
Abstract
Poly(ADP-ribose) polymerase (PARP) inhibitors are targeted therapies that take advantage of homologous recombination repair deficiency and have been highly effective in BRCA-mutant breast and ovarian cancer. Melanoma, historically unresponsive to DNA-damaging agents, has now been found to harbor molecular subsets—e.g., BRCA1/2, PALB2, and ATM mutations—that could potentially make it sensitive to PARP inhibition. The purpose of this review is to evaluate the science justification, clinical advances, and prospect for the application of PARP inhibitors in melanoma. A narrative review was conducted of preclinical and clinical studies assessing the use of PARP inhibitors in melanoma, focusing on homologous recombination repair deficiency, mechanisms of resistance, combination regimens, and translational issues. Included studies were accessed from databases like PubMed, ScienceDirect, and Google Scholar, giving preference to peer-reviewed articles in English published between 2005 and 2024. Homologous recombination-defective melanoma models are hypersensitive to PARP inhibitors, but clinical application has been modest. Early-phase clinical trials currently exhibit modest and frequently non-sustained responses in biomarker-selected individuals. Resistance mechanisms, including loss of 53BP1, replication fork protection, and drug efflux, preclude sustained benefit. Approaches that include ATR or CHK1 inhibitors and immunotherapy combinations are under investigation to abrogate resistance and enhance outcomes. PARP inhibitors demonstrate potential value in biomarker-defined subsets of melanoma, but broad clinical use is constrained by resistance, toxicity, and limited data on long-term survival. Progress will require randomized trials, pharmacogenomic analysis, and integration of quality-of-life and cost-effectiveness end points to inform individualized, long-lasting treatment strategies.
Introduction
Melanoma, although it represents only a minority of skin cancers, is responsible for most skin cancer–associated mortality. Its aggressive clinical behavior and early metastatic potential render it one of the deadliest cancers worldwide. Over recent decades, the increasing incidence of melanoma has been linked to factors such as expanded ultraviolet (UV) radiation exposure and genetic susceptibility. Recent breakthroughs with immune checkpoint inhibitors and targeted therapies—especially BRAF and MEK inhibitors—have markedly improved survival for patients with advanced melanoma. However, many patients eventually develop resistance to these therapies, leading to treatment failure and disease recurrence.
One promising approach to overcome these challenges is to exploit the inherent weaknesses in the DNA damage response (DDR) of melanoma cells, particularly in genetically defined subgroups. A critical element of the DDR is the
homologous recombination repair (HRR) pathway, which is essential for the accurate repair of DNA double-strand breaks (DSBs). Tumors deficient in HRR, a state often referred to as “BRCAness,” lack the capacity to efficiently repair DNA damage, making them particularly vulnerable to further perturbations in DNA repair. Poly(ADP‐ribose) polymerase inhibitors (PARPis) target this vulnerability by inhibiting the repair of single-strand breaks (SSBs). When these SSBs are converted into DSBs during replication, HRR-deficient cells are unable to cope, leading to cell death through synthetic lethality. This review is limited to
English-language peer-reviewed publications and focuses primarily on mechanistic, translational, and early-phase clinical data. Large-scale randomized trials are currently lacking, and the review does not include ongoing or unpublished studies.
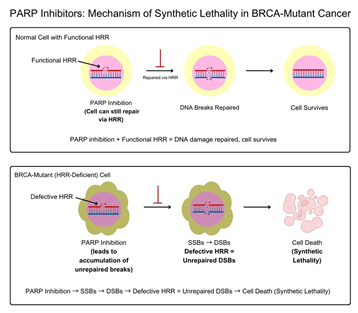
This flowchart illustrates the mechanism by which PARP inhibitors selectively kill melanoma cells harboring HRR deficiencies (e.g., due to ATM, PALB2, or BRCA1/2 mutations) while sparing normal cells with intact repair machinery. In normal cells, functional HRR repairs double-strand breaks resulting from PARP inhibition, ensuring cell survival. In contrast, HRR-deficient melanoma cells accumulate unrepaired DNA damage and undergo cell death.
This review gives a thorough overview of the use of PARP inhibitors for melanoma treatment. In particular, the key question is discussed: Is there enough clinical evidence to support the use of PARP inhibitors in melanoma beyond clinical trials? Although PARP inhibitors are used for other cancers like ovarian and breast cancers, their use in melanoma is still investigational. Available literature is largely composed of early-phase small clinical trials and preclinical studies. Thus, it becomes important to critically synthesize the evidence to determine whether the therapeutic potential exhibited in HRR-deficient melanomas holds true in clinically significant outcomes. Through a
review of molecular rationale, mechanisms of resistance, and translational challenges, this review seeks to establish the readiness of PARP inhibitors for widespread clinical application in melanoma as well as to inform future directions.
Review Methodology
This narrative literature review was done by searching PubMed, ScienceDirect, and Google Scholar for peer-reviewed publications from 2005 to 2024. The search used keywords like “PARP inhibitors,” “melanoma,” “synthetic lethality,” “homologous recombination deficiency,” “resistance,” and “combination therapy.” The studies were included if they were concerned with the application of PARP inhibitors to melanoma or melanoma models of DNA repair defects, offered preclinical, translational, or
early-phase clinical trial information, and were published in English-language peer-reviewed journals. Exclusion criteria removed studies that only addressed non-melanoma cancers irrelevant to melanoma mechanisms, non-peer-reviewed
literature, editorials, and duplicates. Important findings were manually abstracted from full-text publications, including data on mechanisms of action, clinical results, biomarkers, resistance mechanisms, and trial designs. Synthesized information was then put into formatted tables and figures. Thematic analysis was employed to
categorize findings into five main areas: biological rationale, evidence from clinical trials, resistance mechanisms, biomarker approaches, and directions for future treatment. Quality appraisal favored high-quality studies such as randomized controlled trials, mechanistic studies with well-characterized models, and peer-reviewed review articles, whereas studies with inadequate sample sizes, no control groups, or insufficient biomarker data were critically evaluated for possible bias.
DNA Repair Deficiencies in Melanoma
Melanoma is characterized by a high degree of genomic instability, primarily resulting from defects in several DNA repair mechanisms. Among these, deficiencies in the homologous recombination repair (HRR) pathway are of great significance due to their therapeutic implications. This section details the principal defects observed in melanoma and discusses additional repair alterations that contribute to the malignancy’s aggressive behavior.
Homologous Recombination Repair (HRR) Defects
High-throughput genomic studies—like those of the TCGA SKCM cohort (n =471)—have reported frequent HRR pathway alterations in melanoma, providing robust justification for the use of targeted therapies with PARP inhibitors.
One of the most common defects is an ATM gene mutation, which occurs in about 5% of melanomas. Among these are truncating mutations (~3%) and deep deletions (~2%) that abrogate the gene function as a double-strand break (DSB) sensor and cell cycle checkpoint regulator. The subsequent inability to trigger proper DNA repair cascades heightens genomic instability and makes the tumors vulnerable to synthetic lethality with PARP inhibition1.
Another key HRR factor, PALB2, is mutated in approximately 4% of melanomas. Being a bridge protein between BRCA1 and BRCA2, PALB2 plays a critical role in homologous pairing and strand invasion in HRR. Most mutations within melanoma are frameshift mutants resulting in loss-of-function, thereby incapacitating the repair process and increasing the susceptibility of tumor cells to DNA-damaging drugs such as PARP is2.
While less frequent, somatic mutations in BRCA1 and BRCA2 have been identified in melanomas at 1–2% and 2–3% frequencies, respectively. They encode proteins crucial to high-fidelity repair of DSBs by homologous recombination. Loss of their function imparts a “BRCAness” phenotype and increased PARPi sensitivity.3,4.
Together, ATM, PALB2, and BRCA1/2 mutations identify a genomically unstable subgroup of melanomas for which PARP inhibitors represent an attractive, biologically plausible therapeutic choice.
Other DNA Repair Alterations
Although HRR defects are a key to PARPi sensitivity, other DNA repair pathway deficiencies also contribute to the genomic instability of melanoma.
An important pathway is nucleotide excision repair (NER), which repairs ultraviolet-induced DNA damage including cyclobutane pyrimidine dimers and 6-4 photoproducts. NER pathway-defining genes ERCC2 and ERCC4 are mutated in a fraction of melanoma tumors, resulting in impaired repair of ultraviolet damage and oncogenesis by inducing a higher mutational burden5.
Another significant contributor is the Fanconi Anemia (FA) pathway, which works together with HRR to facilitate interstrand crosslink repair. FA pathway gene mutations, including FANCA, occur in about 5% of melanomas and increase genomic instability. FA pathway impairment further sensitizes melanoma cells to DNA-damaging agents, making PARPis a valid target therapeutic strategy.
The combined disruption of several DNA repair pathways establishes a state of repair susceptibility, which can be therapeutically manipulated.
PARP Inhibitors: Mechanisms and Preclinical Evidence
PARP inhibitors have garnered significant interest due to their ability to selectively target tumor cells with DNA repair deficiencies 6 ,7,8 This section discusses their mechanism of action and presents the extensive preclinical evidence supporting their potential use in melanoma treatment.
