
Abstract
Hyperthyroidism is an endocrine disorder characterized by excessive thyroid hormone production, affecting 1.4 % of the global population. Autoimmune thyroid diseases, particularly Grave’s disease, arise from genetic susceptibility and immune dysregulation that disrupts the hypothalamic pituitary thyroid (HPT) axis controlling the thyroid stimulating hormone (TSH), triiodothyronine (T3), and thyroxine (T4) levels. This review addresses the research question: What genetic factors contribute to susceptibility to hyperthyroidism, in autoimmune thyroid diseases such as Graves’ disease, and how do theydisrupt normal TSH–thyroid hormone regulation? Susceptibility genes are associated with alterations in immune regulation that may contribute to the development of autoantibodies and the dysregulation of thyroid hormone production. This paper demonstrates that hyperthyroidism in Grave’s disease results from the combined effects of genetic variations that promote autoantibody production and thyroid overstimulation. These factors disrupt TSH regulation and interact with demographic variables, showing that hyperthyroidism is a multifactorial autoimmune condition rather than a disorder caused by a single gene. Figures in the paper provide a conceptual summary of how TSH receptor antibodies contribute to thyroid hormone overproduction in Graves’ disease. Visual diagrams are included for illustrative purposes, but they do not serve as direct evidence of the mechanisms involved. Furthermore, studying the genetic factors behind hyperthyroidism can reveal why Grave’s disease develops in some individuals despite similar environmental exposures and help clarify the mechanisms of thyroid hormone disruption explaining the biological mechanisms driving disease onset and progression. Additionally, current and emerging therapies targeting both hormone overproduction and autoimmune mechanisms offer new pathways for managing Graves’ disease, highlighting the translational relevance of genetic and immunological insights. This insight may guide future studies on autoimmune thyroid disorders and enhance understanding of how genetic and immune factors contribute to the disease.
Keywords: Hyperthyroidism; Grave’s disease; Autoimmune thyroid disease; TSH regulation; Genetic susceptibility, Immune dysregulation; Physiological diagrams.
Introduction
Hyperthyroidism is a common endocrine disorder characterized by excessive production of thyroid hormones, primarily triiodothyronine (T3) and thyroxine (T4), and is associated with significant metabolic and cardiovascular effects 1. The thyroid gland plays a central role in maintaining metabolic homeostasis through tightly regulated hormone synthesis and release, allowing the body to adapt to energy demands, growth, and developmental processes. Under normal physiological conditions, thyroid hormone levels are regulated by the hypothalamic-pituitary-thyroid (HPT) axis through coordinated signaling involving thyrotropin-releasing hormone (TRH), thyroid-stimulating hormone (TSH), and negative feedback from circulating T3 and T42. Precise regulation of this system is essential to maintain hormonal balance and metabolic stability.
Graves’ disease is the most common cause of hyperthyroidism and represents an autoimmune disorder in which immune system dysfunction disrupts normal thyroid hormone regulation3. In this condition, thyroid-stimulating autoantibodies activate the TSH receptor, canceling normal feedback control and leading to persistent thyroid hormone overproduction despite suppressed TSH levels 4. This autoimmune overstimulation explains the characteristic hormonal imbalance.
Genetic susceptibility plays a central role in the development of Graves’ disease. Genetic studies have identified variations in immune-regulatory and thyroid-specific genes that predispose individuals to immune dysregulation, autoantibody production, and thyroid overstimulation5. Importantly, Graves’ disease does not arise from a single genetic mutation but from the interaction of multiple susceptibility genes with environmental and biological factors 6.
Although genetics is a critical factor, it does not fully account for disease variability or severity.
Epidemiological studies demonstrate that Graves’ disease occurs more frequently in women, with a female to male ratio of approximately 5:1 to 10:1, and a global prevalence estimated at 1-1.5% of the population and is influenced by demographic characteristics and environmental exposures, highlighting the role.
