
Abstract
Although research has begun to take a more systems-level approach, much of it still overlooks the broader picture of how the tumor microenvironment plays a role in the ovarian cancer’s therapeutic resistance. For example, over 80% of these patients experience recurrence, a fact that has fueled debate over the “right” strategies in targeting the tumor and its metastasis1. This review aims to facilitate that shift by synthesizing findings and considering how cell-cell communication in the ovarian cancer tumor microenvironment (TME) can affect tumor progression, plasticity, and resistance to therapy, ultimately identifying mechanisms that are still poorly understood but proven to be important for improving outcomes. To achieve this, we conducted a systematic literature review of peer-reviewed studies using databases such as Google Scholar, PubMed, and the NCBI. In this paper, we focused on signaling interactions between the various components of the TME and how some of the major pathways, such as TGF-β, IL-6, and Wnt act together to help modulate the tumor-supportive environment. While preclinical evidence suggests therapeutic promise in targeting these pathways, clinical validation remains limited and inconsistent. To address this gap, this paper profiles a wide range of literature in order to identify the overarching trends, highlight key advances as well as challenges, and compare and contrast findings across various experimental models. By synthesizing results from both preclinical and clinical research, it aims to show how cell-to-cell communication affects recurrence and treatment resistance and ultimately where future research can make the greatest impact.
Keywords: Ovarian cancer, tumor microenvironment, cell-to-cell communication, cancer-associated fibroblasts (CAFs), immune cells, cancer stem cells (CSCs), extracellular matrix (ECM), TGF-β signaling, IL-6, Wnt pathway, phenotypic plasticity, immune evasion, metastasis, therapeutic resistance, systematic literature review.
Introduction
Ovarian cancer is one of the most lethal gynecologic cancers, often termed a “silent killer” due to its vague symptoms, like bloating, abdominal pain, and fatigue, which are frequently mistaken for less serious conditions2. Consequently, the disease is typically diagnosed at an advanced, metastatic stage. In the U.S. alone, approximately 20,890 new cases and 12,730 deaths are projected in 2025, underscoring the urgent need for improved therapeutic strategies3. Therefore, addressing this critical challenge requires a comprehensive understanding of ovarian cancer’s underlying biology, which requires us to look beyond the cells themselves and pivot towards the environment that truly supports them.
This disease primarily affects postmenopausal women and is influenced by both genetic and environmental factors. For example, BRCA1 and BRCA2 mutations significantly increase risk, whereas obesity and hormone replacement therapy are also contributors4. Most studies in recent years of ovarian cancer research have focused primarily on intrinsic mutations and chemotherapy resistance of tumor cells. However, the field is shifting to acknowledge the tumor microenvironment (TME) as equally important. The TME is a biological ecosystem that consists of the extracellular matrix (ECM), signaling molecules, specialized immune cells, cancer-associated fibroblasts (CAFs), and cancer stem cells (CSCs)5. The ECM refers to proteins and fibers that surround cells and help regulate their behavior, whereas signaling molecules (i.e., cytokines and growth factors) coordinate communication between different cell types6. Immune cells are specialized cells in the body that recognize foreign substances and protect against infectious diseases. CAFs are fibroblasts that become pro-tumorigenic under the influence of the tumor, which means that they shift from their normal role in wound healing to one that supports cancer growth7, and lastly, CSCs are a unique group of cancer cells that retain some stem-like properties in that they self-renew and are at least partly treatment-resistant8. In healthy tissue, the components of this biological ecosystem are important for regulating repair, maintaining immune balance, and setting the conditions to properly support normal cell growth. But in the instance of ovarian cancer, these same systems will become hostile to the body, whereby fibroblasts are pro-tumorigenic, immune cells are suppressed, and ECM signaling is rewired in ways associated with invasion and survival. These components not only coexist with the tumor, but they also have been correlated with angiogenesis, immune evasion, and drug resistance9.
Despite significant advancements in surgery and chemotherapy, overall survival rates for ovarian cancer remain poor due to frequent relapse and acquired drug resistance10. These issues have become increasingly linked to intercellular TME-mediated cell signaling properties that drive tumor plasticity and immunomodulation. Paradoxically, most of the current treatments still only target the tumor itself. Importantly, the progress being made in ovarian cancer therapeutic strategies, including 3D organoid modeling11 and immunotherapy12, has begun to reveal the complexities of TME signaling at an unprecedented level of detail. This makes the present a crucial moment to revisit how this microenvironment is a main driving factor of this deadly disease.
However, we must also consider that ovarian cancer represents a heterogeneous disease with a variety of histological subtypes, including high-grade serous, clear cell, endometrioid, and mucinous carcinomas, each with distinct genetic alterations and tumor microenvironments. Each subtype has its own genetic alterations and tumor microenvironments, meaning that signaling pathways may not operate in the same way across them. For instance, clear cell carcinoma is typically associated with hypoxia-inducible HIF-1 signaling that is separate from the pathology of high-grade serous disease. This variability helps explain why therapeutic responses differ between patients and reinforces that intercellular communication in the TME plays a critical role in driving recurrence and therapy resistance, though often in subtype-specific ways.
In consideration of these rapidly changing areas of understanding, this systematic review will analyze data concerning intercellular signaling that occurs within the ovarian cancer TME, with a particular focus on its impact in promoting disease progression and resistance. In particular, we focus on TGF-β, IL-6, and Wnt pathway signaling, along with the roles of CAFs, other immune cells (including Tregs and TAMs), CSCs, and ECM remodeling behavior. Our goal is to synthesize and highlight emerging and potential therapies to target the tumor-enhancing properties of the TME.
Studies have thoroughly investigated the contributions of CAFs, immune cells, CSCs, and signaling pathways such as TGF-β, IL-6, and Wnt in ovarian cancer. However, it is still not fully understood how these signals work together as a coordinated network to support phenotypic plasticity, promote immune evasion, and help drive recurrence across ovarian cancer subtypes. Our proposed hypothesis is that relapse is reinforced more by the layered communication within the TME that allows tumors to adapt after therapy than by tumor-intrinsic mutations alone. In this paper, we synthesize evidence of these intercellular communication networks in order to profile how they contribute to therapy resistance. Through examining recent advances in the overall understanding of ovarian cancer TME research, in addition to highlighting existing research gaps identified in the current literature, we ultimately aim to inform future efforts toward more effective, multitargeted interventions with the intent of treating this deadly disease.

This figure illustrates the bidirectional interactions of ovarian cancer cells with the major components of the TME: fibroblasts/stromal cells, immune cells, and the extracellular matrix (ECM). Arrows represent the cytokines and growth factors that can signal in both directions (e.g., TGF-β, IL-6, and VEGF), which together remodel the microenvironment to support invasion, survival, and also metastatic behavior.(https://pmc.ncbi.nlm.nih.gov/articles/PMC11588459/) This figure illustrates how each component has a relationship with one another and can either work together or against each other to modulate the ovarian cancer TME. (https://pmc.ncbi.nlm.nih.gov/articles/PMC11275383/ ) Sources: (Yang 2020 (1); Zhang 2022 (14))
Methods
To explore the mechanisms of intercellular communication within the ovarian cancer TME and how it contributes to disease progression and treatment resistance, a systematic literature review was conducted between November 2024 and June 2025. The systematic review was conducted as part of an independent high school research project aimed at synthesizing mechanistic knowledge on cell signaling within the ovarian TME. Our research began with comprehensive database searches using PubMed, Google Scholar, Web of Science, and the NCBI database. We used Boolean combinations of keywords and concepts in the searches including “ovarian cancer”, “tumor microenvironment,” “cell-to-cell communication,” “immune evasion,” “fibroblasts,” “cancer stem cells,” “TGF-β signaling,” “IL-6,” and “Wnt pathway.” These searches were restricted only to peer-reviewed literature published in English from 2010 until 2025.
Following the initial search, articles were screened by a set of predetermined criteria; all articles that passed this first stage of screening were then read in full text to determine adherence to our inclusion criteria. The studies we included had to have utilized either in vitro or in vivo models to study signaling between CAFs, immune cells, CSCs, and the ECM within the ovarian TME. Relevant clinical studies and comprehensive review articles with a mechanistic focus were also considered.
Our initial search identified approximately 240 publications from the databases. We first briefly screened the titles and abstracts for relevance and then performed full-text reviews on about 120 articles. Articles were included if they 1) focused on ovarian cancer, (2) investigated signaling interactions between components of the TME (CAFs, immune cells, CSCs, ECM), and (3) used either in vitro, in vivo, or patient-derived models. We also included clinical studies and mechanistic review papers when directly relevant to ovarian cancer signaling. Exclusion criteria involved studies that (i) focused solely on other cancer types without clear ovarian cancer relevance, (ii) lacked mechanistic or signaling detail, or (iii) were not peer-reviewed. After applying these criteria, 75 articles were included for synthesis. This step-by-step approach ensured transparency and allowed us to use those studies that were most relevant to the TME communication in ovarian cancer.
From the selected eligible studies, key data and details were extracted to analyze, such as author, year, model system (e.g., mouse, patient-derived sample, human cell line), signaling pathways studied, and relevant biological outcomes. After initial screening, we refined the list to those that best fit our criteria for quality and relevance.
PubMed, Google Scholar, and NCBI were used with the following strings:(“ovarian cancer” AND “tumor microenvironment”) OR (“ovarian” AND “CAF*”) OR (“ovarian” AND “TAM*” OR “Treg*”) OR (“ovarian” AND “exosome*” OR “microRNA” OR “lncRNA”) OR (“ovarian” AND “TGF-β” OR “IL-6” OR “Wnt” OR “PDGF” OR “Hippo” OR “YAP” OR “TAZ” OR “CXCL8/IL-8” OR “HIF-1”)
Given the diversity of the collected data, a specific narrative synthesis approach was chosen over a meta-analysis due to the diverse study types and endpoints encountered. This enabled us to group studies by functional trends, such as IL-6-driven inflammation or Wnt-associated stemness, facilitating the identification of both consistencies and contradictions across different models. Furthermore, studies using patient-derived xenografts, cytokine assays, or gene knockdowns were given a higher preference. Concurrently, limitations like small cohorts or conflicting findings across tumor stages that may impact interpretation were noted as well.
