
Abstract
The development of nanocarriers for cancer therapy faces a persistent trade-off between achieving tumor-specific, pH-responsive drug release and preventing premature leakage under physiological conditions. Here, we report a conjugated polymer nanoparticle platform that integrates high drug-loading capacity with intrinsic fluorescence self-reporting of drug release. Among several drug-loaded systems, a paclitaxel-loaded nanoparticle (PFPTX) exhibited pronounced aggregation-induced luminescence enhancement (AILE), enabling optical tracking of nanoparticle assembly and disassembly. Spectroscopic and computational analyses indicate that drug binding induces a distinct fluorescence shift that correlates with nanoparticle disassembly. PFPTX nanoparticles achieved a high drug-loading capacity (45%) and exhibited minimal drug leakage at physiological pH, while releasing more than 40% of paclitaxel under acidic conditions relevant to intracellular tumor environments. In vitro studies across multiple cancer cell lines demonstrated efficient cellular uptake, pH-triggered intracellular release, and dose-dependent cytotoxicity, with fluorescence imaging confirming lysosomal localization and release behavior. Together, these results demonstrate an AILE-based nanocarrier that couples acid-responsive chemotherapy with built-in fluorescence self-reporting, providing a proof-of-concept platform for monitored drug delivery in cancer therapy.
Keywords: Nanoparticles; Luminescence; aggregation-induced luminescence enhancement (AILE); Paclitaxel; Drug delivery; Cancer therapy
Introduction
The global cancer burden, already immense and growing, reached approximately 19.3 million new cases and nearly 10 million deaths in 20201, with projections warning of a 47% increase to 28.4 million cases by 2040. These trends underscore cancer’s profound public health toll and the urgent need for more effective treatments. Current clinical management primarily relies on surgery and chemotherapy; however, conventional small-molecule chemotherapeutics lack tumor specificity, resulting in systemic toxicity and limited therapeutic precision2,3. Peptidic drugs have attracted considerable interest due to their high efficacy and targeting potential. However, their development depends on the availability of well-characterized molecular targets, which remain relatively scarce in oncology4. In addition, natural peptides exhibit poor in vivo stability, as they are highly susceptible to hydrolysis in biological fluids.
Nanocarrier platforms have emerged as promising solutions to overcome the limitations of conventional drug delivery systems, with several nanoparticle-based therapeutics approved for oncological applications. Among these, acid-responsive nanocarriers commonly employ covalent pH-cleavable linkers to trigger drug release within acidic tumor microenvironments. However, the intrinsic lability of such linkers may compromise carrier stability under physiological conditions. For example, hydrazone-functionalized covalent organic framework(NCOF) nanoparticles conjugated with doxorubicin (DOX) exhibit approximately sixfold higher drug release at pH 5.2 than at pH 7.4, demonstrating effective acid-triggered delivery5. Nevertheless, partial hydrolysis persists under neutral conditions, with ~13.5% of encapsulated DOX released over 72 h at pH 7.4, indicating premature leakage6.
Similarly, polymeric micelles engineered with DOX-conjugated hydrazone linkages and co-loaded docetaxel exploit endosomal acidity, releasing approximately 60% of DOX at pH 5.5 while maintaining relative stability at physiological pH, although minor leakage at neutral conditions remains detectable7. These findings highlight that hydrazone-based acid-cleavable bonds enhance tumor-specific drug release but inherently permit non-negligible drug loss under physiological conditions.
Another representative system is the ZIF-8 metal-organic framework (MOF), which remains stable at the physiological pH of 7.4 but rapidly dissolves in mildly acidic endosomal environments to trigger payload release8. Although amine-modified ZIF-8 has been used to load 5-fluorouracil by exploiting this pH sensitivity, unmodified ZIF-8 often exhibits weak host-guest interactions, leading to low loading efficiency and premature drug leakage prior to acid activation9. Indeed, weak host-guest binding has been linked to significant premature drug escape from ZIF-8 carriers9.
Collectively, these systems reveal a common trade-off: while acid-labile chemistries (e.g., hydrazones, acetals, and MOF linkages) enable tumor-selective drug release, their inherent instability or weak bonding can compromise nanoparticle integrity and cause off-target leakage under physiological conditions. To overcome the intrinsic instability and premature leakage associated with acid-labile covalent linkers, we hypothesized that a supramolecular nanoparticle stabilized by strong yet reversible noncovalent interactions could decouple pH responsiveness from carrier degradation. Specifically, we propose that a conjugated polymer–drug network governed by cooperative hydrogen bonding and π–π interactions would remain structurally intact under physiological conditions, while undergoing controlled disassembly in acidic tumor and endosomal environments. Guided by this hypothesis, we designed a multifunctional nanoparticle based on a conjugated polymer hydrogen-bonded framework, enabling dense drug sequestration without reliance on cleavable linkers. pH-responsiveness arises from protonation-induced weakening of hydrogen-bonding interactions within the supramolecular network, preserving carrier integrity under physiological conditions while enabling controlled disassembly and drug release in acidic tumor and endosomal environments10,11. In parallel, we sought to integrate an intrinsic optical reporting function that directly reflects the drug binding and release state rather than merely nanoparticle localization. To this end, PFOP-NEt₃(+) was selected as the polymer backbone due to its donor–acceptor fluorenone-based structure and cationic side chains, which collectively enable aggregation-induced luminescence enhancement (AILE), high aqueous stability, and strong drug affinity. Unlike conventional fluorophores that suffer aggregation-caused quenching, PFOP-NEt₃(+) remains weakly emissive in the dispersed state but exhibits pronounced fluorescence upon aggregation induced by drug binding. We therefore hypothesized that drug-induced aggregation and pH-triggered disassembly would be directly transduced into distinct luminescence changes. This design uniquely couples ultrahigh drug loading (>80 wt%), physiological stability, and real-time fluorescence reporting within a single polymeric scaffold, distinguishing this system from existing luminescent nanocarriers in which imaging and delivery functions are largely decoupled.
Methodology
Materials & Instruments
PFOP-NEt3(+) was synthesized according to a previously reported procedure12. Gemcitabine, Oxaliplatin, Pemetrexed, Etoposide, and Methotrexate were obtained from Macklin (Shanghai, China), while Paclitaxel Injection was supplied by SL Pharmaceutical (Beijing, China). Dulbecco’s Modified Eagle’s Medium (DMEM) was sourced from Corning (USA). Fetal bovine serum (FBS) and Penicillin−streptomycin were purchased from Gibco (Grand Island, NY, USA). MTT, 4% paraformaldehyde, 10× PBS buffer, trypsin-EDTA solution, and dimethyl sulfoxide (DMSO) were supplied by Beyotime (Shanghai, China). For all experiments, ultrapure water was obtained from a Milli-Q purification system with a resistivity of ≥18.2 MΩ·cm.
UV-vis spectra were recorded on an Agilent Cary 100 UV-vis spectrometer, and fluorescence spectra were measured with a SPEX Fluorolog 3-TCSPC spectrometer. Particle size and ζ-potential of the CPNs were analyzed using a Zetasizer Nano ZS. Cellular fluorescence imaging was performed on an Olympus FV1000-IX81 confocal laser scanning microscope (CLSM).
Synthesis of PFOP-NEt₃(+)
Following the general synthetic route shown in Fig. 1, the intermediate substrate 2 was prepared via a two-step substitution reaction. First, hydroquinone (2.21 g, 20 mmol), 1,6-dibromohexane (7.38 mL, 48 mmol), and 1,4-dioxane (100 mL) were added to a 250 mL round-bottom flask and heated at 80 °C for 24 h. The reaction mixture was diluted with dichloromethane (200 mL), washed with saturated sodium bicarbonate, extracted with water and methylene chloride, and dried over anhydrous magnesium sulfate. The condensation product was obtained by vacuum distillation and purified via flash column chromatography (n-hexane: dichloromethane = 6:1), yielding 7.12 g of a yellow solid (82%). Subsequently, 1,4-bis(5-bromoamyl)oxybenzene (7.12 g, 16.4 mmol) and triethylamine (30 mL) were reacted at 120 °C for 24 h in a 100 mL sealed tube. The product, 5,5′-(1,4-phenyldioxy)bis(N,N,N -triethylpentane-1-ammonium) bromide, was isolated as a white solid (9.39 g, 90% yield) after washing with ethyl acetate.