In this review, genetic factors contributing to hyperthyroidism are examined with a focus on autoimmune thyroid diseases, particularly Graves’ disease, and their effects on TSH and thyroid hormone regulation. This analysis argues that genetic variations affecting immune balance and thyroid function led to the production of autoantibodies that overstimulate the TSH receptor, disrupt normal hypothalamic-pituitary-thyroid axis feedback and that genetic influences interact with sex, family history, and environmental triggers.
Following this introduction, the paper reviews normal thyroid function and TSH regulation to establish a physiological foundation. It then examines the autoimmune pathophysiology of Graves’ disease, genetic susceptibility involving immune-related and thyroid-specific genes, and the influence of demographic and environmental factors on disease development and severity.
By synthesizing findings from genetic, immune, and endocrine research, this narrative review aims to clarify the mechanisms underlying thyroid hormone dysregulation in Graves’ disease.
This review was conducted using Google Scholars to identify relevant peer reviewed articles on Graves ‘disease and hyperthyroidism.
Normal Thyroid Function
The thyroid gland is a vital endocrine organ responsible for maintaining metabolic homeostasis, in the human body, through the production of thyroid hormones. Proper thyroid function depends on tightly regulated hormone synthesis and release, allowing the body to adapt to daily energy demands, growth needs, and developmental human processes (Figure 1).
Understanding this normal physiological framework provides essential context for the understanding of later sections that examine how autoimmune and genetic factors disrupt thyroid hormone regulation in Graves’ disease.
Under normal physiological conditions, the thyroid gland synthesizes and secretes two primaries hormones: thyroxine (T4) and triiodothyronine (T3) 7. Although T4 is produced in a bigger amount, T3 represents the more biologically active hormone and is generated largely through peripheral conversion of T4 in target tissues 8. Thyroid hormones regulate basal metabolic rate, thermogenesis, lipid and carbohydrate metabolism, and oxygen consumption in approximately every tissue of the body 1. In addition to metabolic control, thyroid hormone signaling is critical for normal growth, skeletal development, and neurological maturation, especially during fetal development and early life 9.
Because these hormones affect multiple organ systems, precise regulation of their production is required. Disruption to this balance, even minor ones, can lead to widespread physiological effects, explaining why excessive thyroid hormone production results in the characteristic systemic symptom of hyperthyroidism. The understanding of normal thyroid hormone function provides the foundation for examining how regulatory mechanisms fail in disease states. Normal thyroid function relies on the controlled synthesis and systemic actions of T3 and T4 Hormones. The big effects of thyroid hormones highlight the importance of precise regulatory control and explains the extensive clinical consequences that arise when this vital system is disrupted: Establishing this physiological base is critical for understanding how autoimmune thyroid diseases, particularly Graves’ disease, lead to excessive hormone production and metabolic dysregulation. With this context established, the following section examines normal TSH regulation and the role of the hypothalamic-pituitary-thyroid axis in maintaining thyroid hormone balance.
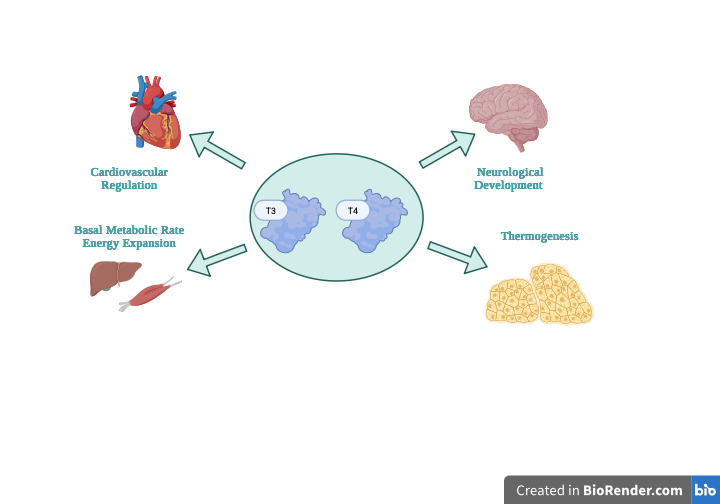
Normal Tsh Regulation And Roles Of T3 And T4 Hormones
Normal thyroid hormone regulation depends on coordinated signaling between the hypothalamus, pituitary gland, and thyroid gland 10. This system ensures that circulatory thyroid hormone levels remain within a precise physiological range to maintain metabolic stability. Examining this system under normal body conditions gives an important context for the understanding of the disruption in hormonal control seen in hyperthyroidism Graves’ disease.