While this review synthesizes evidence from multiple model systems, it is important to note the strength of findings can vary depending on the platform used. For example, mouse-only studies usually demonstrate mechanistic understanding but do not fully include the immune complexity of human disease; however, xenograft models use human tumor tissue in immunocompromised mice, allowing for more direct relevance, but have their own limitations. Similarly, in vitro cell line studies are valuable for isolating specific signaling effects, but they do not fully incorporate the stromal-or immune-interactions which are seen in patient-derived tissues. For these reasons, we have prioritized studies validated in patient-derived xenografts or clinical samples, though sample sizes in human data remain modest, and generalizability is still limited. Recognizing these differences in the strength of different evidence helps contextualize our synthesis and underscores the importance of both integrated and cross-model validation to better characterize ovarian cancer TME-mediated recurrence and resistance. This comprehensive approach allowed for the synthesis of recent research on the ovarian cancer TME, demonstrating both development and highlighting important gaps in knowledge.
Results
Prior to examining individual components of the tumor microenvironment (TME), it is crucial to recognize that this ecosystem is composed of various interacting cell types and structures that interact with each other as illustrated in Figure 1; however, to understand the abnormal environment, we must first consider the normalcy of these aspects and how they react. The following sections will examine how these components, beginning with cancer-associated fibroblasts, promote ovarian cancer progression and treatment resistance.
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) are one of the most abundant stromal cell types in the ovarian tumor microenvironment (TME), and recent research has demonstrated that CAFs play an important role in the progression and metastasis of ovarian cancer13. While normal fibroblasts can exhibit some degree of activation, CAFs remain persistently activated. They can often be characterized by their expression of α-SMA, fibroblast activation protein (FAP), and PDGF receptors14.
Recent single-cell analyses of ovarian tumors revealed multiple CAF subtypes with distinct signaling and spatial patterns that correlate with recurrence and immune suppression15,16. These ovarian-specific datasets confirm that CAF heterogeneity is functionally linked to TGF-β and CXCL12 signaling in the ovarian TME.
CAFs produce many crucial factors that promote epithelial-to-mesenchymal transition, such as TGF-β and HGF, which are associated with increased cell mobility and help cancer cells escape their original location17. Moreover, CAFs produce many matrix metalloproteinases (MMPs), such as MMP-2 and MMP-9, which can degrade the extracellular matrix, and thus may aid tumor cells in their migration, which is particularly important in the metastatic process18.
Beyond their well-established roles in structural remodeling, more recent studies support that CAFs also play critical roles in metabolic crosstalk and immune signaling. Recent literature (2022 – 2024) on CAFs has showcased this perspective even further. When considering CAFs’ role in metabolic crosstalk and immune signaling19, researchers noted that CAFs undergo glycolysis and release lactate and pyruvate to tumor cells, which support oxidative phosphorylation, a process now termed the reverse Warburg effect. This phenomenon has been reported to support tumor cells to thrive even in oxygen-poor environments20. In addition, it was also often noted that CAFs would release IL-6 and CCL2 that stimulated immunosuppressive granulocyte recruitment, either M2 macrophages or regulatory T cells (Tregs). By releasing these cytokines, CAFs ultimately are shown to be contributing to an immunosuppressive environment by suppressing cytotoxic T cell activity and dampening anti-tumor immune responses21.
The various roles of CAFs indicate their promising potential as therapeutic targets; however, clinically translating these discoveries has not been straightforward. Studies have observed that CAF behaviors varied across models, underscoring the need for better in vivo systems22. Several papers reported contradictory results regarding whether CAF depletion was associated with improved overall survival, suggesting that simply eliminating CAFs may have unintended consequences23,24. This inconsistency served to highlight the need for using more physiologically relevant models. It illustrated just how complex and context-dependent CAF signaling may be, reflecting a wider discourse in the field on whether total CAF loss may in fact unintentionally destabilize anti-tumor immunity. Recent single-cell analyses of high-grade serous ovarian tumors revealed that CAFs can form distinct molecular subtypes with unique cytokine and ECM gene signatures specific to ovarian cancer, highlighting greater stromal diversity than previously recognized15,16.
Despite these complexities, the overarching evidence does establish that CAFs are linked to persistence and relapse of ovarian cancer25. Furthermore, these cells have been shown to re-secrete pro-survival cytokines and remodel the ECM post-treatment, and so they could sustain this permissive microenvironment that was conducive to relapse26. As CAF-driven inflammation and immunosuppression reshape the TME, the next section examines how the tumor itself converts immune cells more directly to evade destruction27.
Immune Cells
While the immune system is designed to detect and destroy abnormal cells, ovarian cancer has evolved multiple strategies to evade detection. Our review of the literature indicates that the tumor has been reported to not only suppress immune function but also rewires the immune cell to promote tumor metastasis, thereby promoting tumor survival and long-term therapeutic resistance.
One of the primary mechanisms ovarian cancer uses to evade immune detection involves a variety of techniques to manipulate antigen presentation and T cell activation28. These techniques can include downregulating major histocompatibility complex (MHC) molecules, which are necessary for presenting tumor antigens to cytotoxic T lymphocytes (CTLs). Without this warning signal, CTLs are unaware of how to recognize and destroy cancer cells29. Interestingly, many ovarian tumors upregulate PD-L1, one of the many surface proteins that bind to PD-1 receptors on T cells, and cause the T cells exhaustion. This in return may limit the T cells from mounting a comprehensive anti-tumor response, which occurs even when therapies like checkpoint inhibitors are used30. This mechanism is commonly cited as one of the major limitations of immunotherapies in ovarian cancer compared to other malignancies like melanoma or lung cancer13.
Beyond direct immune cell manipulation, a particularly active area of research is focused on tumor-associated macrophages (TAMs) and their phenotypic reprogramming within the TME. Macrophages are normally flexible cells and can exist in either the pro-inflammatory (M1) or anti-inflammatory (M2) states; however, it appears ovarian tumors have been observed to polarize macrophages towards an M2-like, immunosuppressive phenotype31. It has been demonstrated that these TAMs can secrete IL-10 and TGF-β to inhibit T cell activity and promote immune tolerance32.
Additionally, recent studies with single-cell transcriptomic analysis of the ovarian TME also revealed distinct macrophage populations that were enriched for immunosuppressive markers (e.g., CD163 and MRC1), confirming TAM heterogeneity and immune-checkpoint expression unique to ovarian cancer16. While early studies primarily viewed TAMs as passive producers of cytokines, more recent models suggest they may contribute to post-treatment relapse by remodeling the extracellular matrix and supporting angiogenesis through VEGF secretion33,34. This expanded understanding challenges the previous view of macrophages as passive immune suppressors, indicating their potential role in tumor recovery after chemotherapy. However, the data varied across the different models used, especially between mouse and patient-derived samples, warranting further investigation33.
Regulatory T cells (Tregs), in addition to TAMs, comprise another important immune cell population that contributes to the level of immunosuppression within the ovarian TME.These cells expand within the TME and suppress CTLs through a similar mechanism that involves the secretion of IL-10 and TGF-β, mirroring the effects of TAMs35. Studies indicate that these suppressive effects often overlap, and together they create a layered system of immune evasion that makes it extremely difficult for host immune responses to function effectively, as well as therapeutic interventions to succeed35,36. Again, these shifts do not happen in isolation; cytokines such as IL-6 and TNF-α, secreted by immune and stromal cells, activate subsequent downstream signaling pathways like STAT3 and NF-κB, which in turn signal favorable outcomes for tumor cell survival and dissemination37. Interestingly, while inflammation has traditionally been viewed as an indicator of immune activity, it was consistently associated with worse outcomes in ovarian cancer, including higher recurrence rates. This observation contradicts the conventional views of an ‘active’ immune environment in the context of ovarian cancer progression38.
The immune manipulation described above results in an immunosuppressive, ‘cold’ tumor microenvironment, characterized by limited active CTLs and many immunosuppressive cells. Studies indicate that cold tumors are generally proven to be much less responsive to checkpoint blockade and often relapse easily after chemotherapy39,40. Furthermore, newer therapies(i.e., immune checkpoint inhibitors, CAR-T cells) may be promising and show potential, but it remains unclear whether reactivating exhausted T cells alone is solely sufficient, or if they require combination of the therapies to overcome these deep layers of suppression41. With this uncertainty comes the question of whether future immunotherapies should not only deplete suppressive cells like TAMs and Tregs but also reprogram them, which is an issue that has not been resolved yet in clinical studies.
Adipocytes, Mesothelial Cells, and Omental Immune Cells
In addition to fibroblasts and the traditional immune cells, other populations of omental stromal cells also support ovarian cancer metastasis. These additional cells are being explored because their place of occurrence in the omentum is the most common site of ovarian cancer metastasis42. Adipocytes, or fat cells, can supply fatty acids, which have been associated with fueling tumor growth and secreting cytokines that promote invasion43. Mesothelial cells, which line the peritoneum and omentum, also serve as the first layer that ovarian cancer cells encounter when metastasizing and can be altered by tumor signals to possibly promote adhesion and invasion of ovarian cancer cells. There are also omental immune populations that are in close proximity to clusters of omental immune cells called “milky spots” (small immune hubs in the omentum) that release inflammatory signals to help create a tumor supportive environment for metastasis44. Together, these cells also create a place to allow ovarian cancer to colonize the omentum, but also provide metabolic and immune support linked with promoting therapy resistance.
