
Abstract
Genetic disorders, specifically genetic neurodegenerative disorders, affect the quality of life for many individuals. One example of such diseases is Rett Syndrome, a rare neurodegenerative disorder primarily affecting females and caused by a mutation in the MeCP2 gene, which is essential for proper neuronal function. Affected individuals appear normal at birth but soon after start to regress rapidly. Previously, studies have attempted to treat this disease with drugs to subside symptoms, but there is no actual cure. Recent advancements in gene editing technology show great promise for the future of Rett Syndrome and other genetic diseases. This paper will review the potential of new gene editing technologies in the context of Rett Syndrome, discussing the scientific progress made in research and the ethics of its use in humans for disease treatment.
Introduction
Background of the Disease
Rett Syndrome is an X-linked genetic neurological disorder, meaning that the Rett Syndrome disease is caused by a mutation of a sequence on one of the X-chromosomes. This sequence encodes for a protein called Methyl-CpG binding protein (MeCP2)1, which is essential for proper neuronal function. Rett Syndrome is predominantly found in females, affecting 7.1 out of every 100,000 female births2. Rett Syndrome patients experience a wide array of debilitating symptoms, including seizures, loss of motor skills, speech issues, and respiratory dysfunction. Due to the seriousness of the disease, many have attempted to develop treatments for struggling patients.
Female Predisposition
In females with two copies of the X-chromosome, due to X-inactivation, where each cell randomly chooses an X-chromosome to silence3, the working copy of the MeCP2 sequence is sometimes used. In contrast, in other cells the mutated sequence is expressed. This results in a deficiency of functioning MeCP2 protein, which is why Rett Syndrome is predominantly found in females. As males only have one copy of the X-chromosome, any mutation in the MeCP2 sequence results in a complete absence of a functional protein, making the disease much more consequential for them4.
Existing Forms of Treatment
Rett Syndrome has few treatments besides gene therapy to restore the mutated protein or drugs to help attenuate symptoms4. . On March 10, 2023, the drug Trofinetide was FDA-approved as the first treatment for Rett Syndrome5. Trofinetide works by promoting the growth of the neuronal dendritic tree and improving neuronal plasticity, alleviating symptoms6. Trofinetide is a synthetic analog of glycine-proline-glutamate (GPE), a tripeptide that is naturally produced in the brain through the breakdown of insulin-like growth factor 1 (IGF-1). Caregivers reported improvements in engagement (46.2%), hand use (42.3%), and eye gaze (30.8%). Other medications that provide supportive care like antiseizure, antianxiety, anti-reflux medications may be used7. Gene therapy has also been used in research to observe the efficacy of restoring proper MeCP2 protein. However, gene therapy poses the risk of overexpression of the MeCP2 protein, leading to disease8, and while drugs alleviate symptoms4, drugs are not a permanent treatment. It is important to note that drug treatments do not treat the underlying cause of the disease, rather, focusing on improving the quality of life for Rett syndrome individuals. The expansion of genetic technologies could drastically impact this disease, as gene editing methods may finally provide a long-term solution for people with the disease. This is because they encode for edits that should restore the proper function of the MeCP2 protein, meaning the treatments directly target the cause of the disease and are thus more permanent solutions.
Genetics Background
To understand the genetic technologies used in the studies that will be discussed in this paper, one must have some fundamental knowledge of genetics, such as the differences between DNA and RNA, how the genome creates proteins, and the inheritance pattern in which the Rett Syndrome disease discussed is passed down.
DNA is located in the nucleus, and to create proteins, the genetic sequence must go through two steps: transcription and translation (see Figure 1). DNA can code for proteins through its genetic sequence, which is determined by the order of its nucleotides: adenine, thymine, cytosine, and guanine. First, transcription occurs, where RNA polymerase uses one side of the DNA strand as a template to create RNA9. The RNA polymerase creates a strand of RNA complementary to the original DNA strand. The resulting RNA chain (mRNA) leaves the nucleus and meets a ribosome, a protein that converts RNA to proteins. Reading the RNA in nucleotide groups of three, the ribosome creates an amino acid strand10. That amino acid strand then folds to create a protein. Different proteins are responsible for their own functions, which determine cellular function overall10.
Inheritance patterns are also essential to understanding the mechanics and causes of Rett Syndrome and the reasoning for various treatment methods. Rett Syndrome has a specific inheritance pattern known as an X-linked single-gene disease4. X-linked single-gene diseases are caused by single-gene mutations located on the X-chromosome. Because a specific inheritance pattern is linked to an X-chromosome, which is a sex chromosome, an individual’s sex determines their inheritance of a sex-linked single-gene disease.
Biological females possess two X-chromosomes, so they inherit one X-chromosome from their biological mother and one from their biological father11. Biological males, on the other hand, have one X-chromosome and one Y-chromosome. Biological males receive their X-chromosome from their biological mother and their Y-chromosome from their biological father (see Figure 1). In the case of Rett Syndrome, the inheritance pattern for the affected gene is sex-linked dominant, meaning the presence of a mutant copy of the gene, even if biological females have a normal copy, will still result in Rett Syndrome. Thus, in the case of males with no other X-chromosome, having a copy of the mutant gene will result in a meager survival rate4.
Prevalent Gene Editing Methods
CRISPR-Cas9 Gene Editing
One of the most prominent methods used for treating genetic diseases is Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas9, a highly specific editing system derived from bacterial immune systems12. CRISPR consists of a guide RNA that leads the protein to the correct sequence, and a Cas9 protein that creates specific double-strand breaks in the DNA13. The body’s natural repair mechanisms will then repair the double-stranded break. In some situations, the repair system will utilize a pathway that results in multiple insertions or deletions on the genome, causing a gene to shut off, also known as nonhomologous end joining (NHEJ)12. Less commonly, more precise repair mechanisms will be used, where a template can be provided alongside the genome editor to replace the mutation with the correct sequence. This pathway is called homology-directed repair, or HDR12. In the case of a dominant genetic disorder11, where a mutation in a single allele can cause the disease, the disease-causing gene needs to be knocked out entirely for the disease to disappear. Gene knockout can be achieved using CRISPR-Cas9 and the NHEJ pathway12.
Epigenetic Editing
Other ways to impact gene expression exist, including technologies that do not create a double-stranded break in the genome. Epigenetic editing, or DNA methylation editing, involves the addition or the removal of a methyl group using proteins14. The way the methyl group is bound to the DNA and in which section controls the expression of said genetic sequence14. This method does not directly change the nucleotides or the DNA sequence, but rather, what portion of the DNA is being expressed. In other words, epigenetic editing controls what parts of the genomic sequence are being used to create proteins, as the expression of proteins is what determines cellular function.
RNA Editing
Additionally, RNA editing, or the alteration of nucleotides subsequent to transcription, is a final method that poses a means of treatment for diseases caused by single-point mutations15.. RNA editing does not directly edit the genome that is duplicated for all cellular functions. Instead, only the area that is mutated and is producing a mutated protein is being edited. The specific RNA editing discussed is a point edit of adenosine to inosine. This corrects the Rett-causing mutation, guanosine to adenosine. Inosine is read as guanosine during translation into a protein, correcting the mutation that exists in the parent DNA. This paper outlines the processes of the gene editing technologies, DNA methylation, RNA editing, and CRISPR-Cas9 in preclinical Rett syndrome studies, specifically focusing on the potential of said technologies to address the Rett-causing mutation of the MeCP2 gene, also focusing on the safety, accessibility, and other ethical considerations for their transition to patient care.

Study 1: Multiplex epigenome editing of MECP2 to rescue Rett Syndrome neurons
The study conducted in the paper, “Multiplex epigenome editing of MeCP2 to rescue Rett Syndrome neurons” by J. Qian, et. al attempts to explore the depths that epigenetic modification, also known as DNA methylation, can reach regarding treatment for Rett Syndrome16. The study aimed to see the effects of epigenome editing on the area containing the mutation that causes Rett Syndrome. If successful epigenome editing could be achieved, the resulting pathway of the mutation would change. Then neuronal phenotypic changes may arise, alleviating Rett-like symptoms. If Rett Syndrome symptoms could be alleviated successfully, then epigenome editing may be a permanent solution for Rett Syndrome patients.