In a healthy state, the hypothalamus releases thyrotropin-releasing hormone (TRH), which stimulates the anterior pituitary gland to secrete thyroid-stimulating hormone (TSH) 11. TSH then acts on thyroid follicular cells, activating intracellular signaling pathways that promote iodine uptake, thyroglobulin synthesis, and the synthesis and release of thyroid hormones T3 and T4, making TSH the primary physiological regulator of thyroid function (Figure 2) 8. Within the thyroid, thyroid peroxidase (TPO) catalyzes the iodination of tyrosine residues on thyroglobulin and the coupling of iodothyronines to form T4 (thyroxine) and T3 (triiodothyronine). While T4 is produced in larger amounts, T3 is the more biologically active hormone, and most T3 is generated in peripheral tissues by the deiodinase enzymes (D1 and D2), which convert T4 to T3.
As circulating levels of T3 and T4 rise, they give negative feedback on both the pituitary and hypothalamus, reducing the release of TSH and TRH to maintain hormonal balance 7. In Graves’ disease, this feedback loop at the pituitary is still functional, and TSH is appropriately suppressed 12. However, thyroid stimulation persists because TSH receptor antibodies bind to thyroid receptors and mimic TSH, bypassing pituitary control. This results in continuous thyroid hormone production despite normal negative feedback. When functioning correctly, this system maintains stable thyroid hormone levels, and supports normal metabolic activity between the body and tissues 11.
Disruption in this regulatory circle leads to abnormal suppression of TSH and excessive thyroid hormone production, a highlight of hyperthyroidism. This understanding provides the context for examining how autoimmune mechanisms in Graves’ disease interfere with normal TSH regulation with normal TSH regulation and thyroid hormone feedback. The following section examines the pathophysiology of Graves’ disease and how autoimmune mechanisms disrupt thyroid hormone control.
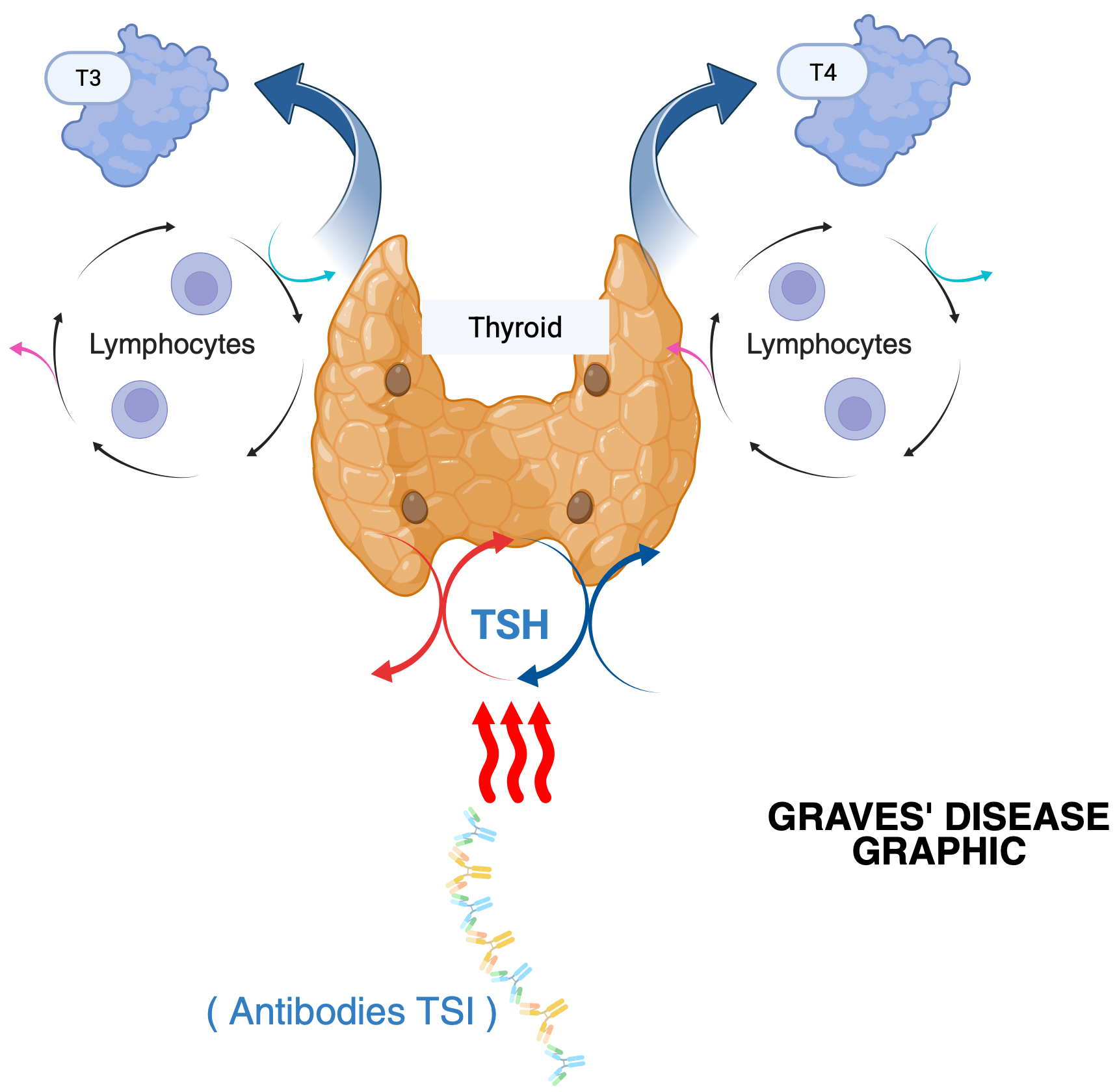
Pathophysiology Of Graves’ Disease
