
Abstract
Non-small cell lung cancer is the leading cause of cancer-related deaths in the U.S. Current chemotherapies lack selectivity, making enzymes over-expressed in cancer cells such as Methylenetetrahydrofolate Dehydrogenase 2 (MTHFD2) promising therapeutic targets. While existing inhibitors selectively bind to MTHFD2 with high potency, their poor permeability and selectivity prevents clinical implementation. This study aims to identify permeable inhibitors that target MTHFD2 through its allosteric site using computational simulations in Schrodinger’s Maestro. Computational screening techniques in Maestro were performed on a library of compounds, prioritizing predicted Caco-2 permeability and strongest binding affinity. The top 10 compounds identified in this study demonstrated high permeability while the control indicated low permeability. While the top compound’s permeability was higher than the control’s (Caco-2 684 nm/sec vs. 0.864 nm/sec, respectively), a two-tailed Mann Whitney U-Test could not detect a statistically significant difference in the binding score between them. However, numerical values suggest that this study’s top compound may be able to enter cancer cells more effectively than current MTHFD2 inhibitors with a slightly stronger level of binding potency (ΔG binding score –72.39 kcal/mol for top compound vs. -71.53 kcal/mol for control, respectively). Drug likeness of these top 11 compounds was evaluated for clinical application, with 2 offering the most promising results. These findings identify computational compounds with predicted properties that could address the permeability barrier of current inhibitors. Further validation of the compounds’ in-vitro potency may contribute to the development of a more effective, targeted non-small cell lung cancer treatment.
Introduction
Lung Cancer Statistics
Lung Cancer is the leading cause of cancer-related deaths in the United States and it accounts for 20% of all cancer deaths. The American Cancer Society estimates 226,650 new cases (110,680 in men and 115,970 in women) and 124,730 deaths (64,190 in men and 60,540 in women) in 2025 alone, with non-small cell lung cancer (NSCLC) accounting for 87% of these cases1. Despite advances in therapies and treatments, Surveillance, Epidemiology and End Results (SEER) data indicates a 5-year survival rate of 37.1% for regional and 9.7% for distant NSCLC2. Current therapies like chemotherapy are non-specific and impact both the cancerous cells and the normal cells. Chemotherapy has a number of potentially lethal side effects due to its chemotoxicity, and some life-threatening toxicities include myelosuppression, febrile neutropenia, severe oral mucositis, and sepsis3. Such statistics highlight the need for more selective therapies for NSCLC. One strategy for achieving such selectivity is targeting metabolic enzymes that are overexpressed in cancer cells but minimally expressed in healthy cells. Among these, MTHFD2 has attracted significant interest.
MTHFD2 as a Cancer Target
One promising metabolic target is methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), a mitochondrial enzyme that supports the folate-mediated one-carbon metabolism. It is a bifunctional enzyme that converts CH2-THF to 5,10-methenyl-tetrahydrofolate through dehydrogenase activity. Then, it converts CH+-THF to 10-CHO-THF through a cyclohydrolase activity. Ultimately, it catalyzes the production of 10-CHO-THF, which is then sent to the next enzyme in the metabolism4. This enzyme has been shown to be overexpressed in a variety of cancers while having low or undetectable expression in normal adult tissues5. Additionally, it has been shown to be involved in the proliferation, migration, and apoptosis of lung cancer cells by mediating MCM4, MCM7, ZEB1, Vimentin, and SNAI1, which contributed to tumor cell growth6. Data confirmed that the MTHFD2 gene and protein was upregulated in lung cancer patients and was correlated with poor survival in lung cancer6. Additional testing on NSCLC cancer cell lines found that the inhibition of MTHFD2 suppressed the progression of the cell lines and induced apoptosis of NSCLC cells7. Although MTHFD2’s role in cancer is known, certain drug design challenges have hindered the effectiveness of current inhibitors.
