
Abstract
Both cancer and autoimmune disease are closely linked to immune system dysregulation: cancer is characterized by the uncontrolled proliferation of cancerous cells due to an underactive immune response, while autoimmune disease involves an overzealous immune system that attacks the patient’s own cells. Small molecule immunotherapy is a promising treatment for both diseases that warrants further research. This study aimed to identify potential small molecule binding candidates to the glucocortocoid-induced tumor necrosis factor receptor-related (GITR) protein, which mediates important T-cell costimulatory pathways. Modulation of GITR may either upregulate antitumor responses or temper an overactive immune system by altering T-cell activity. To discover small molecule candidates, multistep virtual screening was performed on the GITR-GITRL complex: first, binding sites on the GITR protein were identified using geometric (DoGSiteScorer), energy-based (FTSite), and machine-learning (Prankweb) methods to ensure that GITR is a feasible target for small molecule immunotherapy. Pharmacophore-based virtual screening was then used to identify potential small molecule modulators for the GITR protein using Pocketquery and ZINCPharmer. Lastly, the druggability of these compounds was validated using molecular docking (Swissdock), Lipinski’s rule (SwissADME), and toxicity screening (Protox-3.0). After screening a small molecule library containing over 18 million compounds, six promising small molecule candidates and their properties were identified. This study is valuable because it explores the potential applications of small molecule immunotherapy on costimulatory T-cell receptors such as GITR and sets the groundwork for the development of new treatment options for cancer and autoimmune disease patients.
Keywords: Cancer immunotherapy, virtual screening, small molecules, immune checkpoint, costimulatory pathway, autoimmunity, TNFR superfamily, GITR
Introduction
Cancer is a deadly disease that is characterized by a failure of the body’s regulatory mechanisms to manage cell growth and to maintain homeostasis, which results in uncontrolled cancer cell proliferation1. Cancer cells are capable of replicating infinitely, stimulating cell growth independently, evading growth suppressors and apoptosis, manipulating blood vessels to provide them with sustenance, and avoiding eradication by the body’s immune system2. The relentless growth of these cells significantly impairs bodily function through competition with normal cells for resources and space3. Moreover, cancer cells in malignant tumors invade different regions of the body through a process known as metastasis, which results in the formation of tumors in other tissues,which further exacerbates serious health risks posed to patients4.
The prevention and treatment of cancer present one of the most difficult healthcare obstacles to overcome during the 21st century, with nearly 20,000,000 people affected by cancer around the globe in 2022. In the same year, there were almost 10,000,000 deaths caused by cancer, making it the 2nd most prevalent cause of death in the world5. An estimated 2,041,910 new incidences of cancer and 618,120 cancer deaths will occur in the United States by the end of 20256.
Conventional cancer treatments include surgery, radiotherapy, and chemotherapy7. However, these treatments often have harmful side effects and often cannot completely eradicate cancer cells8. Treatments can be classified in two categories: neoadjuvant (administered before main treatment) and adjuvant (administered after main treatment)9. In addition to these conventional cancer treatments, novel approaches like targeted therapy and immunotherapy are gaining traction. Targeted therapy inhibits essential proteins and pathways that facilitate tumor survival, while immunotherapy reinvigorates the body’s immune system and allows it to recognize and kill cancer cells10. Clinical immunotherapy treatment modalities include the use of monoclonal antibodies (mAbs), cell therapy, oncolytic viruses, and cancer vaccines11.
Of these modalities, the use of mAbs is one of the most commonly used treatments. These antibodies are manufactured outside the body and are administered to eliminate cancer cells, both directly and indirectly12. Additionally, the use of small molecule drugs to reactivate immune cells to fight cancer cells is a promising field for ongoing research13. Cell therapy is another viable approach that entails the extraction, replication, and reinjection of patient T-cells14. The use of oncolytic viruses and cancer vaccines as treatments are also under investigation. Genetically modified oncolytic viruses are utilized to selectively lyse tumor cells15, while cancer vaccines aim to facilitate immune system recognition of unique tumor cell identifiers16. Since cancer is a complex disease with a multitude of hallmarks and factors that require consideration17, many of these treatments are currently being administered in combination with each other to treat cancer in a more effective manner18.
One widely successful method to revitalize the immune system to fight cancer cells is the administration of immune checkpoint inhibitors19. Immune checkpoint proteins are found on T-cell surfaces and work to inhibit T-cell activation, which prevents an uncontrolled immune response that could potentially damage the host’s self cells11. However, some cancerous tumors have the capability to manipulate these immune checkpoints and deactivate the immune system, which facilitates tumor growth20. Consequently, immune checkpoint inhibitor drugs have been developed to disrupt attempts to manipulate these inhibitory checkpoints21. The most extensively researched immune checkpoint targets include PD-1 and CTLA-422. Several existing mAb drugs such as Pembrolizumab and Nivolumab block the inhibitory function of the PD-1 protein complex, which reinvigorates T-cell function and fosters a stronger antitumor immune response23.
In the same vein, costimulatory molecule pathways (such as OX40 and GITR) play an essential role in the tumor fighting process by facilitating the activation of T-cells24. The clinical development of T-cell agonists that activate costimulatory pathways and induce a stronger antitumor response is a highly researched topic25. These costimulatory pathways are not only found in cancer research, however. They can also work in the opposite direction by playing a critical role in the treatment of autoimmune diseases26. Autoimmune diseases are serious health conditions characterized by an overzealous immune system that mistakenly attacks the host’s own cells27. Since these costimulatory pathways increase T-cell activation, their inhibition can reduce T-cell activity and hinder an autoimmune response. Thus, the development of therapeutic drugs that inhibit costimulatory pathways is an essential part of autoimmune disease research28.
The GITR/GITRL complex is involved in a T-cell costimulatory pathway that can modulate the immune system’s response strength: the binding of the ligand GITRL to its cognate receptor GITR results in a strengthened immune response by contributing to the invigoration and proliferation of CD8+ and CD4+ effector T-cells (T-effs) and inhibiting regulatory T-cell (T-reg) activity. Thus, GITR agonists that activate this pathway may have potential anticancer applications29. On the other hand, the inhibition of this pathway sustains T-reg cell activity while hindering T-eff cell activation, which can downregulate overactive immune responses caused by autoimmune disease. Therefore, GITR antagonists that block pathway activation may have potential autoimmune disease treatment applications30. These two scenarios are illustrated in detail in Fig. 1.
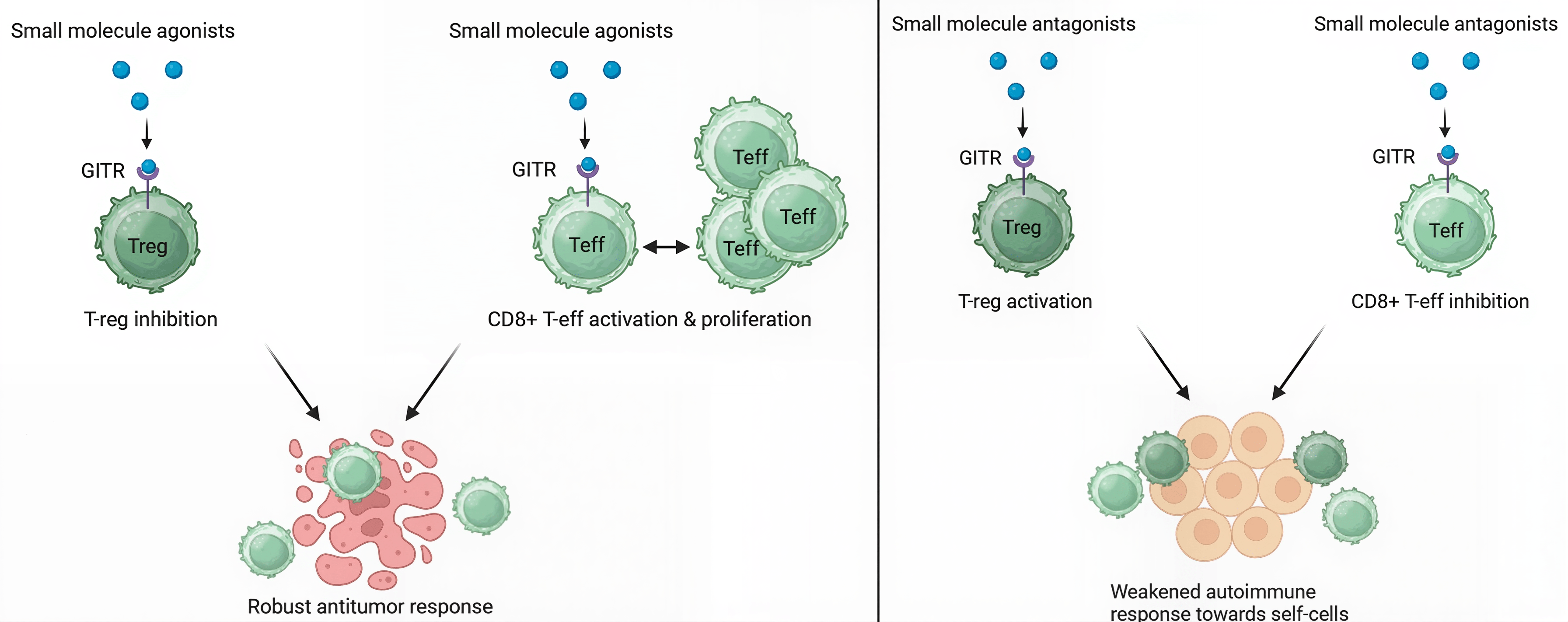
Ongoing research on the modulation of the GITR/GITRL costimulatory pathway primarily focuses on the applications of mAbs as opposed to small molecules. Multiple research studies investigating the role of DTA-1, an agonistic mAb targeting the GITR protein, discovered that the GITR agonist led to the downregulation of T-reg activity and the upregulation of T-eff activation and proliferation, which can contribute to a stronger antitumor response31,32. However, there are no reported attempts, to our knowledge, in the literature to explore the applications of GITR-targeted small molecules for either cancer immunotherapy or autoimmune disease treatment. Small molecule immunotherapy treatments confer several distinct advantages over mAb treatments, since they are administered orally, better penetrate the tumor microenvironment, circulate more quickly, decrease immunogenicity, and are more easily accessible33,34.