The PFOP-NEt₃(+) polymer was then synthesized via the direct arylation polymerization (DARP) method. A 25 mL sealed reaction tube was charged with monomer 1 (0.0518 g, 0.2 mmol), monomer 2 (0.153 g, 0.24 mmol), palladium acetate (0.0023 g, 0.01 mmol), potassium carbonate (0.1104 g, 0.8 mmol), pivalic acid (0.02 g, 0.2 mmol), and a 1,4-dioxane : H₂O mixture (4:1, 1.25 mL). The mixture was stirred at 150 °C for 36 h. After cooling, the reaction was diluted with ethyl acetate (15 mL), and insoluble palladium residues and potassium salts were removed by filtration. The filtrate was dissolved in deionized water (50 mL) and 1,4-dioxane (5 mL), then purified by dialysis (regenerated cellulose membrane, 8 kDa MWCO) and concentrated under reduced pressure to afford the crude polymer product.
Synthesis of PFOEP-SO₃(-)
In a 25 mL pre-dried screw-cap tube equipped with a magnetic stir bar, 2,7-dibromo-9H-fluoren-9-one (0.0518 g, 0.2 mmol), 6,6′-((2,5-diethynyl-1,4-phenylene)bis(oxy))bis(hexane-1-potassium sulfonate) (0.1351 g, 0.24 mmol), tetrakis(triphenylphosphine)palladium(0) (0.0116 g, 0.01 mmol), potassium carbonate (0.1104 g, 0.8 mmol), and a 1,4-dioxane: H₂O mixture (4:1, 1.25 mL) were combined under a nitrogen atmosphere. The reaction was stirred at 120 °C for 36 h. After cooling, the mixture was diluted with ethyl acetate (15 mL) and filtered to remove insoluble residues. The filtrate was dissolved in deionized water (50 mL) and 1,4-dioxane (5 mL), then purified via dialysis (regenerated cellulose membrane, 8 kDa MWCO) and concentrated under reduced pressure to yield PFOEP-SO₃(-) (0.128 g, 85% yield).
Preparation of Nanoparticles (NPs)
Drug solutions were prepared by dissolving 1 mg of Gemcitabine, Oxaliplatin, Pemetrexed, Etoposide, or Methotrexate in 1 mL of DMSO. For nanoparticle formation, 1 mL of PFOP-NEt3(+) (1.5 mM, 100 μL) was mixed with the drug solution (1 mg/mL), diluted to 1.1 mL, and stirred in the dark at room temperature for 12 h. For paclitaxel-loaded NPs, 1 mL of PFOP-NEt3(+) (1.5 mM, 100 μL) was mixed with 300 μL of paclitaxel injection (6.0 mg/mL), diluted to 1.1 mL, and stirred under the same conditions.
To evaluate drug encapsulation efficiency (EE) and drug loading content (DLC), 0.5 mL of NP suspension was freeze-dried and weighed. The dried sample was dissolved in methanol for high-performance liquid chromatography (HPLC) analysis (Hitachi Primaide 1430) using a Diamonsil C18 column (250 × 4.5 mm, 5 μm; Dikma Technologies, Lake Forest, CA, USA). The mobile phase consisted of acetonitrile/deionized water/tetrahydrofuran/phosphoric acid (85:10:5:0.01, v/v), with a flow rate of 1.5 mL/min and detection at 210 nm. EE and DLC were calculated as:
EE (%) = (weight of loaded drug/weight of drug added) × 100
DLC (%) = [weight of loaded drug/ (total weight of loaded drug + polymer)] × 100
Molecular Docking Simulation
The molecular structures were built in ChemBioOffice 20.0 and energy-minimized using Chem3D with the MM2 force field to obtain low-energy conformations. Docking studies were carried out with MolAICal, using PFOP-NEt3(+) as the receptor and paclitaxel as the ligand13,14,15. A docking grid box (center_x = −0.6, center_y = 1.2, center_z = −0.2; size_x = 30, size_y = 30, size_z = 30) was applied around the receptor. All other parameters were set to default. The five best-scoring conformations (based on binding energy) were selected for further interaction analysis and visualized using PyMOL (Molecular Graphics System, Version 3.0, Schrödinger, LLC).
In Vitro Drug Release
For release experiments, 1 mL of PFPTX suspension was sealed in a dialysis bag (3500 Da cutoff) and immersed in 75 mL of PBS buffer at different pH values at 37 °C. At specific intervals, 2 mL of dialysate was withdrawn, its paclitaxel content determined by UV-vis spectrophotometry at 492 nm, and the aliquot immediately returned. The same protocol was used to obtain the release profiles of all other drug-loaded nanoparticles.
Cell Culture
HeLa, A549, and HepG2 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C under 5% CO2. Cells were harvested from subconfluent cultures (<80%) using trypsin-EDTA and resuspended in fresh medium. Subculturing was performed every two days.
Cell Membrane Imaging of PFPTX
HeLa, HepG2, and A549 cells were incubated with PFPTX (1.0 mg/mL) for 0, 4, and 12 h at 37 °C in a 5% CO2 incubator. After incubation, the cells were washed with PBS (three times), fixed with 4% paraformaldehyde for 10 min, and immediately imaged using confocal microscopy. Excitation for both PFPTX and PFOP-NEt3(+) was set at 405 nm.
Cytotoxicity Assay
Cells (10,000 per well) were seeded into 96-well plates with 100 μL of complete medium and allowed to attach overnight. The next day, cells were treated with varying concentrations (0, 0.5, 1, 1.5, 2, 2.5 μg·mL⁻¹) of NPs for 48 h. After treatment, the medium was replaced with 10 μL MTT solution mixed with 90 μL medium per well, and the plates were incubated for 4 h at 37 °C in 5% CO2. Cell viability was determined by measuring absorbance at 490 nm using a microplate reader.
Result
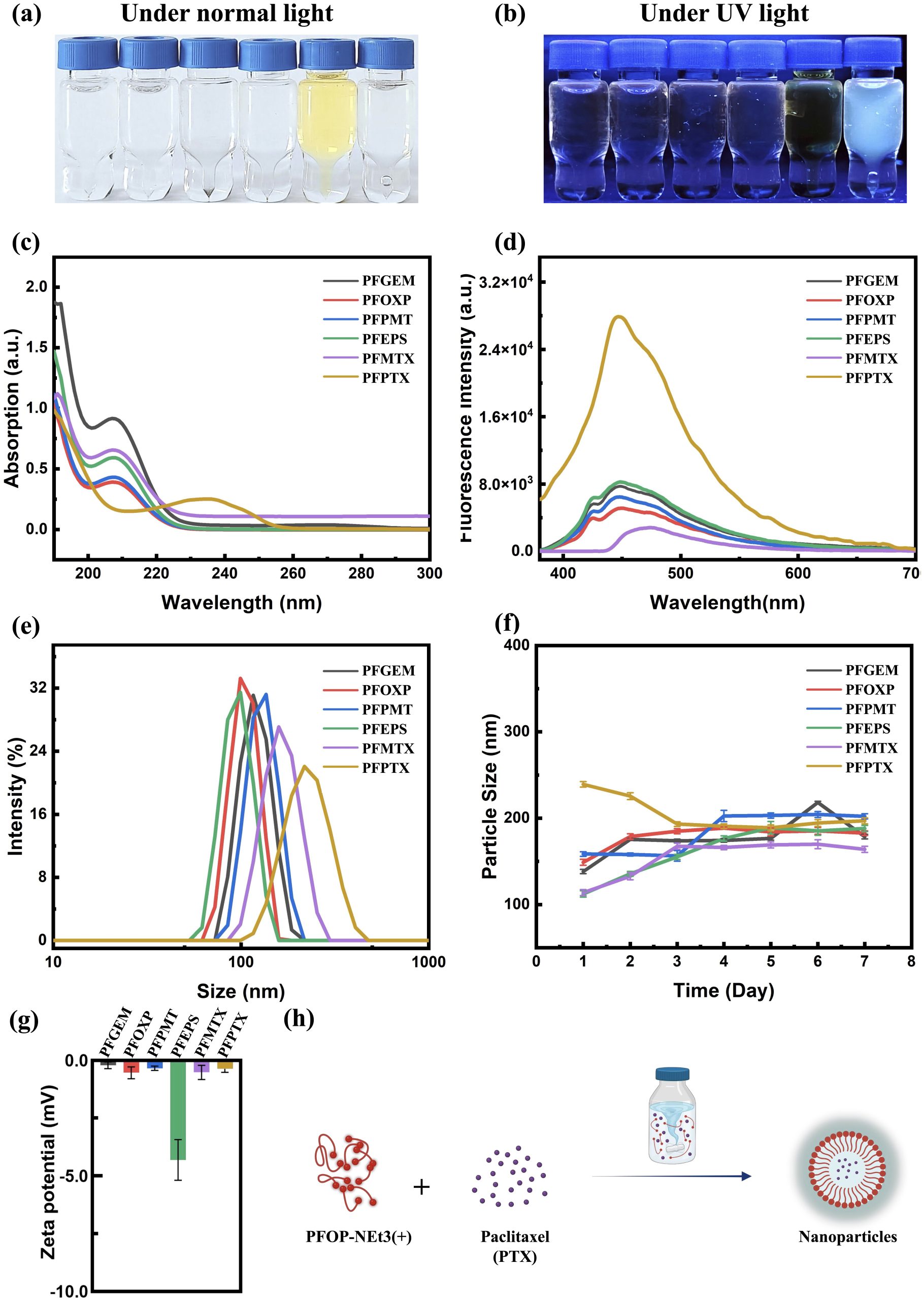
Building upon earlier studies of AILE observed in fluorenone (FO)-based conjugated polymers (CPs) interacting with small hydrogen-bond donor molecules, we investigated whether a similar luminescence response could be extended to drug molecules possessing multiple hydrogen-bonding groups and relatively larger molecular weights. For this purpose, FO-based CPs were combined with a series of clinically used anticancer drugs, gemcitabine, oxaliplatin, pemetrexed, etoposide, methotrexate, and paclitaxel, yielding six types of drug-loaded nanoparticles (NPs) via a self-assembly process (Figure 1a, b, h).
