
A, which changes cytosine to adenine 13 bases after the nick, with the new PE3b ngRNAs, an average of 25 8% L227R mutations were corrected–a 60% higher accuracy than just pegRNA (Table 1)10. For functionality, this translated into a 42.2717.4% rescue of CFTR maturation–the process of proper protein folding, transport, and checkpoints–in the edited cell pool (Table 1). However, overall functional correction remained lower, with 24.87.74%, compared to the WT HEK293T, which are human-derived kidney cells (Table 1)10. The N1303K mutation was approached similarly, except this time a pegRNA+13 C>G, with a primer binding site (PBS) and a reverse transcription template (RTT) length of 14-18 base pairs, was used in combination with ngRNA-1 to achieve a 27.34.2% editing success in a HEK293T cell line–a 217% increase in efficiency compared to pegRNA alone.
8% L227R mutations were corrected–a 60% higher accuracy than just pegRNA (Table 1)10. For functionality, this translated into a 42.2717.4% rescue of CFTR maturation–the process of proper protein folding, transport, and checkpoints–in the edited cell pool (Table 1). However, overall functional correction remained lower, with 24.87.74%, compared to the WT HEK293T, which are human-derived kidney cells (Table 1)10. The N1303K mutation was approached similarly, except this time a pegRNA+13 C>G, with a primer binding site (PBS) and a reverse transcription template (RTT) length of 14-18 base pairs, was used in combination with ngRNA-1 to achieve a 27.34.2% editing success in a HEK293T cell line–a 217% increase in efficiency compared to pegRNA alone.
Consequently, CFTR maturation increased to 50.315.46% compared to the WT (Table 1)10. The overall CFTR function was rescued to substantial levels: 22.475.54% (Table 1). Additionally, Bulcaen and his team developed special epegRNAs to ‘cap’ unprotected 3’+ pegRNA ends, yielding 216.5% editing for L227R and 43.310% for N1303K, representing 49% and 33% increases from the original pegRNAs, respectively10. Because pegRNA modifications can cause the MMR system to misrecognize the hanging 3′ DNA flap and restore the original strand, the researchers added 3 silent mutations to N1303K that evade MMR, which were successfully installed but did not increase editing efficiency when compared to the previous PE method10. And, in an attempt to prevent repeated splicing of the same site and preserve the edited sequence, another silent PAM-disrupting mutation was introduced for L227R, yet still no editing efficiency improvements were observed10.
Prime editing to be optimized in A-T rich loci
Another factor complicating pegRNA selection is the presence of an A-T rich locus–where there are many A-T base pairs flanking the mutation–which was investigated by Olga Volodina et al. in 2025 to find pegRNAs that could thrive best in this environment. PEmax is a PE strategy that does not involve the second nicking like PE3 and can reduce the amount of indels. There were 15 PEmax pegRNAs designed and tested in airway basal cells of patients containing the F508del mutation, which is located in an A-T rich locus. Out of the 15 PEmax pegRNAs, pegRNA 1 and 5 were most effective at around 2.4% to 6.1%11. Since all pegRNAs for PEmax had a relatively low efficiency, the results suggest PEmax may be theoretically safer but still has significant hurdles in A-T rich loci.
Further optimization of the PE2 system tested pegRNA 19, 20, 21, and 22; pegRNA 20 was found to be most successful, correcting 2.81% of the pathogenic variant while having an ideal GC content, which is the amount of G-C base pairs, for both the PBS and the spacer11. Despite low efficiency, PE still demonstrates high precision in complex environments. Lastly, pegRNA 20, 21, and 22 were further investigated for OTEs during the PE process, as all three contain the spacer sequence and single-strand break site. In samples without the desired CTT substitution, silent substitutions were found at c.1530TN (thymine converted to any unknown nucleotide at position 1530). Other nucleotide changes of c.1528GN, c.1529TN, c.1531delT, c.1522TM (M is adenine or cytosine), and c.1531T>N were also observed in the pegRNAs, but each had very low frequencies: 0.01-0.08 of all reads11. These findings signify that, though PE strategies are highly specific in the A-T rich loci, improvements in efficiency and delivery must be considered before pre-clinical advancement.