The study targeted the MeCP2 gene on the inactive X chromosome (Xi), which possessed a working copy of the MeCP2 sequence. The researchers used demethylation editing, a form of chemical genetic editing where a methyl group is removed from a molecule14. Using this method, researchers edited the MeCP2 gene in order to restore production of the missing MeCP2 protein. The study used the treatment on both wild-type neurons, as a control, and RTT, or Rett Syndrome, neurons. An overview of the process of demethylation done in the study is shown in Figure 2.
The epigenome editing tools consisted of a catalytically dead CRISPR-Cas9 protein (dCas9-Tet), demethylation enzymes, and single-guide RNAs (sgRNA). The sgRNA guides the genome editing tools to the mutated sequence, the inactive CRISPR-Cas9 protein allows for binding to the intended sequence without cutting, and the demethylation enzymes allow for the removal of a methyl group, which could alter gene expression. Ten different sgRNAs were designed to lead the CRISPR-Cas9 protein to the promoter region of MeCP2. The different sgRNAs and dCas9-Tet were delivered into human embryonic stem cells, and results showed that methylation percentages were reduced to about 7% from baseline levels between 30-50%. Thus, the desired demethylation was achieved.
To address the possible off-target effects, researchers analyzed the genome to see where additional DNA methylation was occurring. The highest enrichment occurred at the MeCP2 region, with little demethylation occurring at unintended sites, showing that the sgRNAs resulted in specific editing. As few undesired edits occurred and the treatment usually reached the MeCP2 sequence, the DNA methylation editing used was relatively accurate and had a low chance of mistakes.
To see the effects of the demethylation editing on the Rett neurons, they derived Rett human embryonic stem cells, which have a wild-type MeCP2 allele silenced. Researchers analyzed the gene expression of the MeCP2 region, which demonstrated that the allele had been reactivated through targeted methylation. To understand if this restoration of gene expression could also be shown at the protein level, researchers used Western blot tests, which revealed the MECP2 protein. This demonstrated that, on a molecular level, the intervention achieved MeCP2 protein restoration, a crucial first step to alleviating Rett Syndrome symptoms. Next, to see if this restoration correlated with any improvement in phenotype, the authors looked at treated neurons that previously exhibited Rett Syndrome-like phenotypes.
Researchers then wanted to know if normal wild phenotypes would be restored. Small soma size is a common symptom of Rett Syndrome. To study the soma size differences, they examined wild-type neurons, Rett neurons, and treated Rett neurons. Using staining treatments, the researchers outlined the neurons’ somas, allowing them to view the neuronal processes. They found MeCP2 protein in the wild-type neurons as well as the treated Rett neurons. The authors found that the soma size in treated Rett syndrome neurons was significantly increased, using a one-way ANOVA with Bonferroni connection and a sample size of more than 20 neurons and 2 biological replicates for each condition. This indicates that the difference observed between the treated and non-treated Rett syndrome neurons was significant, as indicated by the statistical test. The edited Rett neurons’ soma size was restored to the natural phenotype, demonstrating the efficacy of the treatment. The authors found that their treatment not only restored the function of the MeCP2 protein but also made a difference in the phenotypes of the Rett neurons. Due to the successful restoration of both protein and wild-type phenotypes, the researchers can conclude that epigenetic editing may be a possible way to treat Rett Syndrome and can continue research regarding DNA methylation and Rett Syndrome.
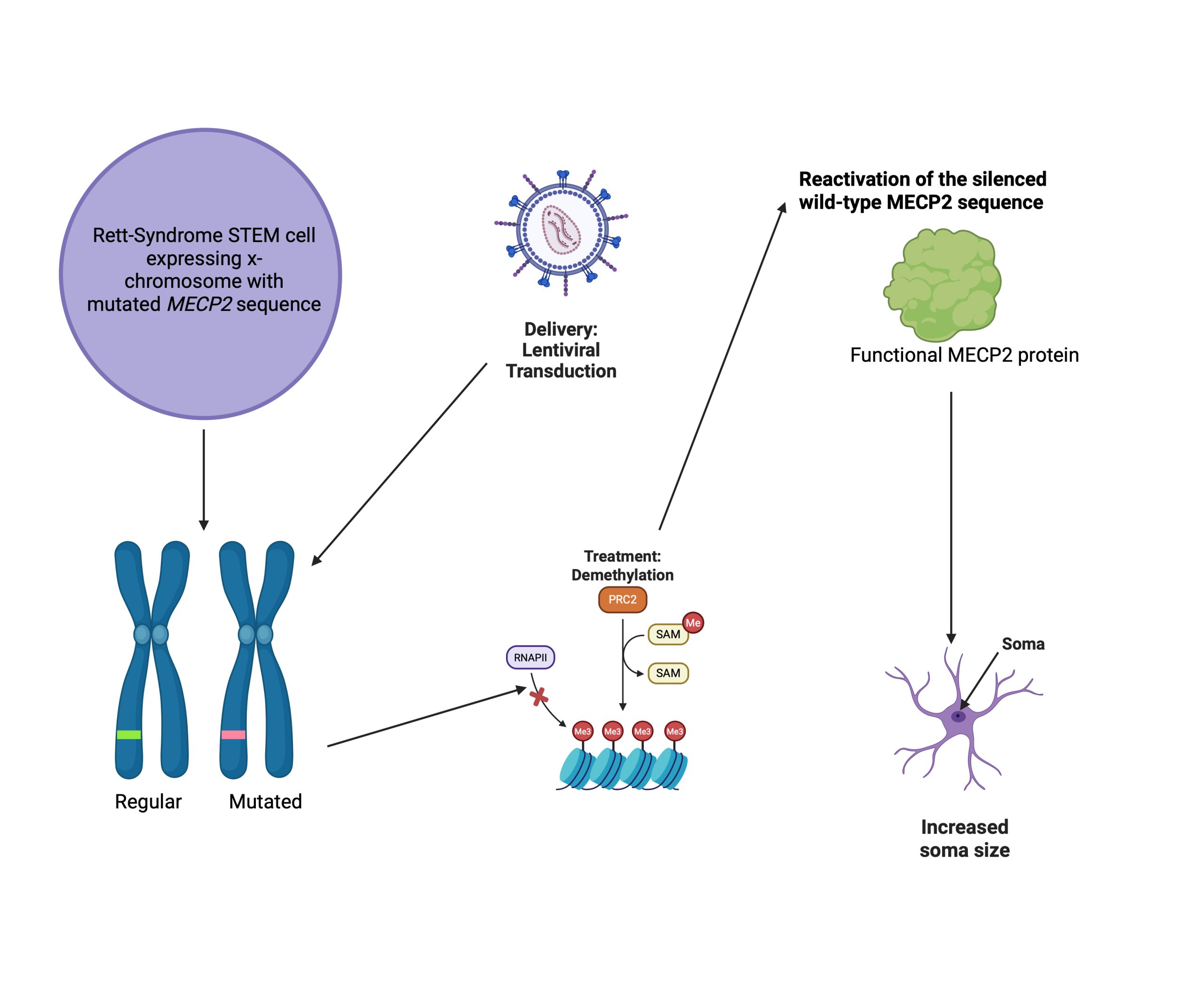
The aim of the study “Multiplex epigenome editing of MeCP2 to rescue Rett Syndrome Neurons” was to restore proper neuronal function through the removal of a methyl group at the MeCP2 sequence. The results of this study show promise in the ability of epigenome editing to cause a breakthrough in the quest to treat Rett Syndrome. This goal was not only achieved but also with lower levels of risk. One notable limitation of this study is the fact that it was tested in human embryonic STEM cells, whereas the Rett syndrome mutation manifests in the mature Rett
syndrome neurons of a patient. The study delivered the epigenetic treatment into undifferentiated cells that they then induced, which is not an accurate portrayal of the biological systems at work in an actual patient. However, using human embryonic stem cell-derived or induced pluripotent stem cell-derived neurons, may model the early neurodevelopmental stages affected by the MeCP2 mutation and can be invaluable in research settings. In the future, this method of treatment would likely have to be performed in mature Rett syndrome neurons or an animal model to more closely resemble and observe how a patient may respond to epigenetic editing in a clinical trial. In addition, the study does check off-target effects by observing the methylation in other regions. However, this analysis was not incredibly comprehensive, only observing the off-target effects at 27 other sites on the genome, rather than an analysis of genome-wide off-target effects.
