
Abstract
Obesity and insulin resistance have significant influence on the onset and progression to Type 2 Diabetes (T2D). This study examines the correlation between obesity, insulin resistance, and the onset and progression of T2D, aiming to clarify the mechanisms linking them and to understand insights that could inform effective preventive and therapeutic strategies grounded in those mechanisms. The review also highlights other factors that modulate T2D risk, including the duration and severity of obesity, and other factors such as genetics, age, sex, and ethnicity. A thorough literature review was conducted to examine these relationships, using data across various studies. The evidence identifies core mechanismschronic low-grade inflammation, adipokine dysregulation, altered lipid metabolism, and ectopic fat depositionand demonstrates that prolonged and severe obesity greatly increases the risk of developing insulin resistance, which further increases the likelihood of progressing to T2D. Additionally, it indicates that variables such as age and sex act as effect modifiers, leading to heterogeneous outcomes under comparable conditions. These insights underscore the demand for prevention methods and effective therapeutic approaches that target obesityespecially sustained weight loss and reduced insulin resistanceto reduce T2D prevalence. Adding on, this review discusses opportunities to optimize therapy and community based prevention programs, thereby reducing diabetes burden and curtailing development from obesity to T2D.
Keywords: Insulin resistance, Insulin sensitivity, Obesity, Mechanisms, Therapeutics, and Type 2 Diabetes (T2D)
Introduction
Diabetes is a rapidly growing crisis, affecting over 830 million people globally1. This number has quadrupled in just the past 35 years and is estimated to have caused upwards of 3.4 million deaths in 2024, meaning that one person is dying from this illness every nine seconds2. According to the CDC, 38.4 million people lived with diabetes in 2021, representing 14.7% of all adults in the United States alone3. Diabetes mellitus is a metabolic disorder that can be characterized by chronic hyperglycemia due to insulin resistance and/or inadequate insulin secretion4 and can lead to extreme complications such as blindness5 and lower-limb amputationaround 80% of which are caused by diabetes6. Due to the severity of these potential consequences, its crucial to understand the underlying causes.
Obesity is considered one of the most modifiable risk factors for type 2 diabetes (T2D)7’8’9. Over the same timeframe that diabetes rates quadrupled, adult obesity doubled and adolescent obesity (ages 519) quadrupled10. Despite playing a pivotal role in T2D pathogenesis, many studies have only examined a narrow range of mechanisms in depth, leading to non-holistic analyses. This review seeks to fill that gap by investigating the biological, physiological mechanisms linking obesity to T2D, while considering other influential demographic factors such as genetics, age, sex, race, socioeconomic status, as well as comorbidities and the duration and severity of obesity. The goal is to lend thorough insight into how these many factors interact to influence disease onset and progression and to explorepossible implications for prevention and therapy.
To accomplish this goal, this review addresses the following research questions:
- What primary biological and physiological mechanisms ofobesity contribute to the progression of T2D?
- How are genetics, demographics, socioeconomic conditions, and particularly obesity severity and duration relatedto T2D risk?
- How might understanding these mechanisms help shapetargeted prevention design and treatment strategies?
It is hypothesized that higher levels of obesity for longer durations will accelerate the onset or progression of T2D and worsen its severity. Identifying these mechanisms will allow individuals to receive more effective, tailored interventions.The scope of this review is limited to peer-reviewed studies, with a focus on recent research to ensure relevance to current scientific understanding. Articles that primarily focused on other diseases, even when mechanisms overlapped with T2D, or articles limited to gestational diabetes were not considered for the purpose of this review. Methodologically, this is a narrative synthesis integrating epidemiological, clinical, and mechanistic evidence to form a multi-angle perspective.
Methods
This literature review combines findings from peer-reviewed studies examining links between obesity and T2D. Relevant literature was identified through structured searches of Google Scholar and PubMed for studies published in English, prioritizing referencing more recent work when feasible. Search terms included “obesity and diabetes,” “obesity and insulin resistance,” “insulin sensitivity and insulin resistance,” “brown adipose tissue” and “gut microbiota.” These searches were made between July through November 2024, with a final update in early August 2025. Eligibility criteria included peer-reviewed studies reporting mechanistic outcomes related to insulin sensitivity, insulin resistance, beta cell (β-cell) function, or T2D pathophysiology. Studies focused solely on gestational diabetes or primarily on other diseases such as cardiovascular disease, even when overlapping mechanisms, were excluded from the search. Information extracted included study design, population, mechanistic endpoints related to insulin sensitivity and insulin resistance, and stated limitations. Findings were synthesized to identify convergent patterns and inconsistencies, with comparable quantitative outcomes contrasted where appropriate. This review incorporates previously published research and does not require Institutional Review Board approval.