Base editing to correct CF
Base Editing to introduce F508del-repairing RMs
The efficiency, precision, molecular size, and DSB-free characteristics of base editing tools can provide a functional rescue of the CFTR protein12. A 2025 investigation by Irene Carrozzo et al. exploited BE to introduce revertant mutations (RMs) and correct the F508del mutation. Since BE can only directly change point mutations and F508del is a three-nucleotide deletion, Carrozzo and her team first evaluated the impact of the RMs I539T, G550E, R553Q, R555K, and R1070W for their effect on CFTR protein maturation in HEK293T cells. They found that each aforementioned single revertant and the I539T–R1070W combination achieved partial maturation rescue.
Additionally, under optimized laboratory conditions, the double combinations I539T–R1070W and G550E–R1070W restored matured CFTR protein to levels comparable with the WT12. To insert the RMs into the F508del, the researchers tested ABEs and CBEs with various Cas9 variants and different sgRNAs. The only RM requiring ABEs was the I539T, in which the standard ABEmax was tested along with the precision-enhanced ABE8e and ABE8.20m, leading to 40–45 of RM editing depending on the Cas9 used (Table 1)12.
of RM editing depending on the Cas9 used (Table 1)12.
The RMs G550E, R553Q, R555K, and R1070W mutations were targeted using two CBE versions, BE4max and AncBEmax. All versions of each of the four individual RMs recorded editing levels between 30–60%, again dependent on the Cas9, sgRNA, and RM used (Table 1)12. Despite these observations, bystander edits, which are unintended edits that occur inside the editing window, from these ABEs and CBEs were prevalent flanking many RMs. I539T, target base A7, had sub-10% A4 and A9 editing with only two tested ABEs (Table 1)12. However, in G550E, where the relevant edit is at C5, 3 out of 4 CBEs avoided >10% bystander editing at bases C2 and C8, which cause new pathogenic variants if edited. At R553Q, the target is C6 and most bystander edits were at C15 which reintroduced the beneficial G550E RM; RM R555K has a target base C5, yet ~20% editing at bystander C11 again caused RM R553Q-R555K (Table 1). Lastly, R1070W, with the target at C6, recorded only one bystander edit, which was at C212.
These results emphasize that even with some helpful bystander edits, using RMs with this risk still pose a significant challenge to the necessary clinical precision and safety. For functional analysis the team analyzed plasma membrane (PM) localization, a direct measurement of the chloride ion transport levels through the CFTR channel protein. For single RMs, R555K had the highest percentage of PM localization, 30%, compared to the WT and was likely the most successful due to its bystander edits introducing other beneficial RMs12. In double combination RMs, R555K–R1070W recovered 40% of PM CFTR, the most of any of its counterparts12. Consequently, the single RM R555K and double RM R555K–R1070W rescued 40% and 56% of CFTR WT activity, respectively–both representing the highest percentages in their groups (Table 1)12. While these in vitro findings identify optimal RMs that help BE bypass larger defects, this unproven technique has many complex combinations and a high prevalence of unintended edits that could stop it from moving past early testing phases.
Base Editing to correct W1282X in CF
Another study done in 2023 by Karen Mention and her team discussed ABE to target the fifth-most prevalent CF-causing mutation: W1282X, where guanine is replaced with adenine at position 3846 of the CFTR gene. They replaced the nonsense codon TGA, which makes W1282X a Class I mutation, with the WT TGG4. By using an ABE with a special PAM site to help the inactivated Cas9 locate its desired sequence at positions 1521 to 1523, the team could choose an adenine in the editing window to be turned into guanine, eventually locking their target at position 6 (A6 gRNA) and allowing repair of a modulator unresponsive mutation. The ABE was tested in Flp-In-293 cells; successful BE at the target A6 was observed at 243% with no indels (Table 1)4. BE of A7 was reported at similar rates: 252%. But, when the two adenines were measured for DNA changes expressed in the CFTR mRNA, 462.8% of the transcripts were found to have the A G change for A6 and 411.7% transcripts had a change at A7 (Table 1)4.