The physicochemical properties of the resulting nanoparticles were systematically characterized, including their appearance under visible and ultraviolet light (Figure 1a, b), optical absorption spectra (Figure 1c), fluorescence emission spectra (Figure 1d), particle size distributions (Figure 1e), temporal size stability over seven days (Figure 1f), and ζ-potentials (Figure 1g). All six nanoparticle systems exhibited highly similar UV–vis absorption profiles (Figure 1c), comparable hydrodynamic sizes (Figure 1e), stable particle diameters over time (Figure 1f), and consistent surface charges (Figure 1g). Despite these similarities, all drug-loaded nanoparticles displayed marked fluorescence enhancement accompanied by a pronounced blue shift relative to the free polymer (Figure 1b, d), indicating the occurrence of AILE. Among the systems investigated, PFPTX exhibited the most significant fluorescence change, characterized by a strong enhancement in emission intensity and a substantial blue shift (Figure 1b, d). This observation motivated further investigation into the molecular origin of the fluorescence change and its potential relationship with drug release behavior.
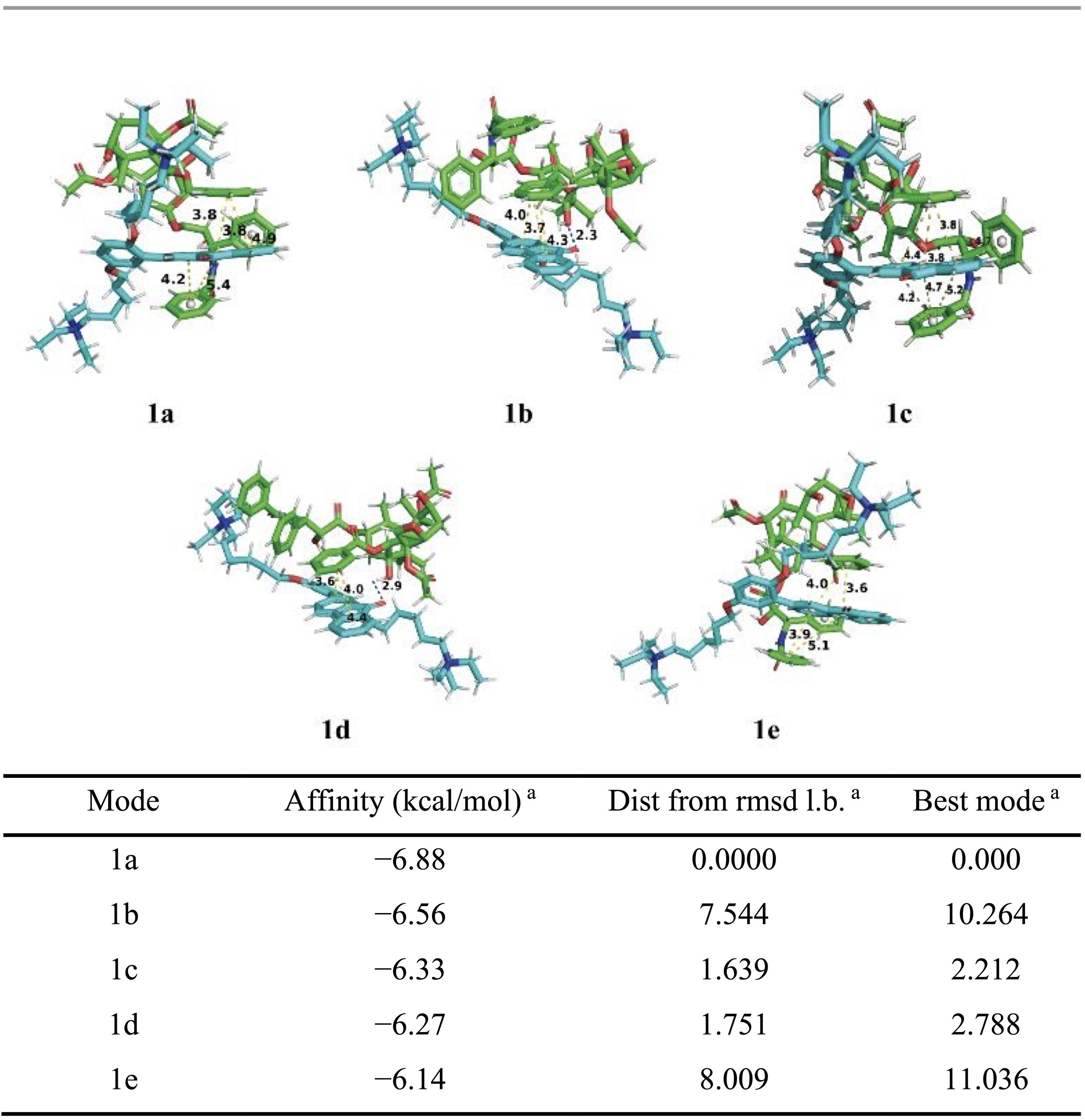
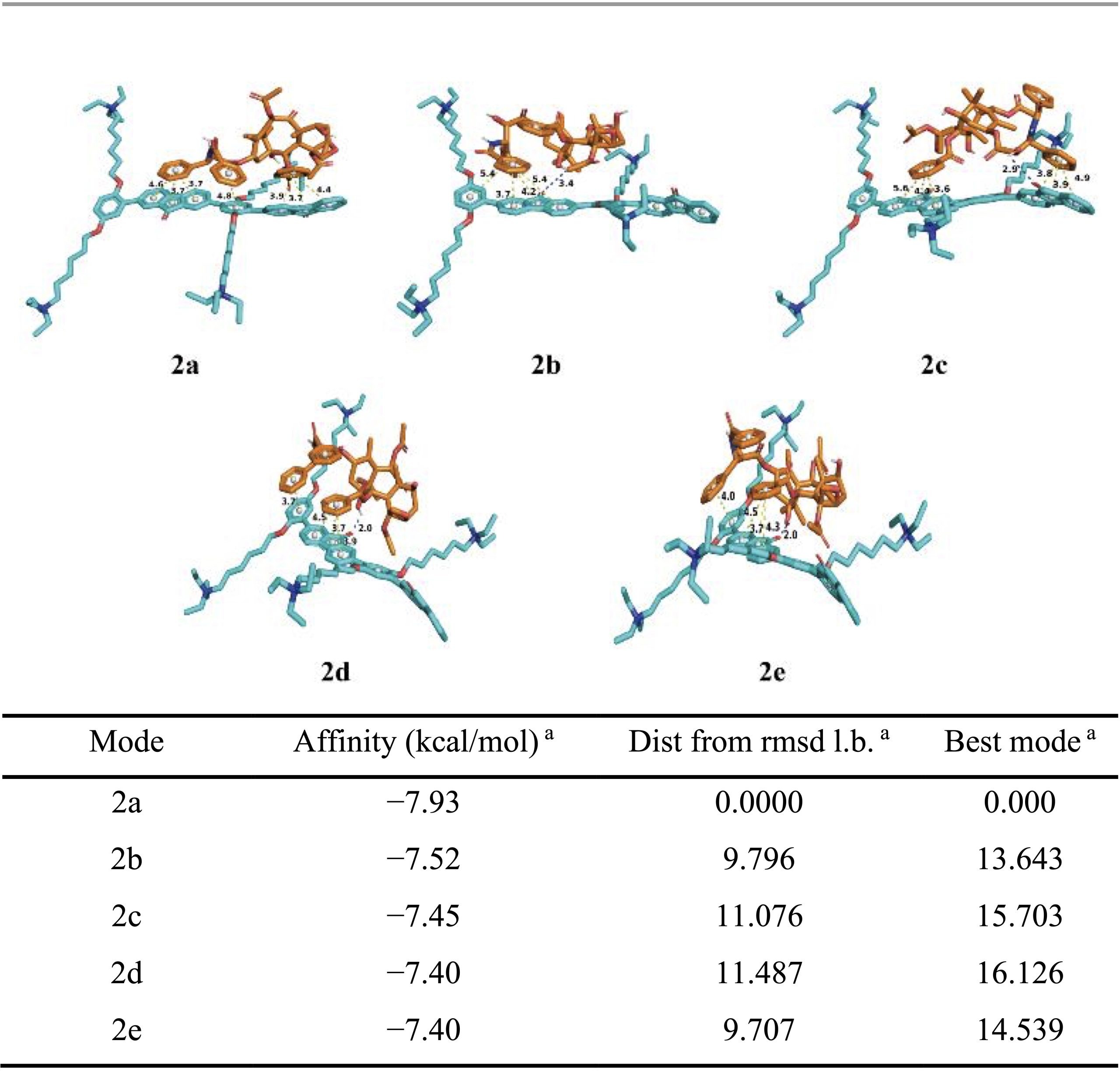
To gain molecular-level insight into these interactions, molecular docking simulations were performed between PFOP-NEt₃(+) and paclitaxel. When a single repeating unit of the polymer was considered, several low-energy binding conformations were identified, with calculated binding energies ranging from −6.14 to −6.88 kcal mol⁻¹ (Table 1). In these structures, π–π stacking interactions were consistently observed, whereas hydrogen bonding occurred only in certain conformations (Figure 2). Structural visualization revealed that paclitaxel promotes partial planarization of the polymer repeat unit through these noncovalent contacts (Figure 2). Extending the analysis to a dimeric polymer model consisting of two repeating units revealed stronger binding interactions, with binding energies ranging from −7.40 to −7.93 kcal mol⁻¹ (Table 2). In contrast to the monomer model, hydrogen