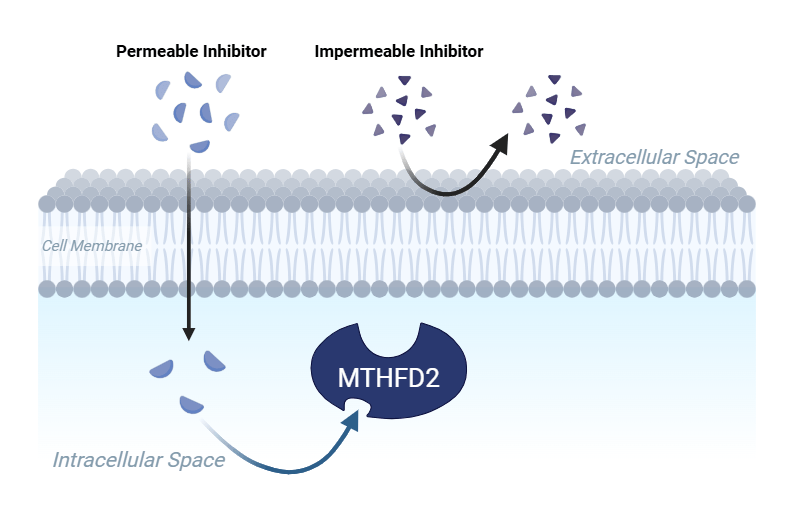
Problem Statement and Rationale
Currently, MTHFD2 inhibitors have been identified, but their lack of permeability limits their cellular efficacy. Additionally, the enzyme lies within two membrane-bound organelles which makes the transport of drug into both the mitochondria and the nucleus important for clinical implementation8. TH9619, an identified inhibitor for MTHFD2, was found to be highly potent (IC50 = 47 nM), but had poor passive permeability due to its low lipophilicity (cLogP: -2.25) and large polar surface area (239 Å2)9. The same inhibitor targeted MTHFD1, a similarly structured protein that does not have the unique expression pattern, indicating low selectivity (and potential off-target effects)10. Because of this, TH9619 inhibits both MTHFD2 and the DC domain of MTHFD1, which results in thymidine depletion and subsequently the suppression of leukemic tumor growth10. A 2024 field review by Ramos et al. noted that TH9619 is not being transported into the mitochondria and it prevents the inhibitor from binding to mitochondrial MTHFD210. This characteristic creates a cellular uptake challenge that prevents TH9619 from being implemented clinically as it is incapable of permeating into the mitochondria. To overcome selectivity barriers, Ma et al. crystallized the structure of MTHFD2 with xanthine derivative 15. This compound was found to bind to MTHFD2 through an allosteric site (a unique site on the protein) which inhibited MTHFD2 and exhibited no activity against MTHFD111. Previous studies have performed pharmacophore-based virtual screenings of natural products that target MTHFD2’s allosteric site, but they overlook cell permeability and optimization of the compounds12. This study also targets MTHFD2’s allosteric site but performs an early-stage permeability screening, uses a chemically diverse library beyond natural products, and applies bioisosteric modifications to improve the potency and permeability. Addressing these challenges could identify inhibitors that are biologically and clinically effective.

Significance and Purpose
An inhibitor that is selective to MTHFD2 and permeable addresses two problems associated with current MTHFD2 inhibitors: off-target effects and limited clinical viability. If confirmed by in-vitro and in-vivo studies in the future, the compounds identified could become starting points for a selective therapy for non-small cell lung cancer.
Objectives
This study aims to identify cell-permeable, allosteric MTHFD2 inhibitors through permeability focused virtual screening. The investigation intends to generate a suitable list of top compounds for biochemical validation. Research Question: How do computationally identified MTHFD2 inhibitors with high cell permeability compare to current inhibitors in permeability and potency? Hypothesis: MTHFD2 inhibitors identified with a permeability-driven virtual screening will have higher Caco-2 permeability values and more favorable ΔG binding scores compared to current inhibitors. To ground these objectives in context, it is important to define the scope of the work and the limitations of the study.
Scope and Limitations
This study is entirely computational and limited to in-silico discovery. It covers protein preparation of a crystal structure of MTHFD2, structure based virtual screening of 140,000 compounds, SwissADME, and human colorectal adenocarcinoma cell (Caco-2) permeability predictions, and MM-GBSA calculations. It excludes all wet-lab validation. All information is limited to the140,000 compounds in the Blumberg Ligand Library. Because the values were calculated using simulations and estimations, they will require biochemical assays, cell-base testing, and in-vivo studies to validate the results. With these boundaries established, the methodology was designed to identify and evaluate promising compounds.
Methodology Overview
140,000 compounds from the Blumberg ligand library were run through a virtual screening. The compounds were screened in several phases to ensure they were permeable, potent, and selective. First, the hits with optimal Caco-2 values and fit in the Lipinksi Rule of 5 were prioritized. Later, the permeable compounds were screened based on their binding affinity to MTHFD2’s allosteric site. Finally, the compound with the strongest binding interaction was optimized using Bioisosteric enumeration and the ADME properties of the top compounds were measured. Detailed protocols can be found in the methods section.
Results
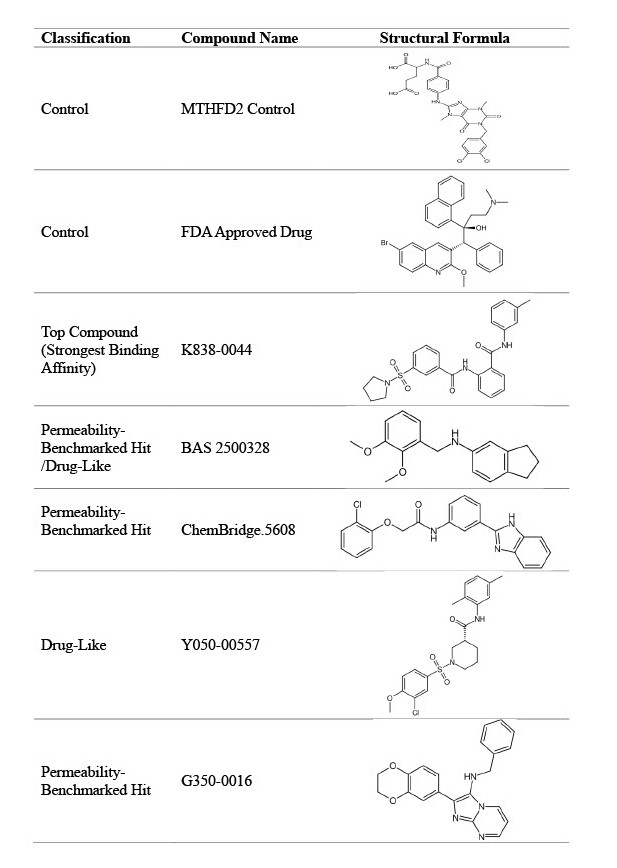
Among the compounds that were run through the virtual screening, 11 candidates with the strongest binding affinity and high permeability were selected for presentation. These compounds were labeled as the “top compounds”. The Lipinski Rule of Five values and percent human oral absorption of the top 11 compounds, FDA approved control, and MTHFD2 control are shown in Table 1. These values were considered during the pre-screening process. The top compounds fit within the constraints of the Lipinski Rule of 5 and had logP values ranging from 2.5 to 4.7 (within ideal range of 0 to 6). Although, 4 compounds exhibited a percent human oral absorption lower than the FDA approved control, all top 11 compounds had significantly higher percentages than the MTHFD2 control.