Previous research on small molecule cancer immunotherapy mainly targets immune checkpoint inhibitors instead of costimulatory pathways. For instance, small molecule inhibitor drugs for the PD-1 immune checkpoint are currently being tested and developed, such as PDI-1, which has been shown to induce antitumor effects through the inhibition of PD-1 immune checkpoints, with effects comparable to mAb-based immune checkpoint inhibitors35. Although limited in scope, some studies have previously reported on the applications of small molecule immunotherapy on costimulatory pathways. A research study on small molecule agonists of the OX40-OX40L complex (a costimulatory molecule with mechanisms similar to the GITR-GITRL complex) discovered that small molecules are capable of stimulating this costimulatory pathway and contributing to increased T-eff cell activity while suppressing T-reg cells36. This demonstrates that it is feasible to identify small molecule compounds that modulate GITR-GITRL and have the potential to treat cancer or autoimmune diseases.
In an effort to address the lack of research on small molecule modulators for GITR, a T-cell costimulatory receptor, we set out to identify binding candidates to the human GITR protein with multi-step virtual screening. We hypothesized that utilizing in-silico screening of the GITR protein would allow us to efficiently identify small molecule candidates for further testing. Computer-based screening was selected because it is a powerful technique that has the potential to save a significant amount of time and money in the initial steps of drug discovery, whereas the alternative, physical screening in the laboratory, is both time-consuming and costly37. Our virtual screening identified six promising small molecule compounds as binding candidates to the GITR protein. This study is of therapeutic relevance because it provides a proof-of-concept for the discovery of small molecule candidates that may modulate costimulatory pathways, and sets the groundwork for the development of cancer immunotherapy or autoimmune disease treatments.
Methods
Target protein structure
In this experiment, we utilized the GITR-GITRL complex structure with the PDB ID 7KHD to complete all subsequent virtual screening steps. We chose to screen the combined complex instead of the individual GITR protein and GITRL ligand to more accurately determine the chemical characteristics that small molecule binders should emulate, since ligand-protein conformations can shift slightly when binding to each other. In order to conform to virtual screening requirements, a fixed protein crystal structure was utilized.
Analysis of binding sites in GITR
Firstly, potential binding sites were located on the GITR protein to verify its nature as a promising protein target for small molecule immunotherapy. In this study, binding sites for the GITR protein were identified using three methods: geometric analysis with the help of DoGSiteScorer, energetic analysis with the help of FTSite, and machine learning analysis with the help of Prankweb. Multiple computational techniques were utilized during binding site identification to ensure that there was strong evidence of GITR’s potential to bind to small molecules before proceeding with further screening.
Geometric method (DoGSiteScorer):
DoGSiteScorer is a computational tool that identifies potential binding sites on protein targets by analyzing the protein’s geometric and physical structure38. To utilize this tool, we navigated to the Proteins Plus website and entered the PDB code 7KHD, which corresponds to the GITR-GITRL complex. Binding site analysis with DoGSiteScorer then proceeded with the C and D chain of the complex, which corresponds to the GITR protein.
Energetic-based method (FTSite):
FTSite is an online tool that discovers potential binding sites on protein targets via energy calculations and the theory that small organic molecules can also interact with ligand binding sites39. To use this tool, we navigated to FTSite and entered the PDB code of 7KHD. The application was then run with chains C and D of the GITR complex.
Machine learning method (Prankweb):
Prankweb is a virtual technique that identifies potential binding sites on a certain protein target via P2Rank, a machine learning algorithm40. To utilize this method, we navigated to the Prankweb website and entered the PDB code of 7KHD. The application was then run with chains C and D of the GITR complex selected.
Pharmacophore-based virtual screening
A pharmacophore is a 3-D map that depicts different types of chemical features vital to ligand-receptor interactions, the frequency of each feature, and the distance between each feature through the use of colored spheres41. These pharmacophores then undergo virtual screening to identify potential small molecule candidates that match the pharmacophore map42. Virtual screening with pharmacophore maps is a very powerful tool that can be utilized as an alternative to or in conjunction with in vitro screening. In this study, the computational tools PocketQuery and ZincPharmer were employed to generate two pharmacophore maps for the GITRL portion of the GITR-GITRL complex. 18 million small molecule compounds were then imposed on these maps to assess the compatibility of potential small molecule binders to the GITR protein. PocketQuery and ZINCPharmer were chosen for their accessibility, ease of use, and online availability.
Target cluster identification (PocketQuery)
PocketQuery is an online tool that examines ligand-protein interactions (GITR and GITRL) and identifies groups of amino acid residues that show promise as potential models for small molecule therapy43. In this experiment, key amino acids on the GITRL ligand that play a crucial role in binding interactions with the GITR protein were utilized for pharmacophore map generation. To utilize this tool, we navigated to the PocketQuery website and uploaded the protein structure for the GITR-GITRL complex. In this case, the PDB ID of 7KHD was not used because it was not analyzed yet by PocketQuery. Instead, the protein structure was manually uploaded and assigned the PocketQuery ID 5SKH9. After initial analysis, we selected an amino acid cluster with a high druggability score from both chain A and chain B of the GITR-GITRL complex, which correspond to two different regions of GITRL with similar sequences. Subsequently, we exported the two clusters identified to ZINCPharmer for further analysis.
Virtual screening (ZINCPharmer)
ZINCPharmer is an online tool that matches over 18 million small molecules from the ZINC virtual library to a given pharmacophore map to identify the most promising potential binders to a specified target44. Starting from a pharmacophore map exported to ZINCPharmer, we hid the ligand and receptor residues present in the viewing window. We then submitted the query to view potential small molecule candidates and sorted them according to their Root Mean Square Deviation (RMSD) values. Promising small molecule candidates that deviate minimally from the pharmacophore are assigned lower RMSD scores, while less promising candidates are assigned higher RMSD scores. The top ten small molecule compounds from each of the two clusters screened were selected for further testing.
Molecular Docking
Molecular docking is a technique that simulates binding interactions between small molecules and target proteins. These simulations then display top clusters that highlight optimal binding alignments and evaluate the binding affinity of these conformations45. In this experiment, we used Swissdock to simulate potential docking site interactions between twenty small molecules and the GITR protein target. Swissdock primarily employs the Attracting Cavities method for small molecule docking simulations. This technique converts the rough protein target into a smooth landscape and employs geometric transformations and energy calculations to identify and score docking sites46. These geometric and energetic calculations provide valuable insight when identifying the most promising small molecule binding candidates for the target protein, and are displayed as SwissParam scores.
Docking with attracting cavities (Swissdock)
Swissdock is an online tool that displays interactions between a given small molecule and a given target protein and sorts the viability of these interactions using their Gibbs free energy values47. Gibbs free energy values dictate the spontaneity of chemical interactions, with negative values corresponding to more favorable binding interactions between the selected small molecule compound and the GITR protein. To utilize Swissdock, we navigated to their website and pasted the Simplified Molecular Input Line Entry System (SMILES) code for each small molecule being tested, which can be found through ZINC15 database searches. Then, we prepared the ligand for screening and entered the PDB ID 7KHD. We chose to keep Chains C and D of the complex, which correspond to the GITR protein. Next, we prepared the protein target without keeping any heteroatoms. After that, we entered the numbers (-60, 2, 48) to set the search box center and entered the numbers (51, 51, 51) to set the search box size. We set the number of Random Initial Conditions to 1, checked the parameters, and began docking simulations. The top six promising small molecule compounds with the most negative SwissParam scores were then selected for further screening to verify druglikeness. In order to quickly validate results derived from virtual docking simulations, we also conducted tests on the aspirin molecule, a common over-the-counter drug expected not to bind to the GITR protein.
ADME and Lipinski’s rule
Small molecule drugs must demonstrate four crucial pharmacokinetic properties to be effective and safe for oral administration: Absorption, Distribution, Metabolism, and Excretion (ADME). Due to the importance of ADME to drug discovery, a multitude of computational models predicting small molecules’ ADME properties with well-established metrics have emerged48. Lipinski’s Rule of Five is one such example of a metric that evaluates small molecules’ ADME properties to determine their druglikeness. Lipinski’s rule requires a given compound to have no more than five hydrogen bond donors, a calculated LogP value less than or equal to five, a molecular mass no more than 500 daltons (500 g/mol), and less than or equal to ten hydrogen bond acceptors49. In this study, Lipinski’s Rule of Five was used to evaluate the druglikeness of the top six small molecule candidates with the help of the computational tool SwissADME.