Results: Mechanisms linking obesity to T2D
Relationship between Insulin Resistance, Insulin Sensitivity and T2D
Insulin resistance describes a reduced response of skeletal muscle, adipose tissue, and liver to physiological insulin, limiting insulin mediated control of glucose metabolism and requiring higher insulin levels to achieve the same metabolic effects11’12’13.
Insulin sensitivity refers to how effectively the body’s cells respond to insulin, the hormone that regulates blood glucose. High insulin sensitivity means that only a small amount of insulin is needed to control blood sugar. Conversely, low sensitivity means the same amount is less effective14. Persistently low sensitivity can progress to insulin resistance. As shown by Figure 1, lower insulin sensitivity and higher insulin resistance are linked to a higher probability of T2D onset and progression. Chronic hyperinsulinemia can further impair metabolic function by driving excess glucose uptake, with surplus being converted to fat. This leads to ectopic fat deposition, hypothalamic inflammation, fatty acid breakdown products, and reduced fat oxidation — disrupting insulin signaling and worsening insulin sensitivity15.
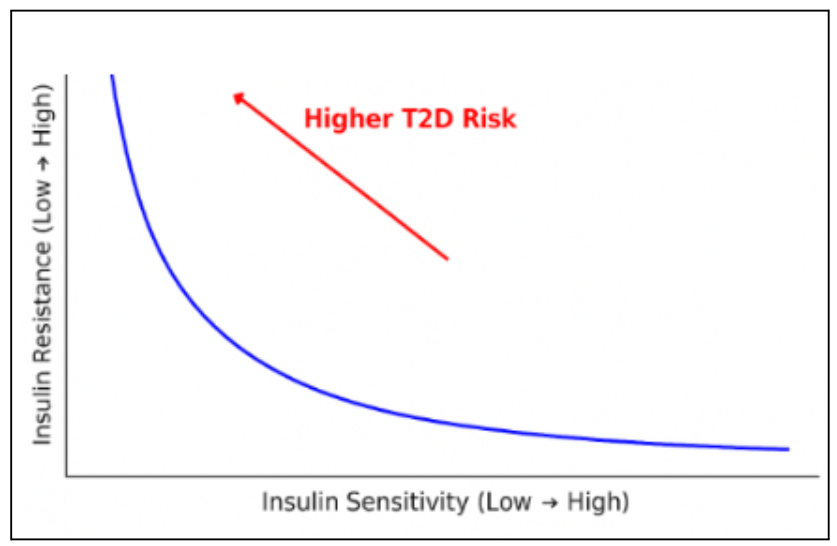
Insulin resistance plays a pivotal role in the onset and progression of T2D and other metabolic disorders, including hypertension, certain cancers, and nonalcoholic fatty liver disease. Many factors such as genetic susceptibility, obesity, aging, coexisting health conditions, and medication effects can influence the development of insulin resistance. Mechanistically, even slight changes in the insulin signaling pathway — including insulin receptor defects, inflammation, hypoxia, lipotoxicity, immune dysregulation, and impaired liver or organ metabolism — can lead to insulin resistance16. As insulin resistance worsens, more insulin is required to maintain normal glucose levels, increasing stress on pancreatic β-cells and accelerating their decline.
When lifestyle changes or oral agents aren’t enough to maintain glycemic control, insulin therapy becomes necessary17. Though there are admittedly positive effects, long-term insulin use can gradually lower insulin sensitivity, especially as dosages increase. For people without prior insulin resistance, prolonged exposure to exogenous insulin can trigger its onset, while in those already insulin-resistant, it can further worsen complications. Long-term intensive insulin therapy can exacerbate these effects by increasing the risks of hypoglycemia, mortality, and weight gain18. Prolonged use can also suppress natural insulin production, leading to treatment dependence within just 10–15 years.
Given these limitations, it is essential to understand the progression from obesity to insulin resistance and ultimately T2D. Understanding how fat build up can change insulin sensitivity and insulin resistance can help create prevention and intervention strategies, reducing T2D prevalence, easing healthcare burdens, and improving long-term patient outcomes16.