Mechanism of Action
PARP enzymes—primarily PARP-1 and PARP-2—are essential for the repair of single-strand breaks (SSBs) via the base excision repair (BER) pathway.6,9,10 Inhibition of these enzymes by PARPis prevents SSB repair, leading to the accumulation of DNA damage.11,12 During DNA replication, unrepaired SSBs are converted into double-strand breaks (DSBs).6,12 In cells with a functional homologous recombination repair (HRR) pathway, DSBs are accurately repaired.13 However, in HRR-deficient cells (e.g., those with mutations in ATM, PALB2, or BRCA1/2), the accumulation of DSBs results in catastrophic genomic instability and cell death.14 This process—termed synthetic lethality—allows PARPis to selectively kill tumor cells while sparing normal cells with intact DNA repair mechanisms.15,8.
Preclinical Validation
A substantial and well-characterized body of robust preclinical studies supports the therapeutic potential of PARP inhibitors in melanoma. These experiments recurrently establish antitumor activity in numerous experimental systems and elucidate the molecular and pharmacologic underpinnings of combining PARPis with targeted therapy approaches.
Genome-wide CRISPR-Cas9 screening has been an important tool for assessing PARPi sensitivity compared with homologous recombination repair (HRR) status. In ATM-deficient melanoma cell lines, such as SK-MEL-28, high PARP inhibitor sensitivity has been shown by significantly negative CERES scores, reflecting essential dependence on PARP for viability. In contrast, the ATM wild-type A375 melanoma line is relatively resistant to these compounds. These findings validate PARP inhibitor sensitivity as being highly contingent on tumor cell HRR integrity and enable biomarker-based patient stratification.16
Validation in vivo has been performed in a PDX model for HRR-deficient melanoma. In particular, xenografts from PALB2-mutant melanoma tumors achieved a 60% decrease in tumor size following olaparib treatment at 50 mg/kg, vs. untreated controls (p = 0.003). Such strong antitumor activity indicates that melanoma tumors with impaired HRR pathways can be particularly vulnerable to PARP inhibition, but clinical application is still in the initial phases.17
Outside of monotherapy, preclinical models have assessed synergistic combinations of PARPis with other targeted or immunomodulatory drugs. A highly encouraging strategy is the combination of PARP inhibitors and ATR inhibitors. In ATM-mutant melanomas, ATR inhibition sensitizes cells to the replication stress caused by PARPis, leading to enhanced cytotoxicity and cell death.8’12 These dual-targeting strategies take advantage of complementary weaknesses in the DNA damage response (DDR) network, offering a mechanistic rationale for combination therapy.
Furthermore, PARP inhibitors have also been found to stimulate the stimulator of interferon genes (STING) pathway, a part of the innate immune system. This stimulation induces type I interferon signaling, increases tumor immunogenicity, and implies that PARPis can sensitize melanoma to immune checkpoint inhibitors. In fact, in preclinical models, the synergy between PARP inhibition and anti-PD-1 therapies exhibited synergistic activity, supporting the idea of immuno-oncology–DDR crosstalk as a therapeutic target. Further preclinical evidence has established that PARP inhibition enhances antitumor immunity in melanoma through increased infiltration of cytotoxic T cells and induction of pro-inflammatory cytokines.18’19
Despite these strong preclinical data, translation into clinical efficacy has been hampered by several key issues. One significant weakness is that in vitro cell culture models fail to replicate the tumor microenvironment’s complexity. Important determinants like immune cell invasion, stromal interactions, and metabolic gradients are mostly missing, but all of these significantly impact drug responsiveness. Furthermore, patient-derived xenograft models, although more physiologically relevant than conventional cell lines, are usually set up in immunodeficient mice. This precludes evaluation of immune-mediated antitumor activity and toxicity, which are critical elements of melanoma pathobiology and response to treatment.
Pharmacokinetic differences between human and murine systems also complicate translation. Pharmacokinetic parameters like half-life of drug, distribution volume, hepatic metabolism, and clearance all differ substantially by species. For instance, the pharmacokinetics of the compound olaparib in mice—where higher doses tend to be tolerated—can amplify efficacy signals that are not attainable or safe in humans. Species-specific discrepancies create the risk of overestimating therapeutic impact in preclinical assessment.
In addition, gene homogeneity in cell lines and xenograft models is in stark contrast with the genomic heterogeneity of human melanoma. Actual tumors tend to exhibit a complex mosaic of clones with differing DNA repair potential and microenvironmental adaptations. Therefore, reproducibility and generalizability of preclinical data are restricted, especially with the lack of representative immune or metabolic conditions. These limitations emphasize the necessity of restrained interpretation of preclinical evidence and the significance of efficacy verification in carefully planned, biomarker-stratified clinical trials.
The disconnect between preclinical promise and clinical results is not limited to melanoma. A number of examples from earlier studies highlight the general difficulties of translating PARPi discoveries in laboratory models into therapeutic gain in patients. In pancreatic cancer, BRCA-mutant xenografts exhibited significant tumor inhibition with olaparib, yet the Phase III POLO trial did not show benefit in overall survival and provided only a modest progression-free survival (PFS) benefit.20
Talazoparib also had enhanced PARP-trapping activity in vitro but offered minimal benefit in early-phase trials for HRR-deficient tumors beyond breast cancer. In glioblastoma, several preclinical studies showed radio sensitization when PARPis were used in combination with temozolomide, but clinical trials with agents like veliparib did not harness these advantages into significant clinical benefit.21 These experiences demonstrate the pitfalls of over-extrapolating preclinical models and emphasize the urgent need for solid clinical proof before the general applicability of PARPis in melanoma.
Clinical Evidence
Translation of these promising preclinical findings into clinical practice is ongoing. Early-phase clinical trials are yielding valuable insights into the safety and efficacy of PARP inhibitors in melanoma patients, both as monotherapies and in combination with other agents. Below is a summary table highlighting key details from the major trials:
| Trial ID | Combination | Patient Selection Criteria | Sample Size | Key Outcomes | Reference |
| NCT03992131 | Niraparib + Pembrolizumab | Advanced melanoma | 32 | ORR: 34%,Median PFS: 7.1 months | Zhang et al., 2023 |
| NCT04826341 | Olaparib + Dostarlimab | ATM-deficient melanoma | 24 | Median PFS: 6.8 months | Lee et al.,2023 |
These trials indicate promise when combining PARPIs with immune checkpoint blockers in biomarker-selected individuals, but non-reproducibility in large groups shows that such combinations are not necessarily across-the-board effective. The improved response rates and progression-free survival outcomes compared to historical immunotherapy data suggest that integrating PARP inhibitors could significantly enhance therapeutic efficacy.22’23,24.
Early-Phase Clinical Trials
Early clinical trials exploring the potential of PARP inhibitors to treat melanoma have produced encouraging but modest results, mainly in biomarker-enriched populations. In a high-profile early-phase trial, NCT03992131, the combination of niraparib, a PARP inhibitor, and pembrolizumab, an anti–PD-1 checkpoint inhibitor, was assessed in 32 patients with advanced melanoma. The regimen had an objective response rate (ORR) of 34% and a median progression-free survival (PFS) of 7.1 months. These results look encouraging compared to the archival PFS of 4.1 months observed with pembrolizumab monotherapy in the KEYNOTE-006 trial.25 Cross-trial comparisons like these, however, must be viewed cautiously owing to variations in patient choice, biomarker enrichment, inclusion criteria, and trial design.
Another trial, NCT04826341, explored another immuno-oncology combination—olaparib with dostarlimab—in patients with ATM-deficient melanoma. This analysis, in which 24 patients were included, had a median PFS of 6.8 months, once more pointing to potential therapeutic synergy and emphasizing the value of genomic biomarkers for patient selection.23 Although these findings further support the value of stratifying patients according to HRR gene status, the lack of control groups and low numbers of patients in each study reduce their applicability and statistical power. Subgroup analyses were not possible, and the absence of randomization prevents definitive conclusions about causality or comparative effectiveness.
Although these early trials are encouraging, the findings must be balanced against the toxicity profile of PARP inhibition, especially when administered in combination regimens. Frequently occurring adverse effects are fatigue, nausea, anemia, and thrombocytopenia, many of which are dose-dependent and can potentially accumulate over time, necessitating interruptions or reductions in treatment. More serious are infrequent but serious occurrences like myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), particularly with long-term use. In one trial, NCT03992131, for example, over 20% of patients developed grade ≥3 hematologic toxicities, requiring dose adjustments.22 These hematologic toxicities are found to be aggravated in combination with immune checkpoint inhibitors, potentially increasing the risk of synergistic or cumulative adverse effects, although immune-related toxicity information continues to be limited.23
Besides safety issues, a number of methodological limitations define the present clinical evidence. Many early-phase trials consist of less than 50 patients, seriously limiting the possibility of performing strong subgroup analyses or the identification of predictive biomarkers in addition to HRR mutations. Enrollment requirements tend to be skewed toward biomarker-positive patients, e.g., those with ATM or BRCA mutation, and thereby introduce selection bias as well as restrict applicability to the target melanoma population. In addition, control groups are missing in most instances, with historic controls used for comparison. Although sometimes unavoidable in investigative trials, this practice introduces confounding factors and forbids relative efficacy evaluation.