Druglikeness evaluation (SwissADME)
SwissADME is an online tool that predicts the ADME properties of small molecules and evaluates their druggability potential with a variety of benchmarks, including Lipinski’s Rule of Five50. To use this tool, we navigated to the SwissADME website and pasted the SMILES code for each small molecule being tested, which can be found through ZINC15 database searches, and ran the simulation.
Toxicity Screening
Screening for potential toxicities in small molecule compounds is essential to ensure patient safety, but experimental testing is both costly and time-consuming. Consequently, several computational approaches have been developed to predict the toxicity of these compounds in an efficient manner51. These tools predict small molecule toxicities using a variety of evaluation metrics, including median lethal dose (LD50) values. Moreover, computational models screen for potential organ toxicity, toxicological endpoints, and toxicological pathways52. In this study, Protox-3.0 was used to predict the toxicity values of the most promising small molecule compound that passed Lipinski’s rule to gauge its druggability potential.
Oral toxicity prediction (Protox-3.0)
Protox 3.0 is an online tool that predicts the potential toxicity of a given small molecule compound with several benchmarks, including the LD50 test52. To utilize this technique, we navigated to the toxicity prediction section of the Protox 3.0 website. Then, we pasted the SMILES code for the small molecule being tested, which can be found through ZINC15 database searches. Finally, we selected all the toxicity prediction models and proceeded with examination.
A summary of the entire virtual screening process can be found in Fig. 2.

Results
Analysis of binding sites in GITR
Small molecule binding sites on GITR were analyzed using a collection of computational tools, including investigations of the geometric and physical properties of binding sites to assess their potential, the identification of areas of high binding affinity based on energetic calculations, and the use of machine learning algorithms such as P2Rank. This analysis was conducted on the combined GITR-GITRL protein complex represented by PDB ID 7KHD, which contains 4 chains, labeled A, B, C, and D. Chains A and B correspond to GITRL, while Chains C and D correspond to GITR. Since the purpose of this study is to identify potential small molecule binders to the GITR protein, we performed binding site identification on Chains C and D only. All three methods identified multiple potential binding sites on the GITR protein for small molecule modulators, demonstrating that the GITR protein is a feasible target for small molecule immunotherapy. This step allows further screening for potential small molecule binding candidates to occur. The detailed results of binding site identification can be found below.
Geometric method (DoGSiteScorer)
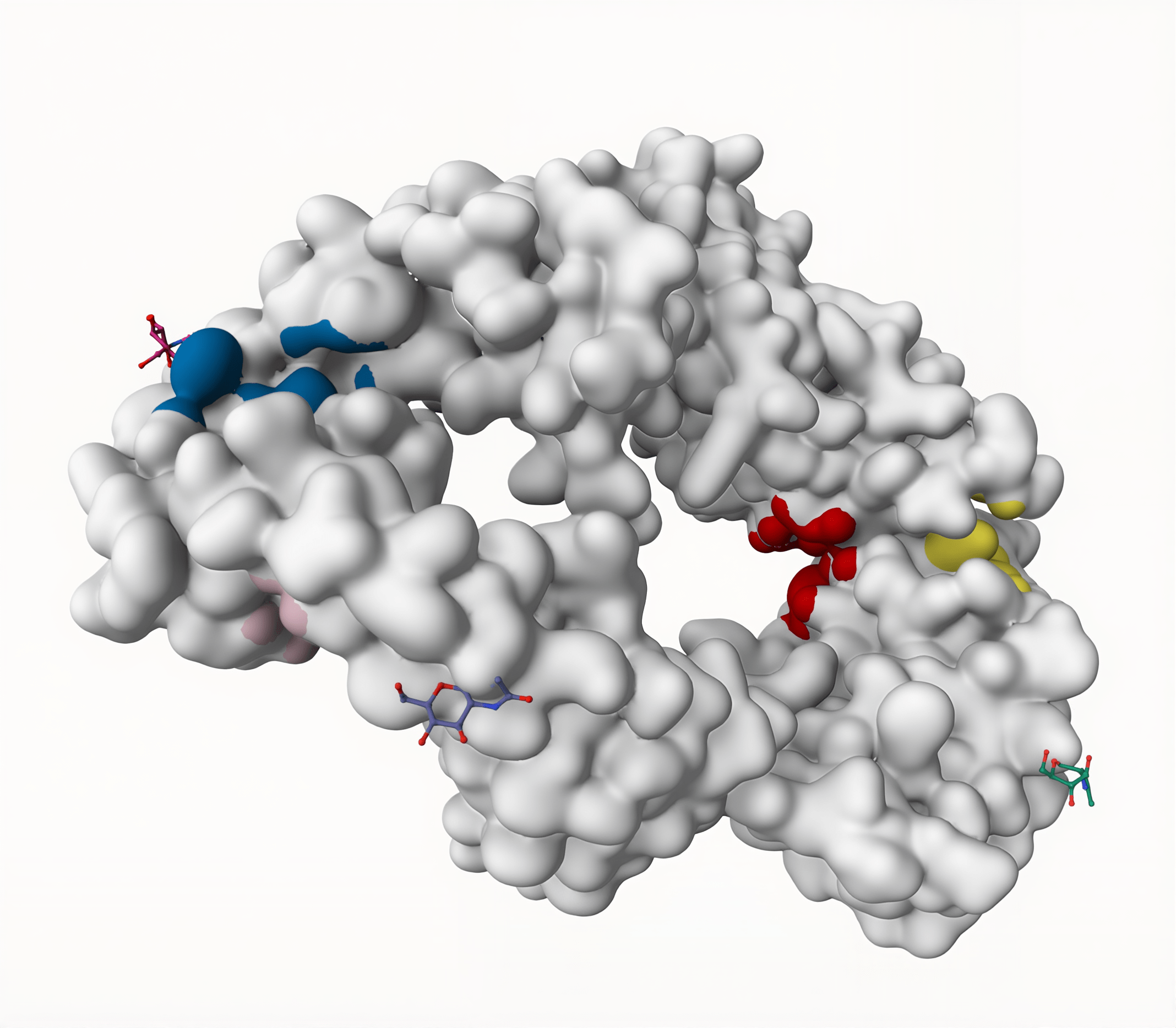
DoGSiteScorer identified 5 potential binding sites for the GITR protein using the geometric method: two are located on Chain C, and three are located on Chain D. The binding site with the largest volume, surface area, and drug score is P_0 (located on Chain C), with a volume of 580.74 ų, a surface area of 960.32 Ų, and a drug score of 0.78. The binding site with the second largest volume, surface area, and drug score is P_0 (located on Chain D), with a volume of 494.02 ų, a surface area of 650.41 Ų, and a drug score of 0.74 (Table 1). From these results, it appears that higher binding site volumes and surface areas correspond to increased likelihoods of becoming a drug target. Diagrams of binding sites identified on the GITR protein by DoGSiteScorer can also be found in Fig. 3.
| Name | Volume (ų) | Surface Area (Ų) | Drug Score |
| P_0 (Chain C) | 580.74 | 960.32 | 0.78 |
| P_1 (Chain C) | 128.58 | 331.07 | 0.29 |
| P_0 (Chain D) | 494.02 | 650.41 | 0.74 |
| P_1 (Chain D) | 193.79 | 349.29 | 0.43 |
| P_2 (Chain D) | 103.94 | 263.07 | 0.21 |

Energetic-based method (FTSite)
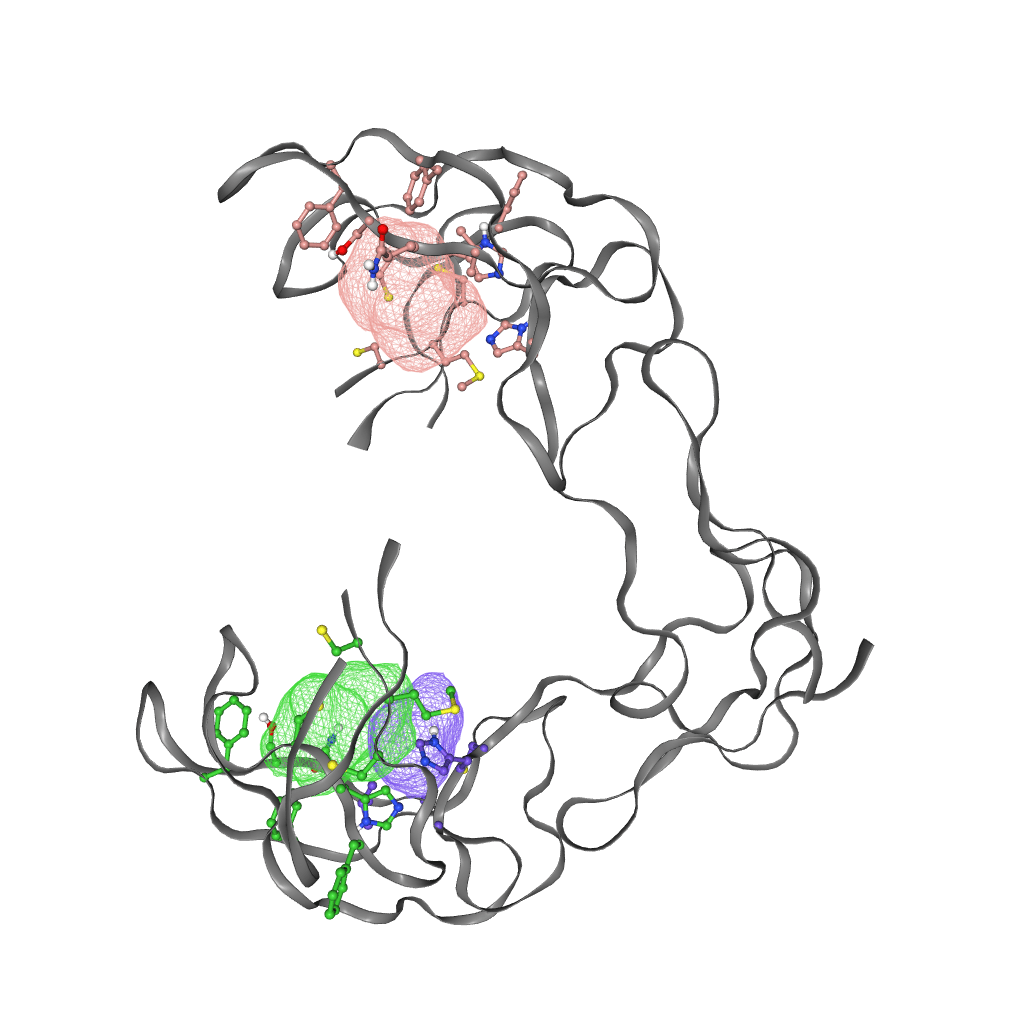
FTSite identified 3 potential binding sites for the GITR protein using the energetic-based method. Pictures of binding sites identified on the GITR protein by FTSite are shown (Fig. 4).