bonds were consistently present in all low-energy dimer conformations, and the polymer backbone adopted a more planar configuration (Figure 2). These results suggest that longer polymer chain segments increase the likelihood of cooperative π–π and hydrogen-bonding interactions with paclitaxel, thereby stabilizing a planarized conjugated structure.
Although the calculated binding energies fall within a moderate range, they are consistent with values reported for stable yet reversible noncovalent drug encapsulation. Previous studies have shown that hydrogen-bonding and π-stacking interactions with binding free energies on the order of −3.6 to −6.8 kcal mol⁻¹ are sufficient to maintain nanoparticle integrity while enabling stimulus-responsive drug release through a binding–unbinding mechanism16. The slightly stronger binding observed in the dimeric polymer model, therefore, supports efficient drug loading under physiological conditions and controlled release under acidic conditions. Taken together, the docking results and spectroscopic data support an AILE-based luminescence mechanism for PFPTX.
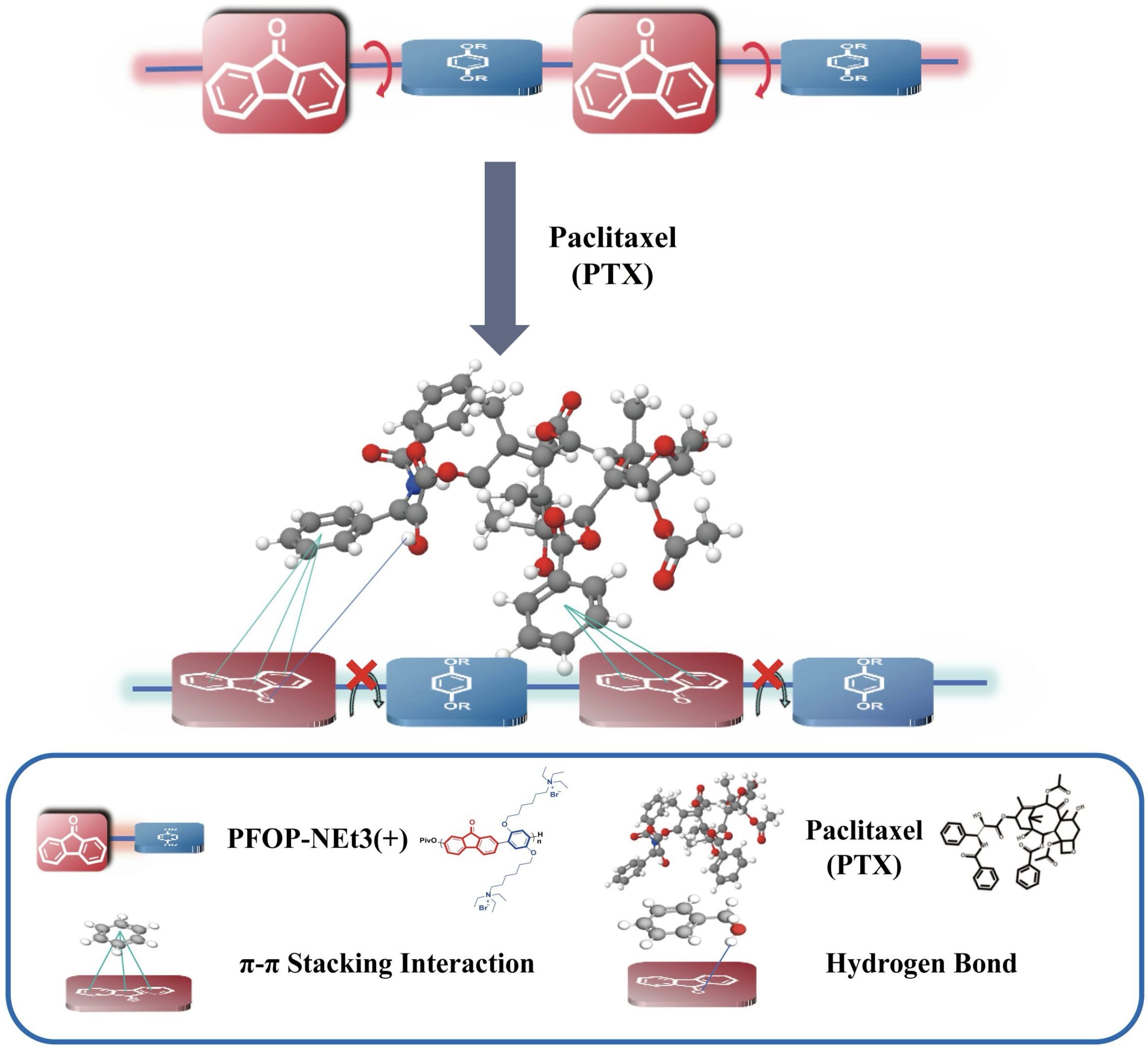
Strong π–π and hydrogen-bonding interactions between PFOP-NEt₃(+) and paclitaxel restrict the torsional motion of the fluorenone units, enforcing a more planar polymer backbone (Figure 2). At the molecular level, the positively charged NEt₃⁺ side chains engage in electrostatic interactions with the electronegative hydroxyl and carbonyl groups of paclitaxel, while the aromatic π-system of the fluorenone units participates in π–π stacking with paclitaxel’s benzene rings. In addition, carbonyl (C=O) and hydroxyl (–OH) groups along the polymer backbone act as hydrogen-bond acceptors and donors, forming multiple hydrogen bonds with paclitaxel’s abundant hydrogen-bonding sites. These cooperative electrostatic, π–π, and hydrogen-bonding interactions stabilize the polymer to drug assemblies and enable dense drug packing. Accordingly, drug-loading content determined by HPLC analysis revealed that PFPTX achieved a drug-loading capacity of 45% and an encapsulation efficiency of 80–86%.