Mechanisms of Insulin Resistance
Adipose Tissue Inflammation
Adipose tissue, once thought to be simply a passive fat store, has now been established as an active endocrine organ that regulates metabolism19. Obesity leads to chronic low-grade inflammation in adipose tissue, which elevates signals from immune cells such as macrophages. In obese fat tissue, macrophages shift toward a pro-inflammatory (M1) state and release more cytokines including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and resistin19’20’21. These cytokines interfere with insulin signaling, progressively reducing insulin sensitivity20. TNF-α and IL-6 turn on certain “switches” in the cell, called signaling pathways, including c-Jun N-terminal kinase (JNK) and NF-κB. Once these switches are activated, they add a small chemical tag (phosphate) to the wrong spot on insulin receptor proteins (serine phosphorylation), which makes it harder for insulin’s message to be passed along. These cytokines can also make special proteins called SOCS, which attach to insulin receptor proteins and mark them to be broken down, further reducing insulin’s ability. This diminished responsiveness, known as insulin resistance, means the body needs more insulin to achieve the same blood sugar–lowering effect22’23.
In healthy individuals, fat tissue secretes beneficial adipokines such as adiponectin, which improve insulin sensitivity by enhancing glucose uptake and fatty acid oxidation. In obesity, however, adiponectin levels decrease while pro-inflammatory signals rise, further hindering insulin action19’24. This shift not only worsens blood sugar control but also increases the workload on the pancreas, requiring it to produce more insulin.
Adipose tissue also releases other bioactive molecules that influence metabolic health. Among these, leptin and adiponectin are particularly important. Leptin regulates hunger and energy balance, but in obesity, its levels remain chronically elevated, leading to leptin resistance—causing appetite suppression to be blocked, promoting further weight gain and insulin resistance25. Meanwhile, reduced adiponectin levels remove an important anti-inflammatory and insulin-sensitizing influence, compounding the metabolic dysfunction24.
As illustrated in Figure 2, adipose tissue expansion/dysfunction initiates inflammation and adipokine shifts (increased resistin and leptin, decreased adiponectin) and promotes ectopic lipid accumulation, collectively lowering insulin sensitivity and increasing insulin resistance, increasing the likelihood of progression to T2D.
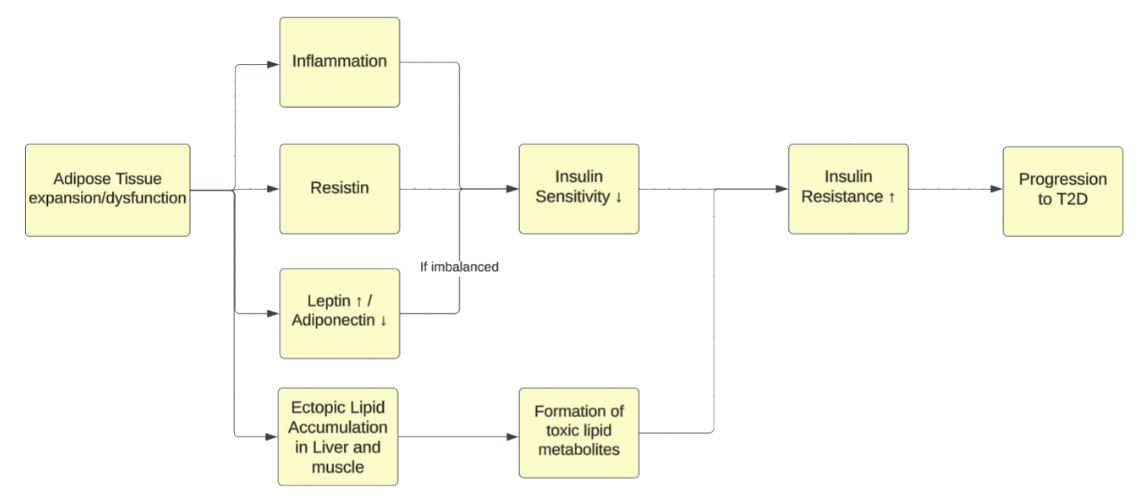
Over time, when obesity sustains this chronic low-grade inflammatory state, it creates a self-perpetuating cycle where inflammation worsens insulin resistance and insulin resistance further fuels inflammation20. Macrophage infiltration, elevated pro-inflammatory cytokines, and reduced anti-inflammatory adipokines become rooted, forming an “inflammatory memory” at the tissue level that can still persist even after moderate weight loss (approximately 2–7% of baseline weight). This makes it difficult, and in some cases nearly impossible, to restore insulin sensitivity in the advanced stages of metabolic dysfunction. With ongoing metabolic stress, insulin sensitivity in muscle, liver, and adipose tissue declines progressively — muscular GLUT4 translocation is reduced, hepatic gluconeogenesis becomes unrestrained, and lipolysis becomes even more prominent in adipose tissue — raising circulating glucose levels and free fatty acids (FFAs)26. Clinically, this stage often corresponds to the transition from prediabetes to overt T2D, with postprandial hyperglycemia progressing to fasting hyperglycemia as β-cell compensation fails.