Machine learning method (Prankweb)
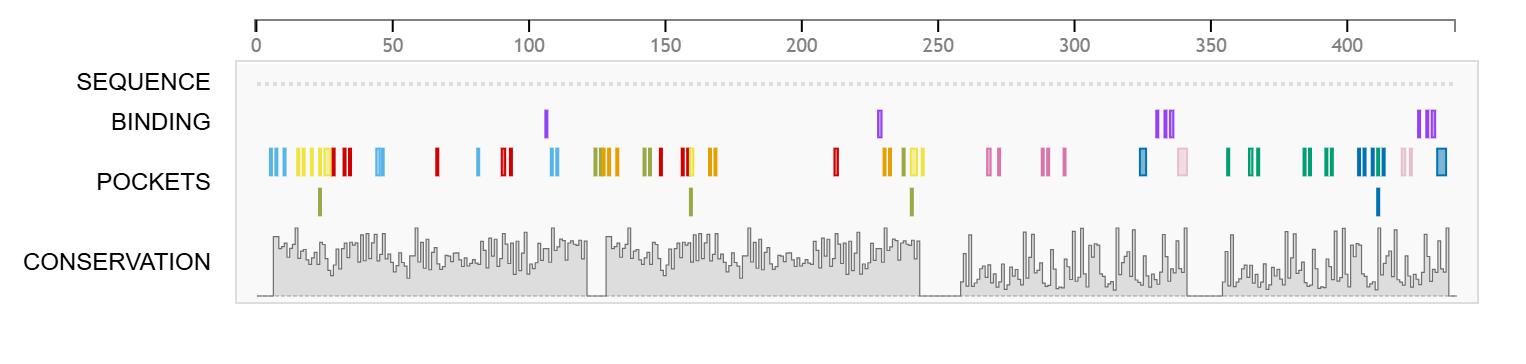
Prankweb identified 9 potential binding sites for the GITR protein using the machine learning method. The binding site on GITR that shows the most promise has a Prankweb score of 4.82, a probability of 0.225, and contains 12 amino acid residues. The second most promising binding site has a score of 3.99, a probability of 0.168, and contains 13 amino acid residues (Table 2). Diagrams of binding sites identified on the GITR protein by Prankweb are shown in Fig. 5 and Fig. 6.
| Pocket Rank | Score | Probability | Amino Acid Residues |
| 1 | 4.82 | 0.225 | 12 |
| 2 | 3.99 | 0.168 | 13 |
| 3 | 3.64 | 0.142 | 7 |
| 4 | 3.26 | 0.117 | 9 |
| 5 | 2.81 | 0.088 | 9 |
| 6 | 2.12 | 0.048 | 12 |
| 7 | 1.47 | 0.020 | 6 |
| 8 | 1.11 | 0.009 | 8 |
| 9 | 0.89 | 0.004 | 7 |
Pharmacophore-based virtual screening
After we conducted binding site analysis of the GITR protein, we examined the GITRL protein portion of the combined GITR-GITRL complex using PocketQuery. Results yielded two pharmacophore maps highlighting portions of the ligand’s chemical structure that are essential to binding interactions with the GITR protein. Then, millions of small molecule compounds were overlaid on these pharmacophore maps with the help of ZINCPharmer to identify promising candidates. We selected the top twenty promising small molecule compounds (ten from each pharmacophore) to move on to the next stage of screening. Virtual screening was conducted on key amino acid clusters on the GITRL ligand instead of GITR because potential small molecule candidates should emulate the chemical properties of the GITRL’s binding regions to effectively target the GITR protein receptor.
Target cluster identification (PocketQuery)
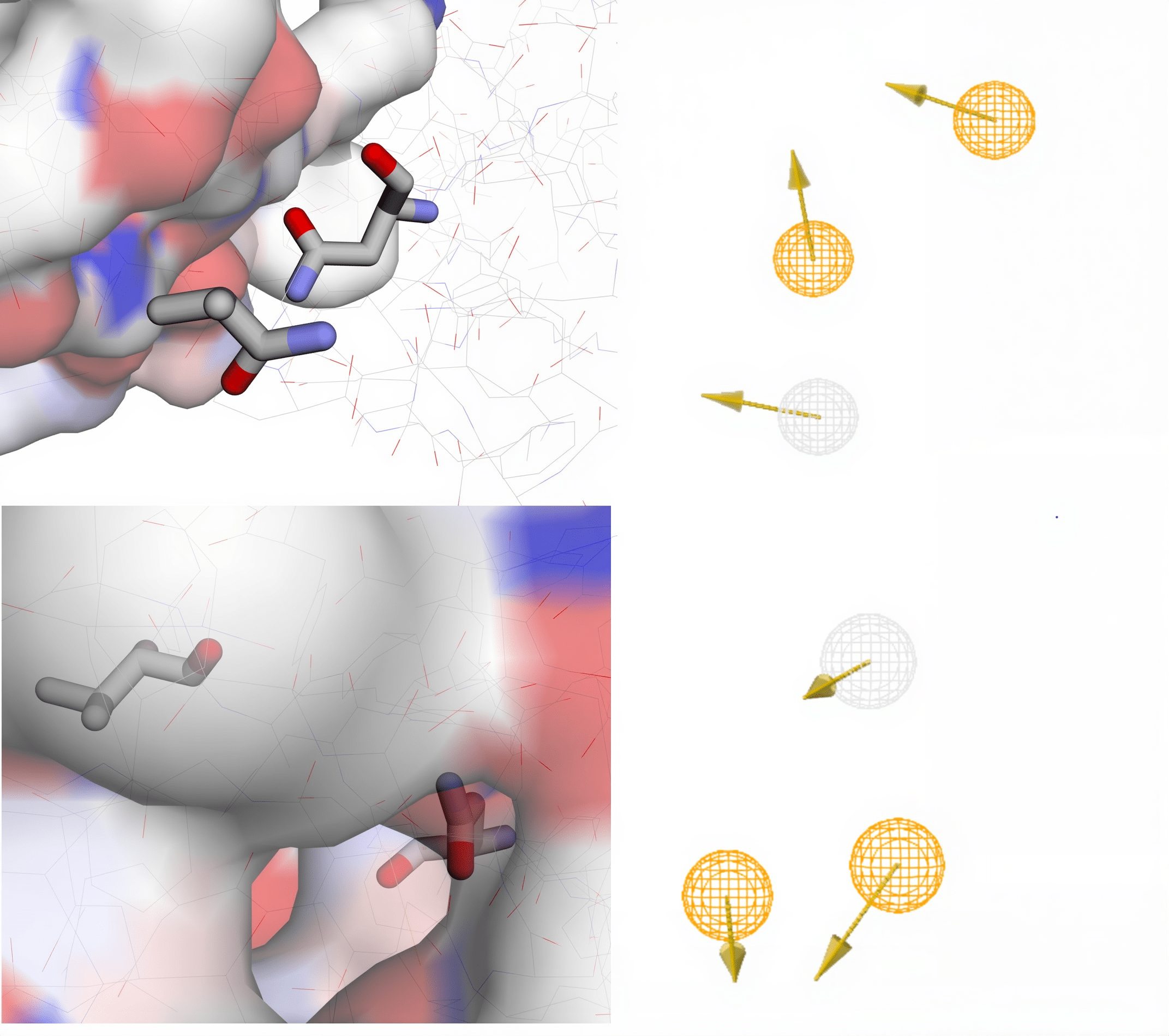
The first pharmacophore map identified using PocketQuery is located on Chain A, contains 2 amino acid residues, has a maximum distance of 9.5558 Å between each residue, and a druggability score of 0.730097. The second pharmacophore map identified using PocketQuery is located on Chain B, contains 2 amino acid residues, has a maximum distance of 9.4921 Å between each residue, and a druggability score of 0.6838 (Table 3). A higher druggability score indicates that the pharmacophore map is more promising than a pharmacophore map with a lower druggability score. These scores range from 0 to 1. However, it is important to note that values generated by computational methods are not perfectly precise and provide a general estimate of each pharmacophore map’s druggability score. Diagrams of each amino acid cluster and their respective pharmacophore maps generated can be found in Fig. 7. On the pharmacophore maps, the gray circle corresponds to a hydrogen acceptor region, while the yellow circles correspond to hydrogen donor regions.