Study 2: Targeted RNA editing in brainstem alleviates respiratory dysfunction in a mouse model of Rett Syndrome
The research described in the paper “Targeted RNA editing in brainstem alleviates respiratory dysfunction in a mouse model of Rett syndrome” by J. R. Sinnamon et. al successfully restored the production of the missing MeCP2 protein through RNA editing17. Similarly to the first study discussed, this study aimed to correct the Rett-causing mutation on the MeCP2 gene. However, this study did not change the DNA but rather the messenger RNA. As a result, only the gene product would be affected, lowering the risks of unintended edits. If restoration of the MeCP2 protein could be achieved through purely RNA editing, then Rett Syndrome patients may have a means of treatment with less chance of unintended consequences.
RNA editing research may be a significant step in the search to treat Rett Syndrome. Unlike the formerly described approach, these authors sought to edit the RNA directly for more precise control of MeCP2 expression, as overexpression can also lead to disease8. RNA editing is where the nucleotides are altered after transcription to the messenger RNA. The method used was the swapping of adenosine to inosine, which would be read as guanosine during translation of the MeCP2 protein. This specific nucleotide change is applicable to a MeCP2 mutation in which Rett Syndrome is caused by guanosine to adenosine single point mutation: the mutation exhibited by the mice in the study. However, the RNA editing method as a whole applies to Rett Syndrome cases caused by single nucleotide changes. Furthermore, point mutations constitute the majority of Rett Syndrome mutations, at 65%18.
An adeno-associated virus expressing both a catalytic ADAR2 domain and a MeCP2 sgRNA was inserted into the hippocampus of their mouse models (see Figure 3). Results showed that three different populations of neurons in the hippocampus had efficient repair of the MeCP2 RNA. To see if their RNA editing could work on nonsense mutations in addition to missense, they used the same treatment in a mouse model expressing the equivalent mutation to the human causing mutation, MeCP2W104X. The mouse equivalent is a mutation called MeCP2G311A which results in an abnormally short MeCP2 protein lacking binding and repressor domains. The process used during experimentation is shown in Figure 3.
As a control, researchers also inserted a catalytic domain with a green fluorescent protein (GFP) sgRNA as a control. The MeCP2 targeting treatment had a 76% editing efficiency compared to the control. The test used for this was a two-tailed Student’s t-test. To maximize chances of on-target editing, they added three copies of a nuclear localization signal, a short peptide which directs the treatment to the nucleus19. In analysis, researchers saw the presence of MeCP2 protein was restored in the mice treated with the MECP2 targeting treatment. This demonstrated that the previously mutated MeCP2 sequence was corrected, allowing for functioning MeCP2 protein to be produced. This is an essential step in treating Rett Syndrome, as the Rett Syndrome phenotypes are caused by the abnormal MeCP2 protein.
To see the effects of the treatment on a phenotypic level rather than just a cellular level, researchers analyzed the effect of their treatment on some of the symptoms of Rett Syndrome. Rett, treated Rett, and wild-type mice were all compared after the treatments. Using a Kaplan-Meier survival analysis, they observed the survival rate of treated Rett mice in comparison to untreated and wild-type mice. Rett Syndrome mice tended to live only 11 to 12 weeks, but after treatment, mice lived to 16 weeks, and sometimes even longer. The researchers also analyzed the change in respiratory function for treated Rett syndrome mice using a Kruskal–Wallis Test and Dunn’s multiple comparisons test. As for the breathing abnormalities, Rett mice had high apneic frequencies and prolonged breathing pauses for longer than one second, but after treatment, the apneic frequency significantly decreased. As the lifespan was prolonged and the apneic frequency decreased in treated Rett syndrome mice, the researchers could conclude that their MeCP2 targeting treatment alleviated Rett-like symptoms and restored wild-type phenotypes.
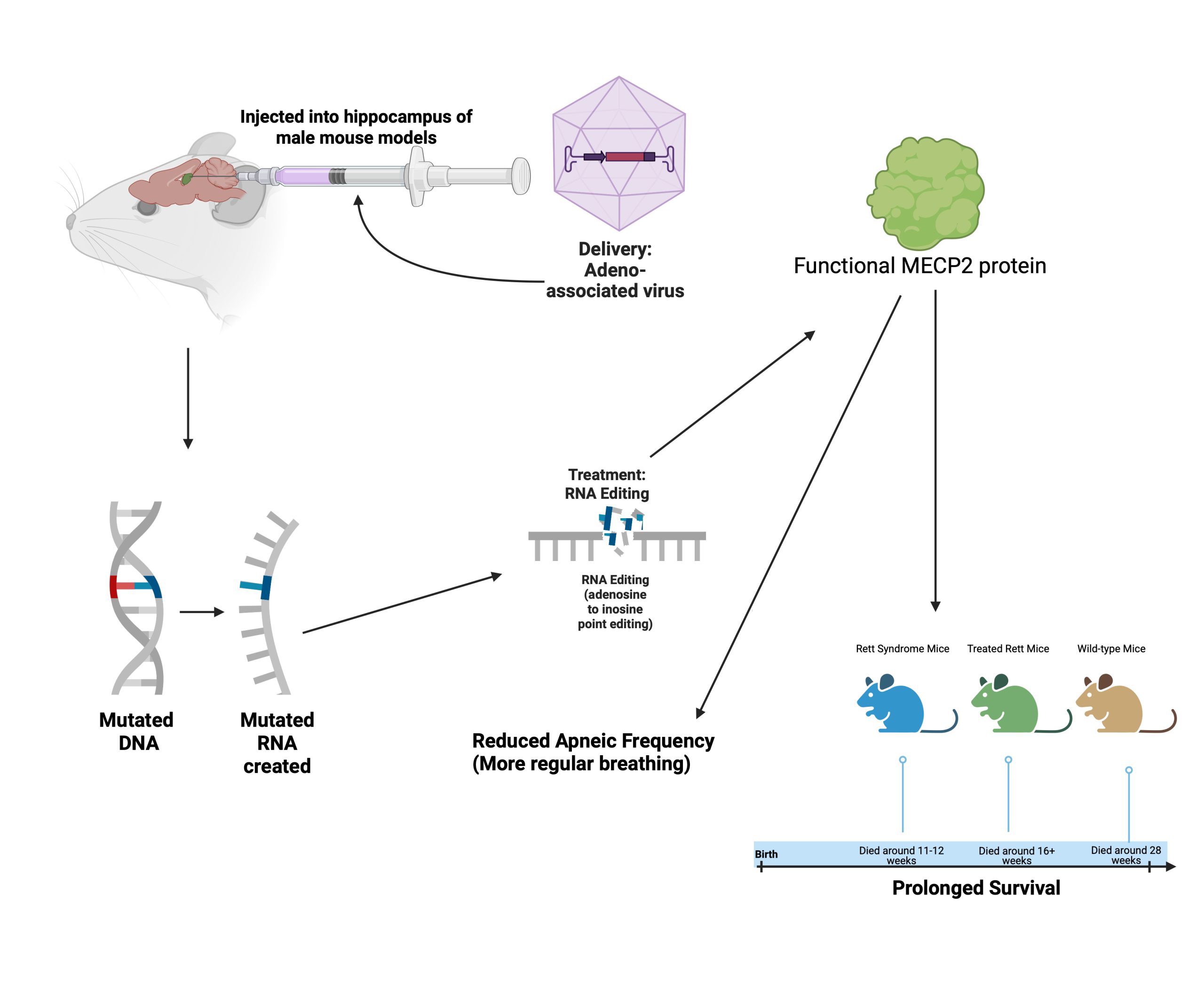
Since they were able to restore MeCP2 protein function and see a change in behavior and lifespan in the Rett Syndrome mice, researchers can conclude that their treatments may be able to improve the lifestyle of individuals with Rett Syndrome. The study only shows the change in editing efficiency in the hippocampus, while Rett Syndrome affects multiple regions of the brain. This raises the question of whether the treatment would truly be able to treat all problematic areas. More delivery methods may be needed to target all these locations. Furthermore, they only attempted to treat one specific point mutation. While this is one Rett causing mutation, Rett Syndrome can arise from many different kinds of mutations in the MeCP2 gene. Future studies would likely need to focus on different Rett-causing mutations to determine whether it is truly a promising means of treatment. Future studies would also need to assess long-term effects of this treatment on living Rett models. In the study discussed, the researchers only analyzed lifespan for 28 weeks and did not thoroughly discuss the potential long-term effects or immune responses that come with this means of treatment and viral delivery.