The Graves’ disease is the most common cause of hyperthyroidism in humans and represents a disorder in which immune system dysfunction specifically affects thyroid hormone regulation13. Instead of responding to normal regulatory signals, the thyroid gland becomes overstimulated caused by autoimmune activity. Understanding the pathophysiology of Graves’ disease is essential and remarkable for the understanding of how normal TSH regulation is disrupted.
In Graves’ disease, the immune system produces autoantibodies that bind to and stimulate the thyroid-stimulating hormone receptor (TSHR) on thyroid follicular cells 6.
These thyroid-stimulating immunoglobulins bind the TSH receptor on thyroid follicular cells, activating G-protein coupled signaling and increasing cAMP levels6. This stimulates thyroid hormone synthesis via thioperoxides- mediated iodination of thyroglobulin. The gland produces excess T4, which is converted to the more active T3 by deiodinase enzymes, leading to systemic metabolic acceleration despite suppressed TSH 1. Autoantibodies targeting the TSH receptor are therefore central to disease pathogenesis, as they interfere with the normal feedback mechanisms of the hypothalamic-pituitary-thyroid axis 14.
Clinically, this hormonal excess manifests as symptoms such as weight loss, heath intolerance, tachycardia, nervousness, tremor, and goiter, with some patients also developing ophthalmopathy characteristics of Graves’ disease 4. This persistent stimulation explains why thyroid hormone production remains elevated despite low TSH levels, distinguishing Graves’ disease from other causes of hyperthyroidism. The next section examines genetic susceptibility in Graves’ disease, focusing on immune-related and thyroid specific genes that increase the disease risk.
Genetic Susceptibility In Graves’ Disease (Immune Related Genes And Thyroid Specific Genes)
Graves’ disease arises from the interaction of multiple susceptibility genes and environmental triggers rather than a single genetic mutation. Variants in HLA class II genes increase the presentation of thyroid antigens to helper T cells, promoting autoantibody production. Similarly, CD40 polymorphisms enhance B-cell activation, further amplifying the production of TSH receptor-stimulating antibodies5. These genetic factors contribute to immune dysregulation and the disruption of normal TSH-thyroid hormone regulation, ultimately promoting abnormal thyroid stimulation and sustained hormone overproduction.