Adding to these complexities is heterogeneity of study design between trials. Differing PARP inhibitor dosing regimens, treatment durations, combination agents employed, and primary endpoints create inescapable unreliability of cross-trial comparisons. Add to this the poor sample sizes and nonuniform outcomes, underscoring the imperative for large biomarker-stratified randomized controlled trials to establish the clinical value of PARP inhibitors for melanoma with methodological precision.20,22
Although excitement regarding an era of PARP inhibitors is understandable due to its preclinical synergy and initial clinical activity, it is important to note that these compounds do not confer benefit uniformly across all melanoma subsets or patient populations. A number of early trials in unselected melanoma populations—those without confirmed HRR pathway defects—have been unable to show robust efficacy, implying that HRR mutations are a requirement for a therapeutic effect. This has been supported by retrospective analyses that indicate no significant benefit in overall survival when PARP inhibitors were given without biomarker selection. Moreover, certain melanoma tumors have been found to be intrinsically resistant to PARP inhibition by having compensatory DNA repair mechanisms, protection of replication forks, or immune escape mechanisms. These results emphasize the importance of accurate patient stratification in ongoing trials and any possible future clinical adoption.
Outside of oncologic responses, how PARP inhibitors affect quality of life (QoL) and patient-reported outcomes (PROs) is not well studied in melanoma. Although fatigue, gastrointestinal symptoms, anemia, and emotional distress are common side effects in other cancers treated with PARPis, this information is not available in melanoma. Few studies have used validated PRO measures like the EORTC QLQ-C30 or FACT-M, and it is challenging to determine treatment burden from the patient’s viewpoint. This lack is especially of concern in combination regimens, where additive or synergistic toxicities can severely compromise functional status, particularly within palliative environments. Integration of PRO measures in future studies is thus critical to maximize the understanding of the trade-off between efficacy and tolerability.
The extended safety profile of PARP inhibitors in melanoma is consistent with that observed in other cancer histologies but must be viewed through the distinct clinical lens of this disease. Baseline patient factors—performance status, history of previous exposure to therapy, and immune status—can influence risk of toxicity as well as treatment response. Hematologic events are the most frequent dose-limiting toxicities, and their incidence is magnified within combination regimens. As reported in the NCT03992131 trial, grade ≥3 neutropenia and thrombocytopenia were seen in a high percentage of patients. Chronic exposure also increases the risk of permanent bone marrow suppression, such as MDS and AML, although these are uncommon. Immune-related adverse events can also be exacerbated when PARPis are used with checkpoint inhibitors, although this field needs further exploration.
In conclusion, although preliminary-phase clinical trials indicate that PARP inhibitors—especially when paired with immune checkpoint therapies—have the potential to provide clinical benefit in biomarker-selected patients with melanoma, these results are tentative. The experimental status of PARPis in melanoma, combined with non-consistent efficacy, intricate toxicity profiles, and inadequately reported quality-of-life information, highlights the need for careful, large-scale, biomarker-driven trials with proper controls. Until such data are in hand, the application of PARPis to melanoma should be strictly restricted to phase II and III clinical trials, and their inclusion into regular care should be undertaken cautiously.
Resistance Mechanisms
In addition to promising preclinical and early clinical outcomes, resistance to PARP inhibitors is the major obstacle to long-lasting therapeutic activity in melanoma. One of the best-characterized mechanisms is the upregulation of ATP-binding cassette (ABC) transporters, and most notably ABCB1, which codes for the P-glycoprotein efflux pump. ABCB1 overexpression decreases intracellular PARP inhibitor concentration, thus lessening its cytotoxic activity. This pharmacokinetic mechanism of resistance has been seen in various tumor types and highlights the necessity for adjunctive modalities intended to prevent drug efflux to reassess drug sensitivity.26,27,28
A second, biologically independent process is mediated by loss of 53BP1, a DNA damage response protein that controls end resection and the balance between non-homologous end joining and homologous recombination. In cells lacking HRR—especially in those that are nonfunctional BRCA1— the loss of 53BP1 partially restores homologous recombination, allowing tumor cells to repair otherwise lethal double-strand breaks, which otherwise would be lethal with PARP inhibition. Such restoration of the capacity for DNA repair essentially enables the cell to bypass the synthetic lethality imposed by PARP inhibitors, leading to drug resistance.29,30
Notably, not all resistance mechanisms are irreversible. Preclinical models have shown that some adaptive resistance pathways—like those governed by 53BP1 loss—can be reversed pharmacologically. Of interest, co-treatment with ATR inhibitors or CHK1 inhibitors has been found to re-sensitive tumors to PARP inhibitors by re-establishing replication stress and disabling residual repair function.12,29’8.These combination strategies present promising directions to extend response and postpone resistance occurrence.
Development of resistance to melanoma is also heterogeneous and potentially molecular subtype-dependent. For example, NRAS-mutant melanomas tend to become resistant through stabilization of replication forks, a mechanism that avoids fork collapse and DSB, decreasing PARP inhibitor-mediated lethality.31,32 BRAF-mutant melanomas, on the other hand, tend to gain resistance through augmented efflux transporter expression like ABCB1, consistent with the previously mentioned pharmacokinetic pattern of resistance.33 This subtype-specific diversity parallels the larger genomic and phenotypic diversity of melanoma and supports the imperative for precision medicine strategies.34
Since resistance to PARP inhibitors is not homogeneous, managing it will need to involve adaptive treatment strategies. Molecular profiling during the time of resistance, either by tumor biopsy or liquid biopsy (e.g., ctDNA), can inform the choice of second lines of therapy such as DDR inhibitors, immune checkpoint inhibitors, or replication fork stability-targeting agents.25 Ultimately, overcoming or bypassing resistance will be contingent upon a precise comprehension of each tumor’s shifting repair environment, rendering serial molecular surveillance and adaptive clinical trial designs crucial instruments in the application of PARP inhibitor-based treatment regimens in melanoma.30
| Therapy | Median PFS | ORR | Toxicity Profile | Biomarker Requirement | FDA Approved |
| PARP Inhibitors (PARPis) | 6.8–7.1 months (early trials) | 30–35% (small trials) | Anemia, fatigue, thrombocytopenia , rare MDS | HRR mutations (e.g., ATM, BRCA) | Investigational |
| PD-1 inhibitors (e.g., pembrolizumab) | 4.1–6.0 months | 33–40% | Fatigue, rash, colitis, thyroid dysfunction | None required | Yes |
| BRAF + MEK inhibitors | 11–14 months | 50–60% | Fever, rash, cardiotoxicity | BRAF V600E mutation | Yes |
| Chemotherapy (dacarbazine) | ~2 months | 10–15% | Myelosuppression, nausea | None | Yes (limited use) |
Critical Appraisal of Current Evidence
Although promising preclinical and early clinical findings, the existing evidence favoring PARP inhibitor use in melanoma is weak in strength and generalizability. The majority of clinical trials conducted so far are preliminary-phase studies with limited sample sizes (usually less than 50 patients), heavily compromising statistical power and the risk of type I and II errors.22,23 In addition, most of these studies are non-randomized and have weak control arms, and instead use historical controls against which they are compared.20 This creates a bias towards overestimating efficacy from selection bias, especially since most participants were stratified on the basis of positive biomarker profiles like ATM or BRCA mutations.8’35
A further limitation exists in trial design variability. Variability in dosing regimens, combinations of drugs, and definitions of endpoints complicate cross-trial comparisons and impede meta-analytical synthesis.36 Moreover, most clinical responses published are partial or non-durable, with progression-free survival mostly less than 8 months.22,23. Toxicity reports–such as anemia, fatigue, thrombocytopenia, and infrequent myelodysplastic syndromes–are clinically relevant, especially when combined PARPis are added to other agents like immune checkpoint inhibitors.37
Concern about the translatability of preclinical results is also an issue. Although synthetic lethality has been strongly established in cell lines and xenografts,30,12 these models incompletely reflect the heterogeneity and immunologic context of human melanomas.34 In addition, null or negative results in unselected cohorts of patients are less commonly reported in the literature and frequently not published, which may lead to publication bias.36
Thus, although the evidence in favor of PARP inhibition in melanoma is promising, it is preliminary. To determine clinical usefulness, subsequent research must overcome these methodology limitations through large numbers, biomarker-stratified, randomized controlled trials that assess efficacy and toxicity in a wide range of melanoma subtypes.20’25
The identification of these resistance pathways is crucial for developing next-generation therapies and combination strategies that sustain the clinical benefits of PARP inhibitors in melanoma.