Study 3: Magnetic Nanoparticle‐Assisted Non‐Viral CRISPR‐Cas9 for Enhanced Genome Editing to Treat Rett Syndrome
The research in the study “Magnetic Nanoparticle‐Assisted Non‐Viral CRISPR‐Cas9 for Enhanced Genome Editing to Treat Rett Syndrome” by H. Y. Cho et. al was successfully able to correct the mutated MeCP2 sequence and restore wild-type phenotypes of the neurons20. The purpose of the study was to successfully restore the function of the MeCP2 protein through CRISPR-Cas9 editing. If achieved with little to no off-target effects due to the new delivery method, Rett Syndrome patients may be able to receive CRISPR treatment without the dangers of unintended consequences.
A Magnetic Nanoparticle-Assisted Genome (MAGE) platform was used for delivery of the CRISPR-Cas9 treatment. This MAGE platform is more accurate and efficient for the CRISPR-Cas9 technology. MAGE uses a magnetic core-shell nanoparticle delivered with magnetic force. MAGE also played a significant role in the efficiency of the treatment. Using nanoparticles as a delivery method in contrast to traditional viral capsules as delivery packages is a benefit, as fewer immune responses are triggered, and there is less risk of off-target effects, such as the viral genome randomly inserting itself into a patient’s DNA.
To deliver the treatment, the researchers inserted the nanoparticle with a Cas9 encoding sequence, sgRNA, and a donor copy of the correct MeCP2 sequence. This platform was inserted into patient-derived stem cells. They then induced the differentiation of stem cells to neurons and observed the expression of the MeCP2 protein. Results showed a 36% increase in the expression of MeCP2 protein in edited Rett neurons. The expression of the correct protein demonstrated the ability of the treatment to edit the mutated spot in the genome. To see if the restoration of the protein made a difference on a cellular level, researchers then observed characteristics of Rett Syndrome neurons and wild-type neurons.
To observe the effect of the repaired MeCP2 expression on Rett phenotypes, the researchers quantified data on four neuronal phenotypic aspects. They observed the soma size, dendritic tree complexity, neurite length, and number. All these characteristics are significantly lower and less complex in Rett Syndrome neurons. However, when treated Rett neurons were analyzed, researchers found that soma size was increased by 22.8% and was closer to wild-type sizes. Additionally, Rett neurons had a dendritic tree morphology similar to that of wild-type neurons. Upon examining the neurites, researchers found that treated Rett neurons had a 76.2% increase in neurite length compared to Rett neurons, and the neurite number also increased by 16.7%. The researchers used a t-test analysis, with p values less than 0.5 considered as statistically significant. Due to the significant increase in numbers, the researchers concluded that their treatment was making a difference in the phenotypes of Rett Syndrome neurons. While the researchers were able to increase MeCP2 expression by 36%, this is still a very modest number. Would this increase truly be able to make
a significant impact on the behavioral phenotypes of Rett Syndrome individuals? The study also only focuses on STEM cells that are then differentiated into neurons. This does not accurately depict the phenotypes of a human with Rett Syndrome. Future studies would need to perform this same method in an organism with a more complex system, such as mice. This would help determine whether the treatment would actually be applicable to humans.
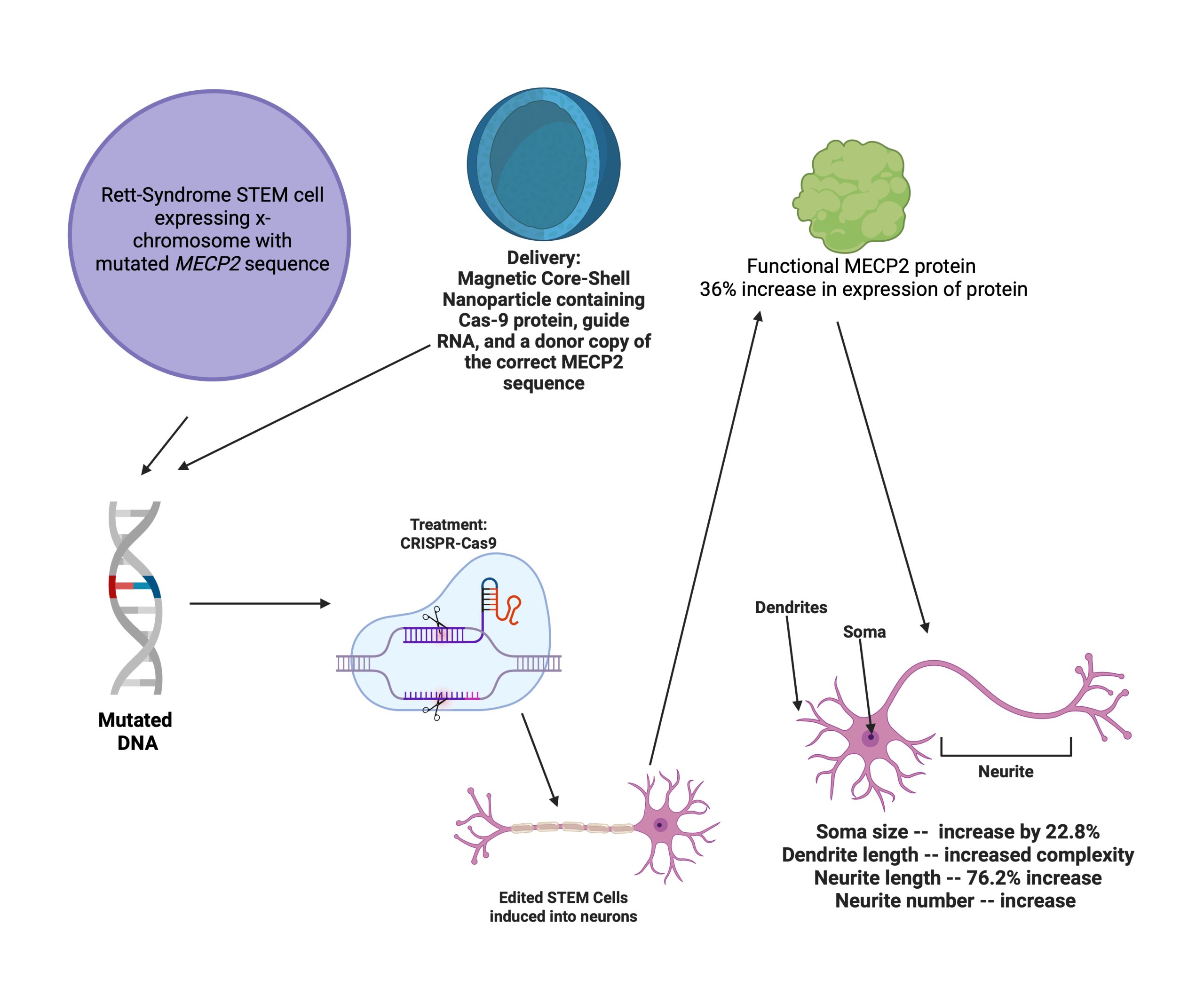
Because researchers saw that their treatment successfully restored MeCP2 protein and wild-type neuron phenotypes, they could infer that CRISPR-Cas9 editing has the ability to correct the mutated MeCP2 sequence successfully. In addition, by observing the improved accuracy and efficiency due to their new delivery package, MAGE, they can conclude that MAGE may be an efficient means of delivery for future genetic treatments.