| Cluster | Chain | Amino Acid Residues | Distance | Score |
| 1 | A | 2 | 9.5558 | 0.730097 |
| 2 | B | 2 | 9.4921 | 0.6838 |
Virtual screening (ZINCPharmer)
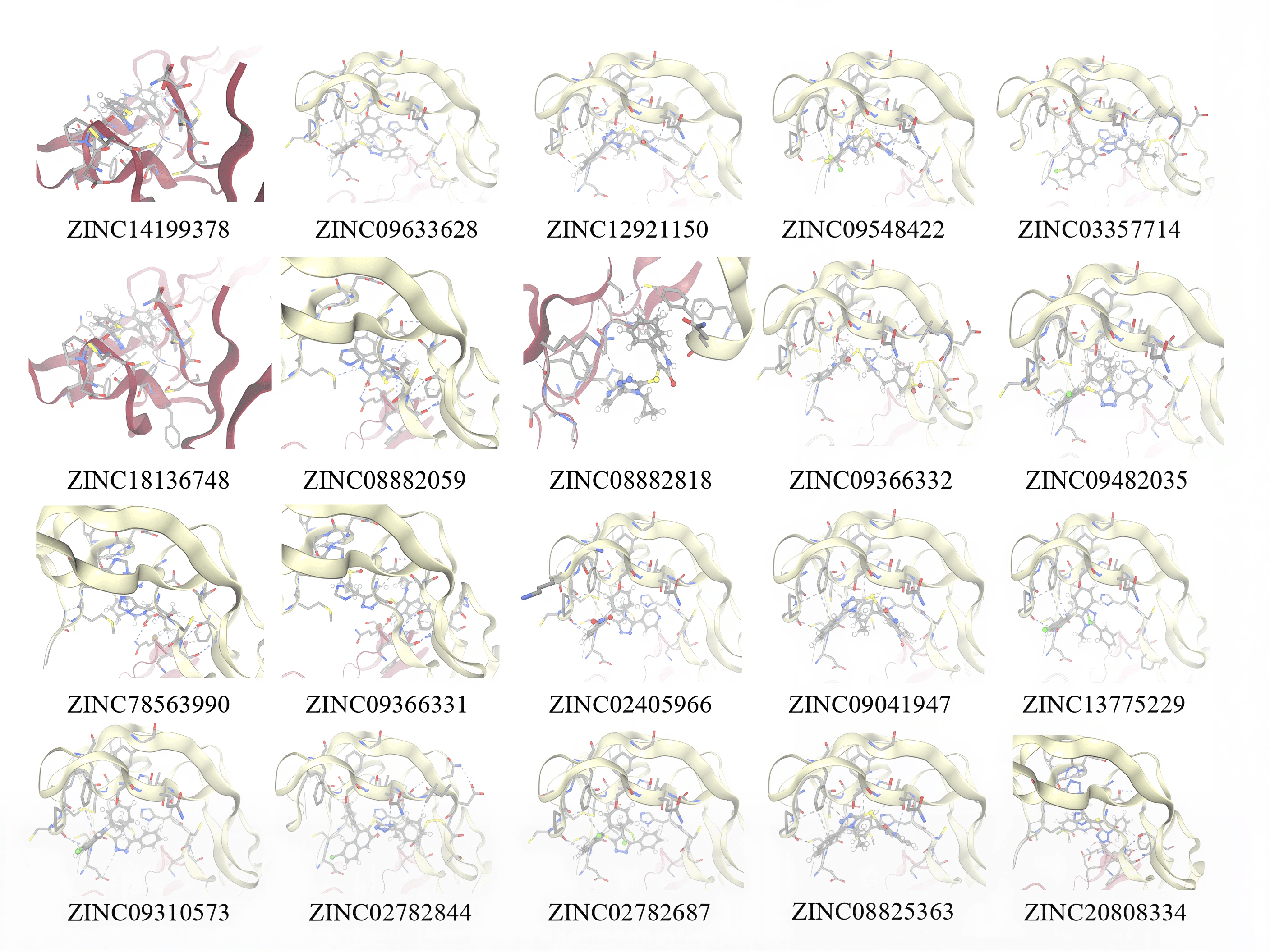
Utilizing a library of 18 million compounds, ZINCPharmer analyzed similarities between each small molecule compound and the pharmacophore maps generated by PocketQuery. The most similar small molecule candidate for the first model cluster in GITRL is ZINC14199378, which has a RMSD score of 0.013 and a molar mass of 418 g/mol. The second most favorable small molecule binder for the first model cluster in GITRL is ZINC09633628, which has a RMSD score of 0.013 and a mass of 442 g/mol. The most similar small molecule binder for the second model cluster in GITRL is ZINC78563990, which has a RMSD score of 0.008 and a mass of 332 g/mol. The second most favorable small molecule binder for the second model cluster in GITRL is ZINC09366331, which has a RMSD score of 0.013 and a molar mass of 446 g/mol. A small molecule with a low RMSD value is more promising than a small molecule with a high RMSD value. These error scores range from 0 to 1. These computationally generated RMSD values provide a useful estimate of each molecule’s fit to the pharmacophore maps, but results should nonetheless be verified using laboratory validation. More information on each small molecule compound for each cluster can be found below (Table 4 and Table 5). In addition, pictures of each small molecule compound overlaid on each cluster’s pharmacophore map can be found in Fig. 8 and Fig. 9, and each molecule’s SMILES code as well as their 2D structure can be found in Fig. 10.
| Name | RMSD (Error) | Mass (g/mol) |
| ZINC14199378 | 0.013 | 418 |
| ZINC09633628 | 0.013 | 442 |
| ZINC12921150 | 0.013 | 429 |
| ZINC09548422 | 0.014 | 403 |
| ZINC03357714 | 0.014 | 460 |
| ZINC18136748 | 0.015 | 431 |
| ZINC08882059 | 0.015 | 327 |
| ZINC08882818 | 0.015 | 438 |
| ZINC09366332 | 0.015 | 446 |
| ZINC09482035 | 0.016 | 472 |

| Name | RMSD (Error) | Mass (g/mol) |
| ZINC78563990 | 0.008 | 332 |
| ZINC09366331 | 0.013 | 446 |
| ZINC02405966 | 0.014 | 471 |
| ZINC09041947 | 0.014 | 470 |
| ZINC13775229 | 0.015 | 493 |
| ZINC09310573 | 0.015 | 459 |
| ZINC08825363 | 0.015 | 425 |
| ZINC02782844 | 0.015 | 459 |
| ZINC02782687 | 0.015 | 493 |
| ZINC20808334 | 0.016 | 432 |


Molecular docking
After pharmacophore map generation with PocketQuery and virtual screening with ZINCPharmer, we performed small molecule docking simulations between the GITR protein and each of the top twenty small molecule compounds virtually via Swissdock. The top six small molecule candidates with the most favorable interactions with the GITR protein were selected to move on to the next stage of virtual screening. Swissdock simulates molecular interactions between the small molecule and the GITR protein and determines if each conformation is geometrically and energetically favorable. These simulations are conducted within a search box specified by the user. In this case, we highlighted the entire GITR protein as the search region for docking simulations (Fig. 11). This method yields valuable predictions of each small molecule binder’s efficacy while forgoing the need for laboratory testing.