To summarize, immune manipulation within the TME does not only influence tumor growth but also critically contributes to recurrence. By undermining immune surveillance and the downregulation of recovery mechanisms post-treatment in the ovarian TME, relapse would inherently arise at a greater likelihood. A comprehensive understanding of this immune behavior is therefore essential for developing therapeutic strategies aimed at preventing long-term recurrence.
Cancer Stem Cells
Cancer stem cells (CSCs) are a subgroup of tumor cells that have the ability to self-renew and differentiate into multiple cancer cell types. This adaptability is shown to allow them to survive treatment and evade therapy, which ultimately leads to tumor regrowth. Several papers that often utilize patient-derived samples or xenograft models45, in fact, cite various markers to distinguish between CSCs from the larger, aggregate tumor cell population, such as CD133, CD44, and ALDH1. Recent spatial transcriptomic studies have further shown that CSC-like populations cluster near CAF-dense and hypoxic niches within ovarian tumors, emphasizing that stromal context can potentially help in driving stemness and resistance16.
A definitional property, in combination with CSC’s inherent capacity to modulate, which contributes to these tumors recurring, is their phenotypic plasticity, especially in regard to their ability to undergo epithelial-to-mesenchymal transition (EMT)46. By transitioning into an EMT state, CSCs acquire a temporary resistance to chemotherapy while also being able to subsequently revert back to a tumor-initiating state47. For example, in one 3D model, it demonstrated that CSCs could reinitiate tumors after a transition through EMT even after chemotherapy48. This research adds to the still ongoing discussion of CSC’s role in the occurrence of new tumors and relapse due to the intrinsic plasticity’s ability to develop into states of dormancy. Previously, 2D models could not address these types of bidirectional signaling and communication; rather, the newer organoid work demonstrated that CAFs will react to the CSC- derived cytokines by enhancing ECM remodeling11. Additionally, there is clear evidence that challenges this idea of CSCs being characterized as a dormant population, as they actively participate and co-modulate with their microenvironment. Some papers we reviewed examined different aspects of the development of immune suppression due to IL-6 secretion from CSCs and activation of STAT3 signaling, which enhanced their own stemness but also represented loss of local immune surveillance49. Traditionally, IL-6 is established as a molecule that promotes immune cell recruitment to the tumor needs; however, in recent findings, it also supports cancer stem cell (CSC) maintenance through STAT3 activation, revealing a broader and previously underappreciated role for this cytokine in regulating tumor development. This dual role challenged earlier models that were narrowly focused on cytokine signaling as primarily immune-related. Using patient-derived organoids, a study demonstrated that CSCs were shown to remodel the extracellular matrix in response to treatment and not only adapted to the environment (which is usually how the development of resistant cancer phenotypes is interpreted)11. This remodeling effort of the tumor environment seems to enable other resistant cells to persist as well.
Even though not a main focus in our review of targetable cancer immune evasion, we should also address that in addition to providing important autocrine function, CSCs are also able to establish and engage in bidirectional interactions with CAFs. As already established in several studies, CSCs typically secrete CXCL12 and other factors that then recruit and stimulate CAFs, which in turn promote ECM remodeling and immunosuppressive signaling50. Although there is strong evidence linking CSCs as a significant factor for recurrence, therapeutic targeting of CSCs regarding their targetable forms of immune evasion and/or signaling pathways has produced mixed results51.
Numerous clinical trials have targeted the inhibition of CSC signaling pathways, in particular Notch, Wnt, and Hedgehog, but the results have largely not produced sustainable remission and typically resulted in relapse within six months of treatment duration, suggesting a need for additional therapy and strategies52. Some papers we reviewed provided evidence that targeting CSCs alone may not be enough for a successful treatment intervention, especially if the TME remains supportive53. One area of ongoing debate is whether CSC-targeted therapies need to be combined with other strategies that inhibit stromal signals. For instance , one study showed that simultaneous CSC and stromal inhibition reported improved outcomes in mouse models, while others reported increased toxicity in a similar approach. While these findings generate uncertainty, it further highlights the need for more research when it comes to the ovarian TME51,54.
These results challenge the initial assumption that CSCs are merely drug-resistant survivors. Instead, the reviewed literature suggests that they too contribute to relapse through their ability to interact with and often alter the TME. Ultimately, understanding how CSCs’ communication is subject to change in state, and also how these cells modulate their environment, is crucial for developing long-term therapeutic approaches that prevent recurrence.
In this section, it is crucial that we should understand that many of the resistant behaviors associated with CSCs, are not intrinsic features of the cells themselves but rather the result of their interactions with the microenvironment. This connection between CSCs and the signals exchanged with adjacent stromal and immune cells sets the stage for a further understanding of how direct cell-cell communication is a potential driving factor of phenotypic plasticity in ovarian cancer.
Cell-to-Cell Communication and its Impact on Phenotypic Plasticity
Although the role of chemical signaling in the tumor microenvironment (TME) has been fairly studied, we found that direct physical communication between the cells also contributed substantially to the plasticity and treatment resistance of ovarian cancer. Junctional proteins and adhesion molecules have more substantial purposes than just holding tissue together; tissue consists of individual cells that communicate in a way that allows tumor cells to modify their identity and survive treatment, much like a web of dynamic feedback.
One of the critical usages of direct intercellular communication is mediated by gap junctions, particularly through Connexin 43 (Cx43). Gap junctions are cellular channels that allow the diffusion of ions and small molecules across adjacent cells and therefore coordinate behaviors like growth and migration55. Several ovarian cancer studies that used in vivo and in vitro models reported that loss of Cx43 disrupted communication and coordination between cells, which increased the likelihood of epithelial-to-mesenchymal transition (EMT), a main representation of phenotypic plasticity56. Although gap junctions have often been seen as structural components, newer evidence suggests that loss of gap junctions promotes increased cellular invasiveness and resistance to chemotherapy, highlighting their regulatory function in tumor progression. This challenged the earlier view of Cx43 as just a passive barrier and pointed to it as a more regulatory component57. Like gap junctions, adhesion molecules also emerged as important mediators of plasticity and survival in the cell.
E-cadherin promotes proliferation of epithelial cells by maintaining strong cell-cell adhesion; however, in ovarian cancer, it is often lost. In one recent model, E-cadherin downregulation was correlated to the appearance of EMT and associated with cancer cells being able to spread more easily after chemotherapy58. Integrins, on the other hand, interact with ECM proteins and transmit mechanical signals to the cells. Structural proteins like integrins were found to clearly influence signaling pathways like PI3K/AKT and FAK; pathways that had been previously characterized in cancer stem cell studies, indicating that they were active in regulating tumor cell behavior59. Several models have also shown that integrin signaling has been linked to pro-stemness and pro-survival in post-treatment conditions59,60. Despite their distinct molecular roles, the consequences that come from the disruption of both gap junction and adhesion molecule functions ultimately arrive at one common endpoint: enhanced phenotypic plasticity.
At first, it seemed like each of these molecules operated in separate pathways, but across numerous models including 3D organoids58 and mouse xenografts, evidence indicates that whether the disruption came from adhesion loss or altered ECM sensing, the outcome was consistently associated with cells becoming more mesenchymal and actually more likely to survive treatment. These various molecules, although different in structure and function, ultimately contributed to a more plastic, therapy-resistant state.
While targeting these physical communication pathways presents significant therapeutic promises, there is still uncertainty regarding their selective modulation without disrupting healthy tissue. For instance, there are some mouse (in vivo/vitro ) studies that agree on the premise about how restoring Cx43 has been reported to contain and reduce EMT and slow relapse, while others61 report that Cx43 overexpression interferes with normal repair of healthy, non-cancerous tissues. This raised a question of whether these pathways can be modulated without disrupting healthy tissue function.
Overall, direct cell-to-cell communication is not only important for maintaining tissue integrity; it functions as a critical promoter of plasticity that allows for ovarian cancer cells to survive, adapt, and recur following therapy. Understanding how these physical interactions influence downstream signaling pathways provides a better understanding of the relationship between structural changes in the TME and the underlying mechanisms responsible for phenotypic switching and long-term therapeutic resistance.
Chemical Signals and Their Effects on Ovarian Cancer
Unlike direct physical interactions, chemical signals in the ovarian tumor microenvironment function at a distance and coordinate the behaviors of cancer, stromal, and immune cells with interconnected signaling pathways as shown in Figure 2. Chemical signals can be soluble factors, which include cytokines, growth factors, and chemokines. These signals work together to not just promote phenotypic plasticity but also, in turn, help cancer cells survive and adapt during treatment62.
One of the most studied chemical signals in ovarian cancer is transforming growth factor-beta (TGF-β) found in the TME. In early cancer stages, TGF-β suppresses cell growth, whereas in advanced disease stages, it seems to do the opposite63. In addition,TGF- β also plays a role in promoting epithelial-to-mesenchymal transition (EMT), thus helping tumor cells lose adhesion and gain mobility and invasiveness. Multiple studies reported this64. For example, one study indicated that TGF-β has been shown to be a driving factor of EMT even in chemotherapy-treated cells, which was notable because it suggests this process occurs even after being exposed to the drugs65. These results contrasted with earlier in vitro findings, where they examined the role of TGF-β with respect to proliferation control and excluded findings related to plasticity or immune evasion, etc. In respect to immune cells, TGF-β also can inhibit immune cell activity, particularly T cells, creating a TME that favors an immune-evasive environment66.
While TGF-β is extensively used in the TME, interleukin-6 (IL-6) represents another key signaling molecule with diverse effects on ovarian cancer progression. Throughout our review, we had mostly seen IL-6 mentioned as an inflammation-related cytokine; however, recent papers are describing how it also supports cancer stem cell (CSC) survival and therapy resistance, for example, by activating the JAK/STAT3 pathway67. This finding signified that IL-6 operates at the intersection of stemness and inflammation, linking two processes that were previously considered distinct in ovarian cancer biology. However, some mouse studies have indicated that blocking IL-6 inhibits normal wound healing, thus raising concerns about whether selective inhibition of this cytokine in patients is conceivable68. After the thorough exploration of recent studies on IL6, the findings raise uncertainty of whether IL-6 could be safely targeted or if doing so would disrupt normal tissue recovery69?