These inflammatory changes in adipose tissue are closely linked to altered secretion of bioactive molecules, which further influence systemic metabolism. These obesity-driven secretory changes are detailed below.
Obesity-Induced Secretory Factors: Adipokines, Free Fatty Acids, and Cytokines
Obesity increases insulin resistance through dysfunctional adipose tissue, which alters its secretory profile to release excess FFAs, pro-inflammatory cytokines, and hormones such as leptin and resistin27. These changes, as well as mitochondrial dysfunction, ectopic lipid accumulation, and hypoxia, impair insulin signaling and reduce insulin sensitivity, speeding up metabolic dysfunction.
Among adipokines, leptin and adiponectin are important regulators of metabolism. Leptin, a hormone that regulates energy and hunger balance, is frequently seen at elevated levels in individuals with obesity, leading to leptin resistance, where appetite suppression is blunted and further weight gain and insulin resistance are promoted25. Adiponectin, an anti-inflammatory adipokine that enhances glucose uptake and fatty acid oxidation, is typically reduced in obesity, and low levels are closely associated with poor insulin sensitivity24.
Elevated FFAs, commonly seen in people with obesity, contribute to lipid accumulation in cells (lipotoxicity) and inhibit insulin signaling. Excess FFAs can be converted into other fat-related molecules called lipid intermediates, including diacylglycerols (DAGs) and ceramides. DAGs switch on an enzyme called PKCε, which makes insulin receptors work less effectively, while ceramides block a crucial step for AKT to move glucose transporters (GLUT4) to the surface of the cell so sugar can enter. These pathways converge to suppress insulin-stimulated glucose uptake. FFAs activate protein kinase C (PKC), which interferes with insulin receptor substrates (IRS) and disrupts the signaling cascade28. Studies confirm that higher circulating FFAs are correlated with reduced glucose uptake, further reinforcing their role in insulin resistance29.
Chronic low-grade inflammation in obese adipose tissue also increases the production of pro-inflammatory cytokines such as TNF-α and IL-6. These cytokines can also turn on cell “switches” known as the JNK and NF-κB pathways. When this happens, they add a chemical tag to insulin-related proteins (serine phosphorylation) and increase the production of SOCS proteins, making it harder for insulin signals to properly work. TNF-α has been shown to inhibit IRS-1 phosphorylation, a critical step in insulin signaling22. In comparable mouse models, deletion of TNF-α preserves insulin sensitivity even on a high-fat diet30. While human studies22 link higher TNF-α levels with insulin resistance, direct experimental evidence in humans remains limited. Mouse experiments display a IRE1α–XBP1  ER-stress pathway arriving at insulin resistance, whereas human studies are equipped with observational links between subclinical hypothyroidism and insulin-resistance metrics. The coherence across species is possibly extrapolatable but the need for human mechanistic confirmation does persist.
ER-stress pathway arriving at insulin resistance, whereas human studies are equipped with observational links between subclinical hypothyroidism and insulin-resistance metrics. The coherence across species is possibly extrapolatable but the need for human mechanistic confirmation does persist.
Excess visceral fat, stored deep in the abdominal area, can be especially harmful. It promotes the secretion of pro-inflammatory adipokines and cytokines19’31, which have damaging effects on the liver, muscle, and pancreas, disrupting glucose and lipid metabolism. This ongoing metabolic imbalance increases the chance of developing insulin resistance and, ultimately, T2D7.
Ectopic Lipid Deposition and Disruption of Insulin Signaling
Ectopic fat deposition refers to the abnormal storage of lipids in non-adipose tissues such as the liver, skeletal muscle, pancreas, and heart. This phenomenon is increasingly understood to be a central driver of insulin resistance, independent of overall obesity levels32’33. When subcutaneous adipose tissue reaches its storage capacity, excess circulating fatty acids are redirected toward insulin-sensitive organs. This lipid overflow disrupts intracellular signaling pathways through the accumulation of diacylglycerols and ceramides, which activate protein kinase C isoforms and impair insulin receptor substrate function34’35. These defects in insulin signaling lead to reduced glucose uptake in skeletal muscle, impaired suppression of hepatic glucose output, and β-cell stress in the pancreas32,35.