Body Immune-Related Genes
Immune-related susceptibility genes play an important and critical role on Graves’ disease by disrupting immune regulation and promoting the development of thyroid-stimulation autoantibodies 15. Variants in human leukocyte antigen (HLA) genes, particularly HLA-DR3, HLA-B8, and HLA-DQA1, are among the strongest genetic risk factors for Graves ‘disease, as they influence antigen presentation and T-cell activation, increasing the probability that the immune system will recognize thyroid antigens as foreign, leading to thyroid dysregulation 5. In addition, polymorphisms in immune regulatory genes such as CD40 contribute to Graves’ disease by influencing its expression on antigen-presenting cells. These variants enhance B-cell activation and promote the production of thyroid-stimulating autoantibodies, thereby increasing susceptibility to autoimmune responses against the thyroid gland3. CD40 risk variants are relatively common in the general population, but Graves’ disease develops in only a subset of individuals, most often during adolescence or early to middle adulthood, indicating that genetic susceptibility alone is insufficient for disease onset 16. Together with HLA variants that impair antigen presentation and immune tolerance, these immune-related genes promote immune dysregulation rather than direct thyroid dysfunction.
Body Thyroid-Specific Genes
Thyroid-specific genes, particularly those encoding the thyroid-stimulating hormone receptor (TSHR), influence the severity and the course of hyperthyroidism in Graves’ disease 14. Genetic variations and activating mutations in the TSHR gene increase the sensitivity of thyroid follicular cells to stimulation, leading to excessive hormone production 17. In Graves’ disease, thyroid-stimulation is primarily driven by autoantibodies that activate the TSH receptor (TSHR). In contrast, genetic alterations in the TSHR gene, such as activating mutations, can also result in persistent receptor activation and hyperthyroidism; however, this represents a distinct non-autoimmune condition 2. These thyroid-specific genetic factors demonstrate and explain why thyroid overstimulation persists despite normal negative feedback natural mechanisms are still working. When combined with immune-related susceptibility genes, TSHR variations contribute to the elevation of T3 and T4 and the clinical consequences of hyperthyroidism. In combination, immune-related and thyroid-specific genes demonstrate that Graves’ disease results from genetic interactions that affect both immune regulation and thyroid activity. Genetic susceptibility in Graves’ disease arises from combined effects of immune-related and thyroid-specific genes that disrupt normal immune tolerance and thyroid hormone balance. Immune genes such as HLA and CD40 promote autoantibody production, while thyroid-specific genes like TSHR increase thyroid gland response and overstimulation 17. These genetic factors explain why Graves’ disease develops only in certain individuals and why hyperthyroidism persists despite suppressed TSH levels. Future research should further investigate how these genetic pathways interact, add and align with environmental triggers to influence disease development and severity. The next section discusses how demographic and environmental factors, including sex, family history, and environmental exposures, modify genetic risk in Graves’ disease Hyperthyroidism.
Influence Of Demographic And Environmental Factors
Although genetic susceptibility plays a crucial role in the development of Graves’ disease, genetics alone doesn’t fully explain disease development or variability in severity.
Epidemiological studies consistently demonstrate that Graves’ disease occurs more frequently in specific demographic groups and is often triggered or worsened by environmental exposures.
Understanding how these factors interact with genetic and immune mechanisms is essential for explaining why some genetically susceptible individuals develop hyperthyroidism while others remain unaffected. This section explores how demographic and environmental influences contribute to immune dysregulation and disruption of normal thyroid hormone regulation to cause hyperthyroidism.