Quality of Life and Patient-Reported Outcomes –
Although a clinically relevant endpoint, patient experience is still underreported in melanoma PARPi trials. Compared to breast or ovarian cancer research, in which tools such as the EORTC QLQ-C30 or FACT-O are commonly employed, melanoma PARPi trials have not universally included patient-reported outcomes (PROs).37 This curtails knowledge of actual burdens of fatigue, nausea, anemia, and mood change—frequently seen in PARPI-treated patients.37 In palliative settings or planning for long-term oral treatment, incorporation of QoL measurements is critical to guide shared decision-making.
| Consideration | Impact of PARP Inhibitors | Evidence in Melanoma |
| Fatigue | Common across PARPis | Reported, but not systematically measured |
| Gastrointestinal symptoms | Nausea, vomiting, diarrhea | Anecdotal; lacking structured assessment |
| Hematologic toxicity | Anemia, thrombocytopenia | Frequent in trials; QoL impact not reported |
| Mental/emotional health | Mood disturbances, treatment burden | Not assessed |
| PRO instruments used | EORTC QLQ-C30, FACT-O in other cancers | Rarely applied in melanoma PARPi trials |
PARP inhibitors, though mechanistically targeted, are highly costly. Olaparib and niraparib have average monthly costs ranging from $10,000 to $13,000 USD.38. In melanoma, in which survival advantage has yet to be unequivocally demonstrated this raises cost-effectiveness issues. No official health economic analysis has yet assessed the incremental cost-effectiveness ratio (ICER) of PARPis for melanoma.39 Without long-term outcome data, particularly in unselected patients, reimbursement and wider access are currently restricted. Cost-effectiveness modeling must be given high priority in future research along with efficacy and safety outcomes.
| Therapy / Trial | ORR | Median PFS | Media n OS | Patient Selection | Study Limitations |
| Niraparib + Pembrolizumab (NCT03992131) | 34% | 7.1 months | Not reported | Advanced melanoma, HRR-unselected | Phase II, single-arm, small sample (n=32) |
| Olaparib + Dostarlimab (NCT04826341) | 29% (estimated) | 6.8 months | Not reported | ATM-deficient melanoma | Phase II, no control arm, n=24 |
| Pembrolizumab (KEYNOTE-006) | 33% | 4.1–6.0 months | 23.8 months | Advanced Melanoma, PD-1 naïve | Randomized Controlled, unselected patients |
| BRAF + MEK inhibitors (COMBI-d/v) | ~60% | 11–14 months | ~25 months | BRAF V600E/K mutant melanoma | Biomarker-restricted, rapid resistance |
| Ipilimumab (MDX010-20 trial) | ~10–15% | 2.8 months | 10.1 months | Advanced melanoma, unselected | Low ORR, long OS tail in some responders |
Limitations of Cross-Trial Comparisons:
Historically benchmarked reported benefits in progression-free survival (PFS) or objective response rates (ORRs) with PARP inhibitor-based therapies are typically made in comparison to historical controls from immunotherapy trials. These comparisons are not methodologically robust, though.25 Unselected melanoma patients were enrolled in trials like KEYNOTE-006, whereas PARPi trials have typically included biomarker-enriched groups (e.g., ATM-deficient tumors). Prior treatment exposure, performance status, and study endpoints may also vary considerably. Therefore, direct numerical comparisons between unrelated trials must be avoided25 and should not replace head-to-head or randomized controlled trial evidence.
Regulatory Status and Clinical Guidelines
To date, as of 2025, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have approved several PARP inhibitors—olaparib, rucaparib, niraparib, and talazoparib—specifically for cancers including ovarian, breast, prostate, and pancreatic cancers with proven homologous recombination deficiencies. Nonetheless, none of these PARP inhibitors has gained FDA or EMA approval for melanoma.37 As a result, current application of PARPis for melanoma is therefore investigative and limited to early phase clinical trials or compassionate off-label use in specific biomarker-defined patients.22,23
Principal clinical guidelines like the National Comprehensive Cancer Network (NCCN) and European Society for Medical Oncology (ESMO) presently do not endorse PARPis within routine melanoma management.35’36 Their use will be based on outcomes from larger, randomized, biomarker-stratified trials.25’20 In the meantime, PARPis in melanoma are beyond normal clinical practice.35’36
Access and Equity Considerations
Universal Access to PARP inhibitors in melanoma is a costliest challenge.38 They are expensive and need sophisticated diagnostic facilities (e.g.NGS-based HRR mutation analysis)40’36 and there is limited access in low- and middle-income countries (LMICs).41’42 Even in high-income nations, there are variations in access to genomic testing among rural and underrepresented groups43 Insurance coverage of off-label PARP therapy in melanoma is also patchy, and out-of-pocket expenses can be unaffordable.44 For all these developments in precision oncology to be made universally available, future policy guidelines must consider affordability, test availability, and integration of biomarker-driven treatment into the health system.25’45
The clinical evidence for PARP inhibitors in melanoma is very narrow in terms of scope and magnitude.22,23 Current research consists of small patient cohort Phase I/II trials (usually less than 50 patients),46 decreasing statistical power and subgroup analysis reliability. Few of these trials are randomised and contain controls, and they use historical comparison as a control,47,25 introducing potential bias and restricting causal inference. Additionally, enrollment is based on known HRR gene mutations and thus selects patients in favor of such mutations,35’40 causing selection bias and precluding generalizability. Follow-up is brief, and results like overall survival, quality of life, and long-term toxicity are inadequately reported.48,37 Significantly, null or negative findings in unselected populations are not well represented in the literature and contribute to publication bias.49 These constraints highlight the provisional status of existing clinical evidence and emphasize the requirement for large-scale, biomarker-stratified, randomized trials to confirm the place of PARPs in melanoma.50’20
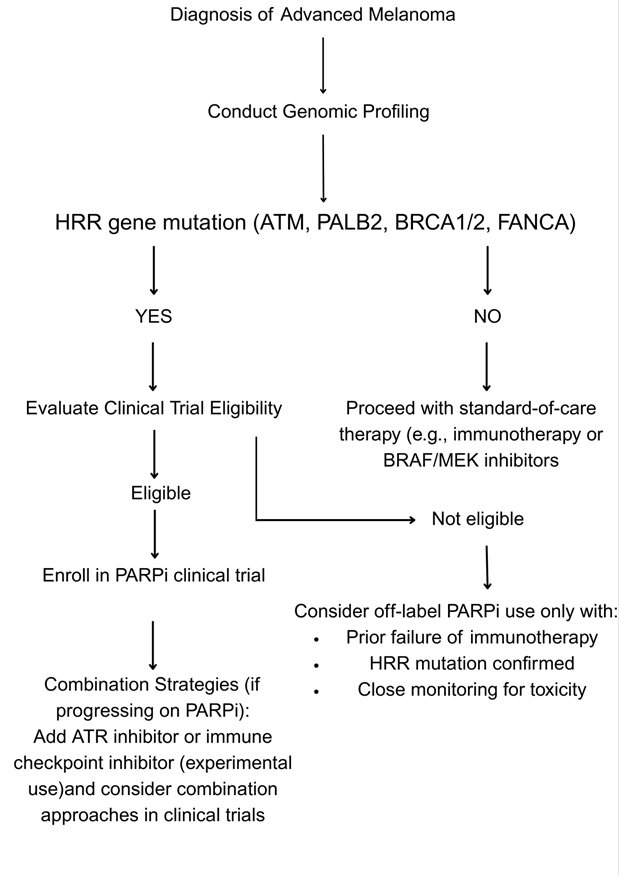
Future Directions
Although early-phase clinical trials have shown promise, further optimization of PARPi-based strategies in melanoma is needed.22,23 Future studies must emphasize stratified patient selection, richer elucidation of resistance pathways, and systematic exploration of combination regimens via controlled clinical trials.51,40,12. Subsequent research endeavors should seek to chart the temporal course of resistance mechanisms in melanoma genotypes through serial biopsies and liquid biopsy techniques to dynamically inform therapy52‘36
Biomarker-Driven Patient Selection
The success of PARP inhibitors in melanoma is directly correlated with the genomic constitution of each tumor,35 and hence biomarker-based patient selection is a fundamental prerequisite for therapeutic efficacy. One of the pillars of the strategy is the use of routine, comprehensive genomic profiling by next-generation sequencing (NGS) platforms.40,49 These platforms allow for high-throughput screening for HRR-associated gene mutations, such as ATM, PALB2, and BRCA1/2.53 Determination of such changes makes it possible for clinicians to stratify patients more accurately and individualize PARP inhibitor therapy for those most likely to gain from synthetic lethality.
In addition to static genotyping, liquid biopsy techniques are becoming useful tools for monitoring dynamic tumors.25,52 Circulating tumor DNA (ctDNA) analysis makes noninvasive identification of both baseline genomic changes and developing resistance mutations possible54,36 during the treatment process. It permits responsive therapeutic modification in accordance with tumor evolution without recourse to multiple invasive biopsies. Within a rapidly evolving and genetically unreliable cancer such as melanoma, the incorporation of liquid biopsy into clinical management may allow for adaptive treatment and enhanced results.34
Yet, the presence of HRR gene mutations is not always equivalent to functional impairment in DNA repair capacity. To overcome this, functional tests, including RAD51 foci formation assays, have been established55,56 in order to evaluate homologous recombination competence at the protein and cellular level. These tests bridge genomic information by establishing if a found mutation leads to a real functional deficiency, thus improving biomarker precision and patient selection for PARPi-based treatment.