Importantly, the risk associated with ectopic fat is not merely quantitative but qualitative, involving the type of lipids stored and their subcellular localization33’35. Even in adolescents with similar BMI(Body Mass Index)s, those with greater visceral fat or intrahepatic triglyceride content exhibit significantly worse insulin sensitivity compared to peers with predominantly subcutaneous fat distribution34,35. Lifestyle interventions that reduce ectopic fat—particularly hepatic and intramyocellular lipids—have been found to improve insulin sensitivity without large fluctuations in body weight, underscoring the causal role of fat distribution rather than total adiposity35,36.
Organ-Specific Manifestations of Ectopic FatInduced Insulin Resistance
Ectopic fat accumulation affects insulin-sensitive organs in distinct yet interconnected ways, amplifying whole-body metabolic dysfunction. In the liver, excess triglyceride storage causes hepatic insulin resistance, enhancing gluconeogenesis and increasing very-low-density lipoprotein (VLDL) secretion, contributing to systemic dyslipidemia33’36. In skeletal muscle, lipid intermediates such as diacylglycerols can fix insulin-stimulated glucose transport channels and glycogen synthesis, weakening the tissue’s role as a large glucose sink32’34. In the pancreas, lipid infiltration promotes β-cell lipotoxicity, causing insulin secretion to slow down and the progression toward T2D to speed up32’35. Adipose tissue, particularly dysfunctional visceral depots, releases excess free fatty acids and pro-inflammatory adipokines that further damage insulin resistance across many different organs. Brown adipose tissue (BAT) dysfunction—marked by reduced thermogenic capacity—further reduces energy expenditure, promoting lipid overflow into ectopic sites35’36.
These organ-specific effects rarely occur in isolation. Hepatic and muscular insulin resistance can act synergistically, with the liver increasing glucose output while muscles reduce glucose uptake, collectively worsening hyperglycemia35. These interplays between inflamed visceral adipose tissue and ectopic lipids in organs perpetuate chronic low-grade inflammation and oxidative stress even further34’35. Understanding the interconnected systems at play is essential for identifying intervention points where targeted fat reduction or functional restoration in one organ could lead to disproportionately positive improvements in systemic insulin sensitivity35’36.
Gut Microbiome–Intestinal Barrier Axis and Insulin Resistance
The gut microbiome–intestinal barrier axis plays a pivotal role in regulating insulin sensitivity and the pathogenesis of T2D. In healthy states, a diverse and balanced microbial ecosystem—enriched with butyrate-producing bacteria such as Faecalibacterium prausnitzii, Roseburia spp., and Akkermansia muciniphila—maintains intestinal barrier integrity through mucus layer reinforcement, tight junction protein upregulation, and short-chain fatty acid (SCFA) production. SCFAs, particularly butyrate and propionate, enhance epithelial function and systemically to promote GLP-1 and PYY secretion, regulate appetite, improve glucose uptake, and reduce inflammation. In dysbiosis, typically found with obesity and T2D, microbial diversity declines, SCFA-producing taxa are depleted, and Gram-negative, LPS-producing bacteria proliferate. This shift weakens the strength of the barrier, allowing lipopolysaccharides (LPS) to translocate into systemic circulation, where they activate Toll-like receptor 4 (TLR4)–NF-κB signaling, driving chronic low-grade inflammation and inducing serine phosphorylation of insulin receptor substrate proteins, thereby impairing insulin signaling in liver, muscle, and adipose tissue37’38’39’40’41’42’43
Additionally, microbial metabolites and host–microbiota metabolic interactions influence systemic insulin sensitivity via multiple pathways. SCFAs inhibit hepatic gluconeogenesis, stimulate glycogen synthesis, and enhance fatty acid oxidation, while also upregulating GLUT4 expression in muscle and adipose tissue through AMPK activation42’43. Conversely, T2D-associated taxa such as Prevotella copri and Bacteroides vulgatus produce branched-chain amino acids (BCAAs) and imidazole propionate, which activate mTORC1 signaling and interfere with insulin action39’40. Dysregulated bile acid metabolism exacerbates insulin resistance by altering FXR and TGR5 signaling41. Certain interventions including high-fibre diets, targeted probiotics, prebiotics, and microbiota-modifying pharmacotherapies such as metformin can restore SCFA-producing populations, reduce LPS-producing taxa, rebalance bile acid signaling, and reinforce intestinal barrier function, offering a multifaceted therapeutic approach to alleviating insulin resistance and preventing T2D progression44’38’39’45’41’42’43.