Docking with attracting cavities (Swissdock)
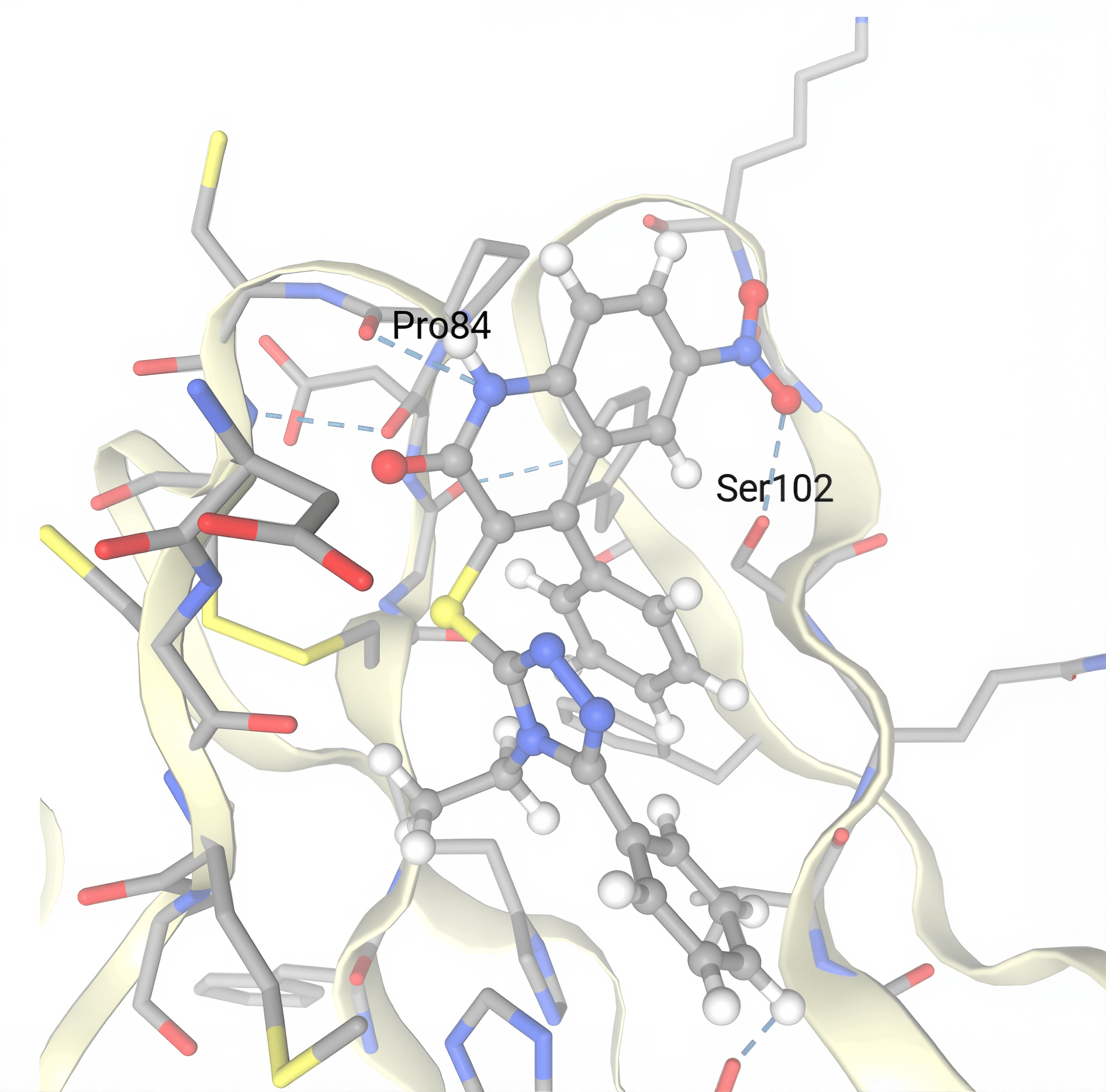
The six small molecule compounds with the most negative SwissParam scores were ZINC09041947, ZINC12921150, ZINC09366332, ZINC09482035, ZINC20808334, and ZINC02405966. The top small molecule candidates were selected using this metric because a more negative SwissParam score signifies a more favorable and efficient binding interaction between the small molecule compound and the GITR protein. ZINC09041947’s most favorable cluster was number one, with one member and a SwissParam score of -7.9363. ZINC12921150’s most favorable cluster was number one, with one member and a SwissParam score of -7.9264. ZINC09366332’s most favorable cluster was number zero, with one member and a SwissParam score of -7.8711. All three of these small molecules show very strong potential to bind with the GITR protein efficiently. In addition, ZINC09482035, ZINC20808334, and ZINC02405966 also show promise, with SwissParam scores more negative than -7.84 each. Additional information on each small molecule compound (Table 6) and pictures of each molecule overlaid on the GITR protein can be found in Fig. 12. A more in-depth diagram is also provided for the most promising small molecule, ZINC09041947, due to its highly negative SwissParam score. The blue dashes represent hydrogen bonds and gray dashes represent hydrophobic contacts. Pro84 and Ser102 were identified as two amino acid residues on chain D of the GITR protein that play an important role in hydrogen bonding between ZINC09041947 and GITR (Fig. 13).
Moreover, to validate docking simulation accuracy, we ran a negative control (aspirin) with ZINC ID ZINC53. Results indicate that ZINC53’s most favorable cluster was number two, with one member and a SwissParam score of -5.9745, which is notably less negative than the top small molecule compounds. Additional information on this test trial can be found in Table 7. These results suggest that randomly selected small molecules are less likely to be strong binding candidates to GITR when compared to those identified via pharmacophore-based screening, which confirms the reliability of our approach.

| Molecule ID | Cluster Number | Cluster Member | SwissParam Score |
| ZINC09041947 | 1 | 1 | -7.9363 |
| ZINC12921150 | 1 | 1 | -7.9264 |
| ZINC09366332 | 0 | 1 | -7.8711 |
| ZINC09482035 | 0 | 1 | -7.8669 |
| ZINC20808334 | 1 | 1 | -7.8599 |
| ZINC02405966 | 0 | 1 | -7.8469 |
| ZINC09633628 | 3 | 1 | -7.8348 |
| ZINC18136748 | 2 | 1 | -7.8330 |
| ZINC14199378 | 1 | 1 | -7.8123 |
| ZINC02782687 | 0 | 1 | -7.7870 |
| ZINC08825363 | 1 | 1 | -7.7859 |
| ZINC09366331 | 2 | 1 | -7.7804 |
| ZINC03357714 | 0 | 1 | -7.7748 |
| ZINC09310573 | 0 | 1 | -7.7246 |
| ZINC13775229 | 2 | 1 | -7.6457 |
| ZINC02782844 | 1 | 1 | -7.5973 |
| ZINC09548422 | 0 | 1 | -7.5029 |
| ZINC08882818 | 7 | 1 | -7.2719 |
| ZINC08882059 | 2 | 1 | -7.1899 |
| ZINC78563990 | 2 | 1 | -7.1608 |
| Molecule ID | Cluster Number | Cluster Member | SwissParam Score |
| ZINC53 | 2 | 1 | -5.9745 |
ADME and Lipinski’s Rule
We screened the top six small molecule binding candidates to the GITR protein for druglikeness using Lipinski’s rule. Lipinski’s rule tests each small molecule for essential properties, including its capabilities for absorption, distribution, metabolism, and excretion. This step works to ensure that the small molecule has strong druggability potential and can efficiently permeate the tumor environment when administered to patients while also being metabolized and excreted relatively quickly to ensure patient safety. We then selected the most promising small molecule that passed Lipinski’s rule as a final candidate for toxicity screening.
Druglikeness evaluation (SwissADME)
All six of the compounds screened followed Lipinski’s Rule, meaning that they all show strong drug potential (Table 8). The compound with the lowest SwissParam score, ZINC09041947, has one hydrogen bond donor, a calculated LogP of 2.76, a molecular mass of 469.52g/mol, and five hydrogen bond acceptors. The second most favorable compound identified using Swissdock, ZINC12921150, has two hydrogen bond donors, a calculated LogP of 2.67, a molecular mass of 429.47g/mol, and four hydrogen bond acceptors. The third most favorable compound identified using Swissdock, ZINC09366332, has two hydrogen bond donors, a calculated LogP of 1.84, a molecular mass of 445.56g/mol, and five hydrogen bond acceptors.
| Compound | Hydrogen Bond Donors | Calculated LogP | Molecular Mass (g/mol) | Hydrogen Bond Acceptors | Druglikeness (Lipinski’s Rule) |
| ZINC09041947 | 1 | 2.76 | 469.52 | 5 | Yes |
| ZINC12921150 | 2 | 2.67 | 429.47 | 4 | Yes |
| ZINC09366332 | 2 | 1.84 | 445.56 | 5 | Yes |
| ZINC09482035 | 1 | 2.85 | 471.96 | 4 | Yes |
| ZINC20808334 | 1 | 3.63 | 431.90 | 4 | Yes |
| ZINC02405966 | 1 | 2.47 | 470.50 | 6 | Yes |
Toxicity Screening
Lastly, we checked the most promising small molecule binder to the GITR protein for potential toxicity using the online tool Protox-3.0. The purpose of this software is to assess the potential organ toxicities, toxicological endpoints, and toxicological pathways associated with each small molecule. This step confirms each binder’s druggability potential by ensuring that small molecule candidates identified using virtual screening are safe for oral administration and produce minimal toxic side effects in patients.