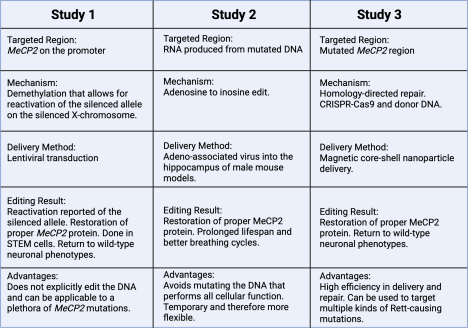
An overview of all three studies can be seen in Figure 5. This figure demonstrates the different gene editing processes and their corresponding outcomes, allowing for comparison of their varying capabilities.
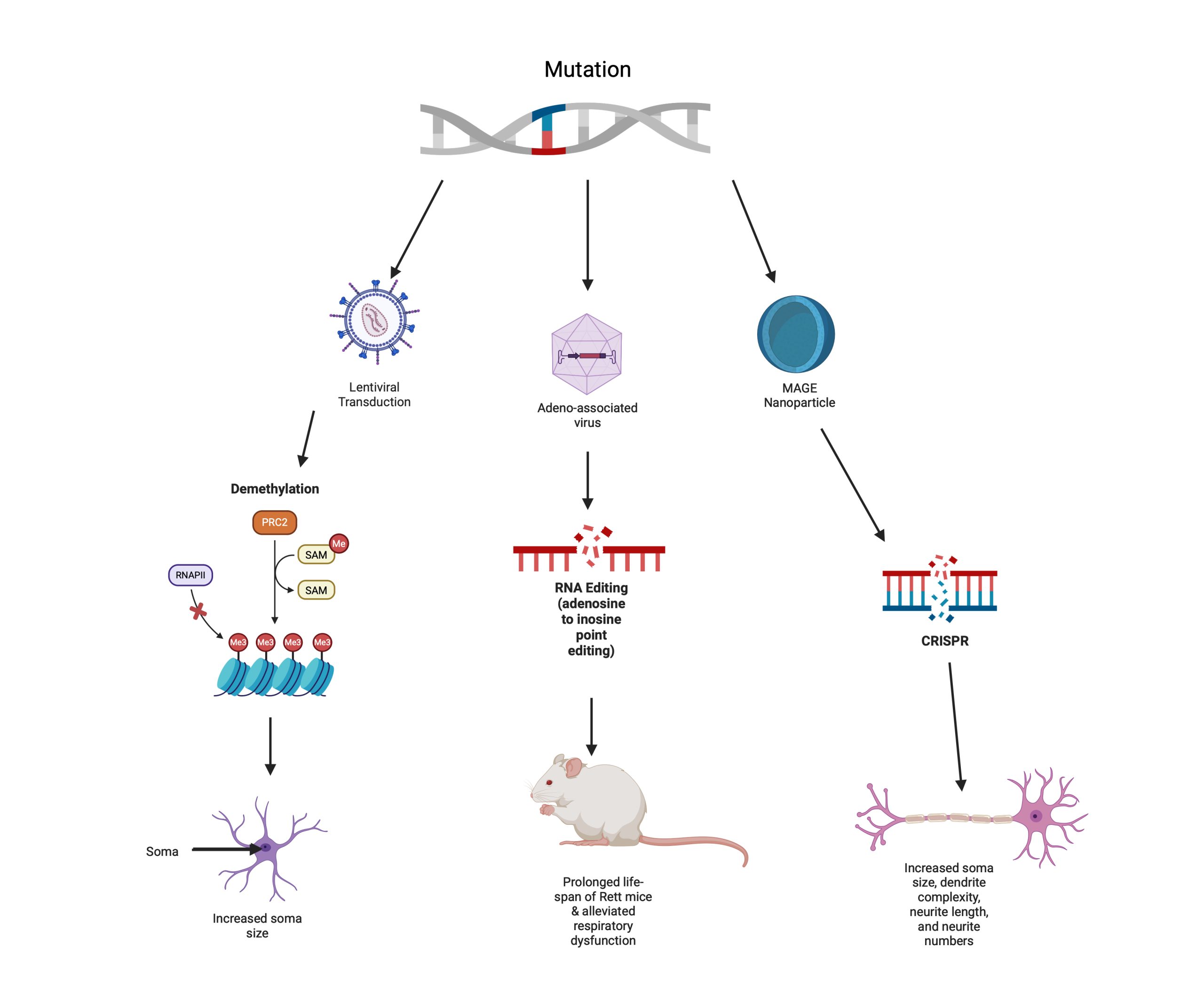
core-shell nanoparticle, resulting in neurons closer to wild-type neurons. Characteristics that were restored included an increased soma size, increased dendrite complexity, increased neurite length, and larger neurite numbers. Created in https://BioRender.com
Discussion
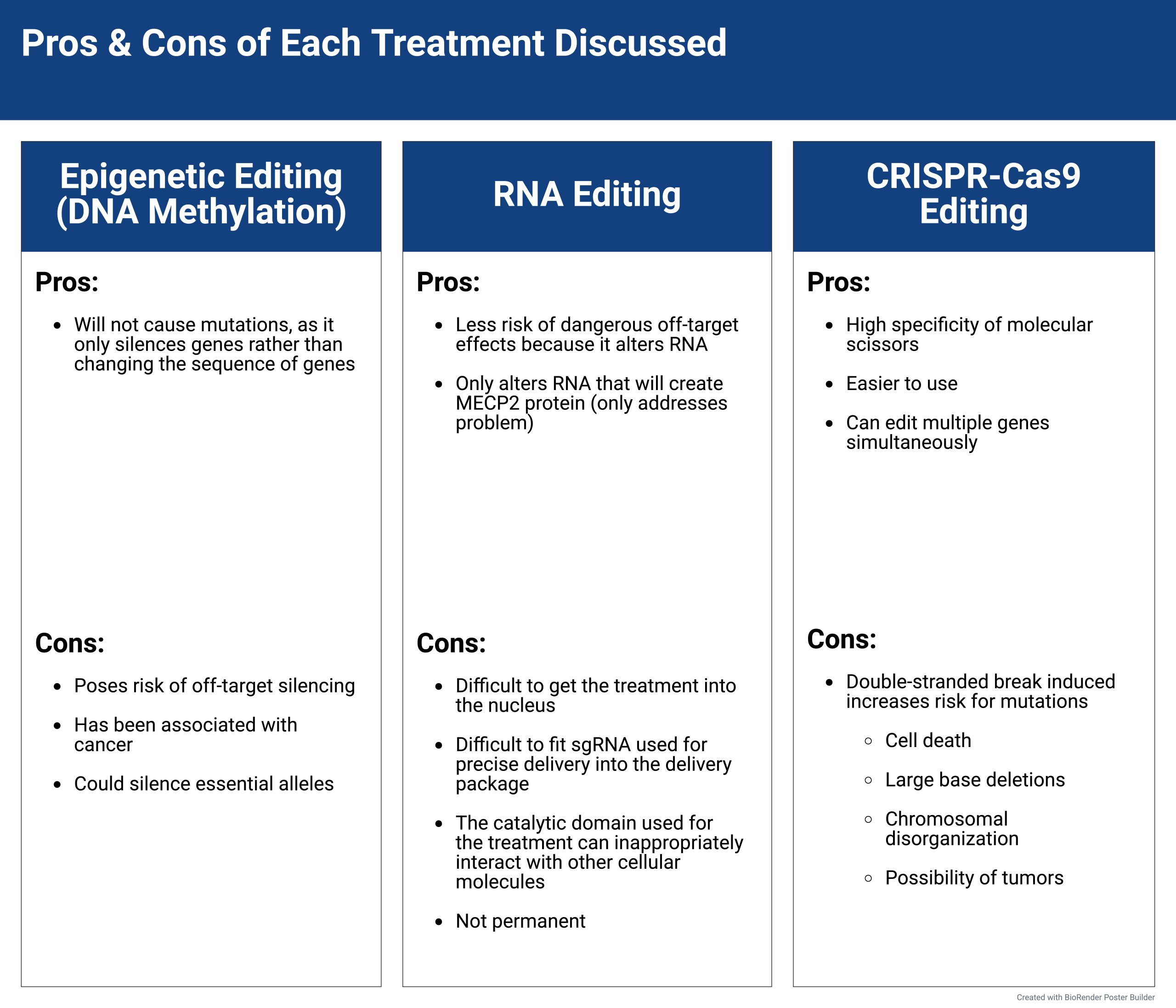
While the gene editing methods discussed may be a significant step in treating Rett Syndrome patients, there are still various factors that patients and their families must consider. Gene editing technologies have substantially progressed since the early 2000s21, but there are still many risks associated with each individual treatment. Furthermore, gene editing treatments are extremely costly, and for such a rare disease as Rett Syndrome, the problem of equity of access becomes a larger concern. Additionally, future generations of Rett Syndrome patients will be affected if they do any type of gene editing treatment, and the concern regarding the impact is significant. These pros and cons of gene editing, along with ethical considerations, are characterized in Figure 6.