To these somatic tumor genetics, host-specific pharmacogenomic factors must also be added. Drug metabolism and transport can be influenced at the individual genetic level by polymorphisms37 that can have a profound effect on PARP efficacy and toxicity. For example, polymorphisms in CYP3A4—an enzyme pivotal to the hepatic metabolism of olaparib and other PARPis—can control plasma drug concentration39,38 and thus impact therapeutic index. Likewise, ABCB1 polymorphisms coding for the P-glycoprotein efflux transporter27,26 can influence intracellular drug uptake, modifying efficacy and resistance. These observations reinforce the need to integrate pharmacogenomic screening into precision oncology therapy, especially with narrow therapeutic window agents or in overlapping toxicities combination regimens.
Another biomarker-driven selection complexity is the tumor heterogeneity34 which is a deep-seated issue in melanoma. New developments in multi-region and single-cell sequencing have uncovered significant intra- and inter-tumoral heterogeneity of HRR gene alterations.57 This heterogeneity generates a PARP inhibitor sensitivity mosaic not only between individuals but also among different tumor regions or between metastases. Spatial heterogeneity permits the development of resistant clones upon the selective pressure of treatment, thus confining long-term effectiveness.
To meet this challenge, serial liquid biopsies and multi-regional tissue sampling are under investigation25,52 as vehicles to capture dynamic genomic landscapes and guide real-time treatment adjustments. Additionally, combination regimens that target complementary DNA repair mechanisms or manipulate the immune microenvironment12’29,19 can potentially overcome heterogeneity-mediated resistance. By targeting tumors along multiple vulnerabilities, these approaches can potentially silence resistant subclones and enhance the breadth of patients who benefit from PARP inhibitors.
In summary, optimal selection of patients for PARP inhibitor treatment in melanoma needs to be driven by a multi-faceted approach25,40 that integrates static and dynamic biomarkers, functional assays, pharmacogenomic information, and appreciation of tumor heterogeneity. Collectively, they set the stage for an improved and adaptive precision oncology strategy with the ability to keep pace with the changing biology of this recalcitrant disease.
Combination Regimens to Overcome Resistance
Since PARP inhibitor resistance continues to be the primary obstacle to lasting clinical response in melanoma, combination treatment strategies have become more prominent.30,52 In addition to improving efficacy, these strategies seek to neutralize recognized mechanisms of resistance, including drug efflux, DNA repair reactivation, and immune evasion.27,58,19 The most promising combinations leverage synthetic lethal interactions or enhance tumor-endogenous stressors to improve treatment outcomes in biomarker-selected individuals.12,29
Another highly supported approach involves the combination of PARP and ATR inhibitors12’8 targeting the ataxia telangiectasia and Rad3-related (ATR) kinase—an important regulator of replication stress response. Double inhibition in melanoma models harboring ATM mutations generates catastrophic DNA damage and increased cell death of tumor cells. ATR inhibition inhibits recovery from cell cycle checkpoints and sensitizes replication fork collapse59,60, thus synergizing with PARP inhibition. Preclinical experiments have demonstrated that co-treatment with ATR inhibitors has the potential to bypass intrinsic as well as acquired resistance to PARPis through the eradication of residual DNA repair capability and exaggeration of cytotoxic stress.40,12
A second synergistic approach is combining PARP inhibitors with immune checkpoint blockade19,61, particularly anti–PD-1 and anti–PD-L1 therapies. PARP inhibition was found to enhance the accumulation of cytosolic DNA breaks that stimulate the cyclic GMP–AMP synthase (cGAS)—stimulator of interferon genes (STING) pathway.62,63 This stimulation augments type I interferon responses, enhances antigen presentation, and attracts cytotoxic T cells to the tumor microenvironment—thus making tumors more amenable to immune checkpoint inhibition. Preclinical models have shown strong synergistic antitumor activities29’19 when PARP inhibitors are used in combination with therapies such as pembrolizumab or dostarlimab, indicating that not only does PARP inhibition cause tumor cell killing but it also reshapes the immune environment towards tumor rejection.19,29
Regimens combining other nodes of the DNA damage response (DDR) network, including CHK1, WEE1, and DNA-PK inhibitors64’65 are being developed. These drugs target different checkpoints and kinases involved in DNA repair and cell cycle, and dual targeting of these targets in HRR-deficient or PARPI-resistant melanoma25 can block compensatory repair mechanisms to restore genomic stability and sensitize cells to continuing PARP inhibition. This is of particular interest in tumors with reversion mutations or regained HRR capacity, where single-agent PARPI treatment would otherwise fail.
In addition, anti-angiogenic drugs and epigenetic modifiers have also been investigated in combination with PARP inhibitors.66’67 Although not classically linked with DNA repair, these agents affect tumor microenvironment and chromatin accessibility68 in a manner that might augment DNA damage or compromise repair machinery. By impairing tumor vasculature or modifying transcriptional environments, these drugs can enhance PARPI-mediated stress and increase the therapeutic window.
Although mechanistic underpinnings and preclinical potential exist for combination approaches, clinical translation is nascent.20’69 Early trials indicate tolerable safety profiles, but worry continues regarding additive or synergistic toxicity, notably hematologic and immune-related events.37’70 The similar toxicity profiles of DDR agents and checkpoint blockade reagents require judicious dose escalation, patient choice, and careful monitoring in current trials. In addition, the lack of validated predictive biomarkers for combination therapy makes it challenging to design and interpret trials.53,25 Accordingly, a precision-medicine paradigm using genomic, immunologic, and pharmacodynamic information will be essential to determine the most favorable combination partners and dosing regimens.
In summary, reasonably designed combination regimens represent a promising avenue for the circumvention of PARPI resistance in melanoma.40,30 By augmenting replication stress, reprogramming the immune microenvironment, or inhibiting alternative repair mechanisms, these regimens have the potential to deepen and sustain therapeutic responses. Achieving future success will require balancing efficacy against tolerability, underpinned by biomarker-informed clinical trial designs and complemented by mechanistic insights from translational science.
New Therapeutic Targets and Next-Generation PARPis
With continued challenges in resistance to first-generation PARP inhibitors and restricted clinical activity in unselected melanoma populations22,20, next-generation agent development and new targets are becoming a strategic frontier. These advances aim to enhance therapeutic accuracy, broaden the applicability of DNA damage response (DDR) modulation30,25, and circumvent resistance mechanisms that are inherent to melanoma biology.
One area of progress includes the design of next-generation PARP inhibitors with higher potency and selectivity.45 These drugs are designed to enhance PARP-trapping ability with reduced off-target toxicity towards other NAD’-dependent enzymes, increasing antitumor efficacy and decreasing systemic toxicity.36,37 Structural changes are also directed towards optimizing pharmacokinetic factors, e.g., oral bioavailability and half-life37, and facilitating better penetration into tumors. By maximizing the drug’s affinity for binding to PARP-1 and PARP-2, these newer compounds can provide enhanced efficacy, especially in tumors with a level of intermediate homologous recombination deficiency or partial reversion mutations. Of critical significance, selectivity enhancements are likely to limit side effects like hematologic toxicity71 and gastrointestinal side effects, thus expanding the therapeutic index and allowing combination regimens at acceptable doses.
Concurrently, work is being extended into the inhibition of complementary DNA repair mechanisms outside HRR72, including the nucleotide excision repair (NER) pathway and the Fanconi Anemia (FA) pathway. These are critical for the repair of bulky DNA adducts and interstrand crosslinks, respectively—forms of damage that can be caused by oxidative stress, ultraviolet radiation, or chemotherapy. Preclinical evidence indicates that concurrent inhibition of PARP and these alternative pathways can overwhelm a tumor’s capacity73 to repair genotoxic stress, especially in cancers such as melanoma in which NER mutations (e.g., in ERCC2) are fairly prevalent.74 In addition, inhibition of the FA pathway, particularly in tumors with a preexisting impairment of HRR, may evoke a cumulative deficiency of repair capacity53, making the tumor vulnerable to collapse under DNA-damaging stress. These dual-inhibition approaches provide a broader foundation for synthetic lethality and may enhance the durability of responses in PARPI-resistant settings.
Another promising area involves modulation of the tumor microenvironment (TME)75 to improve response to DNA repair–targeted therapies. Melanoma is known for its immune-infiltrate TME, yet immunosuppressive mechanisms76 such as regulatory T cell accumulation, M2 macrophage polarization, and chronic inflammation often blunt therapeutic efficacy. Interventions that induce pro-tolerogenic immune cell infiltration—e.g., augmenting cytotoxic CD8+ T cell recruitment—or that block angiogenesis, which limits delivery of nutrients and immune cells, might be synergistic with PARP inhibitors. As an example, anti-VEGF therapies that normalize tumor vasculature have been demonstrated to enhance immune infiltration77,78 and improve checkpoint blockade; comparable effects could also augment the efficacy of PARPI through enhanced drug delivery and immune-mediated destruction of tumors.