Demographic Factors: Sex And Family History
Graves’ disease shows a strong demographic pattern, specifically with sex and familial prevalence. Thyroid disorders, including hyperthyroidism, are significantly more common in women than in men, with prevalence estimates suggesting a five to ten times higher risk in females 9. This sex pattern is thought to be influenced by hormonal and immunological differences, as estrogen has been shown to enhance immune reactivity and autoantibody production, increasing susceptibility to autoimmune diseases. In addition, family history is a strong risk factor for Graves’ disease, reinforcing the role of heritable genetic susceptibility 5. Twin studies further demonstrate higher rates for autoimmune thyroid diseases in monozygotic twins compared to dizygotic twins, meaning identical twins compared to fraternal twins, showing that shared biological traits significantly influence disease risk while still requiring environmental triggers for development 18.
Together, sex and family history influence immune responsiveness and modify genetic risk, helping explain patterns of disease prevalence and severity 19.
Environmental Factors: Triggers And Immune Activation
Environmental factors play a critical role in initiating or triggering Graves’ disease in genetically susceptible individuals. For example, excessive iodine intake has been associated with increased incidence of autoimmune hyperthyroidism by enhancing thyroid antigenicity 20. Smoking has been shown to increase the risk of graves’ disease and is particularly linked to the development of Graves’ ophthalmopathy 21. Additionally, psychological stress can modulate immune function, potentially precipitating disease onset in predisposed individuals 21. Finally, viral and bacterial infections may act as environmental triggers by promoting autoimmune responses through molecular mimicry 1. These environmental modifiers interact with genetic susceptibility to influence both the cause and severity of disease expression. This gene environment interaction model explains why exposure to similar environmental factors do not lead to disease in all individuals.
Integration Of Demographic, Enviormental, And Genetic Risk
The interaction between demographic characteristics, environmental exposures, and genetic susceptibility highlights Graves’ disease as a multifactorial autoimmune disorder 20. Environmental factors may act as initiating events that trigger immune dysregulation in genetically predisposed individuals, while demographic variables such as sex influence, immune responsiveness and disease expression 2. These combined influences lead to the production of thyroid-stimulating immunoglobulins, persistent activation of the TSH receptor, and disruption of normal hypothalamic-pituitary-thyroid axis regulation. This integrated framework emphasizes that hyperthyroidism in Graves’ disease cannot be caused by a single cause but instead arises from complex interactions between genetic, immune, and environmental factors. Demographic and environmental factors significantly influence the risk and severity of Graves’ disease by working and interacting with genetic susceptibility and immune regulation. Female sex and family history increase vulnerability to autoimmune thyroid disease, while environmental triggers such as iodine intake, smoking, stress, and infections can initiate or intensify hyperthyroidism in susceptible individuals 21. These factors collectively contribute to immune activation, autoantibody production, and persistent thyroid overstimulation 6. Future research should focus on identifying how specific environmental exposures interact and influence individual genetic backgrounds to influence disease progression and development. Understanding these interactions may improve risk prediction and avoid specific environmental exposures and triggers to take preventative strategies for autoimmune thyroid diseases.
Current And Emerging Treatments For Graves’ Disease
Treatment of Graves’ disease focuses on reducing thyroid hormone overproduction and managing the directly autoimmune process. The three primary treatment approaches include antithyroid medications, radioactive iodine therapy, and surgical thyroidectomy. Antithyroid drugs such as methimazole and propylthiouracil inhibit thyroid hormone synthesis by blocking thyroid peroxidase activity, thereby reducing T3 and T4 production 1. Radioactive iodine therapy works by selectively destroying overactive thyroid tissue, leading to a gradual reduction in hormone production, while thyroidectomy provides a definitive treatment option in severe cases in different organisms 6.
In addition to these conventional treatments, recent advances in therapeutic approaches are targeting the immune mechanisms underlying Graves’ disease. Because the disease is driven by TSH receptor autoantibodies, emerging strategies aim to reduce autoantibody production or in some cases to modulate immune system activity. Therapies targeting B-cell activation and immune signaling pathways are being investigates as potential methods to decrease autoantibody levels and restore the normal immune tolerance 6. These approaches reflect an improvement toward the treatments that address the autoimmune mechanisms rather than only controlling hormone excess.