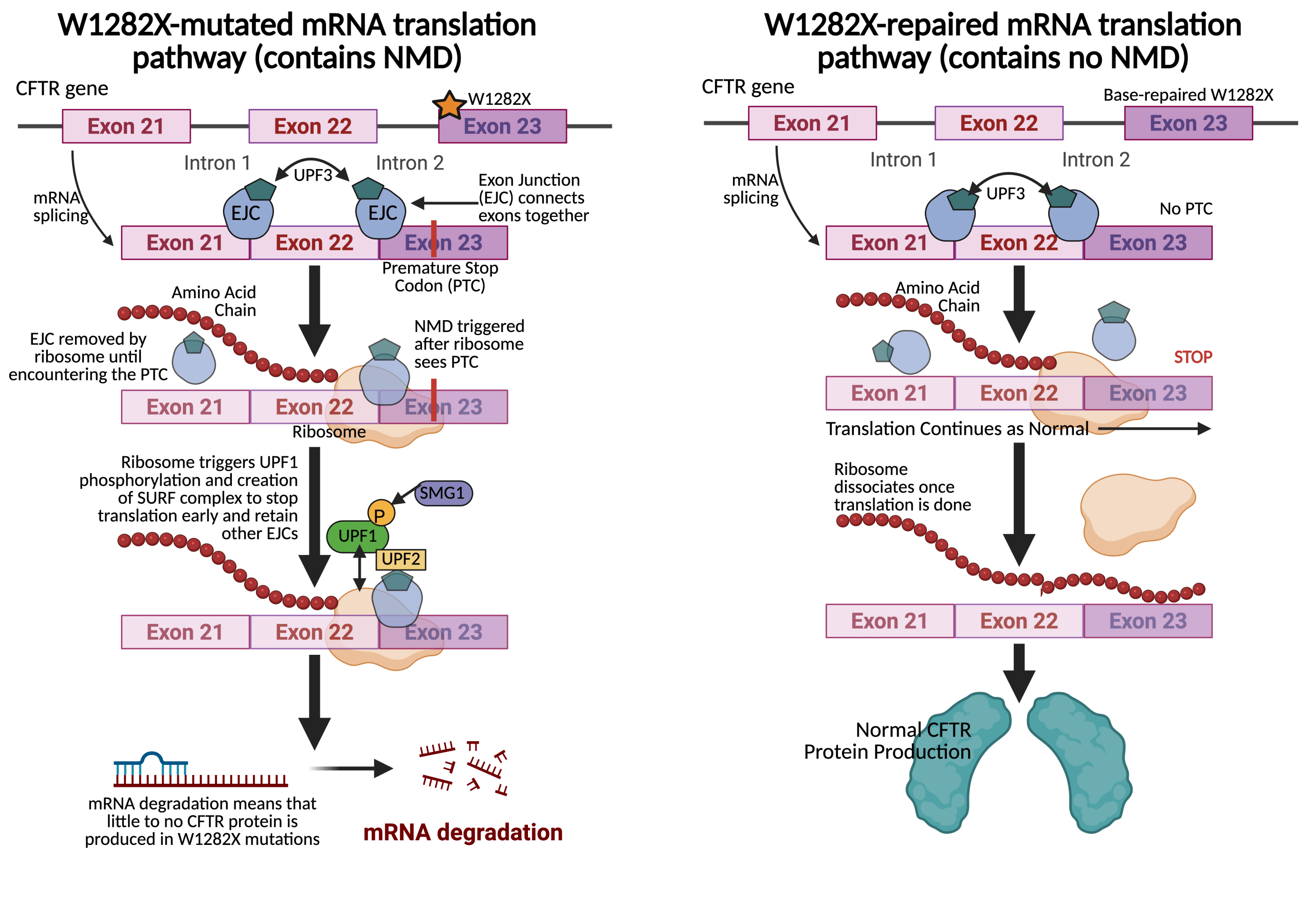
These numbers represent a notable 80% increase in editing frequency compared to the DNA editing, suggesting the edited mRNA bypassed nonsense-mediated decay (NMD) and its strand degradation (Fig. 2)4. The near identical editing levels at A6 and A7 for both the DNA and mRNA implied DNA linkage between the two adenines. To test this, 82 clonal cells were generated, with 13 containing the A > G change in the target window4. 10 out of the 13 correctly edited A6 but also the bystander A7, and only one clone edited A7 alone while none edited A6 alone (Table 1)4.
The bystander edit A7 > G created another mutation R1283G. So, even though the full-length mature glycosylated bands over full-length immature glycosylated bands ratio was higher and were clearly visible in the pool of Flp-In-293 W1282X edited cells compared to untreated W1282X cells, the R1283G prohibited mature/immature levels from reaching near the WT. Rather, protein processing levels remained comparable with Class II CF mutations, suggesting that even NMD-evaded mutations still may need additional gene therapy and modulator treatment to be fully corrected4.