Medical Concerns & Benefits
Gene editing treatments pose many concerns about safety for Rett Syndrome patients, especially the risk of off-target effects. There is always an unknown risk associated with gene editing, as off-target effects could result in unintended harm to the patient22.. In the case of CRISPR-Cas9, the double stranded break that the treatment induces can lead to numerous mutations23. . While Study #3 addresses the likelihood of off-target effects by delivering the CRISPR-Cas9 treatment via a magnetic core-shell nanoparticle rather than in vivo methods; it does little to address the concern of double-stranded break-induced mutations in the genome. They analyzed off-target effects caused by a high concentration of Cas9 in the cell, but concluded from the gene cleavage detection (GCD) assay that little off-target effects occurred after homology directed repair (HDR). Ultimately, however, they do little to minimize off target effects. Consequences of double-stranded break mutations can be more dangerous than the original disease, with outcomes including cell death, large base deletions, chromosomal disorganization, and the possibility of tumors24. In the case of RNA editing treatments, cons include the struggle of getting the treatment to the nucleus, which is where the mRNA is created, getting the gRNA to fit correctly inside the delivery package, and the concern about the catalytic domain inappropriately interacting with other cellular molecules15. Its transience can also pose an issue, as continuous treatment may not be sustainable for patients. Study #2 fails to address the delivery and logistical challenges that come with RNA editing, merely analyzing the efficacy of the treatment in a
single point mutation. Additionally, epigenetics poses the risk of off-target silencing25 and has been associated with cancer26. The ability of DNA methylation to silence important alleles is a large concern, as the mechanisms of how to deliver DNA methylation enzymes to the correct site are still being researched. For many Rett patients, these long-term side effects may feel more detrimental than the disease itself. In addition, epigenetic editing has been shown to be unstable over time. Study #1 fails to analyze the stability of the treatment over time, only looking at short-term effects of the treatment on MeCP2 production and neuronal phenotypes. The debate remains about whether the risks of detrimental side-effects are more precarious than everyday living with the Rett Syndrome disease. Any treatment on the human body, especially regarding the brain, the center of function, poses immense risk on critical function and cognitive ability. This is a case that requires deep consideration for Rett Syndrome patients and their families.
In addition, delivery methods for these different gene editing treatments pose a large concern regarding the safety of Rett Syndrome patients. Treatments for neurological diseases like Rett Syndrome must cross the blood-brain barrier (BBB), which restricts the flow of plasma proteins, water soluble molecules, and leukocytes from an individual’s blood to their central nervous system (CNS)27. Various viral and non-viral methods have been used to do this. Certain types of adeno-associated viruses, such as those with the myelin basic protein (MBP) promoter have a high efficiency at crossing the BBB, and they have a high specificity.
However, factors like the BBB limit gene therapeutics from efficiently reaching the CNS, such as in the case of lentiviral transduction. In contrast, the nonviral delivery method, MAGE, discussed in Study #3 aims to avoid immune responses and other risks associated with the viral methods previously discussed. The magnetic-nanoparticle delivery of the MAGE platform allows it to achieve a high multi-plasmid delivery of 99.3%20, which may be a promising delivery method in the future of gene editing.
On the other hand, as shown in the studies above, research is constantly done to improve the safety and the efficacy of these Rett Syndrome treatments. Furthermore, many of the gene editing treatments discussed earlier have characteristics that remain very beneficial for Rett Syndrome patients. CRISPR-Cas9 has the benefit of high specificity due to its molecular scissors, the Cas9 endonuclease, and its guide RNA, which allows it to identify the precise location to which a cut must be made28. This is extremely important for Rett Syndrome, since treatments will only endeavor to edit mutated sequences. CRISPR-Cas9 is also much easier to use than historical gene editing tools12, which could make CRISPR treatments more accessible. This treatment can be translated to many types of genetic diseases. CRISPR also is able to edit multiple genes simultaneously29, which could be useful for Rett Syndrome cases in which the disease is caused by mutations in multiple genes or areas. DNA methylation editing does not directly edit the DNA, and instead controls gene expression14. In this way, DNA methylation editing decreases the risk of causing mutations. This is a simple yet significant detail, as safety could be a considerable concern of patients suffering from genetic diseases. DNA methylation treatment could likely be applied to various Rett-causing mutations, due to the fact that it is merely expressing the functional gene, rather than trying to correct the mutated one. RNA editing also offers benefits, such as how it poses less risk of dangerous off-target effects because it alters messenger RNA rather than the DNA that codes for all cellular functions30. Thus, if unintended edits were to be introduced to the RNA, they would likely not be permanent, as RNA quickly degrades after use30. This would be extremely beneficial to Rett Syndrome patients, as it would enable effective editing, while reducing their chances of long-term consequences that could result in hindrances to neuronal function. This may be a more popular choice among Rett Syndrome patients due to its transience. As shown in the study above17, RNA editing works through point editing, and therefore may only be able to correct Rett Syndrome patients that possess the disease because of single point mutations.
Even despite the benefits of a temporary treatment, it leads to the question of whether Rett patients will want to spend money on a treatment that does not completely eliminate their mutation. A comprehensive table of all 3 studies can be seen in Figure 7.