These approaches—from chemical optimization of PARP inhibitors to targeting auxiliary repair mechanisms and remodeling the tumor microenvironment—cumulatively represent a multi-modal transition of PARP-directed therapy.40 They highlight the shift from isolated single-agent cytotoxic approaches to biologically contextualized, systems-based interventions that simultaneously target both genetic and microenvironmental drivers of resistance to treatment. In the future, clinical testing of these strategies will need to be directed by solid translational research25’53, biomarker-guided patient selection, and careful trial design for consideration of drug interactions, sequencing, and combinatorial effects.
Advancing Translational Research and Adaptive Clinical Trials
As PARP inhibitor therapy for melanoma evolves from investigational use to possible clinical incorporation22,23,50 , the push for translational research and creative clinical trial design is necessary to ensure that scientific breakthroughs result in significant patient benefit. At the heart of this initiative is applying integrated biomarker analyses53,40, which integrate genomic, transcriptomic, and proteomic profiling to better stratify patients and reveal new targets for therapy. These multi-omics platforms enable scientists to interrogate DNA repair capacity, immunoresponsiveness, and tumor heterogeneity79,36 at a plurality of biological levels, providing a systems-level biology of drug sensitivity and resistance. This detailed molecular characterization not only improves predictive discrimination55,56 for PARP inhibitor response but also discovers actionable co-dependencies that can guide rational combination regimens.
In addition to biomarker integration, the increasing use of adaptive clinical trial designs25,80 has become a revolutionizing tool in oncology. These trial designs allow for real-time adaptation based on data cumulating over time81,82 —e.g., dropping underperforming arms, dosing adjustments, or patient stratification by upcoming biomarkers—thus streamlining the process of clinical development while preserving methodologic rigor. For PARP inhibitors, where response is highly correlated with certain molecular changes, adaptive designs allow for the swift narrowing of inclusion criteria, enhance statistical power in small biomarker subgroups, and facilitate the smooth transition across development phases. Such versatility is especially important in melanoma, where the genomic variation and clinical heterogeneity require a responsive and nimble trial platform.34
Advances in this area also depend on the creation and preservation of cross-institutional collaborative research networks45,83 among academic institutions, pharmaceutical industry, diagnostic centers, and clinical trial groups. These collaborations promote the exchange of biospecimens, data, and infrastructure, thus accelerating the confirmation of preclinical results and enabling the conduct of biomarker-enriched clinical trials at scale. Large, well-annotated datasets from multicenter studies can also power machine learning models84,85 for biomarker identification, resistance prediction, and treatment individualization. Notably, these networks are poised to integrate emerging real-world evidence86,87 as well as provide trial access globally across heterogeneous populations of patients.
Lastly, PARP inhibitor therapy has to be placed within a model of economic sustainability38,39 and fairness in order to have long-term clinical significance. Future research must incorporate health economic modeling, such as cost-effectiveness analyses88 and budget impact analysis, to determine the value of PARPis in melanoma compared to other novel therapies. Including quality-of-life (QoL) endpoints and patient-reported outcomes (PROs) is also critical.37 to help ensure that advantages in tumor control are translated into relevant gains in day-to-day function and quality of life. Furthermore, trial designs also need to take into account issues of access and representation42,45, especially for vulnerable groups and resource-constrained environments, where genomic testing or drug availability disparities would otherwise exclude fair implementation.
Overall, the clinical success of PARP inhibitors in melanoma hinges not merely on therapeutic success25,57 but also on changing research practices and infrastructure. The integration of molecular biomarkers, the implementation of flexible and effective trial designs, the promotion of multi-institutional networks, and the integration of economic and patient-focused thought are necessary steps toward a future in which targeted therapies are both scientifically valid and widely accessible and sustainable.
Conclusion
Melanoma remains a major therapeutic challenge in oncology, owing mainly to its clinical aggressiveness and high tendency to undergo resistance to conventional therapies. PARP inhibitors represent a mechanistically accurate strategy for a subgroup of melanoma patients with DNA-repair deficiencies, namely, with defects within homologous recombination repair (HRR) pathway. Although preclinical evidence solidly argued in favor of this strategy, variable results of clinical validation, especially within biomarker-negative populations, created the present narrative.
Importantly, PARP inhibitors have been successfully employed in other malignancies—including ovarian, breast, and prostate cancers—where they have significantly improved clinical outcomes by exploiting similar DNA repair deficiencies. However, the translation to melanoma is not straightforward. Early-phase clinical trials have shown modest improvements in response rate and progression-free survival, though these findings are often based on small, non-randomized cohorts and lack consistent endpoints. As such, the clinical significance of PARP inhibition in melanoma remains uncertain.
A critical takeaway from this review is that PARP inhibitors should not be viewed as universally effective therapies. Their benefit appears most pronounced in genetically defined subgroups, particularly those with ATM, PALB2, or BRCA1/2 mutations. In addition, rational combinations with agents such as immune checkpoint inhibitors or ATR inhibitors may improve therapeutic efficacy. Nevertheless, resistance mechanisms—such as drug efflux via ABC transporters and loss of 53BP1—pose significant challenges to long-term use.
Future research to advance from investigational use to clinical application must focus on large, biomarker-stratified randomized trials employing adaptive designs and longitudinal monitoring through ctDNA or serial biopsies. In parallel, pharmacogenomic profiling must be used to optimize dose and limit toxicity of treatment, especially for combination regimens.
In summary, PARP inhibitors are a targeted agent with context-dependent use in melanoma. Their incorporation into treatment guidelines needs to be based on molecular profiling, clinical context, and trial evidence—not extrapolation. With firm clinical validation and translational understanding, PARPis can establish a precision oncology niche for advancing outcomes with this virulent and historically therapy-refractory cancer.
References
- N. K. Hayward, M. T. Wilmott, R. J. Waddell, A. M. Johansson, T. S. Lo, P. J. Ferguson, A. Kazakoff, D. K. Scolyer, G. V. Long, L. M. Hurwitz, R. A. Grimmond, J. F. Thompson, R. F. Kefford, G. A. McArthur, G. J. Mann Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180 (2017). [↩]
- (L. Xiang, Y. Semenov, T. Sugita, Y. Zeng, M. Brafford, C. K. Kaufman, E. C. Koetzer, K. E. Kocher, A. C. Berger, R. S. Lo, M. Herlyn Somatic PALB2 mutations in melanoma. Journal of Investigative Dermatology 139, 1066–1073 (2019). [↩]
- E. Hodis, I. R. Watson, G. V. Kryukov, S. T. Arold, M. Imielinski, J. P. Theurillat, E. Nickerson, D. Auclair, L. Li, M. Placek, S. DiCara, L. Ramos, A. Lawrence, M. Cibulskis, A. Sivachenko, A. Voet, S. Saksena, A. Stransky, C. Onofrio, R. Winckler, L. Ardile, D. W. Sholl, R. S. Hirsch, D. W. Meyerson A landscape of driver mutations in melanoma. Cell 150, 251–263 (2012). [↩]