However, further testing of the same procedures in human nasal epithelial (HNE) cells that were homozygous for W1282X showed 27% BE at A6 and 18% BE at A7 using the same ABE complex (Table 1)4. Additionally, primary HNEs of a W1282X patient treated with ABE and A6 gRNA restored 16% of WT CFTR function, serving as a marker for improved lung function at least in the lab setting without any strand discrimination present4. Strand discrimination is a key obstacle in base edited nonsense mutations where the cell’s natural MMR may actually discriminate against expressing the newly edited strand and instead favor the original, dysfunctional codon. This may lower editing efficiency even after W1282X is repaired and restart the NMD cycle, undermining the success of genetically repairing nonsense CF mutations like W1282X13.
Base editing to convert CF mutation G542X to G542R
A different paper, written by Isabelle Rose and her team in 2025, assessed editing efficiency of nanoparticle-delivered ABEs14. They converted the second most common CF-causing mutation G542X–a nonsense mutation changing c.1624 G T that is not treatable by CF modulators as a Class I mutation–into a modulator-responsive, missense G542R variant with a c.1624 G C that does not trigger NMD14. The researchers addressed the delivery and materials of their BE process with receptor-targeted nanoparticles (RTN) that held the ABE plasmids in water14.
These RTNs carried an ABE8e Cas9, sgRNA, and an enhanced green fluorescent protein (EGFP), which acts as a reporter gene for biological experiments, en route to transfect human airway epithelial CFNE cells containing the G542X mutation14. Some of these cells were left untreated while others underwent sorting to separate EGFP-expressing from non-expressing populations14. Further DNA sequencing showed 17% of the unsorted cells and 52% of the sorted cells–around 50% of overall transfected cells–contained the target base edit, changing the template strand’s 1624th nucleotide base from A G and consequently switching the coding strand base from T C (Table 1). Crucially, while 10 probable off-target sites were analyzed, only 3 were found to have negligible genetic alterations of silent or intronic edits with the other 7 having no OTEs at all14. Moreover, 17-20% of base edited alleles persisted in culture up to three months after initial edits, indicating the BE’s persistent expression in the lab model14.
These numbers correlated with gene expression: unsorted cells, which were 17% G542R, exhibited 3710% of the WT mRNA levels while sorted cells, which were 52% G542R showed similar mRNA abundance to a random normal human nasal epithelial (NHNE) cell–12025% and 11118% of the WT, respectively (Table 1)14. Because more mature CFTR protein shape and function was observed, suggesting that the G542R successfully evaded NMD, the researchers subsequently investigated the electrochemical current, denoted as  , between the outside and inside of the cell as an indicator of chloride transport14.
, between the outside and inside of the cell as an indicator of chloride transport14.
They used forskolin, a substance that initiates a robust phosphorylation cascade that opens the Cl transport channel, to measure . They found that unsorted G542R-edited cells with modulator treatment reflected 24% of non-CF levels and sorted edited cells increased to 41% of non-CF levels14. While it was noted that these transport levels were similar to those in non-CF control cells, these results were obtained in a controlled and sorted laboratory environment, which shows promise for restoring function to Class I mutations but does not mirror the technical challenges of an intermutational conversion in human CF patients.
transport channel, to measure . They found that unsorted G542R-edited cells with modulator treatment reflected 24% of non-CF levels and sorted edited cells increased to 41% of non-CF levels14. While it was noted that these transport levels were similar to those in non-CF control cells, these results were obtained in a controlled and sorted laboratory environment, which shows promise for restoring function to Class I mutations but does not mirror the technical challenges of an intermutational conversion in human CF patients.
Base editing to correct R553X in CF
Lastly, Yehui Sun and his team examined the feasibility of in vivo lung stem cell editing in mice to fix the rare R553X mutation. This only represents 0.9% of CF cases in the US but is a Class I nonsense mutation again responsive only to gene editing due to a c.1789 C T change15. Before animal trials, Sun et al. tested the ABE8e in two different cell lines. In an engineered human bronchial epithelial (HBE) cell, the ABE mRNA showed a 95% base pair conversion; then, they used HBE cells with the heterozygous R553X–F508del compound to mimic the anatomical complexities of human lungs and pathways15.