Social Concerns
Rett Syndrome treatments lead to a concern regarding equity of access and treatment prices. Rett Syndrome is an extremely rare disease, and due to the already expensive costs of gene editing technologies, treatments will be nowhere near cheap. Equity of access is a significant concern, as many of these methods can be expensive and require much research31. For patients living in countries where technologies to produce Rett Syndrome treatments are underdeveloped, it leads to the concern about how many patients will actually be helped by these technologies. Furthermore, costs as a whole for treatment will be enormous, and many Rett Syndrome families will have a hard time affording the treatment. A gene editing and therapy program entitled Bluebird Bio focuses on treating specific genetic diseases, yet each costs millions of dollars32. Furthermore, the cost of one treatment of gene therapy for Muscular Dystrophy at Sarepta Therapeutics is $3.2 million33. For even rarer genetic diseases, one can only imagine the price increase34. Rett Syndrome is one such rare disease, meaning that these prices will likely skyrocket. Even though current prices of gene editing treatments such as CRISPR-Cas9 are high35, they may become more accessible as it becomes easier to manufacture. Manufacturing costs currently account for a large portion of the overall cost of a genetic treatment36, but, likely, gene-editing treatments will remain exorbitantly expensive even as these costs go down, as the amount of resources needed to conduct initial experiments and clinical trials which can cost upwards of $1943 million37 will need to be recuperated. Current data shows that gene editing treatments are highly encumbering, and with the unique nature of the Rett Syndrome disease, prices for treatments could likely be too much for families to handle. Furthermore, treatments will begin at clinical trials as they have only been tested on animal models thus far. This means that families will likely have to spend millions of dollars on a
merely experimental treatment. Again, this poses the concern of whether treatments are worth their prices. These concerns are all listed in Figure 6a. In addition, given that many Rett Syndrome possess cognitive impairments, this gives rise to the concern of informed consent to such new and potentially negative treatments38.
Public Opinion
Rett Syndrome patients may also not be open-minded towards genetic treatments, due to the concern of changing their natural composition39. Patients must decide how they would like to cope with their symptoms: to eliminate them completely with gene editing treatments, or to take a more “natural” pathway and rely on symptom-easing drugs. However, the argument may be made that humans have been changing the natural course of nature for centuries, one instance being selective breeding. Furthermore, by making the decision to have a gene editing treatment, Rett Syndrome patients will also be making decisions for their descendents. With the chance of reproduction, gene editing may also impact future generations22. Additionally, current views towards gene editing show a grim acceptance of gene editing treatments. This could reflect on the values that individuals with Rett Syndrome, as well as their families, have towards a treatment that alters their genetic makeup. A considerable amount of the public is still hesitant to accept gene therapy, with a survey done in 2022 reporting that only 49% of Americans said they would be open to gene editing to reduce disease risk40.
Final Assessment
All in all, there are many things that Rett Syndrome patients must consider when seeking out gene editing treatments. However, in contrast to existing treatments, gene editing treatments seem to reach the root of the disease effectively. Existing drug treatments such as Trofinetide merely alleviate symptoms, whereas the methods discussed all target the problematic gene, therefore eliminating symptoms altogether. Furthermore, as discussed in the introduction, gene therapies lead to the risk of overexpression of certain proteins, in this case MeCP2, which can lead to other neurological issues. In the case of CRISPR, the treatment is able to edit the problematic gene rather than adding another gene that may cause transcriptional errors. This therefore is also likely more effective than drug treatments because it eliminates the symptom-causing gene. As for epigenetic editing, the activation of the already existing gene reduces risk of off-target effects, in contrast to gene therapy, which introduces foreign genes to the body. Similarly to CRISPR, it also addresses the root cause of the issue, allowing for direct improvement of symptoms. Finally, with RNA editing, there are less off-target effects than gene therapy due to the transience of the treatment, and it directly addresses the gene, therefore being more impactful on symptoms than drug treatments. These technologies pose a lot of benefits in comparison to existing treatments. For RNA and epigenetic editing, there are currently no approved uses of them in clinical trials. Technically, these treatments are very complex and therefore the barrier to entry into the clinic is high. The research in the studies discussed are very preliminary, being done in simple cell models and mice. One thing that may be done to continue the research for gene editing in Rett Syndrome is to use larger animal models who more closely resemble humans. Even now, CRISPR-Cas9 treatments for sickle-cell anemia have just been approved, so there is currently still a lack of implementation regarding gene editing treatments. Perhaps trials in primates should be the next step so that researchers may be able to see how humans would react to these treatments, and identify issues before clinical trials. There have been current clinical trials using gene editing technologies like CRISPR to treat various genetic diseases, one being successful treatment of sickle-cell anemia41. One thing is for certain: more research must be done if gene editing is to be used for disease treatment, and with those advancements in knowledge, more solutions to ethical concerns may be revealed.

Created in https://BioRender.com
Methods
The National Library of Medicine/PubMed and the Google Scholars search engine were used to search for articles used for this literature review. Key words such as “genetics, Rett Syndrome, gene editing treatment, trials” were used to obtain articles regarding gene editing treatments for the Rett Syndrome disease. To find sources used for the discussion section, key words such as “ethics, gene editing, and pros/cons” were entered into various search engines. Each paper’s references were individually reviewed to check the credibility of old papers cited. Papers with publication dates older than the 1990s were not used. The oldest paper referenced was from 1992. The reason for this choice of filtration was because gene editing treatments have only substantially progressed since the 2000s. Homologous recombination, zinc-finger nucleases, TALENs, and CRISPR all emerged in the early to late 2000s. The reason for papers used earlier than 2000 was to provide context in the area of ethical considerations. After evaluation for various gene editing methods reported in the literature, the decision was made to focus on the technologies of epigenetic editing, RNA editing, and CRISPR-Cas9 editing. These three technologies were selected on the basis of efficacy during trials. For each technology, one paper was selected, resulting in three studies being spotlighted in this review.
References
- I. M. Caballero. MeCP2 in neurons: closing in on the causes of Rett Syndrome. Human Molecular Genetics. 14, suppl_1, 15, R19-R26 (2005). [↩]
- U. Petriti,
D. C. Dudman, E. Scosyrev, S. Lopez-Leon. Global prevalence of Rett syndrome: systematic review and meta-analysis. Systematic Reviews. 12, 1, (2023). [↩]