| Compound Name | Molecular Weight (Da) | LogP o/w | Hydrogen Bond Donors | Hydrogen Bond Acceptors | Percent Human Oral Absorption (%) |
| MTHFD2 Control | 60 | 4.5 | 3.3 | 11.3 | 26.2 |
| FDA Approved Control | 555 | 7.7 | 1.0 | 3.8 | 100 |
| K838-0044 | 464 | 4.4 | 1.0 | 8.5 | 100 |
| BAS 2500328 | 283 | 4.7 | 1.0 | 2.5 | 100 |
| ChemBridge.12766 | 363 | 3.4 | 1.0 | 5.5 | 95.9 |
| ChemBridge.9851 | 410 | 3.8 | 2.0 | 7.5 | 100 |
| ChemBridge.1065 | 329 | 4.1 | 1.0 | 4.0 | 100 |
| L273-0378 | 483 | 3.4 | 2.0 | 9.0 | 96.2 |
| K781-9015 | 403 | 3.1 | 2.0 | 7.0 | 93.8 |
| ChemBridge.5608 | 378 | 4.6 | 2.0 | 4.8 | 100 |
| BAS 1927109 | 340 | 2.5 | 2.0 | 7.0 | 92.8 |
| Y050-0557 | 437 | 4.2 | 1.0 | 7.8 | 100 |
| G350-0016 | 358 | 4.0 | 1.0 | 4.5 | 100 |
The Caco-2 values and ΔG Binding Scores of the top 11 compounds as well as the MTHFD2 control are shown in Table 2. The MTHFD2 control showed low permeability with a Caco-2 value below 25 nm/sec, whereas all the top 11 compounds exceeded the 500 nm/sec cutoff for strong membrane transport. Additionally, three compounds from the top 11 compounds (BAS 2500328, ChemBridge.5608, BAS 1927109) exhibited Caco-2 values higher than the FDA-approved drug. These compounds were labeled as the “Permeability-Benchmarked Hits”. However, their docking scores were substantially weaker than the MTHFD2 control by at least 10.81 kcal/mol (less negative ΔG). The top-binding compound (K838-0044) was the only compound in the top 11 to surpass the MTHFD2 control in binding affinity, but it was only slightly stronger (0.86 kcal/mol). Additionally, a two-tailed Mann-Whitney U Test could not detect a statistically significant difference between the Δ G binding score of the control and K838-0044 (U = 16, n1 = n2 = 6).
| Compound Name | Caco-2 (nm/sec) | ΔG Binding Scores (kcal/mol) |
| MTHFD2 Control | 0.8640 | -71.53 |
| FDA Approved Drug | 1556 | X |
| K838-0044 | 684.3 | -72.39 |
| BAS 2500328 | 8192 | -55.45 |
| ChemBridge.12766 | 553.6 | -68.52 |
| ChemBridge.9851 | 512.6 | -58.44 |
| ChemBridge.1065 | 1194 | -60.86 |
| L273-0378 | 592.8 | -58.32 |
| K781-9015 | 530.5 | -65.45 |
| ChemBridge.5608 | 1562 | -58.21 |
| BAS 1927109 | 710.1 | -35.19 |
| Y050-0557 | 1326 | -56.29 |
| G350-0016 | 2940 | -60.72 |
To further assess the drug-like properties of the top 11 compounds, the ADME properties of the compounds were analyzed. Figure 3 shows the ADME properties of the top 11 compounds. Cells are color-coded based on whether the metric falls within the defined ideal range (green) and outside it (red), detailed in the Variables and Measurements section. Only one permeability-benchmarked hit(BAS 25000328) fulfilled the oral drug-likeness criteria while Y050-00557, not a permeability-benchmarked hit, also fulfilled it. These properties are consistent with oral drug-likeness. Of the candidates that did not fulfill the drug-likeness criteria, eight failed the Fraction Csp3 metric, which indicates a low proportion of sp3 hybridized carbon atoms. Additionally, Table 3 displays the structural formula of the strongest binding compound, permeability-benchmarked hits, and the identified drug-like compounds (candidates of interest). Structurally, K838-0044 contains a moderate molecular weight (463.6 g/mol) and a balanced total polar surface area (104.0 Å2), supports cellular uptake, along with various functional groups (specifically benzenes and amides) which may contribute to stronger binding interactions. In contrast, the MTHFD2 control has a high molecular weight and a large total polar surface area from the multiple carboxylic acids, which limits its permeability. Additionally, BAS2500328 has the lowest total polar surface area (30.49 Å2), and this is likely the reason it is significantly more permeable than the other compounds.