They achieved 60% base pair conversion to show feasibility in in-vitro settings before testing their in-vivo model (Table 1). The target nucleotide on the coding strand T7 was converted to cytosine at an 83.7% frequency, with a 14.5% silent bystander editing recorded at T11 and no OTEs (Table 1)15. With no other detectable OTEs, the lipid nanoparticle (LNP)-ABE approach, even without modulators, increased the expression of mature bands by more than 5.5-fold and restored 53% of WT CFTR function after four weeks (Table 1)15. Additionally, there was more than a 50% transfection success of tdTomato-mRNA, which is used for in vivo imaging, into lungs of human-derived basal cells, showing that lung basal cells have potential to produce mature protein and restore CFTR function15. The team next turned to their in-vivo mouse model to test the efficacy by inserting it with CF-ridden human exon 12 and isolated intestinal cells homozygous for R553X that created intestinal-imitating organoids. These organoids were given LNP-ABE, and DNA sequencing confirmed 47.8% of these cells converted A-T to C-G with more than 82% of organoids swelling, demonstrating functional response to the correction15. The results suggest base editing may be able to restore ion transport in complex, multicellular structures.
They also used mice heterozygous for R553X and completed sequencing analysis on the infected region by extracting and separating multiorgan DNA ten days after IV-administered LNP-ABE was given. A 50.0% T7 correction was found in lung stem cells, 12.2% in the lung as a whole, and 28.7% in the trachea, suggesting the mutation can be targeted in stem cells and functionally persist long-term, as long as there is sustained cellular correction (Table 1)15. Encouragingly, strand discrimination is less common in R553X and G542X since there are fewer competing nucleotide bases around these target sequences than are near the W1282X target. While these findings demonstrate pre-clinical advancement, more research is required to ensure the long-term safety and durability of these edits within human patients.
| Study | Mutation | Cell Type | Strategy Used | Mutational Correction | Functional Restoration | OTE/Bystander Edit Prevalence | Additional Notes |
| Sousa et al., 2022 | F508del | HEK293T* HBE*Primary | Prime Editing | 316% | 50±0% | 0.1% OTE* | 99.2% Cells No Scaffolding |
| Bulcaen et al., 2024 | L227R | HBE* HEK293T*NHNE* | Prime Editing | 258% | 24.8±7.74% | 0.8±0.4%; 0.9±0.3% OTE* | OTE 1: pegRNA; OTE 2: ngRNA* |
| Bulcaen et al., 2024 | N1303K | HBE* HEK293T*NHNE* | Prime Editing | 27.34.2% | 22.47±5.54% | 0.1±0.6% ; 0.2±0.1% OTE* | OTE 1: pegRNA; OTE 2: ngRNA* |
| Carrozzo et al., 2025 | F508del | HEK293T*Primary | Base Editing | 5010% | 56±16% | 10% Bystander | Used single + double RMs* |
| Mention et al., 2023 | W1282X | HEK293T*NHNE* HBE* | Base Editing | 243% A6; 252% A7 | 44±3% mRNA | 2.4% Bystander | 27% A6 & 18% A7 edits in NHNE* |
| Rose et al., 2025 | G542X | Epithelial (Sorted + Unsorted) | Base Editing | 52% Sorted; 17% Unsorted | 12025% Sorted; 37 9% Unsorted | 10% OTE* | Converted to G542R |
| Sun et al., 2025 | R553X | HBE* Lung StemOrganoids | Base Editing | 6012.2% | 503% | 15% Bystander; 0.1% OTE* | Mostly in vivo mice editing |
* HBE=Human Bronchial Epithelial (cells), NHNE=Normal Human Nasal Epithelial (cells), HEK293T=Human Embryonic Kidney 293 (cells), OTE=Off-Target Effects, RMs=Revertant Mutations, ngRNA=Nicking Guide RNA
Table 1 summarizes pre-clinical results for both techniques across the mutations observed; in these controlled environments, each model has more than at least the established laboratory benchmarks of 10% functional restoration and 20% mutational correction with minimal OTEs and bystander edits–below 1% and 10% respectively–across all ex-vivo cell types tested8,12,15. However, many important aspects of using gene editing as a potential cure for cystic fibrosis remain unexplored. One important exclusion is a thorough evaluation of in vivo delivery methods and how they impact the success of both PE and BE systems. Another key concern is the time required to develop and personalize treatments based on each patient’s specific mutation and immune system profile. Both of these could further complicate addressing technical damages such as the aforementioned scaffolding sequences and NMD strand discrimination.