Brown Adipose Tissue Dysfunction in Obesity
Brown adipose tissue (BAT) is a metabolically active organ that contributes to whole-body energy homeostasis through non-shivering thermogenesis, facilitating the uptake and oxidation of glucose and lipids. In obesity, however, BAT’s thermogenic activity and substrate utilization capacity become quite significantly impaired. This dysfunction closely parallels insulin resistance and T2D through multiple mechanisms. Reduced glucose uptake in BAT—demonstrated both in obesity and in T2D—is a particularly critical defect, as it heavily limits the tissue’s contribution to postprandial glucose clearance and systemic metabolic flexibility46’47. Impaired BAT activation also results in reduced clearance of circulating lipids, promoting ectopic fat deposition in insulin-sensitive tissues such as the liver and skeletal muscle, which can exacerbate lipotoxicity and mitochondrial stress. These disturbances disrupt insulin signaling pathways, amplifying systemic insulin resistance and accelerating the onset or progression of T2D48.
Furthermore, obesity-related BAT dysfunction is not always due to a full loss of thermogenic ability; sometimes, fatty acid oxidation and mitochondrial activity are relatively preserved, while glucose handling is selectively impaired46. This defect can explain why cold-induced BAT activation alone is not sufficient in restoring glycemic control in insulin-resistant states47. Pharmacologic strategies that enhance BAT activity through insulin-independent routes—such as SGLT2 inhibitors that promote white adipose tissue turning brown, increase mitochondrial uncoupling, and shift substrate utilization toward lipid oxidation—represent a promising therapeutic direction49. By restoring BAT’s ability to regulate both glucose and lipid metabolism, these approaches may mitigate obesity-related metabolic complications and therefore improve overall insulin sensitivity.
Beta-cell Dysfunction: Overload and Exhaustion
Under normal conditions, the β-cells of the pancreas play a pivotal role in glucose homeostasis; these cells sense blood glucose levels and secrete insulin in a meticulously regulated way50’51. This involves a rapid first-phase insulin release after meals, followed by sustained second-phase secretion to maintain stable glucose levels. β-cells can also personalize their output according to the specific metabolic demand, making sure that even the peripheral tissues receive sufficient insulin for efficient glucose uptake.
However, when there are multiple metabolic stressors occurring simultaneously, this can disrupt this finely managed system. Chronic exposure to elevated blood glucose (glucotoxicity), high levels of circulating free fatty acids (lipotoxicity), and inflammatory cytokines present persistent strain on β-cells51’52. These conditions lead to oxidative stress — an overproduction of reactive oxygen that damages cellular structures — and endoplasmic reticulum (ER) stress, which overwhelms the protein-folding machinery51’52. Because β-cells possess inherently low antioxidant capacity, they are especially vulnerable. Mitochondrial dysfunction impairs ATP production needed for insulin granule exocytosis, while calcium signaling defects, amyloid deposition, and defective autophagy further impact cell viability52.
To combat early functional impairment, β-cells compensate by increasing insulin output, a state known as functional overload. This sustained overactivity gradually accelerates cell exhaustion, leading to a decline in both secretory capacity and beta-cell mass through apoptosis and dedifferentiation. As insulin output falls short of systemic needs, blood glucose levels rise, which in turn exacerbates beta-cell stress, creating a self-reinforcing cycle50’52’53.
This deterioration from normal function to overload and then to exhaustion not only contributes to the onset of T2D but also accelerates its progression in people who are already affected. Preserving beta-cell integrity through early intervention — by taking preventative measures such as reducing secretory demand and mitigating oxidative and inflammatory damage — is therefore critical for delaying or even outright preventing the deterioration of glycemic control.
In conjunction with upstream drivers in Section 3.2, these β-cell alterations complete the sequence that links obesity to T2D. Figure 3 combines the mechanisms from Sections 3.2 and 3.3 into a single cascade from adipose inflammation and ectopic fat to organ specific insulin resistance, compensatory hyperinsulinemia with increased β-cell workload, β-cell dysfunction, and hyperglycemia. This indicates the feedback loops and the modifiers that act across many steps.

Systemic and Population-Level Modulators
Genetics
Genetic predisposition impacts individuals’ insulin sensitivity; this combines with adiposity and other exposures to shape overall metabolic risk16. In human NAFLD, risk variants (e.g., PNPLA3, TM6SF2) can increase hepatic triglyceride content regardless of overall adiposity; importantly, such “genetic NAFLD” is less related to systemic insulin resistance than metabolically driven NAFLD31. Genetic risk can also interact with obesity—frequently amplifying liver disease severity and even dissociating hepatic steatosis from whole-body insulin resistance31. Mechanistic reviews further point to heritable influences on insulin signaling, β-cell function, and lipid handling, and discuss gene–environment interactions with diet and physical activity16. Even genetic differences that ethnicities come with can have impacts: Black women—despite having lower visceral and intrahepatic fat than White women—exhibited greater postprandial hyperinsulinemia, lower hepatic insulin clearance, and, in the postmenopausal group, significantly lower insulin sensitivity (with a tendency toward lower sensitivity premenopausally)54. In certain groups of premenopausal women, Black participants had lower intrahepatic lipid and higher intramyocellular lipid, and the associations between ectopic fat and insulin resistance appeared more pronounced in Black women34.