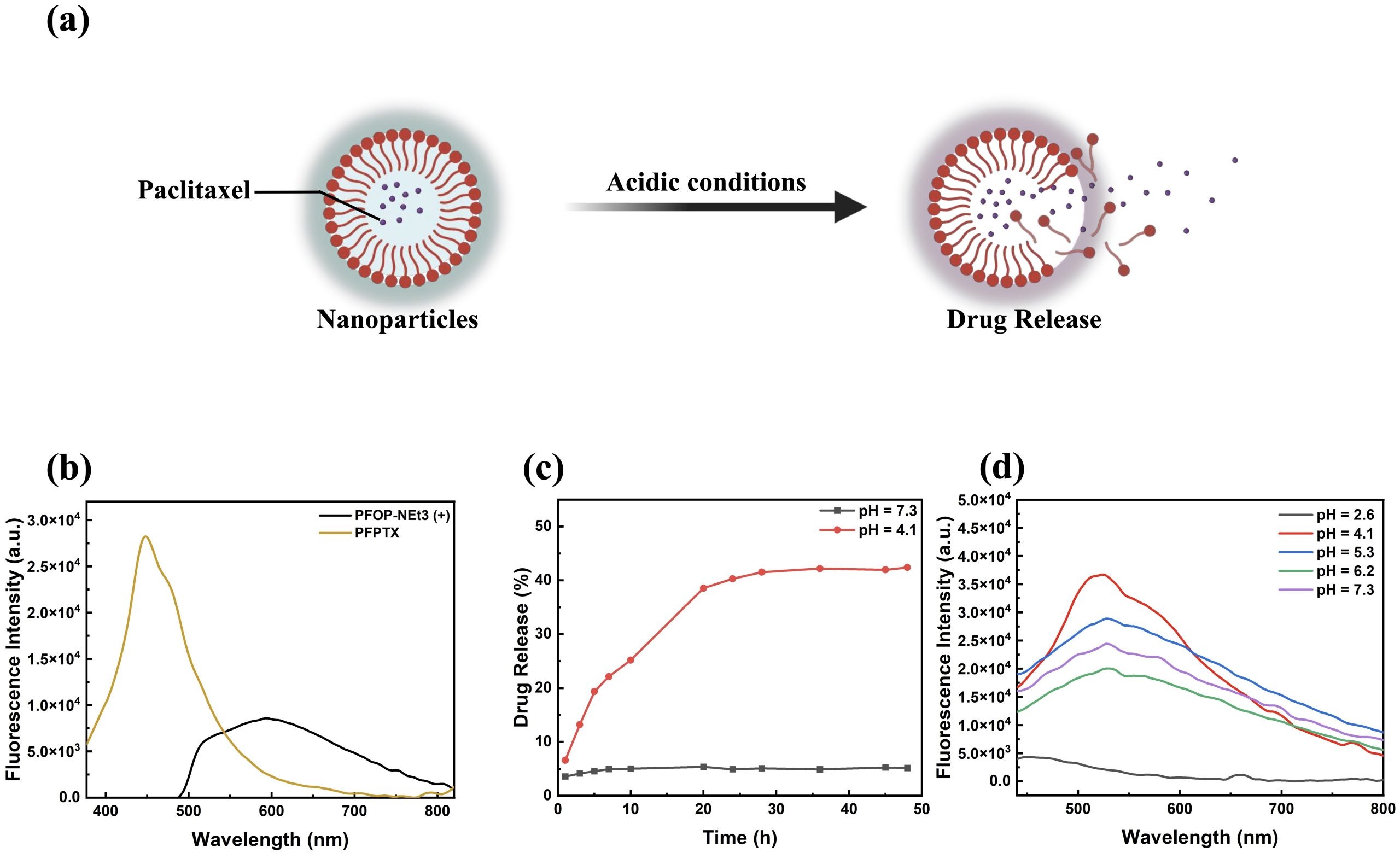
Since PFOP-NEt₃(+) and paclitaxel assemble into nanoparticles through noncovalent interactions, the marked fluorescence transition of blue shift suggested that luminescence might serve as an intrinsic indicator of drug loading and release. To explore this possibility, the pH-responsive release behavior of PFPTX was examined (Figure 3a). In aqueous solution, free PFOP-NEt₃(+) exhibited weak red emission with an emission maximum at approximately 600 nm (Figure 3b). This behavior can be attributed to the twisted conformation of the fluorenone units in the dispersed state, where unrestricted torsional motion reduces the effective π-conjugation length, thereby favoring lower-energy emission. Upon aggregation induced by drug binding, strong π–π and hydrogen-bonding interactions restrict intramolecular torsion, leading to conformational rigidification of the polymer backbone. This restriction enables more efficient π-electron delocalization and enhances radiative decay, resulting in a pronounced blue shift of approximately 146 nm to 454 nm and a substantial increase in fluorescence intensity (Figure 3b). This pronounced red-to-blue transition reflects a fundamental change in the polymer’s electronic structure driven by aggregation-induced backbone planarization and enhanced π-electron delocalization upon drug binding. Drug release experiments demonstrated that PFPTX was highly stable at physiological pH (7.3), with cumulative paclitaxel release remaining below 5% after 24 h (Figure 3c, black curve). In contrast, under acidic conditions (pH 4.1), paclitaxel release increased in a time-dependent manner, exceeding 40% after 24 h (Figure 3c, red curve), consistent with reported values for nanoparticle-based drug delivery systems in the literature17,18,19.
Although the extracellular tumor microenvironment is only mildly acidic (typically pH 6.5–6.8), intracellular lysosomes exhibit a broader and more acidic pH range, approximately pH 4.0–6.520. Multiple nanodrug researchers aim to destroy the function of lysosomes and directly lead to the death of tumor cells21. Since endocytosed nanoparticles are trafficked into lysosomes following cellular uptake, lysosomal acidity represents the most relevant trigger for intracellular drug release21. Therefore, pH 4.1 was selected to simulate the lower bound of lysosomal conditions and to evaluate PFPTX behavior under the most stringent acidic environment. Fluorescence measurements conducted at pH values of 2.6, 4.1, 5.3, 6.2, and 7.3 showed a gradual decrease in fluorescence intensity with increasing acidity (Figure 3d), confirming that disruption of polymer–drug interactions leads to fluorescence quenching and drug release.
Paclitaxel release was quantified indirectly by monitoring fluorescence intensity changes rather than by conventional UV absorption at 227–230 nm. The standard absorption band of paclitaxel overlaps strongly with the intrinsic absorption of PFOP-NEt₃(+), resulting in significant spectral interference and unreliable quantification (Figure 1c). To avoid this limitation, fluorescence intensity at 492 nm, corresponding to PFPTX emission, was used as an indirect measure of paclitaxel release (methodological justification). This approach was validated using calibration curves constructed with known paclitaxel concentrations under identical pH conditions, which showed a strong linear correlation (R² = 0.996) between fluorescence intensity decrease and paclitaxel concentration.