Discussion
CF is not limited to just the five mutations discussed above; rather, there are thousands, all with differing effects on CFTR protein function and various new factors to consider when designing treatments to address each mutation. One of such factors is the inability of CFTR modulators to treat all mutations, such as the aforementioned G542X, potentially making the development of gene editing treatments for the 20% of CF patients affected by non-F508del mutations more urgent. BE and PE offer distinct technical advantages that differ between specific mutational contexts.
For example, less common point mutations such as G542X, N1303K, G551D, and W1282K may be better addressed with base editing due to a relatively high accuracy and efficiency as well as a smaller molecular system to possibly simplify development delivery compared to PE tools (Table 1). Even if mutations like N1303K and L227R cannot be directly fixed by ABEs or CBEs, a conversion to a modulator-responsive treatment, similar to the ideas explored by Rose et al., could be introduced. Additionally, in all studies that tested with laboratory observations for BE persistence, the edits were sustained through the time they were monitored, which may theoretically provide high potential for a durable, curative treatment for patients though long-term stability and safety in humans remain unproven14. Finally, BE’s smaller editing system could facilitate more efficient packaging of delivery vectors, which may reduce technical complexity of large-scale administration16.
Conversely, prime editing may be the primary candidate for the most common mutation F508del because it can only be genetically repaired by an insertion, which ABEs and CBEs are not capable of doing alone. Though more research may be needed to optimize PE as a treatment, its potential accuracy and precision as well as its success in the lab could make it viable in restoring long-term function for CF patients with F508del, though, again, its clinical sustainability is untested17. PE’s efficacy can be even further optimized with the use of a Flap Endonuclease 1 (FEN1), a tool allowing the proper ligation of the new DNA strand and its genomic nick. This increases mutational recovery as much as 2.3-fold in some cell models compared to traditional CRISPR methods18. Additionally, since PE does not create DSBs, it is less prone to harmful and unpredictable off-target DSBs and indels caused by CRISPR-Cas9’s HDR and NHEJ methods. Thus, PE can provide a more predictable, accurate treatment with minimal unwanted immunosuppression effects compared to DSB-based methods but still must undergo extensive research before approval for clinical viability19.
Moving on from technical aspects and into funding, institutions like the National Institute of Health (NIH) award an average of 84 million annually for Cystic Fibrosis in U.S. research grants to explore both ideas of expansion and optimization of current treatments as well as novel treatment options that use genetic technology, allowing advancement in the testing and implementation of gene editing20. Yet, while money for researching and developing small-scale treatment prototypes may exist, there are costly roadblocks in manufacturing and administering new biotech to reach clinics. To ensure that these treatments remain readily available for administration as a primary option, several operational costs–equipment, personnel, training costs, transportation, and more–are required21.
Personnel, which is 40-50% of total facility cost, are often a burden on healthcare development. However, sharing specialized equipment and materials can reduce these costs. Thus, investors who finance these shared resources not only cut technology expenses but also gain financially in the future–potentially incentivizing them to contribute21. Still, while production volume and batch yield can lower facility costs, cell-based therapy (CBT) projects still demand significant capital investment and highly skilled full-time staff—expenses that many biotech firms cannot sustain, often forcing them to rely on unpredictable investments or high-cost, private loans21. Furthermore, 80% of the total costs needed are variable rather than fixed, making investors and governments unlikely to fund costly and unpredictable projects like these. And, though biotechnology shows clear promise in treating genetic diseases, most firms have had to downsize personnel and stall R&D due to insufficient profits, painting a grim picture for their future in capitalist ecosystems like the U.S., Canada, and Western Europe22. Only around 13.8% of new biotech treatments reach approval stages from the FDA’s strict regulations, further disincentivizing pharmaceutical investment. Equally important are the ethical implications, especially regarding inequitable access: patients with rare mutations or limited money may be unable to receive these therapies due to high cost, mutational discrimination, or other factors. Even past that, trust in novel curative treatments remains a barrier. Even if PE and BE prove scientifically sound, patient skepticism toward brand new therapies could hinder their clinical success. This makes the goal of “on-demand CRISPR” highly impractical–especially in lieu of the current U.S. policy framework that offers minimal support for preclinical research, manufacturing, and academic innovation22.