Age
Aging is associated with reduced insulin sensitivity. There are a couple of proposed mechanisms explaining this: adipose-tissue dysfunction and mitochondrial stress. Given the link between insulin resistance and frailty, age-related tissue and cellular changes—such as adipose-tissue dysfunction and mitochondrial stress—may promote insulin resistance and, in turn, raise T2D risk11. In a retrospective cohort analysis using NHANES, being obese from young adulthood throughout midlife was correlated with the highest incidence of diabetes, while losing weight and not being clinically obese after early adulthood was associated with substantially lower 10-year incidence versus stable obesity (HR 0.33, 95% CI 0.14–0.76). Nonetheless, for consistently non-obese adults (HR 1.47, 95% CI 0.65–3.36), risk did not always return, possibly reflecting small numbers in the weight-loss group55. In accordance with the 2012 to 2022 U.S. BRFSS data, adults 65 years or older had roughly a tenfold higher adjusted odds of a diabetes diagnosis than those aged 18 to 24 years (aOR 10.23, 95% CI 9.99 to 10.47) and about fivefold higher than those aged 45 to 64 years (aOR 5.16, 95% CI 5.05 to 5.29), reflecting odds instead of risk; however, it is worth noting that this estimate relates to self-reported diabetes in adults (predominantly type 2) and does not measure prospective incidence or a relative risk56.
Sex
BMI and T2D risk are both significantly impacted by the differences in hormonal activity, muscle mass, fat distribution that are linked to sex. Discussions in the field suggest that even the exact same BMI in men and women can still manifest as vastly differing levels of metabolic risk and excess body fat57. In women for example, there is a similar pattern of obesity worldwide that especially post-menopause, that may suggest T2D burden that is sex-specific58. Furthermore, in the United States between 2013 and 2016, the adjusted population attributable fraction for incident diabetes due to obesity was higher in women than in men within each race or ethnicity, for example non Hispanic White women 53 percent and men 36 percent59. Therefore, the term “metabolically healthy obesity” can encompass a very wide spectrum of cases, with sex-related factors causing a difference in risk level between sexes even at similar BMI levels60.
However, the research on sex based disparities in diabetes differ across studies. Using 2012-2022 BRFSS data from across the US (n≈5.31 million adults), Neupane et al.56 found a higher prevalence in diabetes amongst men than women in the U.S. (12.56% vs 11.56%; adjusted OR 1.15). On the other hand, Boutari & Mantzoros58 used data provided by the WHO and Global Burden of Disease and reported a globally higher prevalence in obesity among women, especially during the midlife period, which may be linked to elevated sex-specific T2D risk that occurs after menopause. However, Sun, H. et al61 reported that sex related prevalence did not account to a meaningful correlation; 10.8% in men and 10.2% in women61. These discrepancies may be caused by differences in outcomes (diabetes vs obesity), data sources (self-reported surveillance vs synthesized epidemiologic estimates), age and menopausal transition, and differences in fat distribution between men and women (greater amounts of fat found in the internal organs of men vs more gluteofemoral fat in premenopausal women), as well as socioeconomic factors.
Race and ethnicity
Race and ethnicity influence both obesity and T2D, but they can yield different outcomes even following similar exposures. Multiple U.S. studies have discovered that non Hispanic Black, Hispanic, American Indian and Alaska Native, and Asian American populations typically have higher rates of T2D than non Hispanic White populations. For instance, Neupane et al.56 found that in logistic regression, as compared with non Hispanic Whites, Hispanics (aOR 1.60, 95% CI 1.57 to 1.64), non Hispanic Asians (aOR 1.67, 95% CI 1.59 to 1.76), and non Hispanic Blacks (aOR 2.10, 95% CI 1.98 to 2.22) had higher odds of reaching a diabetes diagnosis. Specifically, Native American communities experience high prevalence of T2D, with proposed explanations ranging from unidentified genetic factors to a fetal origins framework wherein fetal undernourishment and other intrauterine complications propagate risk across generations62. Unrelated to underlying mechanisms, T2D risk and prevalence show heterogeneous trends across racial and ethnic groups. Although typical BMI and insulin resistance are often lower, greater central adiposity and pronounced beta cell dysfunction contribute to the onset of T2D at lower BMI thresholds, emphasizing that risk levels are tied to ethnicity63. Social norms stemming from race and ethnicity regarding program participation levels are also important considerations for prevention and treatment. Bowen et al.9 reported relatively lower participation by Black, Hispanic, Asian, American Indian and Alaska Native, and Native Hawaiian and Pacific Islander men in the U.S. National DPP lifestyle change program, along with higher rates of cardiovascular comorbidity among Black, Hispanic, and Native American Medicare participants. These differences were hypothesized to be associated with poverty, food insecurity, and place of residence, and studies recommended partnering with trusted community leaders for recruitment and using approaches such as family involved enrollment and workplace technology to bolster engagement. Viewed jointly, these findings support race and ethnicity specific screening approaches, targeted prevention strategies, and community tailored implementation.