Another example is CXCL12, a chemokine secreted by stromal fibroblasts, which also contributes to plasticity in ovarian cancer. This chemokine binds to the CXCR4 receptor on cancer cells and is associated with directing the cancer cells towards supportive niches in the TME and reinforcing stem-like qualities70. One study showed that this signaling loop helped retain the CSC characteristics even in a low-nutrient, post-chemo environments71,8. Though it added another layer to our understanding, it was not just about directing migration under environmental stress but also about preserving adaptability under duress.
Furthermore, vascular endothelial growth factor (VEGF), primarily associated with angiogenesis, also exhibits unexpected roles in promoting cancer cell plasticity72. Models even demonstrated that VEGF-induced hypoxia has been reported to correlate with EMT even without direct TGF-β input73. While VEGF is primarily linked to angiogenesis, new findings suggest it also contributes to phenotypic plasticity by promoting identity shifts in cancer cells. Evidence indicates that when chemical signals are operating outside of their independent signaling roles, they often reinforce each other’s effects on plasticity and survival74.
In order to communicate their signals, ovarian cancer cells make use of not only cytokines and growth factors but also exosomes and non-coding RNA. Exosomes are small vesicles released by cells that carry proteins, lipids, and RNA to other cells and function to deliver packets of information75. In the ovarian TME, tumor-derived exosomes have been shown to possibly inhibit anti-tumor immune responses and transfer drug resistance to neighboring cells. Non coding RNAs (ncRNAs), such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), can regulate gene expression without producing proteins76. Several studies show that exosomal miRNAs can even possibly inhibit chemotherapy-induced cell death, and lncRNAs have yet another mechanism of action by reprogramming immune cells to favor tumor growth77,78. Additionally, in this review, we have specifically examined several central signaling pathways that are most consistently implicated with ovarian cancer progression and resistance to treatment, including TGF-β, IL-6, CXCL12, VEGF, Hippo/YAP/TAZ, PDGF, CXCL8/IL-8, and hypoxia-induced HIF-1 signaling. These pathways were chosen as they demonstrate the key mechanisms of the TME in promoting phenotypic plasticity and even survival under treatment stress79,80. In addition to what has been previously discussed, additional pathways are resurfacing in recent years as recognized regulators in ovarian cancer progression. One of these pathways in the TME is known as the Hippo pathway, which regulates cell growth and division through the effectors YAP and TAZ, protein components that move to the nucleus to activate genes that are also linked with increased stem-like properties and therapy resistance81. Their activity has been associated with the ability of ovarian cancer cells to recur after chemotherapy. Another signal, known as platelet-derived growth factor(PDGF), is secreted by CAF’s to try and stimulate fibroblast growth, angiogenesis, and extracellular matrix remodeling82. These factors all contribute to the development of a supportive microenvironment and contribute to tumor metastasis. CXCL8 or interleukin-8 (IL-8) is another important chemokine that typically recruits immune cells, such as neutrophils; however in ovarian cancer, CXCL8 is also linked with the ability to promote chronic inflammation and even a reduced response to the platinum-based therapies82. The last main key signaling pathway in our search criteria was the protein hypoxia-inducible factor 1(HIF-1) which promotes tumor survival in the low-oxygen settings by activating genes that regulate metabolism and an epithelial-mesenchymal translation. This consequently allows tumors to adapt when stressed by therapy and is associated with recurrence83. All of these pathways highlight how stress, immune, and growth signals support but still reinforce plasticity in the ovarian cancer TME.
Overall, these diverse chemical signals converge on common downstream pathways that can ultimately modulate tumor cell adaptation and resistance. Regardless of their different origins, the downstream effect appears to be similar across the signals in that tumor cells become more adaptable, resistant to treatment, and more immune evasive. This synthesis stood out to us because it showed how the TME supports plasticity not just through one route, but through a system of overlapping mechanisms.
However, the therapeutic targeting of these complex chemical signaling networks presents significant challenges. Some preclinical models suggest that inhibition of VEGF or IL-6 can decrease plasticity, but others report a consistent decrease, or, worse, cite toxicity or unintended immune suppression. For instance, while TGF-β inhibitors show promise, some studies also reported adverse effects in epithelial repair74. This raises the question of whether multi-target strategies might be needed to avoid simply trading one target effect for another.
The central role of chemical signals in shaping identity shifts in cancer cells was a significant finding. What initially appeared as a simple list of ‘growth factors’ transformed into a complex communication system allowing for tumor evolution and survival. This enhanced understanding offers crucial insights into the persistent challenge of treating ovarian cancer and informs potential avenues for future therapeutic intervention.
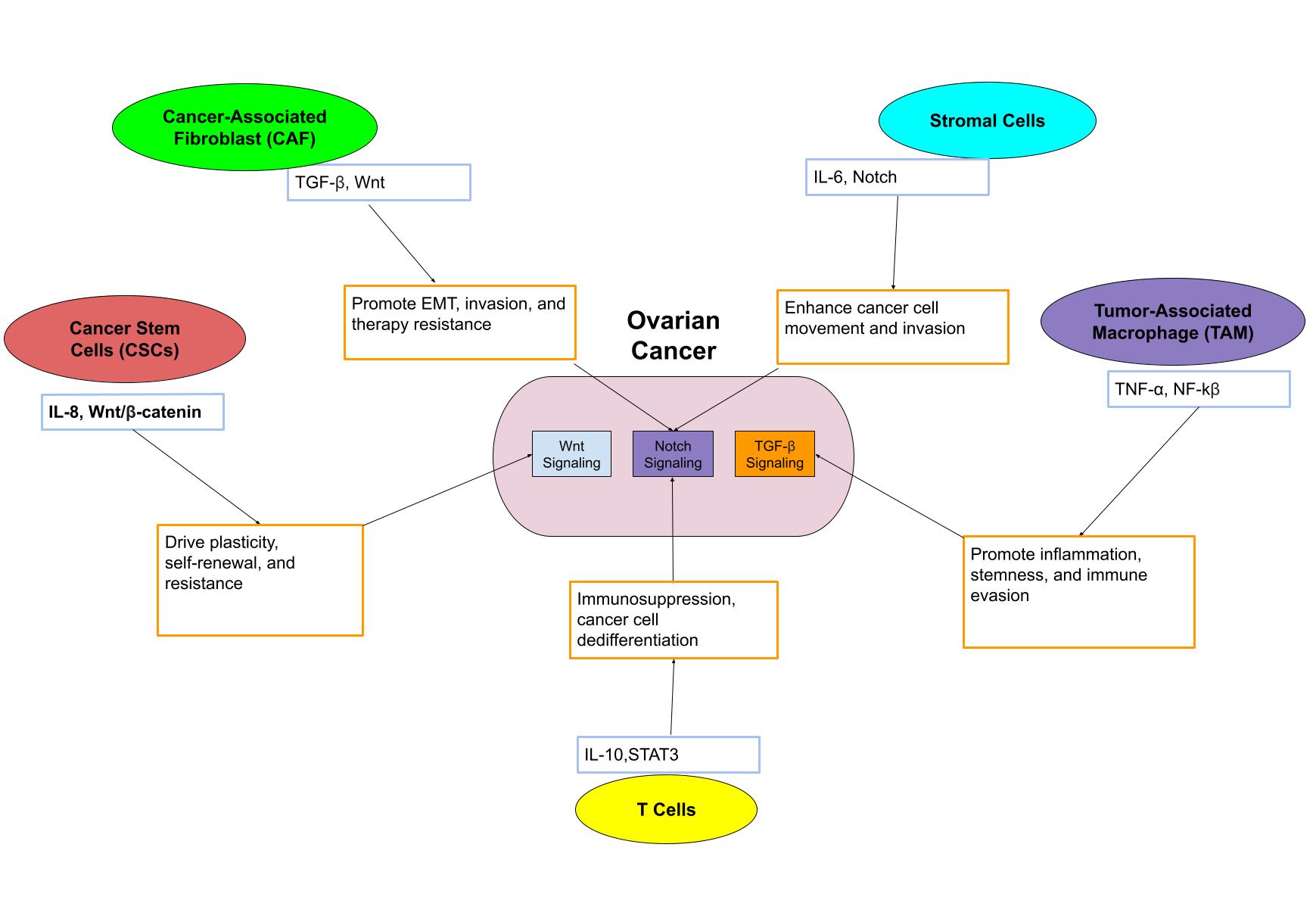
Therapeutic Strategies to Disrupt Cell Communication in Ovarian Cancer
As research has progressed past thinking of ovarian cancer as a disease of dividing cells, new therapeutic strategies are now starting to change and potentially to reflect a systems-level approach of how the tumors are able to manipulate intercellular communication of their microenvironment to survive. Collectively, from the literature, we have seen that targeting cell-to-cell communication, either through soluble signals, ECM interactions, or cell adhesion, has been associated with lowered tumor plasticity and even enhances treatment response.
When reviewing these therapeutic strategies, it is essential to separate findings from preclinical research from findings from human clinical trials. Most of the evidence summarized in this section is drawn from preclinical models, such as xenografts, in vitro assays, and organoid systems, that all shed light on potential mechanisms of signaling disruption within the TME. However, only a few of these approaches, such as TGF-β and Wnt inhibition, have been studied in early-phase human studies where the outcomes have been variable. By distinguishing between these types of evidence, this section illustrates that while there is a great mechanistic promise in preclinical studies, translating this mechanistic promise to consistent clinical benefit remains an ongoing challenge for effective therapy in patients with ovarian cancer.