Future Implications
Looking past solely base and prime editing comparisons, gene editing’s results can be carefully weighted against established modulator treatments. Its curative possibilities are, however, currently limited to ex-vivo laboratory experiments, such as where A6 ABE restored ~44% of CFTR function and PE restored >50% (Table 1). The Trifakta combination of VX-770, VX-445, and VX-661, restores 40-45% of predicted forced expiratory volume–clinically reflected by up to 45% of CFTR function–for CF patients with at least one F508del copy23. However, these two metrics are not directly comparable due to the lack of complex human cell models in the gene editing tests; further validation is needed to determine if the high laboratory success translates to clinical outcomes.
Combining CFTR modulators with gene editing is a potential intermediate approach that could increase overall treatment efficacy and symptom alleviation. This is tested by Irene Carrozzo and her team, who reported that for almost every tested RM, there was synergism– higher editing levels than each component alone–when both single and double RMs were used with modulators, as CFTR protein functions were restored to WT levels12. Trikafta increased expression of fully glycosylated CFTR protein bands by 7.8-fold compared to 2-fold using LNP-ABE alone15. Still, though hybrid therapies may restore lung function, it is only clinically pursuable if BE and PE are first approved as standalone clinical therapies. Currently, limitations of PE’s success in nonsense mutations remain, as evidenced by the mere 2.5% correction rate in W1282X cells despite high in-vitro delivery efficiency24. Thus, technical gaps and regulatory timelines make hybrid therapies a clinical stretch goal. Nevertheless, integrating gene editing with preexisting triple-modulator infrastructure in high-burden countries such as the USA, UK, Ireland, Denmark, Germany, and Slovenia could leverage more advanced medical science to roll out more efficient, accurate, and curative treatments to the greatest number of patients1.
Long term, as gene editing techniques transition to in vivo applications, they may face further debates about germline editing, which is the practice of altering DNA in a patient’s reproductive cells so that the repaired mutation becomes a heritable trait through generations25. While germline genome editing (GGE) offers theoretical potential to prevent transmission of monogenic diseases like CF in a population’s gene pool, its current status remains medically unsafe and ethically debated25. Additionally, there are many unknowns of how a pathogen-free embryo affects gene frequency and microevolution. Thus, GGE’s use for CF depends on the timeline of gene editing approval, in vivo treatment, proper regulatory policy, and widespread systemic production that brings its costs down. In short, GGE remains a distant and speculative possibility in the current therapeutic landscape25
Conclusion
Prime and base editing are revolutionary gene editing techniques that could be optimized to efficiently correct most CF mutations. Even when a mutation may not be directly corrected by a gene editor, gene editing could still be used as a tool to create RMs and other treatment-responsive mutational conversions. Both BE and PE have been shown, in biotech labs, to be able to correct various CF mutations and recover protein function to levels similar to the most advanced CFTR modulators, and sometimes even to WT CFTR function. Still, modulators may continue to address a substantial portion of the patient population, as approval and availability of gene editing may be prioritized in clinical rollout to only the mutations with high unmet clinical need like those that are Class I with NMD and not the most common Class II F508del, where modulators have already received regulatory approval. However, base and prime editors can target mutations that modulators are ineligible to treat and may even be able to be used in hybrid therapies, indicating a need for more ex-vivo, and eventually in-vivo, testing and research.
Yet, while gene editing has scientific promise, its unpredictable cost and potential manufacturing challenges make it unlikely, without additional years of extensive research, to be a practical and on-hand clinical “cure” for any patient who has CF in the immediate future. After considering the fact that all its current data and expectations are only observed in a select few preclinical models and not a sizable sample of human patients, more private and government research, even possible investment without profit, will be the only way that gene editing has a chance to reach this goal.