Oral toxicity prediction (Protox-3.0)
Protox 3.0 predicted a LD50 score of 1000mg/kg and a toxicity class of four for ZINC09041947, the most promising small molecule binder to GITR (Table 9). The toxicity class scores range from one to six, with one being the most toxic and six being the least toxic, demonstrating that ZINC09041947 is a relatively safe compound. In addition, the inactive types of toxicity for the compound far outnumbered the active types of toxicity. The darker colored circles represent predictions with confidence scores over 70%, while the lighter colored circles represent predictions with confidence scores under 70% (Fig. 14). Furthermore, the toxicity radar chart for ZINC09041947 demonstrates that the small molecule binder has lower toxicity values for nearly all toxicity types when compared to the average toxicity levels of similar active molecules, showing that it has a high druggability potential (Fig. 15). It is important to note that virtual screening results for toxicity testing should be validated with further laboratory testing due to a relatively low accuracy score of 54% (Table 9).
| Predicted LD50 (mg/kg) | Toxicity Class | Average Similarity (%) | Accuracy (%) |
| 1000 | 4 | 44.86 | 54.26 |


Discussion
In this study, we aimed to discover potential modulators of the GITR protein for cancer immunotherapy and autoimmune disease treatment applications. Many GITR-targeted immunotherapy treatments currently being tested utilize mAbs, but none, to our knowledge, investigate the applications of small molecules. Small molecule treatments provide distinct advantages over mAb treatments because they are administered orally, penetrate the tumor microenvironment more efficiently, circulate more quickly, reduce immunogenicity, and have greater accessibility33,34. Consequently, we utilized in-silico screening methods to identify promising small molecule candidates binding to the GITR protein, a crucial first step in the small molecule drug discovery process. First, DoGSiteScorer, FTsite, and Prankweb were used to verify that GITR proteins have suitable binding sites for small molecule compounds. After that, pharmacophore-based virtual screening was performed using Pocketquery and ZINCPharmer to identify twenty small molecules with the potential to interact with binding sites on GITR. The drug potential of these small molecule candidates was then evaluated using free energy calculations determined by Swissdock. The top six small molecule candidates were then screened using Lipinski’s Rule to determine their ADME properties as identified by SwissADME. Lastly, toxicity screening was conducted on the most promising small molecule compound using Protox 3.0 to verify that it is safe for oral administration. Results indicate that ZINC09041947 is the most promising small molecule binder to the GITR protein because it has a SwissParam score of -7.9363, adheres to Lipinski’s Rule of 5, and has a LD50 value of 1000mg/kg. Moreover, ZINC12921150, ZINC09366332, ZINC09482035, ZINC20808334, ZINC02405966 also exhibit strong binding tendencies (SwissParam scores of -7.84 and lower) and follow Lipinski’s Rule. Thus, this study identifies six promising small molecule candidates that are worth considering for future testing.
These small molecule candidates that emulate the chemical properties of GITRL have the potential to either activate or to inhibit the GITR costimulatory pathway through the modulation of the GITR protein. These properties are clinically relevant because small molecule agonists of this pathway contribute to increased and enhanced T-eff cell activation and reduced T-reg cell activity, which serves to reinvigorate the immune system to produce a robust antitumor response in cancer patients. Moreover, small molecule antagonists of this pathway contribute to increased T-reg cell activity and reduced T-eff cell activity, which works to temper the immune system to mitigate the impacts of an overzealous immune response in cases of autoimmune disease. Thus, using virtual screening methods to identify potential small molecule modulators of the GITR costimulatory pathway is of therapeutic relevance because these small molecules can eventually be used to treat either cancer or autoimmune disease, which pose serious threats to human health and wellbeing.
One limitation of this experiment is that small molecule screening was conducted through a completely virtual process because computational methods are restricted in scope and cannot fully validate small molecule binding candidates when used alone. For instance, a fixed crystal protein structure of the GITR-GITRL complex was utilized during computational screening, which may neglect to account for dynamic changes in the proteins during binding. It is known that virtual screening techniques exhibit strong predictive properties, which save scientists a significant amount of time and money in the initial stages of the drug discovery process37. However, it is important to note that numerical values generated by computational techniques, such as molecular docking scores, provide approximate indicators of each molecule’s binding affinities. Thus, computational methods must be supplemented with rigorous laboratory testing in order to confirm the efficacy and safety of small molecule modulators. Potential future steps include physical lab testing with bioassays to verify ZINC09041947’s ability to bind to GITR while determining if it acts as an agonist (cancer treatment applications) or antagonist (autoimmune disease treatment applications).
Furthermore, the structure-activity relationship of the most promising small molecule compounds should be investigated to optimize its efficacy and safety for oral administration. Moreover, because combination treatments are of increasing relevance, each small molecule’s potential for serving as bifunctional agonists or antagonists for GITR and another pathway should be investigated further53. Many current bifunctional treatments are for proteins and mAbs, but the identification of bifunctional small molecules is a feasible goal, as demonstrated by a 2023 study that discovered bifunctional small molecule inhibitors of PD-L1 and CXCL12 for cancer immunotherapy treatment54.
In addition to the limitation of utilizing virtual screening with fixed protein structures, another potential concern is that the isolated GITR or GITRL structures may differ from the structure of the GITR-GITRL complex, which may make it challenging for the small molecules identified in this study to bind to GITR in the absence of GITRL. However, a 2021 study on GITR-GITRL complex structure revealed that the GITR protein undergoes minimal conformational changes when binding to GITRL55, so this factor likely will not impair the ability of promising small molecule candidates to bind to GITR.
Moreover, it is important to note that the GITR-GITRL protein complex is unique because GITRL exhibits trimeric properties and typically binds to three GITR proteins simultaneously. These activated GITR-GITRL complexes then combine to form a branched hexamer network, which plays an essential role in GITR stimulation and contributes to a more robust antitumor response55. One potential challenge associated with developing small molecule agonists is the difficulty in finding a multivalent binder that is capable of stimulating and clustering GITR proteins together into these hexameric networks while also maintaining the decreased molecular size required by small molecule immunotherapy. This feature may be a strong reason why many GITR agonists being tested and developed utilize multivalent mAbs or multimeric GITRL proteins instead of small molecules53’56. Nevertheless, the benefits of small molecules over mAbs mentioned above make the search for GITR-targeted small molecules a worthwhile endeavor. Nevertheless, small molecule agonists exhibiting these properties have been found for OX40, another costimulatory pathway belonging to the same tumor necrosis factor receptor (TNFR) superfamily36. Therefore, finding an effective small molecule agonist for the GITR protein is feasible. In terms of finding small molecule antagonists of the GITR protein to treat autoimmune diseases, the unique clustering properties of the GITR protein may serve as an advantage. Specifically, these properties open up the possibility of screening for small molecules that can prevent the formation of either the hexameric networks or the GITR-GITRL activated trimer, which can temper overzealous autoimmune responses. Moreover, it is important to note that this study focused primarily on small molecule screening for compounds using the interactions displayed by the structure of the activated GITR-GITRL complex as a model. Future studies could also investigate crystal structures depicting interactions between GITR and GITR-specific mAbs currently showing efficacy during testing to potentially find alternate binding sites that may work better for GITR modulation.
Overall, this study is of significance because it is the first to our knowledge to explore the discovery of small molecule modulators of the GITR-mediated costimulatory pathway and sets the groundwork for future research on the development of new treatment alternatives for cancer and autoimmune disease patients.
Acknowledgements
I would like to thank my mentor, Dr. Moustafa Gabr from Cornell University, for introducing me to these virtual screening technologies and guiding me throughout the research process as I collected my research data and prepared this manuscript.