One of the most extensively studied approaches involves inhibiting the TGF-β signaling pathway. In xenograft models, TGF-β receptor blockade demonstrated decreased EMT markers and showed slowing of metastasis71. The goal of these inhibitors of TGF-β is to interrupt the signaling that allows tumor cells to escape immune detection and modify phenotypes post-chemotherapy84. Still, several other trials have reported immune-related side effects, suggesting dose and context may matter85,74.
Another target that has been the focus of many new therapeutic approaches is Wnt/β-catenin signaling due to its frequent upregulation in ovarian CSCs. One in vitro study demonstrated that Wnt inhibition restored chemosensitivity in platinum-resistant cells, reducing viability by over 50%86, while human trials were ineffective and have struggled with toxicity, especially in normal gut and skin stem cells, highlighting a major challenge in finding a usable approach87.
In addition to targeting soluble signaling pathways, researchers are also studying both biological and physical therapies by disrupting the tumor’s physical interactions with its surroundings. For example, integrins help cells sense mechanical tension in the ECM and respond to circumstances that trigger survival pathways under stress88. In a mouse xenograft study, it was found that blocking integrin αvβ3 reduced EMT and slowed tumor progression. However, these studies were not able to produce these same results in patient-derived organoids, possibly due to different stromal composition or signaling feedback loops89,90.
Targeting CAFs offers another promising avenue to treating ovarian cancer. Inhibiting CAF-derived signals like IL-6 and CXCL12 can destabilize the supportive TME; however, some CAF depletion models led to impaired tissue repair and inflammation rebound, which raised some concerns about long-term safety and the potential for unintended immunosuppressive effects91.
Similarly, attempts to modify ECM composition through studies that attempted to lessen stiffness or collagen cross-linking have shown some success through preclinical findings. For example, the effects of ECM softening in 3D cultures significantly decreased invasive migration, as demonstrated in this study92. These physical interventions may decrease the structural cues promoting plasticity, especially following chemotherapy.
While traditional chemotherapy continues to be a first-line treatment, its success is increasingly understood to be tied to the TME’s variable response. Several studies demonstrate that the chemotherapy-resistant cells will try to utilize their cell-cell signaling to reinitiate tumor growth. In one such study, the CSCs upregulated CXCR4 to migrate into protective stromal niches93. Thus, it is conceivable that we can at least formulate a working hypothesis that traditional therapies might work best when combined with agents that block all of the post-chemo reprogramming back to the TME during this time.
Across these strategies, regardless of whether it targeted soluble signaling, adhesion, or mechanical interactions, the common theme we found was that blocking communication resulted in tumors being less able to shift or survive the treatment stress. Still, this field does present many unanswered questions. While some interventions ( TGF-β inhibition) produced decreased metastasis effects in animal studies, other forms were not able to translate clinically, implying just how complex it is to selectively target the TME without damaging healthy tissues.
| Therapy Target | Mechanism of Action | Supporting Evidence | Limitations/Challenges |
| TGF-β Signaling | Blocks signals that promote EMT and immune evasion in advanced tumors. | Mouse studies show decrease in EMT markers and slower metastasis after administration of TGF-β receptor blockade. | Clinical trials have shown possible immune side effects, dependent upon dose and context. |
| Wnt/β-catenin Pathway | Decreases CSC-driven stemness and resistance through interference of the Wnt pathway. | In vitro experiments found that chemosensitivity was restored in resistant cells by over 50%. | Toxicity observed in gut and skin stem cells may lead to limits of safe delivery in patients. |
| Integrin Signaling | Blocks mechanical sensing in tumor cells, which thereby leads to loss of stress survival and preventing EMT phenotype. | Xenograft studies have shown that it slows tumor growth, but organoids didn’t show the same effect. | Effects varied across model systems; benefits in mice were not always seen in organoids. |
| Cancer-Associated Fibroblasts (CAFs) | Disrupts CAF-derived cytokines like IL-6 and CXCL12 that promote tumor growth. | Studies found that after depleting CAFs, there was a decrease in pro-survival signals but sometimes harmed tissue repair. | Pathway reports cited potential for unintended immune suppression or rebound inflammation signal. |
| Extracellular Matrix (ECM) Remodeling | Softens the tumor environment, which decreases migration and plasticity post- chemotherapy | 3D cultures showed that ECM softening reduced cell invasion after chemo. | Needs more validation in human systems; ECM targeting may affect normal repair. |
| Combination with Chemotherapy | Softens the tumor environment | CSC reprogramming via CXCR4 observed after chemo; pairing with inhibitors blocked regrowth. | Still unclear how to time and dose combinations without increasing toxicity. |
This table outlines the six major strategies currently being explored to disrupt the signaling networks that support ovarian cancer progression and recurrence. The table represents a systems-level approach, one that focuses not only on tumor cells but also on their surrounding environment, which is becoming more important in ovarian cancer research. This figure also illustrates why some therapies that looked promising in mice or 3D models still haven’t translated to safe or consistent results in humans and provides a broader insight into the recent study findings that have contributed to ovarian cancer’s treatment.
In order to offer a clearer review of the evidence evaluated in the review, we compiled representative studies across the major components of the ovarian tumor microenvironment. While reviewing some models(2D cell culture, xenografts, or patient-derived organoids) in these studies, we wanted to distinguish between model systems to show where findings are consistent, limited, or even contradictory. This distinction will help assess to what level each of the mechanisms is supported and which biological model system can offer insights into the ovarian cancer progression and therapeutic resistance.
| TME Component/Focus | Representative Studies | Model Type | Main Summative Findings | Assessment/Contradictions |
| Cancer-Associated Fibroblasts (CAFs) | Zhang et al., 2022 (14); Rizzolio et al., 2022 (20) | In vitro & mouse xenograft | CAFs are shown to be associated with promoting EMT, secreting IL-6/CCL2 and remodeling the ECM to support invasion | Preclinical evidence is consistent in supporting a pro-tumor role, but the results of CAF-depletion studies(21,22) show more mixed outcomes with context-dependent effects. |
| Immune Cells (TAMs, Tregs) | Li et al., 2020 (34); Wang et al., 2025 (31) | In vivo (mouse) & patient samples | TAMS polarize to an M2-like state, which then releases IL-10 and TGF-β in order to suppress CTLs. Tregs can also expand to possibly continue supporting immune tolerance. | Patient data confirms an immunosuppressive phenotype, however, some mouse models overestimate the extent of Treg activity so this model requires further clinical validation. |
| Cancer Stem Cells (CSCs) | Chu et al., 2024 (23); Zhang et al., 2021 (44) | 3D organoid & patient-derived xenograft | CSCs express CD133/CD44/ALDH1 and induce IL-6/STAT3 signaling which enhances stemness and chemoresistance. | The organoid data confirm plasticity mechanisms, but the results of the therapeutic clinical trials in CSCs vary widely. (46, 49). |
| Physical Signaling (Connexins, Integrins) | Aasen et al., 2016 (50); Cooper et al., 2019 (54) | In vitro & xenograft | Loss of Cx43 and integrin signaling are correlated with stimulating EMT and even increasing cell survival post therapeutic intervention. | Mouse models demonstrated a mechanistic correlation whereas the organoid results are contradictory demonstrating the differences in systems. (56). |
| Chemical Signals (TGF-β, IL-6, VEGF, CXCL12) | Tian et al., 2010 (58); Bharti et al., 2016 (62); Guo et al., 2016 (65); D.R. Senger et al., 2017 (67) | In vitro, xenograft & clinical | These cytokines and growth factors have been shown to enhance plasticity and immune evasion; VEGF and TGF-β inhibition are linked to reduced EMT but can also impair normal repair (70). | Preclinical evidence is strong but human studies report toxicity and limited efficacy; benefits thus are context-dependent |
The reviewed studies consistently demonstrated that the CAF and cytokine signaling (e.g., TGF-β, IL-6) were correlating factors with increased EMT and therapy resistance throughout the studies (14, 20, 58, 62), and in vitro assays supported the mechanistic links but lacked the immune complexity. Several CAF depletion and Wnt-inhibition studies (21, 46) contradicted evidence from the prior studies, as those studies had limited survival benefit or toxicity, thus underscoring context-dependent effects. The patient-derived organoids (11, 78) had the most physiologically relevant evidence, as they captured the stroma-immune axis that was absent in other studies, but their findings were again very limited in scale. In summary, these comparisons show that the preclinical models convincingly support TME-driven resistance; these mechanisms do not necessarily translate to consistent outcomes in the clinical setting. Together, these findings reveal clear research trends within the ovarian cancer TME. As illustrated in Figure 5, chemical signaling and immune-evasion mechanisms were reported most often, whereas physical and exosomal communication remain less frequently explored.