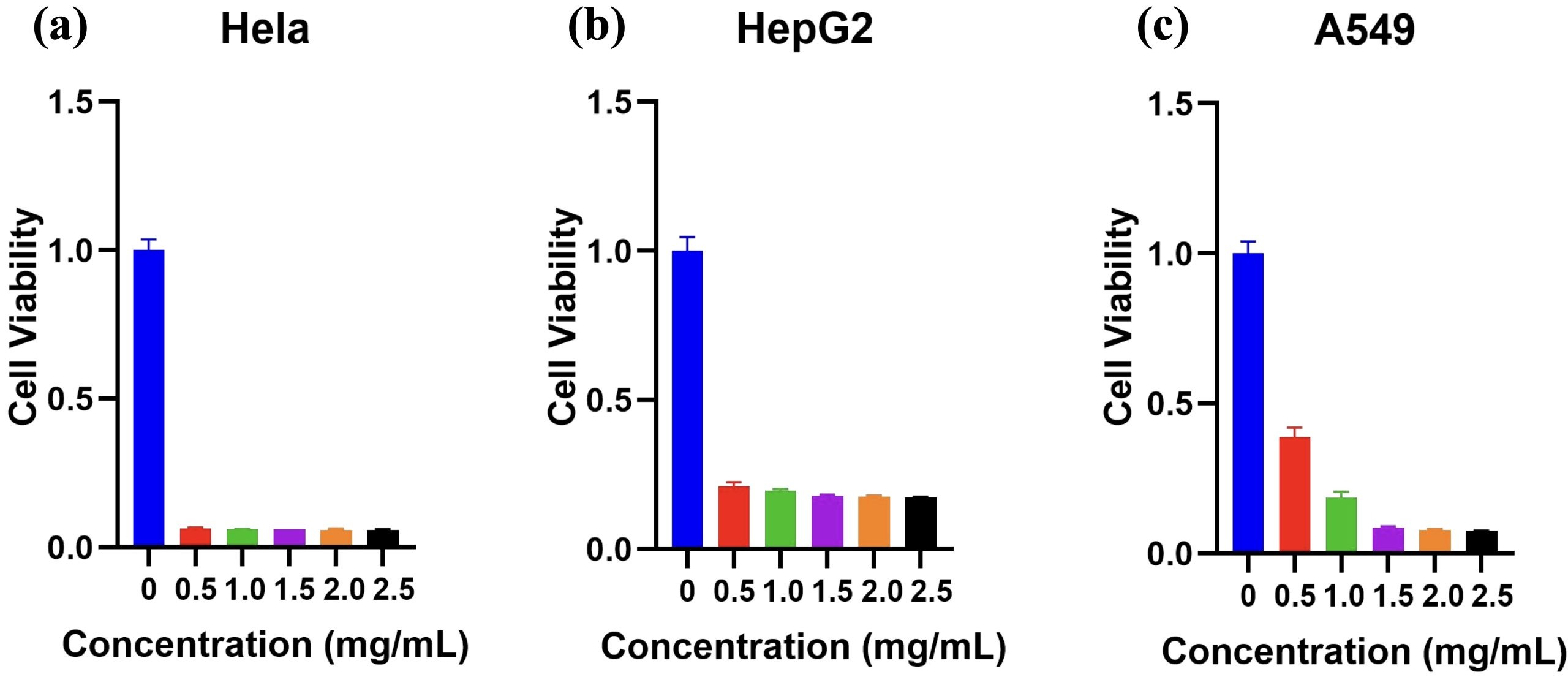
The cytotoxic effects of PFPTX were evaluated in HeLa, HepG2, and A549 cancer cell lines exposed to increasing concentrations of PFPTX (0-2.5 mg/mL) for 48 h at 37 °C in 5% CO2 (n=3). Cell viability assays showed pronounced dose-dependent cytotoxicity across all three cell lines (Figure 4a–c). HeLa cells were particularly sensitive, with 0.5 mg mL⁻¹ PFPTX reducing cell viability to approximately 6%, which shows 94% cytotoxicity (Figure 4a). In HepG2 cells, the same concentration caused 79% cytotoxicity, showing moderate sensitivity, and at 1.5 mg/mL, survival dropped to 18%. At low concentrations, A549 cells showed modest resistance, retaining 37 % and 19 % viability at 0.5 and 1.0 mg mL⁻¹, respectively; however, cytotoxicity surged to 92 % once the concentration reached 1.5 mg mL⁻¹. Statistics show that HepG2 cells showed moderate sensitivity (Figure 4b), while A549 cells exhibited partial resistance at lower concentrations but substantial cytotoxicity at higher doses.
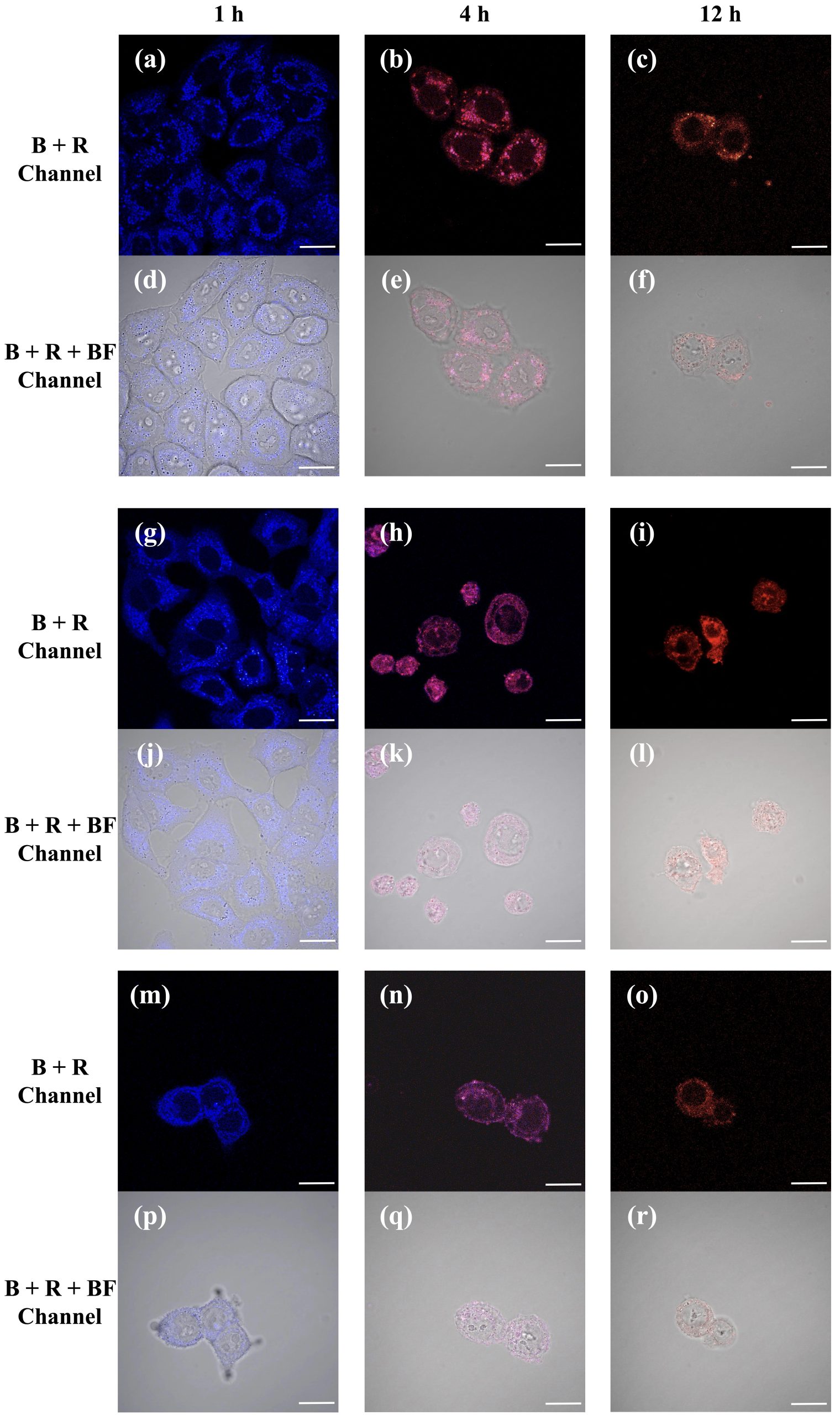
To visualize NP uptake and intracellular localization, fluorescence imaging was performed on HeLa, A549, and HepG2 cells treated with PFPTX (2 mg/mL) for different incubation times (1, 4, and 12 h) (n=3). Dual-channel imaging revealed progressive intracellular accumulation of nanoparticles, with distinct fluorescence signals corresponding to intact PFPTX and released polymer components (Figure 5a–c). Confocal microscopy further showed colocalization of fluorescence signals with lysosomes at all time points (Figure 5), consistent with endocytic uptake and lysosomal trafficking. At early incubation times, fluorescence images were dominated by blue emission, indicating intact nanoparticles (Figure 5a, g, m). At intermediate time points, overlapping blue and red signals produced purple emission, suggesting partial drug release (Figure 5b, e, h). At prolonged incubation times, fluorescence was predominantly red, consistent with extensive paclitaxel release and nanoparticle degradation (Figure 5c, i, o). Together, these results demonstrate that PFPTX functions as both an efficient drug delivery system and a built-in fluorescent reporter capable of tracking intracellular drug release in real time.