Understanding current and emerging treatments highlights how advances in genetic and immunological research are contributing to improved managements for autoimmune diseases like in Graves’ disease and reinforces the importance of targeting both hormonal dysregulation and immune dysfunction in this condition.
Conclusion
This review demonstrates that hyperthyroidism in Graves’ disease arises from complex interactions between genetic susceptibility, immune dysregulation, altered thyroid hormone regulation, and different demographic and environmental factors. Rather than resulting from a single genetic defect or isolated trigger, Graves’ disease reflects a multifactorial autoimmune process in which inherited variations affecting immune tolerance and thyroid responsiveness disrupt normal hypothalamic-pituitary-thyroid axis control, leading to persistent thyroid overstimulation and excessive hormone production 15.
By integrating from thyroid physiology, autoimmune pathophysiology, and genetic research, this review highlights the role of immune-related genes in promoting autoantibody formation in Graves’ disease. Variants in genes such as HLA, CD40, and CTLA4 can alter immune cell activation and tolerance, leading B-cells to produce TSH receptor autoantibodies that continuously stimulate thyroid hormone production. Understanding these mechanisms clarifies how genetic susceptibility contributes to persistent thyroid overstimulation.
Demographic and environmental factors further modify this risk. Female sex, family history, and environmental exposures such as iodine intake and smoking act as influencing variables that influence immune activation and disease severity, reinforcing the concept that genetic predisposition alone is insufficient to cause disease 21. Together, these findings support a gene environment interaction model as the most accurate explanation for Graves’ disease development. However, current research often addresses these factors superficially. Key gaps remain in understanding how specific genetic variants interact with environmental triggers at the molecular level and how these interactions influence disease onset, progression, and severity. Current and emerging treatments for Graves’ disease target both hormone overproduction and underlying autoimmunity. Conventional therapies like antithyroid drugs, radioactive iodine, and thyroidectomy control excess thyroid hormone, while newer approaches focus on reducing TSH receptor autoantibodies by modulating B-cell activity and immune responses 6. Integrating knowledge of genetic susceptibility and immune dysregulation into treatments strategies may improve outcomes, reduce recurrence, and guide personalized therapy.
Integrating population-based genetic studies with endocrine and epidemiological data may improve estimating risk, early identification of susceptible individuals, and the development of preventive strategies for autoimmune thyroid disorders.
Overall, this review emphasizes that understanding Graves’ disease requires a comprehensive approach that considers genetic, immune, endocrine, and environmental influences together while highlighting and thinking about the critical gaps that future research must address to fully clarify autoimmune hyperthyroidism while other autoimmune diseases. Advancing knowledge in this area is essential for improving prediction, prevention, and management of autoimmune hyperthyroidism.
Acknowledgment
I sincerely thank my mentors, Dr. Jorge Ávila and Bre Calhoun, from the Genetic IRIS Research Program, for their guidance and support throughout the project. I also thank my Professor Joel Flores for his valuable advice and mentorship. I appreciate my family and peers for them feedback and encouragement.