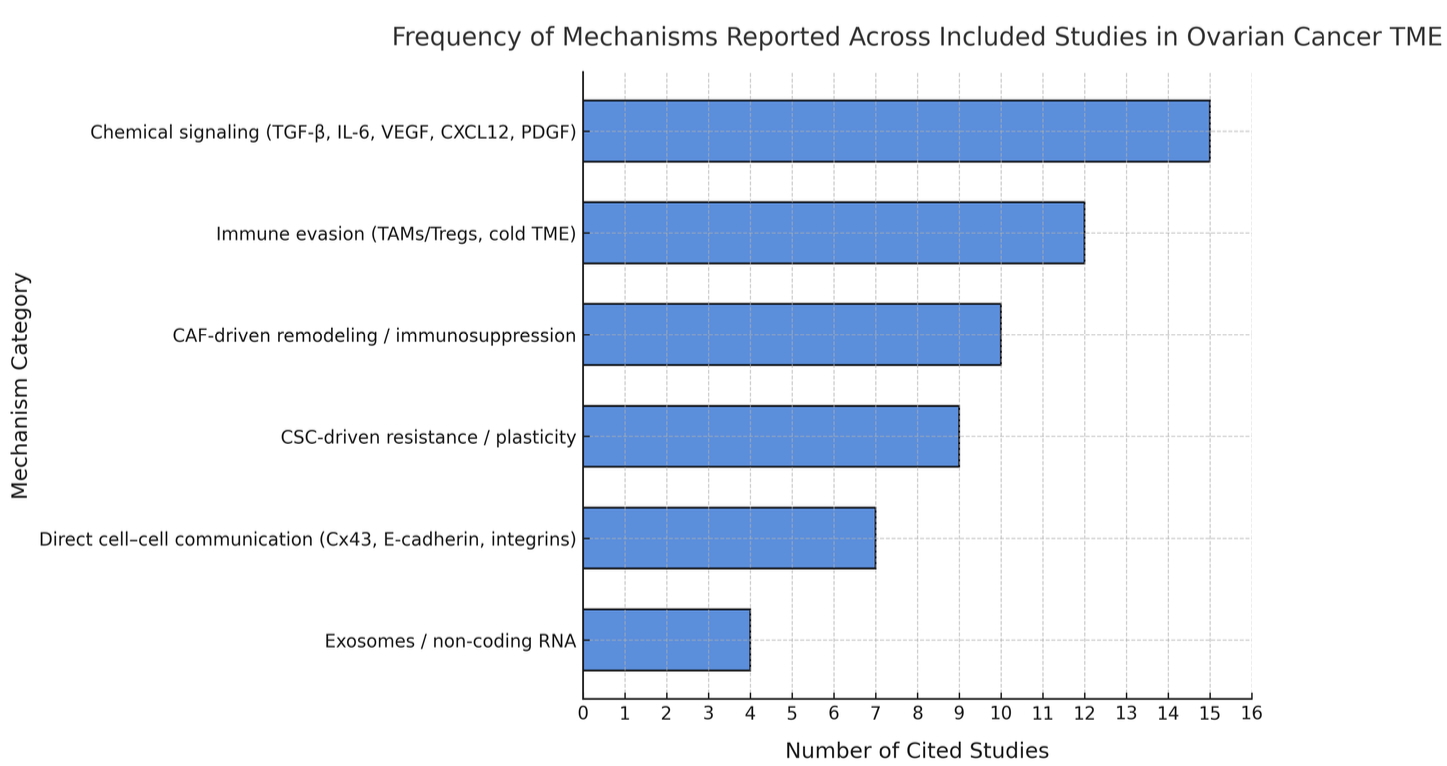
Data: based on coding of mechanisms across the 75 studies reviewed; studies that did not fit predefined categories were excluded from category totals (see Methods)
Discussions
This report examined the role of cell-to-cell communication in the ovarian TME and how it relates to cancer progression, recurrence, and treatment resistance. In all sections of the report, including those focused on immune cells, fibroblasts, stem-like cancer cells, and physical signaling, we observed that the tumor cells adaptations and survival are linked to their local environment. In totality, these findings suggest that relapse is not due to one cell type or communication pathway, but how the layers of communication interrelate with one another and ultimately contribute to recurrence of ovarian cancer.
While it is true that many current therapies, including those discussed in our Therapeutic Strategies section, attempt to target fast-dividing tumor cells, we found that recurrence likely involves the survival of many rare and communication-dependent cells, which are often supported by TME signaling pathways rather than just intrinsic mutations94. This shift in thinking, from treating cancer as a problem of cell division as opposed to understanding cancer as an adaptation issue, promotes the development of treatments that disrupt signaling, adhesion, and immune interactions. The variable outcomes of targeting TGF-β, Wnt, or integrins highlight the difficulty of isolating one pathway in a system where multiple signals overlap74. Nevertheless, some of these techniques produced inconclusive data in organoids or raised safety concerns in animal studies, especially when targeting fibroblasts/strategies that broadly suppress signaling74.
While preclinical models can provide valuable information, there can be many factors involved in direct clinical translation and they often show much inconsistency across different platform systems. Therefore, our research sought to test if an approach to disrupting cell communication could help lessen recurrence from ovarian cancer. While the studies we reviewed support this approach, results seemed to vary by model, emphasizing the importance of testing therapies across systems and better understanding how TME signals interact.
Beyond the variability in experimental models, this systematic review also acknowledges certain limitations. One of the significant limitations to this review was the reliance on preclinical studies, particularly in vitro models and mouse xenograft studies, which do not completely reflect the complexities of signaling heterogeneity and immune dynamics associated with human ovarian tumors. Additionally, several pathways we discussed, such as TGF-β and Wnt, may serve dual roles depending on tumor stage or context, potentially making it harder to target them without interrupting normal tissue repair or immune function.
Looking forward, advancing ovarian cancer treatment necessitates the development of more sophisticated models that integrate a greater complexity of the TME. For example, patient-derived organoids containing fibroblasts, immune cells, and ECM components, as well as more advanced research tools such as single-cell RNA sequencing and spatial transcriptomics, can allow researchers to determine and evaluate what changes in signaling networks occurred after an intervention. More generally, we want to advance therapies that not only prevent elimination of the cancer cells but also limit the recovery of the communication networks that enable tumors to re-establish themselves.
In conclusion, by focusing on how ovarian cancer cells are able to engage and communicate with their environment, we gain a clearer understanding of why recurrence happens and how we may prevent it in the future. Disrupting these communication systems and networks in cancer, rather than simply attempting to target cancer cells in isolation, may offer a more sustainable long-term strategy to prevent relapse. Despite the challenges for translating this knowledge into effective therapies, it will always remain important to characterize the adaptive behaviors of tumors within the TME. This systems-level view of cancer progression will help guide the development of future treatments that respond to not just how the tumor is evolving but also anticipate how it will grow.
- Y. Yang, Y. Yang, J. Yang, X. Zhao, X. Wei. Tumor microenvironment in ovarian cancer:function and therapeutic strategy. Frontiers in Cell and Developmental Biology. 8, 758 (2020). [↩]
- J. Sun, K.M. Bogie, J. Teagno, Y.-H. Sun, R.R. Carter, L. Cui, G.-Q. Zhang. Design and implementation of a comprehensive web-based survey for ovarian cancer survivorship with an analysis of prediagnosis symptoms via text mining. Cancer Informatics. 13, 113–123 (2014). [↩]
- National Cancer Institute. Cancer Stat Facts: Ovarian Cancer. https://seer.cancer.gov/statfacts/html/ovary.html (2023). [↩]
- G. Zheng, H. Yu, A. Kanerva, A. Försti, K. Sundquist, K. Hemminki. Familial risks of ovarian cancer by age at diagnosis, proband type and histology. Journal of Clinical Medicine. 7, 330 (2018). [↩]
- R. Wei, S. Liu, S. Zhang, L. Min, S. Zhu. Cellular and extracellular components in tumor microenvironment and their application in early diagnosis of cancers. Analytical Cellular Pathology(Amst). 2020, 6283796 (2020). [↩]
- B. Yue. Biology of the extracellular matrix: an overview. Journal of Glaucoma. 23 , S20 (2014). [↩]
- D. Yang, J. Liu, H. Qian, Q. Zhuang. Cancer‑associated fibroblasts: from basic science to anticancer therapy. Nature Reviews Molecular Cell Biology. 24, 677–692 (2023). [↩]
- L.T.H. Phi, I.N. Sari, Y.-G. Yang,S-H. Lee, N. Jun, K.S. Kim, Y.K. Lee, H.Y. Kwon. Cancer stem cells(CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cell International. 19, 321-334 (2018). [↩] [↩]
- A. Tiwari, R. Trivedi, S.-Y. Lin. Tumor microenvironment: barrier or opportunity towards effective cancer therapy. Journal of Biomedical Science. 29, 83 (2022). [↩]
- A. Chandra, C. Pius, M. Nabeel, M. Nair, J.K. Visgwanatha, S. Ahmad, R. Basha. Ovarian cancer: current status and strategies for improving therapeutic outcomes. Cancer medicine. 8, 7018–7031(2019). [↩]
- A. Dominijanni, M. Devarasetty, S. Soker. Manipulating the Tumor Microenvironment in Tumor Organoids Induces Phenotypic Changes and Chemoresistance. iScience. 23, 101851 (2020). [↩] [↩] [↩]
- A. Pawłowska, A. Rekowska, W. Kurylo, A. Panczyszyn, J. Kotaski, I. Wertel. Current understanding on why ovarian cancer is resistant to immune checkpoint inhibitors. International Journal of Molecular Sciences. 24, 10859 (2023). [↩]
- K. De Veirman, L. Rao, E.D. Bruyne, E.Menu, E.V.Valckenborgh, I.V. Riet, M.A. Frassantio, L.D. Marzo, A. Vacca, K. Vanderkerken. Cancer associated fibroblasts and tumor growth: focus on multiple myeloma. Cancers. 6, 1363–1381 (2014). [↩] [↩]
- M. Zhang, Z. Chen, Y. Wang, H. Zhao, Y. Du. The role of cancer-associated fibroblasts in ovarian cancer. Cancers. 14, 2637(2022). [↩]
- B. Izar, I. Tirosh, E. H. Stover, I. Wakiro, M. S. Cuoco, I. Alter, C. Rodman, R. Leeson, M.-J. Su, P. Shah, M. Iwanicki, S. R. Walker, A. Kanodia, J. C. Melms, S. Mei, J.-R. Lin, C. B. M. Porter, M. Slyper, J. Waldman, L. Jerby-Arnon, O. Ashenberg, T. J. Brinker, C. Mills, M. Rogava, S. Vigneau, P. K. Sorger, L. A. Garraway, P. A. Konstantinopoulos, J. F. Liu, U. Matulonis, B. E. Johnson, O. Rozenblatt-Rosen, A. Rotem, A. Regev. A single-cell landscape of high-grade serous ovarian cancer. Nature Medicine. 26, 1271–1279 (2020). [↩] [↩]