| Compound Name | XLOGP3 | MW | TPSA | Log S (ESOL) | Fraction Csp3 | Num. Rotatable bonds |
| Control | 2.95 | 603.4 | 177.6 | -5.09 | 0.23 | 11 |
| K838-0044 | 4.14 | 463.6 | 104.0 | -5.20 | 0.20 | 8 |
| BAS 2500328 | 4.18 | 283.4 | 30.49 | -4.32 | 0.33 | 5 |
| ChemBridge.12766 | 2.79 | 363.2 | 66.48 | -3.96 | 0.12 | 4 |
| ChemBridge.9851 | 3.54 | 410.4 | 84.50 | -4.53 | 0.05 | 8 |
| ChemBridge.1065 | 3.94 | 328.8 | 49.41 | -4.48 | 0.22 | 4 |
| L273-9015 | 2.77 | 482.6 | 135.4 | -4.17 | 0.43 | 10 |
| K781-9015 | 3.72 | 403.3 | 83.65 | -4.58 | 0.06 | 7 |
| ChemBridge.5608 | 4.65 | 377.8 | 67.01 | -5.29 | 0.05 | 6 |
| BAS 1927109 | 2.87 | 340.4 | 83.65 | -3.92 | 0.06 | 5 |
| Y050-00557 | 3.73 | 437.0 | 84.09 | -4.81 | 0.38 | 6 |
| G350-0016 | 4.30 | 358.4 | 60.68 | -5.08 | 0.14 | 4 |

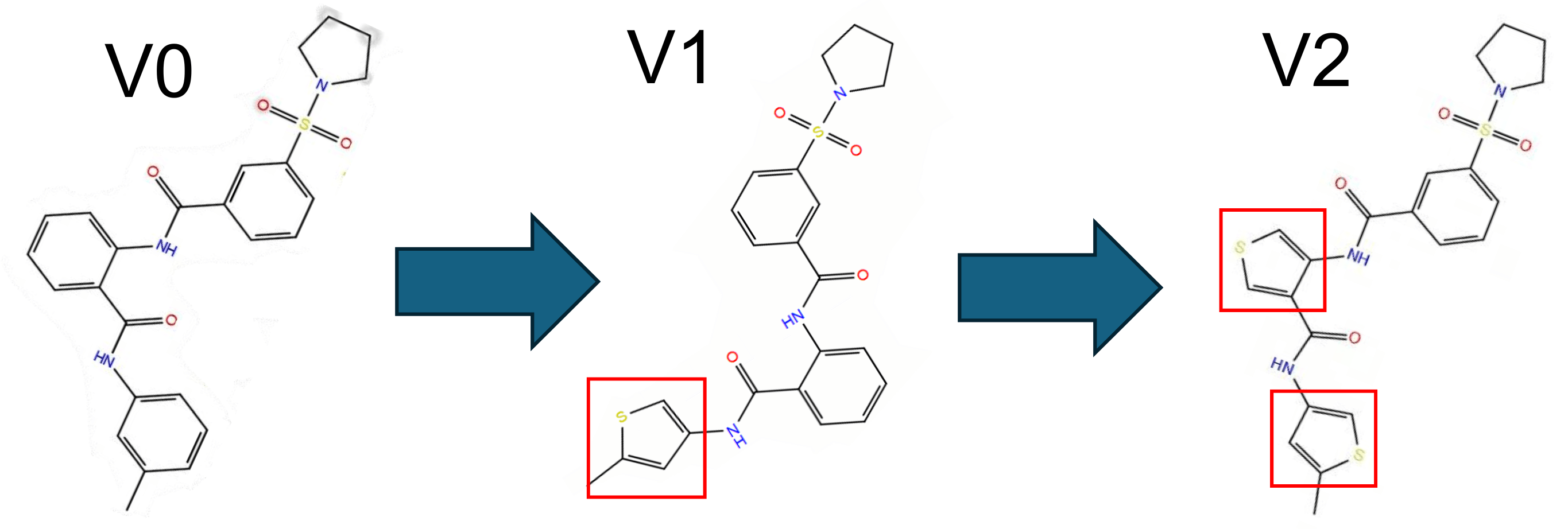
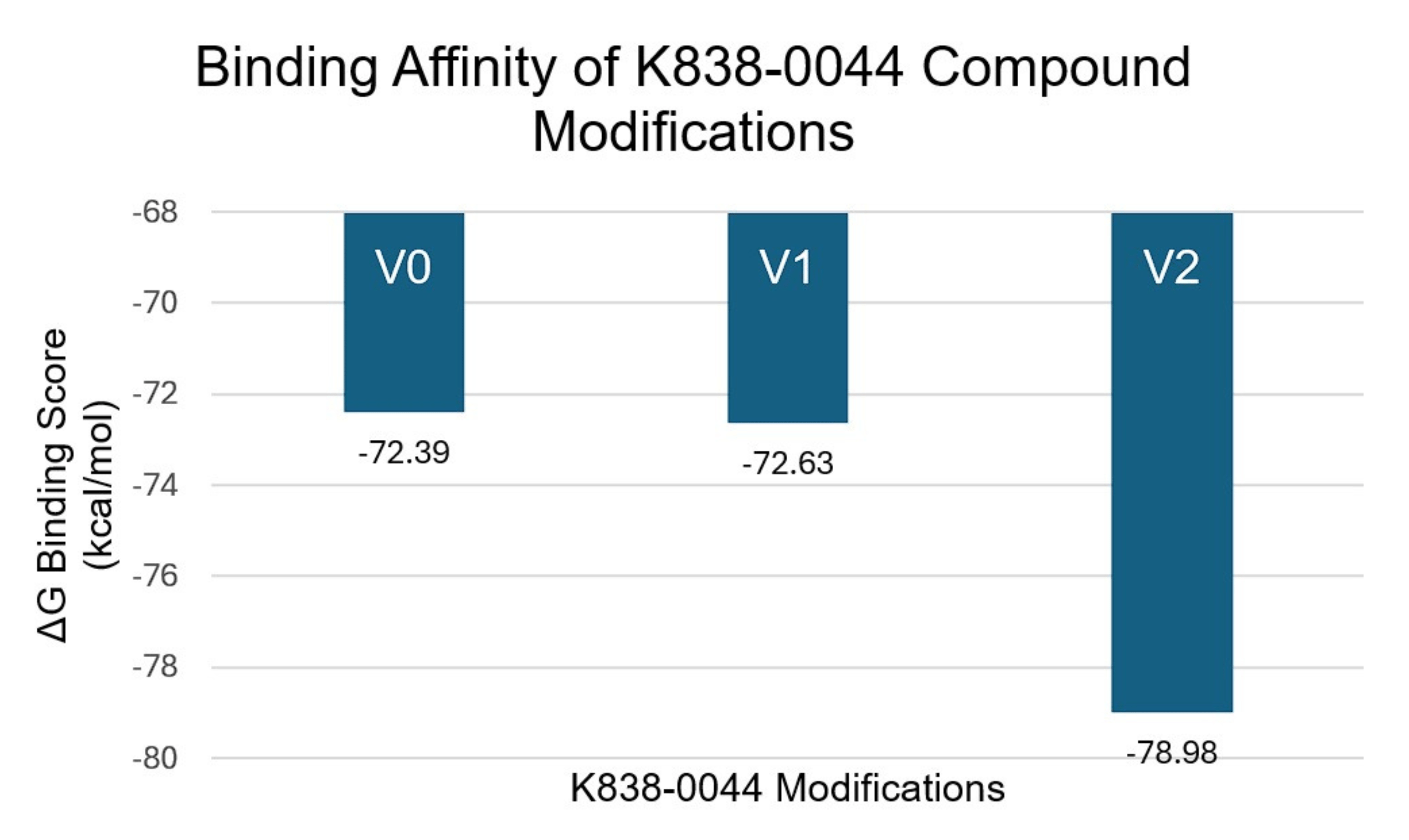
The compound with the strongest binding affinity (K838-0044) was optimized using bioisosteric enumeration to improve the permeability and the binding strength further. Figure 2 shows the aromatic ring modifications made to the compound. Figure 5 shows the ΔG Binding Scores values associated with those modifications. Figure 6 shows the Caco-2 values associated with those modifications. For the ΔG Binding Scores, the 2nd modification (V2) improved the binding affinity the most by -6.59 kcal/mol (from V0 to V2). Additionally, this modification had a stronger binding interaction than the MTHFD2 control and the unmodified K838-0044. For the Caco-2 values, the 1st modification (V1) improved the permeability the most by 128.2 nm/sec (V0 to V1). Although the permeability of the medication was higher than the unmodified K838-0044, it was still lower than the FDA approved control.