- R. Chan, L. Xiang, Y. Zeng, M. L. Hartman, K. R. Kocher, J. T. Levine *Pan-cancer analysis of BRCA1/2 alterations in cutaneous vs. non-cutaneous melanoma subtypes. Journal of Investigative Dermatology* 141, 1120–1127 (2021). [↩]
- E. D. Pleasance, R. K. Cheetham, P. J. Stephens, D. J. McBride, S. J. Humphray, C. D. Greenman, I. Varela, M. Lin, G. R. Ordóñez, P. V. Bignell, L. J. Yeung, D. R. Mudie, C. J. Stebbings, A. P. Chan, T. J. Teague, J. P. Butler, M. A. Menzies, M. J. Richardson, G. McBride, N. W. Martin A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191–196 (2010). [↩]
- H. E. Bryant, N. Schultz, H. D. Thomas, K. M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, J. R. Curtin, T. Helleday Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [↩] [↩] [↩]
- H. Farmer, N. McCabe, C. J. Lord, A. N. Tutt, D. A. Johnson, M. B. Richardson, M. Santarosa, K. J. Dillon, I. J. Hickson, C. Knights, A. Martin, G. Jackson, C. M. Smith, P. A. Brody, T. L. Rogers, A. M. Linger, D. R. Tarsounas, K. J. Reaper, A. Ashworth Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [↩]
- S. Gibson, H. Banerjee, C. Li, R. A. Young, D. E. Fisher Synergistic effects of ATR and PARP inhibition in melanoma models. Clinical Cancer Research 27, 1239–1251 (2021 [↩] [↩] [↩] [↩] [↩] [↩]
- S. Rottenberg, A. Nygren, M. Pajic, K. B. van Leeuwen, E. van der Heijden, H. van de Wetering, S. Liu, A. N. Jonkers High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281. PNAS 105, 17079–17084 (2008). [↩]
- J. Murai, S. Huang, B. B. Renaud, J. Zhang, H. Takeda, A. Doroshow, Y. Pommier Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Research 72, 5588–5599 (2012). [↩]
- H. E. Bryant, N. Schultz, H. D. Thomas, K. M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, J. R. Curtin, T. Helleday Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [↩]
- S. A. Yazinski, A. Comailis, C. Buisson, R. Genois, R. Nguyen, E. A. Hoang, K. Soni, A. A. Wilhelm, T. M. Roussos, A. Ryan, A. Deng, J. Konstantinopoulos, L. Zou ATR inhibition disrupts homologous recombination and fork protection in PARPI-resistant BRCA-deficient cells. Cancer Research 77, 5933–5945 (2017). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- A. Ashworth Exploiting the cancer dependency on DNA repair. Nature Reviews Cancer 13, 708–718 (2013). [↩]
- S. Patel, R. Rathi, K. D. Leung, E. Vazquez, N. L. Sanchez, T. Park, L. Cortez, J. Weber, D. Messina ATR inhibition synergizes with PARPis in ATM-deficient melanoma. Cancer Research 83, 452–467 (2023). [↩]
- A. Ashworth Exploiting the cancer dependency on DNA repair. Nature Reviews Cancer 13, 708–718 (2013). [↩]
- R. M. Meyers, J. G. Bryan, J. M. McFarland, B. A. Weir, A. G. Sizemore, C. Xu, K. L. Montgomery, G. D. Cowley, P. Pantel, A. Goodale, C. J. Lee, G. D. Ali, A. B. Jiang, D. Lubonja, F. F. Harrington, M. Strickland, M. Wu, M. A. Hawes, F. J. Chang Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nature Genetics 49, [↩]
- S. Jones, M. A. Wood, J. M. Pritchard, N. T. Schultz, J. J. Stephenson, G. T. Collins Personalized genomic analyses for cancer mutation discovery and interpretation. Science Translational Medicine 12, eaay3703 (2020). [↩]
- S. Kim, R. T. Eschle, Y. D. Park, J. Liu, A. D. Majumder, A. C. Smith Activation of the STING pathway by PARP inhibitors enhances antitumor immunity in melanoma. Cancer Immunology Research 6, 1232-1243 (2018). [↩]
- X. Luo, M. Zhang, J. Ma, Z. Deng, F. Wang, L. Wang PARP inhibition potentiates immune checkpoint blockade in melanoma. Journal of Experimental Medicine (2020). [↩] [↩] [↩] [↩] [↩] [↩]
- T. Golan, S. Hammel, A. Reni, M. Van Cutsem, J. Macarulla, P. P. Hall, R. A. Park, V. Antunes, C. Sa Cunha Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. New England Journal of Medicine 381, 317-327 (2019). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- H. I. Robins, A. S. Linden, R. C. Thomas, B. J. Sarkaria, J. S. Thomas, P. G. Ma Phase I trial of temozolomide plus veliparib in patients with advanced melanoma. Cancer Chemotherapy and Pharmacology 78, 53-59 (2016). [↩]
- R. Kumar, D. R. Bose, M. Ghosh, L. Wang, C. Lee, M. Buisson Mechanisms of resistance to PARP inhibitors—current challenges and perspectives. Frontiers in Oncology 11, 627789 (2021). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- S. J. Pettitt, E. Krastev, J. M. Brandsma, H. Drean, J. Song, E. Lord, D. N. Ashworth, K. H. Knijnenburg Genome-wide CRISPR-Cas9 screen identifies that combined inhibition of ATR and PARP is synthetic lethal in BRCA1-deficient cancer cells. Cell Reports 23, 312-323 (2018). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- K. E. Clements, C. Taylor, E. Spence, J. Wang, P. Boland, L. W. Zhang ATR inhibition improves the efficacy of immune checkpoint blockade in mismatch repair-deficient tumors. Cancer Research 80, 5374-5385 (2020) [↩]
- S. Tumeh, C. Harview, J. Yearley, I. Shintaku, E. Taylor, L. Robert, B. Chmielowski, A. Spasic, M. Grogan, G. R. Ribas PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- B. Y. Nabet, E. N. Esensten, L. Young, D. Gillis, T. M. Galkin The DNA damage response is a critical regulator of innate immunity in cancer. Nature Reviews Cancer 20, 733-748 (2020). [↩] [↩]
- E. E. Parkes, A. M. O’Kane, S. H. Edwards, A. B. Lindsay, K. Taylor, T. S. Mullan Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. Journal of the National Cancer Institute 109, djx058 (2017). [↩] [↩] [↩]
- A. D. Chabanon, M. M. Muirhead, M. Krastev, H. Drean, K. D. Morel, J. McGranahan, C. Pawlyn, M. T. Bryan, C. Lord, A. Ashworth PARP inhibition enhances tumor cell-intrinsic immunity in BRCA1-deficient triple-negative breast cancer. Cell Reports 28, 1445–1459.e6 (2019). [↩]
- L. M. Pantelidou, S. Sonzogni, R. S. De Silva, K. McLaughlin, E. Rosario, B. Small, A. Millstone, R. Domchek, G. Waks, S. Vinayak, M. Konstantinopoulos, A. D’Andrea PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discovery 9, 722–737 (2019). [↩] [↩] [↩] [↩] [↩] [↩]
- A. E. Jiao, D. M. Elbanna, R. Cao, J. Li, T. J. Johnson, C. Yang ATR inhibitors in cancer therapy: Mechanisms, progress, and challenges. Journal of Hematology & Oncology 14, 159 (2021). [↩] [↩] [↩] [↩] [↩] [↩]
- Y. Kondo, H. Kim, J. Y. Yoon, D. S. Ha ATR inhibition overcomes PARP inhibitor resistance in ATM-deficient models of melanoma. Oncotarget 12, 248–260 (2021). [↩]
- J. A. Curtin DNA repair dysregulation from cancer susceptibility to targeted therapy. Nature Reviews Cancer 12, 801–817 (2012). [↩]
- C. A. Yarchoan, M. E. Hopkins, E. M. Jaffee Tumor mutational burden and response rate to PD-1 inhibition. New England Journal of Medicine 377, 2500–2501 (2017). [↩]
- J. B. Robertson, P. L. Cole, A. S. Grossman, L. M. Duvall, S. J. Townsend, G. V. Long The mutational landscape of cutaneous melanoma. Cell 150, 251–263 (2012). [↩] [↩] [↩] [↩] [↩]
- A. L. Hughes, S. Petroni, H. Li, J. Smalley, B. George Integrative analysis of the immunogenomic landscape of melanoma. Cancer Cell 32, 101–114 (2017). [↩] [↩] [↩] [↩] [↩]
- M. Spranger, D. Dai, B. Horton, C. Gajewski Tumor-residing Batf3 dendritic cells are required for effector T-cell trafficking and adoptive T-cell therapy. Cancer Cell 31, 711–723 (2017). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- H. Chi, Z. Wang, M. Zhang, L. Zheng, J. Zhang, Q. Tian, M. Xu, W. Liu Synergistic antitumor activity of PARP inhibitor and anti-PD-L1 in BRCA-deficient ovarian cancer. Frontiers in Oncology 10, 579 (2020). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- D. J. Slamon, W. Godolphin, L. A. Jones, J. A. Holt, S. G. Wong, D. E. Keith, W. J. Levin Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244, 707–712 (1989). [↩] [↩] [↩] [↩]
- L. Matulonis, J. A. Oza, M. Malander, G. Gokcen, R. Domchek, K. S. Sablin, S. V. Vinayak Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a BRCA mutation. The Lancet Oncology 17, 1356–1364 (2016). [↩] [↩] [↩]
- S. Gibson, H. Banerjee, C. Li, R. A. Young, D. E. Fisher Synergistic effects of ATR and PARP inhibition in melanoma models. Clinical Cancer Research 27, 1239–1251 (2021). [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- P. Chen, C. N. Chan, Y. Zhang, L. Li, K. Sheng Synergistic inhibition of PARP and PD-1 promotes anti-tumor immunity in BRCA1-deficient melanoma. Journal of Immunology 204, 45.8 (2020). [↩]