- C. M. Disteche, J. B. Berletch. X-chromosome inactivation and escape. Journal of Genetics. 94, 4, 591-599 (2015). [↩]
- S. M. Weng, M. E. S. Bailey, S. R. Cobb. Rett syndrome: from bed to bench. Pediatrics and Neonatology. 52, 6, 309-316 (2011). [↩] [↩] [↩] [↩] [↩]
- S. A. Hudu, F. Elmgdadi, A. A. Qtaitat, M. Almehmadi, A. A. Alsaiari, M. Allahyani, A. Aljuaid, M. Salih, A. Alghamdi, M. A. Alrofaidi, Abida, M. Imran. Trofinetide for Rett Syndrome: highlights on the
development and related inventions of the first USFDA-approved treatment for rare pediatric unmet medical need. Journal of Clinical Medicine. 12, 15, 5114 (2023). [↩]
- Z. U. N Mughal, B. Ahmed, B. S. Rangwala, H. S. Rangwala, H. Fatima, M. Ali, A. A. Farah. Trofinetide receives FDA approval as first drug for Rett syndrome. Annals of Medicine and Surgery. 86, 5, 2382 (2024). [↩]
- S. J. Keam. Trofinetide: First Approval. Drugs. 83, 9, 819–824 (2023). [↩]
- A. L. Collins, J. M. Levenson, A. P. Vilaythong, R. Richman, D. L. Armstrong, J. L. Noebels, D. J. Sweatt, H. Y. Zoghbi. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human Molecular Genetics. 13, 21, 2679–2689 (2004). [↩] [↩]
- S. Bhattacharyya, M. F. Go, B. E. Dunn, S. H. Phadnis. Transcription and translation. Helicobacter pylori: Physiology and Genetics. 21290711, Chapter 26 (2001). [↩]
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts, P. Walter. From RNA to protein. Molecular Biology of the Cell. 4, (2002). [↩] [↩]
- H, Chial. Mendelian genetics: patterns of inheritance and single-gene disorders. Nature. 1, 1, 63 (2008). [↩] [↩]
- M. Hryhorowicz, D. Lipiński, J. Zeyland, R. Słomski. CRISPR/Cas9 immune system as a tool for genome engineering. Archivum Immunologiae et Therapiae Experimentalis. 65, 233-240 (2017). [↩] [↩] [↩] [↩] [↩]
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna, E. Charpentier. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 6096, 816-821 (2012). [↩]
- L. D. Moore, T. Le, G. Fan. DNA methylation and its basic function. Neuropsychopharmacology. 38, 1, 23-38 (2012). [↩] [↩] [↩] [↩]
- B. J. Booth, S. Nourreddine, D. Katrekar, Y. Savva, D. Bose, T. J. Long, D. J. Huss, P. Mali. RNA editing: expanding the potential of RNA therapeutics. Cell Press. 31, 6, 1533-1549 (2023). [↩] [↩]
- J. Qian, X. Guan, B. Xie, C. Xu, J. Niu, X. Tang, C. H. Li, H. M. Colecraft, R. Jaenisch, X. S. Liu. Multiplex epigenome editing of MeCP2 to rescue Rett Syndrome neurons. Science Translational Medicine. 15, 679, eadd4666 (2023). [↩]
- J. R. Sinnamon, M. E. Jacobson, J. F. Yung, J. R. Fisk, S. Jeng, S. K. McWeeney, L. K Parmelee, C. Ngai-Chan, S. P. Lee, G. Mandel. Targeted RNA editing in brainstem alleviates respiratory dysfunction in a mouse model of Rett Syndrome. Proceedings of the National Academy of Sciences. 119, 33, e2206053119 (2022). [↩] [↩]
- R. Krishnaraj, G. Ho, J. Christodoulou. Rettbase: Rett syndrome database update. Human Mutation. 38, 8, 899-1032 (2017). [↩]
- J. Lu, T. Wu, B. Zhang, S. Liu, W. Song, J. Qiao, H. Ruan. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Communication and Signaling. 19, 60 (2021). [↩]
- H. Y. Cho, M. Yoo, T. Pongkulapa, H. Rabie, A. R. Muotri, P. T. Yin, J. W. Choi, K. B. Lee. Magnetic nanoparticle-assisted non-viral CRISPR-Cas9 for enhanced genome editing to treat Rett Syndrome. Advanced Science. 11, 24, (2024). [↩] [↩]
- M. Pacesa, O. Pelea, M. Jinek. Past, present, and future of CRISPR genome editing technologies. Cell. 187, 5, 1076-1100 (2024). [↩]
- K. E. Ormond, Y. Bombard, V. L. Bonham, L. Hoffman-Andrews, H. C. Howard, R. Isasi, K. Musunuru, K. A. Riggan, M. Michie, M. Allyse. The clinical application of gene editing: ethical and social issues. Personalized Medicine. 16, 4, 337-350 (2019). [↩] [↩]
- M. Hryhorowicz, D.
Lipiński, J. Zeyland, R. Słomski. CRISPR/Cas9 immune system as a tool for genome engineering. Archivum Immunologiae et Therapiae Experimentalis. 65, 233-240 (2017). [↩]
- L. Thorne. CRISPR gene therapies: current challenges and a promising future. https://www.biocompare.com/Editorial-Articles/609559-CRISPR-Gene-Therapies-Current-Chall enges-and-a-Promising-Future/#:~:text=CRISPR%20Gene%20Therapies%3A%20Current%20C hallenges%20and%20a%20Promising%20Future,-Share&text=Lucy%20Thorne%2C%20Ph.,D.
&text=Since%20its%20discovery%2C%20CRISPR%20and,a%20range%20of%20genetic%20di seases. (2024). [↩]
- T. Phillips. The role of methylation in gene expression. Nature. 1, 1, 6 (2008). [↩]
- M. Borchiellini, S. Ummarino, A. Di Ruscio. The bright and dark side of DNA methylation: a matter of balance. MDPI. 8, 10, 1243 (2019). [↩]
- C. A. Maguire, M. L. Ramirez, D. J. Rodriguez, S. M. Sena-Esteves, B. L. Davidson. Gene therapy for the nervous system: challenges and new strategies. Neurotherapeutics. 11, 4, 817–839 (2014). [↩]
- A. E. Modell, D. Lim, T. M. Nguyen, V. Sreekanth, A. Choudhary. CRISPR-based therapeutics: current challenges and future applications. Trends in Pharmacological Sciences. 43, 2, 151-161 (2022). [↩]
- C. Thomas. What are the pros and cons of CRISPR-Cas9?
https://www.idtdna.com/pages/education/decoded/article/crispr-cas9-what-are-the-pros-and-cons (2023). [↩]
- S. Reardon. Step aside CRISPR, RNA editing is taking off. Nature. 578, 7793, 24-27 (2020). [↩] [↩]
- (K. E. Ormond, Y. Bombard, V. L. Bonham, L. Hoffman-Andrews, H. C. Howard, R. Isasi, K. Musunuru, K. A. Riggan, M. Michie, M. Allyse. The clinical application of gene editing: ethical and social issues. Personalized Medicine. 16, 4, 337-350 (2019). [↩]
- N. Pagliarulo. With $2.8M gene therapy, Bluebird sets new bar for US drug pricing. https://www.biopharmadive.com/news/bluebird-bio-gene-therapy-price-zynteglo-million/629967 /#:~:text=Bluebird%20bio’s%20new%20gene%20therapy,and%20among%20the%20highest%20 globally. (2022). [↩]
- B. Fidler. Sarepta prices Duchenne gene therapy at $3.2M.
https://www.biopharmadive.com/news/sarepta-duchenne-elevidys-price-million-gene-therapy/65 3720/ (2023). [↩]
- D. A. Marshall, B. Gerber, D. L. Lorenzetti, K. V. MacDonald, R. J. Bohach, G. R. Currie. Are we capturing the socioeconomic burden of rare genetic disease? A scoping review of economic evaluations and cost-of-illness studies. PharmacoEconomics. 41, 1563-1588 (2023). [↩]
- D. Gupta, O. Bhattacharjee, D. Mandal, M. K. Sen, D. Dey, A. Dasgupta, T. A. Kazi, R. Gupta, S. Sinharoy, K. Acharya, D. Chattopadhyay, V. Ravichandiran, S. Roy, D. Ghosh. CRISPR-Cas9 system: a new-fangled dawn in gene editing. Life Sciences. 232, 116636 (2019). [↩]
- J. Sterling. Cell and gene therapy manufacturing costs limiting access. https://www.genengnews.com/insights/cell-and-gene-therapy-manufacturing-costs-limiting-acces s/ (2023). [↩]
- M. T. Sabatini, M. Chalmers. The cost of biotech innovation: exploring research and development costs of cell and gene therapies. Pharmaceutical Medicine. 37, 5, 365-375 (2023). [↩]
- L. Wiley, M. Cheek, E. LaFar, X. Ma, J. Sekowski, N. Tanguturi, A. Iltis. The Ethics of Human Embryo Editing via CRISPR-Cas9 Technology: A Systematic Review of Ethical Arguments, Reasons, and Concerns. HEC Forum. 37, 2, 267–303 (2025). [↩]
- E. Lucassen. The ethics of genetic engineering. Journal of Applied Philosophy. 13, 1, 51-62 (1996). [↩]
- L. Rainie, C. Funk, M. Anderson, A. Tyson. Americans are closely divided over editing a baby’s genes to reduce serious health risk. https://www.pewresearch.org/internet/2022/03/17/americans-are-closely-divided-over-editing-a babys-genes-to-reduce-serious-health-risk/ (2022). [↩]
- H. Frangoul, D. Altshuler, M. D. Cappellini, Y.-S. Chen, J. Domm, B. K. Eustace, J. Foell, J. de la Fuente, S. Grupp, R. Handgretinger, T. W. Ho, A. Kattamis, A. Kernytsky, J. Lekstrom-Himes, A. M. Li, F. Locatelli, M. Y. Mapara, M. de Montalembert, D. Rondelli, A. Sharma, S. Sheth, S. Soni, M. H. Steinberg, D. Wall, A. Yen, S. Corbacioglu. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. New England Journal of Medicine. 384, 3, 252–260 (2021). [↩]



{kind=link}