Discussion
Restatement of Key Findings
A virtual screening of more than 140,000 compounds resulted in 11 candidates that had high permeability while the MTHFD2 control exhibited low permeability. Three of those compounds (BAS 2500328, ChemBridge.5608, BAS 1927109) displayed Caco-2 values higher than the FDA approved control (Permeability-Benchmarked Hits). Further testing using SwissADME predictions identified two ligands with oral-drug likeness. 8 out of the 11 compounds failed the “Fraction Csp3” metric, which means that there was a low proportion of sp3 hybridized carbon atoms and a flat molecular structure. This is likely due to the presence of multiple aromatic rings, specifically benzene, in the identified compounds. It indicates that the compounds may have unfavorable absorption/distribution and could be more metabolically unstable in clinical settings. A two-tailed Mann-Whitney U Test could not detect a statistically significant difference between the ΔG Binding Scores of K838-0044, the compound with the strongest binding affinity, and the MTHFD2 control (U = 16, n1 = n2 = 6). However, numerical values suggest that K838-0044 has a slightly stronger binding score (ΔG binding score –72.39 kcal/mol for K838-0044 vs. -71.53 kcal/mol for control, respectively) than the control while being significantly more permeable (Caco-2 684 nm/sec vs. 0.864 nm/sec, respectively). Bioisosteric enumeration of K838-0044 improved interaction strength by -6.59 kcal/mol (from V0 to V2) and Caco-2 permeability by 128.2 nm/sec (from V0 to V1) compared to the unmodified K838-0044.