References
- D. Hanahan, R. A. Weinberg. The hallmarks of cancer review. Cell. 100, 57–70 (2000). [↩]
- D. Hanahan, R. A. Weinberg. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [↩]
- What is Cancer. https://www.cancer.org/content/dam/CRC/PDF/Public/6041.00.pdf (2025). [↩]
- What is Cancer. https://www.cancer.gov/about-cancer/understanding/what-is-cancer (2021). [↩]
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram, A. Jemal. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 74, 229–263 (2024). [↩]
- R. L. Siegel, T. B. Kratzer, A. N. Giaquinto, H. Sung, A. Jemal. Cancer statistics, 2025. CA. Cancer J. Clin. 75, 10–45 (2025). [↩]
- M. Arruebo, N. Vilaboa, B. Sáez-Gutierrez, J. Lambea, A. Tres, M. Valladares, Á. González-Fernández. Assessment of the evolution of cancer treatment therapies. Cancers 3, 3279–3330 (2011). [↩]
- A. Zafar, S. Khatoon, M. J. Khan, J. Abu, A. Naeem. Advancements and limitations in traditional anti-cancer therapies: a comprehensive review of surgery, chemotherapy, radiation therapy, and hormonal therapy. Discov. Oncol. 16, 607 (2025). [↩]
- M. Bilusic. What are the advantages of neoadjuvant immunotherapy over adjuvant immunotherapy? Expert Rev. Anticancer Ther. 22, 561–563 (2022). [↩]
- M. Vanneman, G. Dranoff. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer. 12, 237–251 (2012). [↩]
- C. Liu, M. Yang, D. Zhang, M. Chen, D. Zhu. Clinical cancer immunotherapy: current progress and prospects. Front. Immunol. 13, (2022). [↩] [↩]
- F. Hamdan, V, Cerullo. Cancer immunotherapies: a hope for the uncurable? Front. Mol. Med. 3, (2023). [↩]
- W. G. Kerr, J. D. Chisholm. The next generation of immunotherapy for cancer: small molecules could make big waves. J. Immunol. 202, 11–19 (2019). [↩]
- E. N. Baruch, A. L. Berg, M. J. Besser, J. Schachter, G. Markel. Adoptive T cell therapy: an overview of obstacles and opportunities. Cancer 123, 2154–2162 (2017). [↩]
- J. Santos Apolonio, V. Lima De Souza Gonçalves, M. L. Cordeiro Santos, M. Silva Luz, J. V. Silva Souza, S. L. Rocha Pinheiro, W. R. De Souza, M. Sande Loureiro, F. F. De Melo. Oncolytic virus therapy in cancer: A current review. World J. Virol. 10, 229–255 (2021). [↩]
- M. J. Lin, J. Svensson-Arvelund, G. S. Lubitz, A. Marabelle, I. Melero, B. D. Brown, J. D. Brody. Cancer vaccines: the next immunotherapy frontier. Nat. Cancer. 3, 911–926 (2022). [↩]
- Y. A. Fouad, C. Aanei. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 7, 1016–1036 (2017). [↩]
- S. Singh, D. Barik, A. P. Arukha, S. Prasad, I. Mohapatra, A. Singh, G. Singh. Small molecule targeting immune cells: a novel approach for cancer treatment. Biomedicines 11, 2621 (2023). [↩]
- R. W. Jenkins, D. A. Barbie, K. T. Flaherty. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer. 118, 9–16 (2018). [↩]
- D. M. Pardoll. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 12, 252–264 (2012). [↩]
- P. Darvin, S. M. Toor, V. Sasidharan Nair, E. Elkord. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 50, 1–11 (2018). [↩]
- X. He, C. Xu. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 30, 660–669 (2020). [↩]
- K. C. Ohaegbulam, A. Assal, E. Lazar-Molnar, Y. Yao, X. Zang. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 21, 24–33 (2015). [↩]
- Y. Fu, Q. Lin, Z. Zhang, L. Zhang. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B. 10, 414–433 (2020). [↩]
- Y. Choi, Y. Shi, C. L. Haymaker, A. Naing, G. Ciliberto, J. Hajjar. T-cell agonists in cancer immunotherapy. J. Immunother. Cancer. 8, e000966 (2020). [↩]
- J. Sakowska, L. Arcimowicz, M. Jankowiak, I. Papak, A. Markiewicz, K. Dziubek, M. Kurkowiak, S. Kote, K. Kaźmierczak-Siedlecka, K. Połom, N. Marek-Trzonkowska, P. Trzonkowski. Autoimmunity and cancer—two Sides of the same coin. Front. Immunol. 13, (2022). [↩]
- Autoimmune Diseases. https://www.niehs.nih.gov/health/topics/conditions/autoimmune (2025). [↩]
- S. M. Jung, W.-U. Kim. Targeted immunotherapy for autoimmune disease. Immune Netw. 22, (2022). [↩]
- G. Buzzatti, C. Dellepiane, L. Del Mastro. New emerging targets in cancer immunotherapy: the role of GITR. ESMO Open. 4, e000738 (2019). [↩]
- J. Tian, B. Zhang, K. Rui, S. Wang. The role of GITR/GITRL interaction in autoimmune diseases. Front. Immunol. 11, (2020). [↩]
- D. Coe, S. Begom, C. Addey, M. White, J. Dyson, J.-G. Chai. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol. Immunother. 59, 1367–1377 (2010). [↩]
- K. Narumi, R. Miyakawa, C. Shibasaki, M. Henmi, Y. Mizoguchi, R. Ueda, H. Hashimoto, N. Hiraoka, T. Yoshida, K. Aoki. Local administration of GITR agonistic antibody induces a stronger antitumor immunity than systemic delivery. Sci. Rep. 9, (2019). [↩]
- J. L. Adams, J. Smothers, R. Srinivasan, A. Hoos. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 14, 603–622 (2015). [↩] [↩]
- B. Cheng, J. Lv, Y. Xiao, C. Song, J. Chen, C. Shao. Small molecule inhibitors targeting PD-L1, CTLA4, VISTA, TIM-3, and LAG3 for cancer immunotherapy (2020–2024). Eur. J. Med. Chem. 283, 117141 (2025). [↩] [↩]
- Y. Wang, T. Gu, X. Tian, W. Li, R. Zhao, W. Yang, Q. Gao, T. Li, J.-H. Shim, C. Zhang, K. Liu, M.-H. Lee. A small molecule antagonist of PD-1/PD-L1 interactions acts as an immune checkpoint inhibitor for NSCLC and melanoma immunotherapy. Front. Immunol. 12, (2021). [↩]
- Y. Song, E. Margolles‐Clark, A. Bayer, P. Buchwald. Small‐molecule modulators of the OX 40– OX40 ligand co‐stimulatory protein–protein interaction. Br. J. Pharmacol. 171, 4955–4969 (2014). [↩] [↩]
- D. Giordano, C. Biancaniello, M. A. Argenio, A. Facchiano. Drug design by pharmacophore and virtual screening approach. Pharmaceuticals. 15, 646 (2022). [↩] [↩]
- A. Volkamer, D. Kuhn, F. Rippmann, M. Rarey. DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics. 28, 2074–2075 (2012). [↩]
- C.-H. Ngan, D. R. Hall, B. Zerbe, L. E. Grove, D. Kozakov, S. Vajda. FTSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics. 28, 286–287 (2012). [↩]
- L. Jendele, R. Krivak, P. Skoda, M. Novotny, D. Hoksza. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res. 47, W345–W349 (2019). [↩]
- T. Seidel, O. Wieder, A. Garon, T. Langer. Applications of the pharmacophore concept in natural product inspired drug design. Mol. Inform. 39, (2020). [↩]
- A. Voet, X. Qing, X. Y. Lee, J. De Raeymaecker, J. Tame, K. Zhang, M. De Maeyer. Pharmacophore modeling: advances, limitations, and current utility in drug discovery. J. Recept. Ligand Channel Res. 81–92 (2014). [↩]
- D. R. Koes, C. J. Camacho. PocketQuery: protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res. 40, W387–W392 (2012). [↩]
- 43: D. R. Koes, C. J. Camacho. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. 40, W409–W414 (2012). [↩]
- X.-Y. Meng, H.-X. Zhang, M. Mezei, M. Cui. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided-Drug Des. 7, 146–157 (2011). [↩]
- U. F. Röhrig, M. Goullieux, M. Bugnon, V. Zoete. Attracting cavities 2.0: improving the flexibility and robustness for small molecule docking. J. Chem. Inf. Model. 63, 3925–3940 (2023). [↩]
- A. Grosdidier, V. Zoete, O. Michielin. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 39, W270–W277 (2011). [↩]
- K. M. Honorio, T. L. Moda, A. D. Andricopulo. Pharmacokinetic properties and in silico ADME modeling in drug discovery. Med. Chem. 9, 163–176 (2013). [↩]
- C. A. Lipinski, F. Lombardo, B. W. Dominy, P. J. Feeney. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 64, 4–17 (2012). [↩]
- A. Daina, O. Michielin, V. Zoete. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, (2017). [↩]
- P. Banerjee, A. O. Eckert, A. K. Schrey, R. Preissner. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46, W257–W263 (2018). [↩]
- P. Banerjee, E. Kemmler, M. Dunkel, R. Preissner. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 52, W513–W520 (2024). [↩] [↩]
- S. Chan, N. Belmar, S. Ho, B. Rogers, M. Stickler, M. Graham, E. Lee, N. Tran, D. Zhang, P. Gupta, M. Sho, T. MacDonough, A. Woolley, H. Kim, H. Zhang, W. Liu, P. Zheng, Z. Dezso, K. Halliwill, M. Ceccarelli, S. Rhodes, A. Thakur, C. M. Forsyth, M. Xiong, S. S. Tan, R. Iyer, M. Lake, E. Digiammarino, L. Zhou, L. Bigelow, K. Longenecker, R. A. Judge, C. Liu, M. Trumble, J. P. Remis, M. Fox, B. Cairns, Y. Akamatsu, D. Hollenbaugh, F. Harding, H. M. Alvarez. An anti-PD-1–GITR-L bispecific agonist induces GITR clustering-mediated T cell activation for cancer immunotherapy. Nat. Cancer 3, 337–354 (2022). [↩] [↩]
- B. Cheng, W. Wang, T. Liu, H. Cao, W. Pan, Y. Xiao, S. Liu, J. Chen. Bifunctional small molecules targeting PD-L1/CXCL12 as dual immunotherapy for cancer treatment. Signal Transduct. Target. Ther. 8, 91 (2023). [↩]
- F. Wang, B. Chau, S. M. West, C. R. Kimberlin, F. Cao, F. Schwarz, B. Aguilar, M. Han, W. Morishige, C. Bee, G. Dollinger, A. Rajpal, P. Strop. Structures of mouse and human GITR–GITRL complexes reveal unique TNF superfamily interactions. Nat. Commun. 12, 1378 (2021). [↩]
- D. Davar, R. Zappasodi, H. Wang, G. S. Naik, T. Sato, T. Bauer, D. Bajor, O. Rixe, W. Newman, J. Qi, A. Holland, P. Wong, L. Sifferlen, D. Piper, C. A. Sirard, T. Merghoub, J. D. Wolchok, J. J. Luke. Phase IB study of GITR agonist antibody TRX518 singly and in combination with gemcitabine, pembrolizumab, or nivolumab in patients with advanced solid tumors. Clin. Cancer Res. 28, 3990–4002 (2022). [↩]



{kind=link}