- E. Denisenko, L. de Kock, A. Tan, A. B. Beasley, M. Beilin, M. E. Jones, R. Hou, D. Ó Muirí, S. Bilic, G. R. K. A. Mohan, S. Salfinger, S. Fox, K. P. W. Hmon, Y. Yeow, Y. Kim, R. John, T. S. Gilderman, E. Killingbeck, E. S. Gray, P. A. Cohen, Y. Yu & A. R. R. Forrest. Spatial transcriptomics reveals discrete tumour microenvironments and autocrine loops within ovarian cancer subclones. Nature Communications. 15, Article 2860 (2024). [↩] [↩] [↩] [↩]
- Y. Zhang, N. Lv, M. Li, M. Liu. Cancer-associated fibroblasts: tumor defenders in radiation therapy. Cell Death & Disease. 14, 541 (2023). [↩]
- M. Soundararajan, S. Kannan. Fibroblasts and mesenchymal stem cells : Two sides of the same coin? Journal of Cellular Physiology. 233, 9099-9109(2018). [↩]
- F. Zhang, Y. Ma, D. Li, J. Wei, K. Chen, E. Zhang, G. Liu, X. Chu, X. Liu, W. Liu, X. Tian, Y. Yang. Cancer-associated fibroblasts and metabolic reprogramming: unraveling the intricate crosstalk in tumor evolution. Journal of Hematology & Oncology. 17, 80 (2024). [↩]
- A. Filer, C.D. Buckley. Fibroblasts and stromal cells. Fundamentals of Inflammation(Chapter X), 126-140 (2010). [↩]
- L. Li, R. Yu, T. Cai, Z. Chen, M.Lan, T. Zou, B. Wang, Q. Wang, Y. Zhao, Y. Cai. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. International Immunopharmacology. 88, 106939(2020). [↩]
- S. Rizzolio, S. Giordano, S. Corso. The importance of being CAFs (in cancer resistance to targeted therapies). Journal of Experimental & Clinical Cancer Research. 41, 319(2022). [↩]
- K. Yoshikawa, M. Ishida, H. Yanai, K. Tsuta, M. Sekimoto, T. Sugie. Prognostic significance of PD‑L1‑positive cancer‑associated fibroblasts in patients with triple‑negative breast cancer. BMC Cancer. 21, 239 (2021). [↩]
- M. Haro, S. Orsulic. A Paradoxical Correlation of Cancer-Associated Fibroblasts With Survival Outcomes in B-Cell Lymphomas and Carcinomas. Frontiers in Cell and Developmental Biology. 6, 00098(2018). [↩]
- X. Chu, W. Tian, J. Ning, G. Xiao, Y. Zhou, Z. Zhai, G. Tanzhu, J. Yang, R. Zhou. Cancer stem cells: advances in knowledge and implications for cancer therapy. Signal transduction and targeted therapy. 9, 170 (2024). [↩]
- R. Prakash, C.M. Telleria. Cancer cell repopulation after therapy: which is the mechanism? Oncoscience. 10, 14–19 (2023). [↩]
- F. Zhang, Y. Ma, D. Li, J. Wei, K. Chen, E. Zhang, G. Liu, X. Chu, X. Liu, W. Liu, X. Tian, Y. Yang. Cancer-associated fibroblasts and metabolic reprogramming: unraveling the intricate crosstalk in tumor evolution. Journal of Hematology & Oncology. 17, 80 (2024 [↩]
- R. Gupta, R. Kumar, C.A. Penn, N. Wajapeyee. Immune evasion in ovarian cancer: implications for immunotherapy and emerging treatments. Trends Immunology. 46, 166-181(2025). [↩]
- B.C. Taylor, J.M. Balko. Mechanisms of MHC‑I downregulation and the role in immunotherapy response. Frontiers in Immunology. 13, 844866(2022). [↩]
- A. Dumitru, E-C Dobrica, A. Croitoru, S.M. Cretoiu, B.S. Gaspar. Focus on PD‑1/PD‑L1 as a therapeutic target in ovarian cancer. International Journal Molecular Sciences. 23,12067 (2022). [↩]
- E. Cendrowicz, Z. Sas, E. Bremer, T. Rygiel. The Role of Macrophages in Cancer Development and Therapy. Cancers(Basel). 13, 1946(2021). [↩]
- Y. Wang, C. Ma, X. Li, F. Yang, N. Wang, G. Ji, Q. Liu, H. Zhu, S. Zu,H. Li. Unraveling the role of M2 TAMs in ovarian cancer dynamics: a systematic review. Journal of translational medicine. 23, 633(2025). [↩]
- V. Riabov, A. Gudima, N. Wang, A. Mickley, A. Orekhov, J. Kzhyshkowska. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Frontiers in Physiology. 5, 75(2014). [↩] [↩]
- C. Guo, A. Buranych, D. Sarkar, P.B. Fisher, X-Y. Wang. The role of tumor-associated macrophages in tumor vascularization. Vascular cell. 5, 20(2013). [↩]
- C. Li, P. Jiang, S. Wei, X. Xu, J. Wang. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Molecular Cancer. 19, 116 (2020). [↩] [↩]
- S. Imani, R. Farghadani, G. Roozitalab, M. Maghsoudloo, M. Emadi, A. Moradi, B. Abedi, P. Jabbarzadeh Kaboli. Reprogramming the breast tumor immune microenvironment: cold-to-hot transition for enhanced immunotherapy. Journal of Experimental & Clinical Cancer Research. 44, 131 (2025). [↩]
- E.N. Scott, A.M. Gocher, C.J. Workman, D.A.A. Vignali. Regulatory T cells: barriers of immune infiltration into the tumor microenvironment. Frontiers in Immunology. 12, 702726 (2021). [↩]
- D.S. Benito, E. Vercher, E. Conde, J. Glez-vaz, I. Tamayo, S. Hearvas-stubbs. Inflammation and immunity in ovarian cancer. European Journal of Cancer Supplements. 15, 56-66(2020). [↩]
- P. Ouyang, L. Wang, J. Wu, Y. Tian, C. Chen, D. Li, Z. Yao, R. Chen, G. Xiang, J. Gong, Z. Bao. Overcoming cold tumors: a combination strategy of immune checkpoint inhibitors. Frontiers in Immunology. 15, 1344272 (2024). [↩]
- P. Bonaventura, T. Shekarian, V. Alcazer, J. Valladeau‑Guilemond, S. Valsesia‑Wittmann, S. Amigorena, C. Caux, S. Depil. Cold tumors: a therapeutic challenge for immunotherapy. Frontiers in Immunology. 10, 168 (2019). [↩]
- P. Ouyang, L. Wang, J. Wu, et al. Overcoming cold tumors: a combination strategy of immune checkpoint inhibitors. Frontiers in Immunology. 15, 1344272 (2024). [↩]
- T.-L. Yeung, C. S. Leung, K.-P. Yip, C. L. Au Yeung, S. T. Wong, S. C. Mok. Cellular and molecular processes in ovarian cancer metastasis. American Journal of Physiology – Cell Physiology. 309, C444-C456 (2015). [↩]
- K. M. Nieman, I. L. Romero, B. Van Houten, E. Lengyel. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochimica et Biophysica Acta. 1831, 1533-1541 (2013). [↩]
- K. A. Kenny, C. Y. Chiang, E. A. White, E. M. Schryver, M. Habis, I. L. Romero, A. Ladanyi, C. V. Penicka, J. George, K. Matlin, K. Montag, K. Wroblewski, S. D. Yamada, A. P. Mazar, D. Bowtell, E. Lengyel. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. Journal of Clinical Investigation. 124, 4614–4628 (2014) [↩]
- H.E. Lien, M.E. Hjelmeland, H.F. Berg, et al. Multiplex single‑cell profiling of putative cancer stem cell markers in endometrial cancer. Molecular Oncology. 19, 1651–1667 (2025). [↩]
- T. Celià‑Terrassa, M.K. Jolly. Cancer stem cells and epithelial‑to‑mesenchymal transition in cancer metastasis. Cold Spring Harbor. Perspectives in Medicine. 10, a036905 (2020). [↩]
- R. Paul, J.F. Dorsey, Y. Fan. Cell plasticity, senescence, and quiescence in cancer stem cells: biological and therapeutic implications. Pharmacology & Therapeutics. 231, 107985 (2022). [↩]
- H. Zhang, A. Steed, M. Co, X. Chen. Cancer stem cells, epithelial‑mesenchymal transition, ATP and their roles in drug resistance in cancer. Cancer Drug Resistance. 4, 684–709 (2021). [↩]
- C. Zhang, K. Ma, W-Y Li. IL-6 Promotes Cancer Stemness and Oncogenicity in U2OS and MG-63 Osteosarcoma Cells by Upregulating the OPN-STAT3 Pathway. Journal of Cancer. 10, 6511-6525 (2019). [↩]
- S. Babazdeh, S.M. Nassiri, V. Siavashi, M. Sahlabadi, M. Hajinasrollah, M. Zamani-Ahmadmahmudi. Macrophage polarization by MSC-derived CXCL12 determines tumor growth. Cellular & Molecular Biology Letters. 26 , 30(2021). [↩]
- T-S. Chan, Y. Shaked, K.K. Tsai. Targeting the Interplay Between Cancer Fibroblasts, Mesenchymal Stem Cells, and Cancer Stem Cells in Desmoplastic Cancers. Frontiers in Oncology. 9, 00688(2019). [↩] [↩]
- P.S. Kharkar. Cancer Stem Cell (CSC) Inhibitors in Oncology─A Promise for a Better Therapeutic Outcome: State of the Art and Future Perspectives. Journal of medicinal chemistry. 63, 15279– 15307(2020). [↩]
- H.-R. Sun, S. Wang, S.-C. Yan, P.J. Nelson, H-L. Jia, L-X. Qin, Q-Z. Dong. Therapeutic strategies targeting cancer stem cells and their microenvironment. Frontiers Oncology. 9, 1104 (2019). [↩]
- J. Cooper, F. G. Giancotti. Integrin signaling in cancer: Mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell 35, 347–367 (2019). [↩]