Implications and Significance
These results demonstrate that a permeability focused virtual screening can overcome a large obstacle in MTHFD2 inhibitors, their poor permeability. By integrating Caco-2 calculations along with docking and MM-GBSA scoring, this workflow identified candidates that had strong binding affinity and were considered “orally drug-like” through computational estimations. This study also identifies new compounds that are capable of binding to MTHFD2’s allosteric site, providing a starting point for more structure-based optimization.
Connection to Objectives
The primary objective was achieved as eleven compounds with high permeability were identified. Of them, 3 surpassed the FDA approved control and 2 met the full ADME criteria, potentially making them promising candidates for clinical implementation. A secondary aim was to analyze binding strength, and 1 compound was found to have a comparable ΔG Binding Score to the MTHFD2 Control.
Recommendations for Future Work
In the future, the metabolic stability of the top compounds will be assessed, like investigating the CYP enzyme interactions, to ensure that the compounds are therapeutically effective. Additionally, potential resistance mechanisms, such as identifying MTHFD2 mutations, will also be investigated. Wet lab validation will be performed to validate the in-silico results. This would be in the form of a Caco-2 assay to measure the permeability and an IC50 assay to measure the potency of each compound. Testing on lung cancer cell lines may also reveal whether the compounds identified can be used as a viable treatment. Further bioisosteric enumeration on the other compounds could be performed to improve both binding affinity and permeability. Furthermore, long molecular dynamics simulations could refine the docking score ranking.
Limitations
All the findings are predictive and rely on a single crystal structure which may limit the reliability of the results. One limitation of this study is that the permeability and binding affinity values are computational predictions that have not been validated through in vitro assays. However, the main permeability metric, Caco-2, was predicted using QikProp, which uses a regression model trained on experimental data from 150 unique compounds. From an experiment comparing QikProp to experimental results, the log PCaco-2 predictions had an r2 of 0.71 and a root-mean-square (RMS) error of 0.56 log(nm/sec)13. Other ADME metrics were also predicted using validated models from QikProp. The binding scores in this study were calculated using Prime MM-GBSA. In one study, Chandna et al. (2015) reported that the correlation between the computational predictions and experimental results was high (correlation coefficient up to 0.77) for a set of N-myristoyltransferase inhibitors14. Although MTHFD2 benchmarking has not been performed, these findings support the use of Prime MM-GBSA for identifying promising targets. For the modification made to K838-0044 (V2 change), the predicted binding score improved by 6.59 kcal/mol, which is extremely large for a single ring change. While the computational workflow and parameters were the same for all compounds, this large improvement should be interpreted cautiously. This is because the calculation could not be independently repeated due to loss of software access after an internship. The two-tailed Mann Whitney U-Test was conducted with a small sample size (n=6 for both compounds) which does not generate a p-value and limits the statistical power of the test. Furthermore, no additional controls were used to validate the binding site boundaries beyond Glide’s default ligand-based grid generation. Although a known potent MTHFD2 inhibitor was used as a positive control for MM-GBSA predictions, no benchmarking studies were performed to validate the accuracy of the docking, MM-GBSA, or Caco-2 predictions. Therefore, the accuracy of the computational predictions is not fully validated against experimental data. The Caco-2 permeability predictions estimate a compound’s ability to pass through the cell membrane, but it has limitations for predicting mitochondrial uptake, which is important for MTHFD2 targeting. While computational screening identifies promising candidates, in vitro testing is essential to confirm function inhibition and clinical relevance.
Closing Thought
By showing how a permeability focused virtual screening helps to identify drug-like compounds against MTHFD2, this study illustrates how computational methods can accelerate the development of tumor-selective therapies. Pairing these predictions with experimental validation now works as a tangible path toward the first clinically viable MTHFD2 inhibitor for non-small cell lung cancer.
Methods
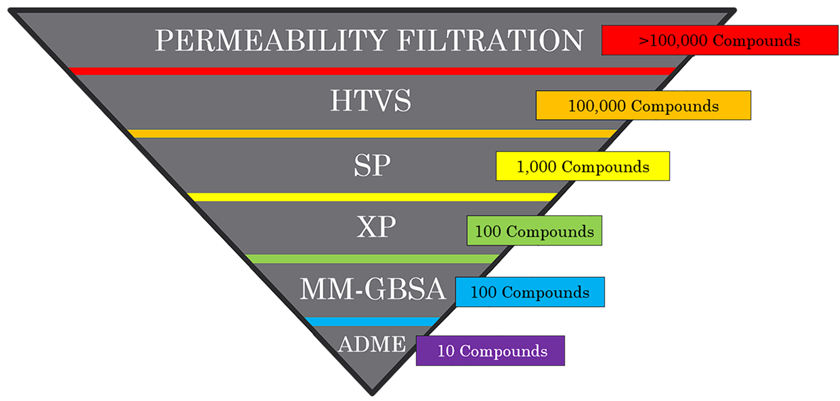
Research Design
This study used an in-silico, screening-based experimental design. It had two main phases: A high-throughput virtual screening focuses on permeability and docking of a chemical library on MTHFD2’s allosteric site. The 2nd phase involves optimizing the ligand with the strongest binding affinity to improve permeability and interaction strength as well as predicting the ADME properties of the top 11 compounds using SwissADME. This approach was designed to prioritize the early identification of drug-like and permeable allosteric inhibitors for MTHFD2.