Discussion
The Summary of Experimental Results
Conjugated polymers combined with clinically used drugs were systematically investigated in terms of their physicochemical properties, luminescence behavior, and biological performance. Among the systems studied, PFPTX exhibited pronounced fluorescence enhancement, a distinct blue shift, high drug-loading capacity, and pH-responsive release behavior. Molecular docking analysis revealed stable interactions between paclitaxel and the polymer backbone, supporting the observed AILE. In vitro experiments demonstrated efficient cellular uptake, time-dependent fluorescence changes correlated with drug release, and pronounced dose-dependent cytotoxicity across multiple cancer cell lines. Collectively, these results indicate that PFPTX functions both as a high-capacity nanocarrier and as an intrinsic fluorescence reporter for drug delivery at the cellular level.
Significance and Impact of the Results
Previous studies have demonstrated that FO monomers interacting with small double hydrogen-bond donors, such as ethylene glycol or ethylenediamine, undergo planarization and π–π stacking, resulting in AILE behavior12. The present study extends this concept to larger, clinically relevant drug molecules containing multiple hydrogen-bonding groups. In the PFPTX system, strong π–π interactions and hydrogen bonding between paclitaxel and the polymer restrict torsional motion of the fluorenone units, leading to pronounced planarization of the polymer backbone. As a result, a substantial blue shift (~146 nm) and an approximately 20-fold increase in fluorescence intensity (~1.96 × 104 a.u.) were observed (Figure 2).
Based on these observations, an updated luminescence mechanism for AILE in drug-loaded FO-based conjugated polymers can be proposed. In aqueous solution, the free polymer adopts a twisted backbone conformation, resulting in weak red fluorescence. Upon interaction with paclitaxel through hydrogen bonding and π–π contacts, the torsional motion of the fluorenone units is restricted, enforcing a more planar backbone configuration. This conformational transition induces a distinct luminescence shift from red to intense blue emission, which represents the characteristic AILE response in this system (Figure 2).
Because PFOP-NEt3(+) and paclitaxel assemble into nanoparticles through noncovalent interactions, a marked fluorescence transition from weak red to bright blue emission was observed. This behavior prompted an investigation into whether fluorescence variation could serve as an intrinsic indicator of drug release. Release studies under different pH conditions showed that PFPTX remained relatively stable at physiological pH, while more than 40% of paclitaxel was released under mildly acidic conditions, consistent with the tumor microenvironment. Notably, the gradual decrease in fluorescence intensity with decreasing pH further supports a direct correlation between fluorescence signaling and drug release behavior.
The high DLC (45%) and encapsulation efficiency (80–86%) reflect strong supramolecular interactions between paclitaxel and the polymer, exceeding those reported for many conventional nanoparticle systems. Given that paclitaxel is a widely used chemotherapeutic agent that disrupts mitotic spindle formation and leads to abnormal DNA segregation, these properties underscore the potential relevance of PFPTX as a delivery platform22,23,24. However, it should be noted that the present evaluation is limited to in vitro performance and does not account for in vivo pharmacokinetics or systemic behavior. Cytotoxicity assays demonstrated that PFPTX effectively reduced cancer cell viability in a dose-dependent manner. Concurrent fluorescence imaging provided real-time evidence of intracellular uptake and drug release. The progressive transition from blue to purple and eventually to red fluorescence over time indicates a correlation between nanoparticle disassembly, drug release, and subsequent cellular apoptosis.
Together, these findings demonstrate that PFPTX functions as an efficient nanocarrier with pH-responsive release and as a fluorescence-based reporter for drug delivery in vitro. While the integration of delivery and optical reporting suggests potential multifunctionality, the current conclusions are restricted to cellular-level studies. Future work will be required to evaluate in vivo biodistribution, pharmacokinetics, and biocompatibility, as well as to determine whether the fluorescence–release coupling observed in vitro can be maintained under physiological conditions. Such studies will be critical for assessing the translational potential of this AILE-based delivery system.
Remaining Questions and Future Studies
Several directions for further investigation emerge from this study. At the mechanistic level, the precise origin of the pronounced fluorescence blue shift needs additional clarification, particularly with respect to the relative contributions of polymer planarization and intermolecular packing. Beyond mechanistic questions, translation of these findings from cell-based assays to in vivo systems remains an essential next step. Small-animal studies, such as xenograft mouse models, would enable evaluation of nanoparticle biodistribution, tumor accumulation, and preliminary safety. These experiments will be necessary to determine whether the pH stability and acid-triggered release observed in vitro are preserved within the complex physiological environment.
Combination therapy represents another potential extension of this platform, but such applications remain speculative at this stage. Future studies could examine the feasibility of co-loading multiple drugs and assess whether distinct release behaviors or fluorescence responses can be reliably distinguished. Importantly, such investigations should be supported by quantitative in vivo validation rather than conceptual extrapolation. Future studies should first focus on in vivo validation of the current system, including quantitative assessment of biodistribution, tumor accumulation, clearance pathways, and systemic biocompatibility. In addition, animal studies examining drug release behavior and fluorescence signaling in physiological environments will be essential to confirm that the proposed mechanism operates reliably beyond in vitro conditions. This stepwise approach will be critical for translating conceptual multifunctionality into experimentally supported performance.
Limitations and Sources of Error
Despite the promising results, several limitations of this study should be acknowledged. First, all experiments were conducted under in vitro conditions using controlled buffer systems and immortalized cancer cell lines. While these models are appropriate for proof-of-concept evaluation, they do not replicate the complexity of living organisms, where factors such as immune interactions, protein corona formation, vascular transport, and tumor heterogeneity may substantially influence nanoparticle behavior. Consequently, the present findings cannot be directly extrapolated to in vivo or clinical settings.
Second, the dialysis-based method used to assess paclitaxel release may introduce experimental bias. Because this approach relies on passive diffusion across a semipermeable membrane, the measured release kinetics may not accurately reflect nanoparticle disassembly or intracellular drug liberation within endosomal or lysosomal compartments. As a result, the true release rate under physiological conditions may be either underestimated or overestimated.
Third, the observed aggregation-induced luminescence enhancement and favorable release behavior appear to be drug dependent. Paclitaxel exhibited the strongest effects, which may arise from its specific molecular structure rather than representing a general property of the polymer platform. Accordingly, the applicability of this fluorescence-reporting mechanism to other drugs with distinct physicochemical characteristics remains to be systematically evaluated.
Finally, although three cancer cell lines (HeLa, HepG2, and A549) were examined, each represents a relatively homogeneous and immortalized population. In contrast, clinical tumors are genetically diverse and influenced by stromal interactions and heterogeneous microenvironments. These factors may alter nanoparticle uptake, intracellular trafficking, and therapeutic response.
Overall Conclusion
In summary, this study demonstrates an AILE-active nanoparticle system that integrates pH-responsive drug release with fluorescence-based tracking under in vitro conditions. By exploiting π–π stacking and hydrogen-bonding interactions between fluorenone-based polymers and paclitaxel, the system achieves high drug-loading efficiency, environmental stability, and effective cytotoxicity in cancer cell models. Importantly, the fluorescence response provides a direct optical proof of nanoparticle integrity and drug release at the cellular level. While these results establish a mechanistic and functional proof of concept, further in vivo studies are required to evaluate biodistribution, pharmacokinetics, biocompatibility, and translational potential.