- T. Aasen, M. Mesnil, C.C. Naus, P.D. Lampe, D.W. Laird. Gap Junctions and Cancer: Communicating for 50 Years. Nature Reviews Cancer. 16, 775-788(2016). [↩]
- M. Sinyuk, E.E. Mulkearns-Hubert, O. Reizes, J. Lathia. Cancer connectors: connexins, gap junctions, and communication. Frontiers in Oncology. 8, 646 (2018). [↩]
- R.E. Strauss, R.G. Gourdie. Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. International Journal of Molecular Sciences. 21, 8355(2020). [↩]
- C.-Y. Loh, J. Y. Chai, T. F. Tang, W. F. Wong, G. Sethi, M. K. Shanmugam, P. P. Chong, C. Y. Looi. The E-Cadherin and N-Cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 8, 1118 (2019). [↩] [↩]
- E. Alsina-Sanchís, A. Figueras, Á. Lahiguera, M. Gil-Martín, B. Pardo, J. M. Piulats, L. Martí, J. Ponce, X. Matias-Guiu, A. Vidal, A. Villanueva, F. Viñals. TGFβ controls ovarian cancer cell proliferation. International Journal of Molecular Sciences. 18, 1658 (2017). [↩] [↩]
- G. Wu, Y. Li. TGF-β induced reprogramming and drug resistance in triple-negative breast cells. BMC Pharmacology and Toxicology. 23, 23 (2022). [↩]
- D.W. Laird, P.D. Lampe. Therapeutic strategies targeting connexins. Nature Reviews Drug Discovery 17, 905–921 (2018). [↩]
- Z-D. Shi, K. Pang, Z-X. Wu, Y. Dong, L. Hao, J-X. Qin, W. Wang, Z-S. Chen, C-H. Han. Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies. Signal Transduction and Targeted Therapy. 8, 113 (2023). [↩]
- M. Tian, J.R. Neil, W.P. Schiemann. Transforming Growth Factor-β and the Hallmarks of Cancer. Cell Signal. 6, 951-962(2010). [↩]
- H.J. Zhu, Y.S. Li, Y.P. Wang, X.H. Hu, X.R. Zhang, L. Qiu, W/F/ He, G.X. Luo. Effects of skin γδ T lymphocytes on wound healing of mice through regulating proliferation and differentiation of mice epidermal cells. Chinese Journal of Burns. 35, 298-305(2019). [↩]
- F. Guo, Y. Wang, J. Liu, S.C. Mok, F. Xue, W. Zhang. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. 35, 816-826(2016). [↩]
- A. Dahmani, J-S. Delisle. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Caners. 10, 194(2018). [↩]
- R. Bharti, G. Dey, M. Mandal. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Science Direct. 375, 51-61(2016). [↩]
- K. Prystaz, K. Kaiser, A. Kovtun, M. Haffner-Luntzer, V. Fischer, A. E. Rapp, A. Liedert, G. Strauss, G. H. Waetzig, S. Rose-John, A. Ignatius. Distinct effects of IL-6 classic and trans-signaling in bone fracture healing. American Journal of Pathology. 188, 474–490 (2018). [↩]
- X. Wei, Y. Chen, X. Jiang, M. Peng, Y. Liu, Y. Mo, D. Ren, Y. Hua, B. Yu, Y. Zhou, Q. Liao, H. Wang, B. Xiang, M. Zhou, X. Li, G. Li, Y. Li, W. Xiong, Z. Zeng. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Molecular Cancer. 20, 7 (2021). [↩]
- H. Ramirez, S. B. Patel, I. Pastar. The role of TGFβ signaling in wound epithelialization. Advances in Wound Care. 3, 482–491 (2014). [↩]
- M. Lahn, S. Kloeker, B. S. Berry. TGF-β inhibitors for the treatment of cancer. Expert Opinion on Investigational Drugs. 14, 629–643 (2005). [↩] [↩]
- D.R. Senger. Vascular Endothelial Growth Factor: Much More than an Angiogenesis Factor. Molecular Biology of the Cell. 21, 3(2017). [↩]
- O. Gonzalez-Moreno, J. Lecanda, J. E. Green, V. Segura, R. Catena, D. Serrano, A. Calvo. VEGF elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (PIN)-like cells via an autocrine loop. Experimental Cell Research. 316, 554–567 (2010). [↩]
- L. K. Huynh, C. J. Hipolito, P. ten Dijke. A perspective on the development of TGF-β inhibitors for cancer treatment. Biomolecules. 9, 743 (2019). [↩] [↩] [↩] [↩] [↩]
- R. Kalluri, V. S. Le Bleu. The biology, function, and biomedical applications of exosomes. Science. 367, eaau6977 (2020). [↩]
- J. C. R. Fernandes, S. M. Acuña, J. I. Aoki, L. M. Floeter-Winter, S. M. Muxel. Long non-coding RNAs in the regulation of gene expression: physiology and disease. Non-coding RNA. 5, 17 (2019). [↩]
- P. Santos, F. Almeida. Role of exosomal miRNAs and the tumor microenvironment in drug resistance. Cells. 9, 1450 (2020). [↩]
- Z. Luo, W. Zhang, Y. Zhang, L. Wang. Exosomal long non-coding RNAs: emerging players in the tumor microenvironment. Molecular Therapy – Nucleic Acids. 21, 546-558 (2020). [↩]
- H. Liu, M. Sun, N. Wu, B. Liu, Q. Liu, X. Fan. TGF-β/Smads signaling pathway, Hippo-YAP/TAZ signaling pathway, and VEGF: their mechanisms and roles in vascular remodeling related diseases. Immunology and Inflammation Diseases. 11, e1060 (2023). [↩]
- S. P. Sayers, P. M. Clarkson. Force recovery after eccentric exercise in males and females. European Journal of Applied Physiology. 84, 122–126 (2001). [↩]
- R. B. Mokhtari, N. Ashayeri, L. Baghaie, M. Sambi, K. Satari, N. Baluch, D. A. Bosykh, M. R. Szewczuk, S. Chakraborty. The Hippo pathway effectors YAP/TAZ-TEAD oncoproteins as emerging therapeutic targets in the tumor microenvironment. Cancers. 15, 3468 (2023). [↩]
- Z. Fang, Q. Meng, J. Xu, W. Wang, B. Zhang, J. Liu, C. Liang, J. Hua, Y. Zhao, X. Yu, S. Shi. Signaling pathways in cancer-associated fibroblasts: recent advances and future perspectives. Cancer Communications (London). 43, 3–41 (2023). [↩] [↩]
- L. K. Kirschner, H. A. Smith, K. N. Reddy, T. D. Wilson, S. M. Patel. Hypoxia-inducible factor-1 regulation and phosphorylation dynamics in stressed cells. Journal of Cell Biochemistry. 117, 267–278 (2016). [↩]
- C.-Y. Huang, C.-L. Chung, T.-H. Hu, J.-J. Chen, P.-F. Liu, C.-L. Chen. Recent progress in TGF-β inhibitors for cancer therapy. Biomedicine & Pharmacotherapy. 134, 111046 (2021). [↩]
- H. Ungefroren. Blockade of TGF-β signaling: a potential target for cancer immunotherapy? Expert Opinion on Therapeutic Targets. 23, 679–693 (2019). [↩]
- A. B. Nagaraj, P. Joseph, O. Kovalenko, S. Singh, A. Armstrong, R. Redline, K. Resnick, K. Zanotti, S. Waggoner, A. DiFeo. Critical role of Wnt/β-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget. 6, 23720–23734 (2015). [↩]
- M. Khan. Can we safely target the WNT pathway?. Nature Reviews Drug Discovery 13, 513–532 (2014). [↩]
- Z. Sun, S. S. Guo, R. Fässler. Integrin-mediated mechanotransduction. Journal of Cell Biology. 215, 445–456 (2016). [↩]
- K. Mitsuyuki, T. Watabe, S. Naka, Y. Liu, M. Tatsumi, E. Shimosegawa, H. Kato. Evaluation of integrin αvβ3 expression in murine xenograft models: [68Ga]Ga-DOTA-C(RGDfK) PET study with immunohistochemical confirmation. International Journal of Molecular Sciences. 20, 4008 (2019). [↩]
- Y.-Q. Chen, H.-Y. Song, Z.-Y. Zhou, J. Ma, Z.-Y. Luo, Y. Zhou, J.-Y. Wang, S. Liu, X.-H. Han. Osthole inhibits the migration and invasion of highly metastatic breast cancer cells by suppressing ITGα3/ITGβ5 signaling. Acta Pharmacologica Sinica. 43, 1544–1555 (2022). [↩]
- Y. E. Lee, G.-Y. Go, E.-Y. Koh, H.-N. Yoon, M. Seo, S.-M. Hong, J. H. Jeong, J.-C. Kim, D. Cho, T. S. Kim, S. C. Kim, E. Jun, M. Jang. Synergistic therapeutic combination with a CAF inhibitor enhances CAR-NK-mediated cytotoxicity via reduction of CAF-released IL-6. Journal for Immuno Therapy of Cancer. 9, e178849 (2024). [↩]
- X. Feng, F. Cao, X. Wu, W. Xie, P. Wang, H. Jiang. Targeting extracellular matrix stiffness for cancer therapy. Frontiers in Oncology. 14, 1393749 (2024). [↩]
- Y. Zhao, M. He, L. Cui, M. Gao, M. Zhang, F. Yue, T. Shi, X. Yang, Y. Pan, X. Zheng, Y. Jia, D. Shao, J. Li, K. He, L. Chen. Chemotherapy exacerbates ovarian cancer cell migration and cancer stem cell-like characteristics through GLI1. British Journal of Cancer. 122, 1638–1648 (2020). [↩]
- N. Moore, J. Houghton, S. Lyle. Slow-cycling therapy-resistant cancer cells. Stem Cells and Development. 21, 1822–1830 (2012). [↩]



{kind=link}