Computational Sample
The dataset used in this study was the Blumberg Ligand library from the PA Biotechnology Center, and it contains 139,588 crystallized structures of compounds. Additionally, a crystal structure of MTHFD2 (PDB: 7EHM) was prepared using Maestro’s Protein Preparation Wizard and used in this study. Using Glide, a receptor grid was generated to define MTHFD2’s allosteric site. The co-crystallized allosteric ligand (from PDB: 7EHM) 15 was selected as the reference, and Glide automatically centered the grid on the ligand. The dimensions of the receptor grid were set by Glide based on the reference ligand.
Variables and Measurements
The programs used in this study were Schrodinger’s Maestro (computational software that simulates the interaction between small molecules and proteins), SwissADME (a free online tool that predicts a compound’s pharmacokinetic properties), and the Protein Data Bank (an archive that provides the crystal structure for proteins).
Variables
- Caco-2 Permeability predicts a compound’s ability to cross intestinal and cell membranes (nm/sec). High permeability (> 500) low permeability ( <25) according to Schrodinger’s QikProp manual. Caco-2 predictions have a root-mean-square (RMS) error of 0.56 log(nm/sec) according to the QikProp Technical Manual13.
- ΔG Binding Score estimates binding free energy to MTHFD2 (kcal/mol).
ADME Properties: measurements help to determine whether a compound has oral drug likeness. Figure 3 was color-coded based on whether the value fell within the ideal range (green) or outside of it (red) for each property.
- XLOGP3 measures lipophilicity. Ideal range (between −0.7 and +5.0).
- MW (Molecular Weight) measures the weight of compound (indicates size). Ideal range (between 150 and 500 g/mol).
- TPSA (Topological Polar Surface Area) indicates the polarity of the compound. Ideal range (between 20 and 130 Å2).
- Log S (ESOL) measures solubility. Ideal range (between –6 and 0).
- Fraction Csp3 is the fraction of carbons in the sp3 hybridization (to determine saturation). Ideal range (between 0.25 and 1).
- Num. Rotatable Bonds indicates the flexibility of the compound. Ideal range (between 0 and 9).
Controls
- FDA Approved Control: Bedaquiline, an FDA approved drug that targets an enzyme in the mitochondria, was labeled as the “FDA Approved Control” in this study. This control was used because of its validated ability to enter the mitochondria (MTHFD2 is in the mitochondria). It acts as a clinically relevant standard for assessing the compounds’ permeability (Caco-2).
- MTHFD2 Control: A current MTHFD2 inhibitor from the PDB 7EHM and it has been identified to inhibit MTHFD2 and was bound to its allosteric site. This was used as a baseline for binding affinity as it has already been proven to bind strongly to MTHFD2, and it was labeled as the “MTHFD2 Control” in this study.
Data Collection and Procedure
The compound library was initially filtered using the Lipinski Rule of Five and logP o/w limits (0 to 6) to remove compounds with poor predicted bioavailability. The remaining compounds were screened for binding strength using Glide:
- High-Throughput Virtual Screening (HTVS) rapidly docks the compounds against MTHFD2’s allosteric site. This method screened through tens of thousands of compounds efficiently, and it identifies the top 1000 compounds in terms of interaction strength (most negative binding scores)
- Standard Precision docking (SP) offers a slower but more accurate binding score than HTVS, and it identified the top 100 compounds in terms of binding affinity.
- Extra Precision docking applied stricter scoring to the 100 compounds than SP to calculate a more accurate binding score, and it created a pose of the compound binding to the allosteric site
- Molecular Mechanics, General Born Surface Area (MM-GBSA) is a free-energy calculation method that generated free energy estimates (a ΔG binding score) of the binding between the compounds and the binding site. Additionally, it accounted for the protein’s movement in the calculation and it scored the top 100 compounds from SP and XP.
ADME Predictions
SwissADME measured the ADME (Absorption, Distribution, Metabolism, Excretion) values of the selected compounds and it determined whether the compounds would make a good oral drug. It generated values such as XLOGP3 (lipophilicity), molecular weight, topological polar surface area, Log S (solubility), Fraction Csp3, and Num. Rotatable bonds.
Bioisosteric Enumeration
K838-0044 (the compound with the greatest binding affinity) was modified to improve its binding and permeability. In the first modification (labeled V1), one of the benzenes was replaced with a thiophene. In the second modification (labeled V2), the first modification was kept, and an additional benzene was replaced by a thiophene. During this phase, multiple aromatic replacements were tested, and the selected modifications (V1 and V2) produced the greatest increase in both binding and permeability.
Mann Whitney U-Test
6 independent MM-GBSA jobs were run for both K838-0044 and the MTHFD2 control to generate 6 ΔG Binding Scores for each compound. These values were compared with a two-tailed Mann-Whitney U-Test (a non-parametric test meant for smaller sample sizes) to determine whether there was statistically significant evidence of a difference between the binding affinity of the compounds. A p-value was not generated due to the size of the sample, but a U-statistic was calculated to determine statistical significance (a value less than 5 would be considered significant). The null hypothesis was “There is no difference between the ranks of each binding score” while the alternative hypothesis was “There is a difference between the ranks of each binding score”.
Data Analysis
A Mann-Whitney U-Test created by hand using the ΔG Binding Scoresgenerated by Maestro. The ΔG Binding Scores and Caco-2 values of the compounds were compared in Maestro. Heatmaps and data visualizations were generated using Excel.