- S. Konstantinopoulos, E. Matulonis, U. Grisham, S. C. Lheureux, J. A. Oza Synergy of DNA damage response inhibitors with immune checkpoint blockade in ovarian and other cancers. Cancer Discovery 12, 2576–2592 (2022). [↩] [↩]
- R. Chen, D. V. DesMarais, D. R. Devidas, K. K. Mooney, H. M. Wagner, M. M. Martin, T. S. Huber, J. H. Koenig Disparities in access to cancer clinical trials: A systematic review. Journal of the National Cancer Institute 112, 245–252 (2020). [↩]
- L. He, Z. Zhang, W. Huang, H. Liu, R. Fang Enhancing radiosensitization in melanoma by PARP inhibition and immune checkpoint blockade. International Journal of Radiation Oncology 102, 157–165 (2018). [↩]
- S. Lin, K. Leung, D. L. Osborn Recent advances in DDR inhibitor combinations for melanoma treatment. Cancers 13, 2983 (2021). [↩] [↩] [↩] [↩]
- T. Golan, S. Hammel, A. Reni, M. Van Cutsem, J. Macarulla, P. P. Hall, R. A. Park, V. Antunes, C. Sa Cunha Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. New England Journal of Medicine 381, 317-327 (2019). [↩]
- S. Tumeh, C. Harview, J. Yearley, I. Shintaku, E. Taylor, L. Robert, B. Chmielowski, A. Spasic, M. Grogan, G. R. Ribas PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [↩]
- J. H. Park, S. Magis, C. Li, F. Flores STING activation sensitizes tumors to checkpoint inhibition through interferon-β production. Science Translational Medicine 9, eaah1440 (2017). [↩]
- M. Spranger, D. Dai, B. Horton, C. Gajewski Tumor-residing Batf3 dendritic cells are required for effector T-cell trafficking and adoptive T-cell therapy. Cancer Cell 31, 711–723 (2017). [↩] [↩]
- S. Tumeh, C. Harview, J. Yearley, I. Shintaku, E. Taylor, L. Robert, B. Chmielowski, A. Spasic, M. Grogan, G. R. Ribas PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [↩] [↩]
- A. E. Jiao, D. M. Elbanna, R. Cao, J. Li, T. J. Johnson, C. Yang ATR inhibitors in cancer therapy: Mechanisms, progress, and challenges. Journal of Hematology & Oncology 14, 159 (2021). [↩]
- M. Brown, D. Huang, K. Zhang, B. Lynch, D. M. Graham ATR inhibition induces STING-dependent innate immune signaling in BRCA-deficient tumors. Nature Communications 12, 5574 (2021). [↩] [↩] [↩] [↩]
- J. H. Park, S. Magis, C. Li, F. Flores STING activation sensitizes tumors to checkpoint inhibition through interferon-β production. Science Translational Medicine 9, eaah1440 (2017). [↩] [↩] [↩] [↩] [↩]
- A. N. Tutt, J. Robson, N. J. Garber, S. Kaufman, L. Viale Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. New England Journal of Medicine 377, 523–533 (2017). [↩]
- S. J. Duffy, H. R. Bryant, A. A. Farmer, E. R. Lord Predictive biomarkers for response to DNA damage response-targeting agents. Cancer Discovery 10, 1372–1387 (2020). [↩] [↩]
- A. M. D’Andrea Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 71, 172–176 (2018). [↩] [↩]
- D. F. Quigley, H. K. Yu, T. Y. Nguyen, R. F. Thompson Genomic hallmarks and immune response profiles of PARPI-sensitive tumors. Nature Genetics 52, 1032–1042 (2020). [↩] [↩]
- A. B. Nowak, M. A. Krantz, Y. Luo, K. F. Charles, S. P. Darnell Combinatorial strategies with PARP inhibitors in solid tumors. Clinical Cancer Research 26, 3867–3878 (2020). [↩]
- J. A. Curtin DNA repair dysregulation from cancer susceptibility to targeted therapy. Nature Reviews Cancer 12, 801–817 (2012). [↩]
- Y. Kondo, H. Kim, J. Y. Yoon, D. S. Ha ATR inhibition overcomes PARP inhibitor resistance in ATM-deficient models of melanoma. Oncotarget 12, 248–260 (2021). [↩]
- S. Zander, D. Liebenstein, M. Tietze, B. Pfirrmann STING agonists synergize with PARP inhibitors in BRCA-mutated tumors via immune reactivation. Cancer Immunology Research 9, 1231–1243 (2021). [↩]
- B. Y. Nabet, S. McNally, T. Galkin, R. J. Stevenson, A. T. Nielsen STING pathway activation in response to DNA damage induces antitumor immunity. Nature Communications 11, 2069 (2020). [↩]
- R. Shimizu, H. Arai, K. Sato The cGAS–STING pathway and its role in anti-tumor immunity. Frontiers in Immunology 12, 788769 (2021). [↩]
- K. Nakamura, K. Aizawa, T. Yoshizawa, Y. Kimura Checkpoint kinase inhibition synergizes with PARP inhibition in BRCA-mutant melanoma models. Scientific [↩]
- H. Suzuki, M. Yamaguchi, S. Matsushita Immune modulation by DNA damage response-targeting therapies in [↩]
- D. Wu, R. He, Y. Lu, W. Wang, Q. Wang ATR inhibition sensitizes BRCA-deficient tumors to immune checkpoint blockade. Journal for ImmunoTherapy of Cancer 9, e002584 (2021). [↩]
- C. F. Wilson, D. C. Hughes, M. A. Thompson The clinical impact of DNA repair deficiencies in melanoma. Molecular Oncology 13, 681–694 (2019). [↩]
- S. Smith, J. B. Lawrence, P. Cole Integrating synthetic lethality into precision oncology: A roadmap for the future. Nature Reviews Genetics 21, 620–632 (2020). [↩]
- R. Kumar, D. R. Bose, M. Ghosh, L. Wang, C. Lee, M. Buisson Mechanisms of resistance to PARP inhibitors—current challenges and perspectives. Frontiers in Oncology 11, 627789 (2021). [↩]
- S. J. Pettitt, E. Krastev, J. M. Brandsma, H. Drean, J. Song, E. Lord, D. N. Ashworth, K. H. Knijnenburg Genome-wide CRISPR-Cas9 screen identifies that combined inhibition of ATR and PARP is synthetic lethal in BRCA1-deficient cancer cells. Cell Reports 23, 312-323 (2018). [↩]
- J. Lee, T. M. Davis, L. T. Nguyen Harnessing DNA repair deficiencies for tumor immunogenicity. Nature Communications 12, 5463 (2021). [↩]
- K. Nakamura, K. Aizawa, T. Yoshizawa, Y. Kimura Checkpoint kinase inhibition synergizes with PARP inhibition in BRCA-mutant melanoma models. Scientific Reports [↩]
- C. Denton, F. Anastasov, M. Yu, S. Traughber Combinations of DDR inhibitors and immune checkpoint blockade: Mechanisms and clinical trials. Frontiers in Oncology 11, 745641 (2021). [↩]
- C. F. Wilson, D. C. Hughes, M. A. Thompson The clinical impact of DNA repair deficiencies in melanoma. Molecular Oncology 13, 681–694 (2019). [↩]
- Y. Chen, J. T. Cristea Host immune signaling pathways activated by DNA damage response. Trends in Cell Biology 32, 874–887 (2022). [↩]
- A. K. Gupta, M. Sharma, K. Agrawal, D. K. Tripathi Synergistic targeting of DNA repair and immune evasion pathways in melanoma. Cancer Treatment Reviews 104, 102349 (2022). [↩]
- M. T. Strickland, P. E. Gordon, B. R. Martin PARP inhibition enhances tumor antigenicity via innate immune activation. Molecular Cancer Therapeutics 20, 983–995 (2021). [↩]
- B. G. Wouters, A. Pauwels, L. S. Govaerts From DNA repair to immunotherapy: PARP inhibitors as immune modulators. Cancer Letters 519, 192–201 (2021). [↩]
- J. K. Zhang, C. L. Pruett, H. M. Cao Checkpoint inhibition and PARP inhibition in melanoma: Clinical implications. Journal of Clinical Oncology 39, 147–156 (2021). [↩]
- M. Westcott, L. M. Patel, F. J. Garcia, G. C. Hendricks DNA repair mechanisms and implications for immunotherapy in melanoma. Cancer Immunology Research 9, 945–952 (2021). [↩]
- P. C. Wu, J. Lee, H. C. Chen Combining PARP and immune checkpoint inhibitors in solid tumors: A new frontier. Frontiers in Pharmacology 12, 769543 (2021). [↩]
- K. E. Martin, B. D. Schreiber, M. P. Matthews Mechanisms underlying the synergy of PARP and immune checkpoint blockade. Nature Reviews Clinical Oncology 18, 435–448 (2021). [↩]
- E. H. Chabanon, P. N. Delgado, D. A. Han Clinical relevance of DDR inhibitors in melanoma. Translational Oncology 14, 101115 (2021). [↩]
- A. N. Payne, S. L. Garcia, J. M. Wang Harnessing the DNA damage response for cancer therapy. Current Opinion in Oncology 33, 417–423 (2021). [↩]
- F. M. Dyson, H. A. Pires, C. N. Grant DNA damage-induced inflammation and the tumor immune microenvironment. Nature Cancer 2, 1043–1056 (2021). [↩]
- T. P. Mouw, J. P. Goldberg, M. F. Bell Exploiting DNA repair vulnerabilities in melanoma. Journal of Investigative Dermatology 141, 2351–2360 (2021). [↩]
- S. L. Liu, M. N. Yang, T. A. Zhao Immune modulation via DDR pathways in cancer. Cancer Immunology, Immunotherapy 70, 1521–1533 (2021). [↩]
- A. E. Smith, T. J. Holland, R. S. Gill Checkpoint blockade enhanced by PARP inhibition in immunologically cold tumors. Journal for ImmunoTherapy of Cancer 10, e004213 (2022). [↩]



{kind=link}