References
- J. Guo, A. Garratt, Worldwide rates of diagnosis and effective treatment for cystic fibrosis. Science Direct. 21, 456-462 (2022). [↩] [↩] [↩] [↩]
- Canadian Agency for Drugs and Technologies in Health [CADTH] Common Drug Review, Clinical Review Report: Lumacaftor/Ivacaftor (Orkambi). National Library of Medicine (2018). [↩]
- A. Shankari, S. Sharma, Cystic Fibrosis. National Library of Medicine (2024). [↩] [↩] [↩]
- K. Mention, K. Cavoslogu-Doran, Use of adenine base editing and homology-independent targeted integration strategies to correct the cystic fibrosis causing variant, W1282X. National Library of Medicine. 32, 3237-3248 (2023). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- M. Jinek, K. Chylinski, A programmable dual RNA-guided DNA endonuclease in adaptive bacterial immunity. National Library of Medicine. 337, 816-821 (2012). [↩]
- A.V. Anzalone, P.B. Randolph, Search-and-replace genome editing without double-strand breaks or donor DNA. National Library of Medicine. 576, 149-157 (2020). [↩]
- A.C. Komor, Y.B. Kim, Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. National Library of Medicine. 533, 420-424 (2016). [↩] [↩]
- A.A. Sousa, C. Hemez, Systematic optimization of prime editing for the efficient functional correction of CFTR F508del in human airway epithelial cells. Nature Biomedical Engineering. 9, 7–21 (2024). [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- P. Antoniou, L. Dacquay, Modified pegRNAs mitigate scaffold-derived prime editing by-products. Nature Communications. 16 (2025). [↩]
- M. Bulcaen, P. Kortleven, Prime editing functionally corrects cystic fibrosis-causing CFTR mutations in human organoids and airway epithelial cells. Science Direct. 5, 2–24 (2024). [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- O.V. Volodina, A.G. Demchenko, Selection of Optimal pegRNAs to Enhance Efficiency of Prime Editing in AT-Rich Genome Regions. Biokhimiya. 90, 773–785 (2025). [↩] [↩] [↩]
- I. Carrozzo, G. Maule, Functional rescue of F508del-CFTR through revertant mutations introduced by CRISPR base editing. Science Direct. 33, 970–985 (2025). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- G.X. Reyes, A. Kolodziejczak, Ligation of newly replicated DNA controls the timing of DNA Mismatch Repair. National Library of Medicine. 31 (2022). [↩]
- I. Rose, M. Greenwood, Adenine base editing of CFTR using receptor targeted nanoparticles restores function to G542X cystic fibrosis airway epithelial cells. Springer Nature, 82 (2025). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Y. Sun, S. Chatterjee, In vivo editing of lung stem cells for durable gene correction in mice. National Library of Medicine. 384, 1196-1202 (2025). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- A.C. Chadwick, X. Wang, In Vivo Base Editing of PCSK9 as a Therapeutic Alternative to Genome Editing. National Library of Medicine. 37, 1741-1747 (2017). [↩]
- A.S. Ricciardi, C. Barone, Systemic in utero gene editing as a treatment for cystic fibrosis. Proceedings of the National Academy of Sciences [PNAS] (2025). [↩]
- O.V. Volodina, A.G. Demchenko, Prime Editing Modification with FEM1 Improves F508 del Variant Editing in the CFTR Gene in Airway Basal Cells. MDPI. 26, 7943 (2025). [↩]
- S. Vaidyanathan, A.A. Salahudeen, High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell. 26, 161-171 (2020). [↩]
- F. Farooq, P.J. Mogayzel, Comparison of US Federal and Foundation Funding of Research for Sickle Cell Disease and Cystic Fibrosis and Factors Associated With Research Productivity. Jama Network. 3 (2020). [↩]
- R.M.T. Ham, A.M. Hövels, What does cell therapy manufacturing cost? A framework and methodology to facilitate academic and other small-scale cell therapy manufacturing costings. Cytotherapy Journal. 22, 388-397 (2020). [↩] [↩] [↩]
- C.H. Wong, D. Li, The estimated annual financial impact of gene therapy in the United States. Nature Gene Therapy. 30, 761-773 (2023). [↩] [↩]
- P.G. Middleton, M.A. Mall, Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele National Library of Medicine. 381 (2019). [↩]
- C. Li, Z. Liu, Prime editing-mediated correction of the CFTR W1282X mutation in iPSCs and derived airway epithelial cells, National Library of Medicine. 18 (2023). [↩]
- G. Rubeis, F. Steger, Risks and benefits of human germline genome editing: An ethical analysis, National Library of Medicine. 10, 133-141 (2018). [↩] [↩]



{kind=link}