Ethical Considerations
The study used only publicly available molecular and structural data. It did not use human, animal, or clinical information and no institutional ethics approval was required.
Acknowledgements
I would like to thank my mentors Dr. Scott Willett and Ryan Ford for their advice and guidance throughout this project. I am also grateful to my adult sponsor Mark Hayden for his guidance over the past year. Finally, I would like to thank my parents for their continued support.
References
- American Cancer Society. (2024, January 29). Lung Cancer Early Detection, Diagnosis, and Staging. American Cancer Society. Retrieved May 16, 2025, from https://www.cancer.org/cancer/types/lung-cancer/detection-diagnosis-staging/survival-rates.html [↩]
- Surveillance, Epidemiology, and End Results. (n.d.). Cancer Stat Facts: Lung and Bronchus Cancer. National Cancer Institute. Retrieved May 16, 2025, from https://seer.cancer.gov/statfacts/html/lungb.html [↩]
- Juthani, R., Punatar, S. & Mittra, I. New light on chemotherapy toxicity and its prevention. BJC Rep 2, 41 (2024). https://doi.org/10.1038/s44276-024-00064-8 [↩]
- Zhu Z and Leung GKK (2020) More Than a Metabolic Enzyme: MTHFD2 as a Novel Target for Anticancer Therapy? Front. Oncol. 10:658. doi: 10.3389/fonc.2020.00658 [↩]
- Nilsson, R., Jain, M., Madhusudhan, N. et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun 5, 3128 (2014). https://doi.org/10.1038/ncomms4128 [↩]
- Mo, J., Gao, Z., Zheng, L. et al. Targeting mitochondrial one-carbon enzyme MTHFD2 together with pemetrexed confers therapeutic advantages in lung adenocarcinoma. Cell Death Discov. 8, 307 (2022). https://doi.org/10.1038/s41420-022-01098-y [↩] [↩]
- Feng Zhou, Ziyi Yuan, Yuyan Gong, Luyao Li, Yanmao Wang, Xian Wang, Chunbo Ma, Lehe Yang, Zhiguo Liu, Liangxing Wang, Haiyang Zhao, Chengguang Zhao, Xiaoying Huang, Pharmacological targeting of MTHFD2 suppresses NSCLC via the regulation of ILK signaling pathway, Biomedicine & Pharmacotherapy, Volume 161, 2023, 114412, ISSN 0753-3322, https://doi.org/10.1016/j.biopha.2023.114412.https://www.sciencedirect.com/science/article/pii/S0753332223002007 [↩]
- Huang, J., Qin, Y., Lin, C., Huang, X., & Zhang, F. (2021). MTHFD2 facilitates breast cancer cell proliferation via the AKT signaling pathway. Experimental and therapeutic medicine, 22(1), 703. https://doi.org/10.3892/etm.2021.10135 [↩]
- Bonagas, N., Gustafsson, N. M. S., Henriksson, M., Marttila, P., Gustafsson, R., Wiita, E., Borhade, S., Green, A. C., Vallin, K. S. A., Sarno, A., Svensson, R., Göktürk, C., Pham, T., Jemth, A. S., Loseva, O., Cookson, V., Kiweler, N., Sandberg, L., Rasti, A., Unterlass, J. E., … Helleday, T. (2022). Pharmacological targeting of MTHFD2 suppresses acute myeloid leukemia by inducing thymidine depletion and replication stress. Nature cancer, 3(2), 156–172. https://doi.org/10.1038/s43018-022-00331-y [↩]
- Louise Ramos, Martin Henriksson, Thomas Helleday, Alanna C. Green; Targeting MTHFD2 to Exploit Cancer-Specific Metabolism and the DNA Damage Response. Cancer Res 1 January 2024; 84 (1): 9–16. https://doi.org/10.1158/0008-5472.CAN-23-1290 [↩] [↩] [↩]
- Lee, L. C., Peng, Y. H., Chang, H. H., Hsu, T., Lu, C. T., Huang, C. H., Hsueh, C. C., Kung, F. C., Kuo, C. C., Jiaang, W. T., & Wu, S. Y. (2021). Xanthine Derivatives Reveal an Allosteric Binding Site in Methylenetetrahydrofolate Dehydrogenase 2 (MTHFD2). Journal of medicinal chemistry, 64(15), 11288–11301. https://doi.org/10.1021/acs.jmedchem.1c00663https://pmc.ncbi.nlm.nih.gov/articles/PMC8389891/ [↩]
- Rana, N., Patel, D., Parmar, M. et al. Targeting allosteric binding site in methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) to identify natural product inhibitors via structure-based computational approach. Sci Rep 13, 18090 (2023). https://doi.org/10.1038/s41598-023-45175-3 [↩]
- Jorgensen, W. L., & Schrödinger. (2006). QikProp Technical Manual. Retrieved August 9, 2025, from https://qikprop.molssi.org/static/pdf/QP30.manual.pdf [↩] [↩]
- Chandna N, Kumari MK, Sharma C, Vijjulatha M, Kapoor JK, et al. (2015) QM/MM Docking Strategy and Prime/MM-GBSA Calculation of Celecoxib Analogues as N-myristoyltransferase Inhibitors. Virol-mycol 4: 141. [↩]



{kind=link}