References
- S. Y. Lee, E. N. Pearce. Hyperthyroidism: a review. JAMA. Vol. 330, pg. 1472, 2023 https://doi.org/10.1001/jama.2023.19052 [↩] [↩] [↩] [↩] [↩]
- A. Hébrant, W. C. G. Van Staveren, C. Maenhaut, J. E. Dumont, J. Leclère. Genetic hyperthyroidism: hyperthyroidism due to activating tshr mutations. European Journal of Endocrinology. Vol. 164, pg. 1–9, 2011 https://doi.org/10.1530/EJE-10-0775 [↩] [↩] [↩]
- A. P. Weetman. Graves’ disease. New England Journal of Medicine. Vol. 343, pg. 1236–1248, 2000 [↩] [↩]
- Jody Ginsberg. Diagnosis and management of graves’ disease. JAMC. 2003 [↩] [↩]
- Y. Tomer. Genetic susceptibility to autoimmune thyroid disease: past, present, and future. Thyroid. Vol. 20, pg. 715–725, 2010 https://doi.org/10.1089/thy.2010.1644 [↩] [↩] [↩] [↩]
- A. Antonelli, S. M. Ferrari, F. Ragusa, G. Elia, S. R. Paparo, I. Ruffilli, A. Patrizio, C. Giusti, D. Gonnella, A. Cristaudo, R. Foddis, Y. Shoenfeld, P. Fallahi. Graves’ disease: epidemiology, genetic and environmental risk factors and viruses. Best Practice & Research Clinical Endocrinology & Metabolism. Vol. 34, pg. 101387, 2020 https://doi.org/10.1016/j.beem.2020.101387 [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- M. A. Shahid, M. A. Ashraf, S. Sharma. Physiology, thyroid hormone. 2018 [↩] [↩]
- G. A. Brent. Mechanisms of thyroid hormone action. The Journal of Clinical Investigation. Vol. 122, pg. 3035–3043, 2012 [↩] [↩]
- M. Moleti, M. Di Mauro, G. Sturniolo, M. Russo, F. Vermiglio. Hyperthyroidism in the pregnant woman: maternal and fetal aspects. Journal of Clinical & Translational Endocrinology. Vol. 16, pg. 100190, 2019 [↩] [↩]
- N. Stathatos. Thyroid physiology. Medical Clinics. Vol. 96, pg. 165–173, 2012 [↩]
- Y. Pirahanchi, F. Toro, I. Jialal. Physiology, thyroid stimulating hormone. 2018 [↩] [↩]
- R. Latif, S. A. Morshed, M. Zaidi, T. F. Davies. The thyroid-stimulating hormone receptor: impact of thyroid-stimulating hormone and thyroid-stimulating hormone receptor antibodies on multimerization, cleavage, and signaling. Endocrinology and Metabolism Clinics of North America. Vol. 38, pg. 319–341, 2009 [↩]
- J. R. Reid. Hyperthyroidism: diagnosis and treatment. 2005 [↩]
- P. Kopp. Human genome and diseases: review¶the tsh receptor and its role in thyroid disease: Cellular and Molecular Life Sciences. Vol. 58, pg. 1301–1322, 2001 https://doi.org/10.1007/PL00000941 [↩] [↩]
- V. Panicker. Genetics of thyroid function and disease. Vol. 32, 2011 [↩] [↩]
- P. S. Ramos, A. M. Shedlock, C. D. Langefeld. Genetics of autoimmune diseases: insights from population genetics. Journal of Human Genetics. Vol. 60, pg. 657–664, 2015 [↩]
- L. Grixti, L. C. Lane, S. H. Pearce. The genetics of graves’ disease. Reviews in Endocrine and Metabolic Disorders. Vol. 25, pg. 203–214, 2024 https://doi.org/10.1007/s11154-023-09848-8 [↩] [↩]
- T. H. Brix, L. Hegedüs. Twin studies as a model for exploring the aetiology of autoimmune thyroid disease. Clinical Endocrinology. Vol. 76, pg. 457–464, 2012 [↩]
- N. M. Y. Journy, M.-O. Bernier, M. M. Doody, B. H. Alexander, M. S. Linet, C. M. Kitahara. Hyperthyroidism, hypothyroidism, and cause-specific mortality in a large cohort of women. Thyroid. Vol. 27, pg. 1001–1010, 2017 https://doi.org/10.1089/thy.2017.0063 [↩]
- P. N. Taylor, D. Albrecht, A. Scholz, G. Gutierrez-Buey, J. H. Lazarus, C. M. Dayan, O. E. Okosieme. Global epidemiology of hyperthyroidism and hypothyroidism. Nature Reviews Endocrinology. Vol. 14, pg. 301–316, 2018 [↩] [↩]
- T. Uchida, M. Shimamura, H. Taka, N. Kaga, Y. Miura, Y. Nishida, Y. Nagayama, H. Watada. The effect of long-term inorganic iodine on intrathyroidal iodothyronine content and gene expression in mice with graves’ hyperthyroidism. Thyroid. Vol. 33, pg. 330–337, 2023 [↩] [↩] [↩] [↩]