Reference
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, F. Bray. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. Vol. 71, pg. 209–249, 2021 https://doi.org/10.3322/caac.21660 [↩]
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang, S. Yang. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduction and Targeted Therapy. Vol. 6, pg. 201, 2021 https://doi.org/10.1038/s41392-021-00572-w [↩]
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu, J. M. Pérez De La Lastra. Cancer chemotherapy and beyond: current status, drug candidates, associated risks and progress in targeted therapeutics. Genes & Diseases. Vol. 10, pg. 1367–1401, 2023 https://doi.org/10.1016/j.gendis.2022.02.007 [↩]
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang, C. Fu. Therapeutic peptides: current applications and future directions. Signal Transduction and Targeted Therapy. Vol. 7, pg. 48, 2022 https://doi.org/10.1038/s41392-022-00904-4 [↩]
- D. Fu, L. Zhong, J. Xu, A. Mo, M. Yang. Hydrazone-functionalized nanoscale covalent organic frameworks as a nanocarrier for ph-responsive drug delivery enhanced anticancer activity. RSC Advances. Vol. 14, pg. 20799–20808, 2024 https://doi.org/10.1039/D4RA01955E [↩]
- D. Fu, L. Zhong, J. Xu, A. Mo, M. Yang. Hydrazone-functionalized nanoscale covalent organic frameworks as a nanocarrier for ph-responsive drug delivery enhanced anticancer activity. RSC Advances. Vol. 14, pg. 20799–20808, 2024 https://doi.org/10.1039/D4RA01955E [↩]
- L. Farhoudi, S. M. Hosseinikhah, A. Kazemi-Beydokhti, L. Arabi, S. H. Alavizadeh, S. A. Moosavian, M. R. Jaafari. PH-sensitive polymeric micelles enhance the co-delivery of doxorubicin and docetaxel: an emerging modality for treating breast cancer. Cancer Nanotechnology. Vol. 15, pg. 37, 2024 https://doi.org/10.1186/s12645-024-00275-1 [↩]
- S. Mirzanejad, M. Bagherzadeh, A. Bayrami, H. Daneshgar, A. Bahrami, M. Mahdavi. Improving the drug delivery performance of zif-8 with amine functionalization as a 5-fluorouracil nanocarrier. Scientific Reports. Vol. 15, pg. 18793, 2025 https://doi.org/10.1038/s41598-025-03542-2 [↩]
- J. Yan, C. Liu, Q. Wu, J. Zhou, X. Xu, L. Zhang, D. Wang, F. Yang, H. Zhang. Mineralization of ph-sensitive doxorubicin prodrug in zif-8 to enable targeted delivery to solid tumors. Analytical Chemistry. Vol. 92, pg. 11453–11461, 2020 https://doi.org/10.1021/acs.analchem.0c02599 [↩] [↩]
- S. Chu, X. Shi, Y. Tian, F. Gao. PH-responsive polymer nanomaterials for tumor therapy. Frontiers in Oncology. Vol. 12, pg. 855019, 2022 https://doi.org/10.3389/fonc.2022.855019 [↩]
- C. Zhang, Q. Yuan, Z. Zhang, Y. Tang. A ph-responsive drug delivery system based on conjugated polymer for effective synergistic chemo-/photodynamic therapy. Molecules. Vol. 28, pg. 399, 2023 https://doi.org/10.3390/molecules28010399 [↩]
- X. Pang, Y. Tan, C. Tan, W. Li, N. Du, Y. Lu, Y. Jiang. One-step construction of fluorenone-based donor–acceptor-type conjugated polymers via direct arylation polymerization for cell-imaging applications. ACS Applied Materials & Interfaces. Vol. 11, pg. 28246–28253, 2019 https://doi.org/10.1021/acsami.9b04630 [↩] [↩]
- Q. Bai, S. Tan, T. Xu, H. Liu, J. Huang, X. Yao. MolAICal: a soft tool for 3d drug design of protein targets by artificial intelligence and classical algorithm. Briefings in Bioinformatics. Vol. 22, pg. bbaa161, 2021 https://doi.org/10.1093/bib/bbaa161 [↩]
- H. Zhou, Y. Zhang, R. Zhang, M. Zhao, W. Chen, Y. Liu, Y. Jiang, Q. Li, Q. Miao, M. Gao. A tumor‐microenvironment‐activatable molecular pro‐theranostic agent for photodynamic and immunotherapy of cancer. Advanced Materials. Vol. 35, pg. 2211485, 2023 https://doi.org/10.1002/adma.202211485 [↩]
- Q. Bai, T. Xu, J. Huang, H. Pérez-Sánchez. Geometric deep learning methods and applications in 3d structure-based drug design. Drug Discovery Today. Vol. 29, pg. 104024, 2024 https://doi.org/10.1016/j.drudis.2024.104024 [↩]
- X. Song, X. Duan, W. Xiang, S. Zhao. Noncovalent interaction thresholds control translocation and cytotoxicity: a combined computational–experimental study. Journal of Medicinal Chemistry. Vol. 68, pg. 15015–15025, 2025 https://doi.org/10.1021/acs.jmedchem.5c01196 [↩]
- J. Sun, J. Li, X. Li, L. Yang, Y. Liu, H. Gao, L. Xiang. Sequentially responsive size reduction and drug release of core-satellite nanoparticles to enhance tumor penetration and effective tumor suppression. Chinese Chemical Letters. Vol. 34, pg. 107891, 2023 https://doi.org/10.1016/j.cclet.2022.107891 [↩]
- S. Shakiba, S. Shariati, H. Wu, C. E. Astete, R. Cueto, E. H. Fini, D. F. Rodrigues, C. M. Sabliov, S. M. Louie. Distinguishing nanoparticle drug release mechanisms by asymmetric flow field–flow fractionation. Journal of Controlled Release. Vol. 352, pg. 485–496, 2022 https://doi.org/10.1016/j.jconrel.2022.10.034 [↩]
- Y. Zhao, Y. Zhou, D. Yang, X. Gao, T. Wen, J. Fu, X. Wen, G. Quan, X. Pan, C. Wu. Intelligent and spatiotemporal drug release based on multifunctional nanoparticle-integrated dissolving microneedle system for synergetic chemo-photothermal therapy to eradicate melanoma. Acta Biomaterialia. Vol. 135, pg. 164–178, 2021 https://doi.org/10.1016/j.actbio.2021.09.009 [↩]
- B. A. Webb, F. M. Aloisio, R. A. Charafeddine, J. Cook, T. Wittmann, D. L. Barber. PHLARE: a new biosensor reveals decreased lysosome ph in cancer cells. Molecular Biology of the Cell. Vol. 32, pg. 131–142, 2021 https://doi.org/10.1091/mbc.E20-06-0383 [↩]
- X. Tian, A. Shi, H. Yin, Y. Wang, Q. Liu, W. Chen, J. Wu. Nanomaterials respond to lysosomal function for tumor treatment. Cells. Vol. 11, pg. 3348, 2022 https://doi.org/10.3390/cells11213348 [↩] [↩]
- J. Škubník, V. Svobodová Pavlíčková, T. Ruml, S. Rimpelová. Autophagy in cancer resistance to paclitaxel: development of combination strategies. Biomedicine & Pharmacotherapy. Vol. 161, pg. 114458, 2023 https://doi.org/10.1016/j.biopha.2023.114458 [↩]
- D. Hao, Q. Meng, B. Jiang, S. Lu, X. Xiang, Q. Pei, H. Yu, X. Jing, Z. Xie. Hypoxia-activated pegylated paclitaxel prodrug nanoparticles for potentiated chemotherapy. ACS Nano. Vol. 16, pg. 14693–14702, 2022 https://doi.org/10.1021/acsnano.2c05341 [↩]
- T. Das, U. Anand, S. K. Pandey, C. R. Ashby, Y. G. Assaraf, Z.-S. Chen, A. Dey. Therapeutic strategies to overcome taxane resistance in cancer. Drug Resistance Updates. Vol. 55, pg. 100754, 2021 https://doi.org/10.1016/j.drup.2021.100754 [↩]



{kind=link}