Socioeconomic Status (SES)
Socioeconomic status (SES) also influences how obesity is distributed in a population and its metabolic impacts. Boutari & Mantzoros58 found that in high-income countries, obesity tends to be more prevalent in lower-SES groups, whereas in many low and middle-income countries it is more prevalent in higher-SES groups. Indeed, Neupane et al.56 discovered that high income (aOR 0.59, 95% CI 0.58 to 0.61) had strong correlations with lower odds of obesity; meaning, had variables in the model been held constant, the odds of a diabetes diagnosis within the high income group were 41 percent lower than that of the reference income group56.
Social determinants of health (SDOH) such as food insecurity, neighborhood safety, access to healthcare, safe community spaces for physical activity, and rurality all have an impact on prevention and metabolic risk. Evidence indicates lower participation in the National DPP lifestyle change program among Black, Hispanic, Asian, Native American, and Native Hawaiian/Pacific Islander men, as well as low first-year participation in DSMES, even though the program is available nationwide. Tailored recruitment through trusted community leaders, family-involved enrollment, and workplace or technology-based delivery models has been suggested to mitigate this issue9.
The effects of this phenomenon are evident among younger populations as well. A global systematic review and meta-analysis spanning 2,033 studies in 154 countries (≈45.9 million participants) estimated obesity at 8.5%, overweight at 14.8%, and excess weight at 22.2%. Considerable variation was seen between countries, ranging from 0.4% in Vanuatu to 28.4% in Puerto Rico, with higher prevalences in high-income and high-HDI settings. Compared to 2000–2011, rates were roughly 1.5 times higher in 2012–202364. As a result, this means that the link between socioeconomic status and obesity/ T2D differs across settings and cannot be explained by a single pattern.
Comorbidities
Other health conditions can worsen insulin resistance and accelerate the development of T2D. For example, even mild hypothyroidism can increase insulin resistance by disrupting normal cell stress pathways (IRE1α/XBP-1), which helps explain the connection between thyroid problems and impaired insulin function30. NAFLD can both result from and contribute to insulin resistance, with liver fat and metabolic dysfunction raising T2D risk beyond what body fat alone explains31. Medications such as glucocorticoids can independently raise hepatic glucose production and impair peripheral insulin sensitivity65. The occurrence of comorbidities such as obesity with hypertension, dyslipidemia, and diabetes all contribute to metabolic stress and complicates disease management66.
Duration and Severity of Obesity as Predictors of T2D Risk
Evidence from multiple populations shows that obesity’s severity and length of duration are strong, independent predictors of T2D and related disorders. Higher BMI categories, particularly the WHO’s obesity class II and III, are associated with a significantly greater likelihood of developing metabolic syndrome, a precursor to T2D, and prolonged exposure to obesity compounds this risk regardless of current metabolic status67. Longitudinal studies in British and U.S. populations show a clear pattern: extended periods of obesity from early life to mid adulthood are linked to more severe HbA1C elevations, even when accounting for obesity severity68. A metric called “obese years,” which accounts for both BMI and duration of obesity, is a stronger predictor of T2D than either on its own. Those with the greatest exposure have about six times greater risk of developing it69.
Overweight and obesity duration both raise the risk of T2D, though the effect is less pronounced when present obesity levels are high, particularly people with long-term obesity70. Studies from the Framingham Heart Study show that prolonged obesity raises T2D risk independently of BMI levels, particularly amongst adults with early onset obesity71. The evidence indicates that diabetes risk and progression is strongly shaped by overall exposure to obesity, whether measured by duration, intensity, or combined metrics. Preventive strategies should be designed with the goal of delaying obesity onset, limit its persistence, and reduce severity to effectively lower lifetime T2D risk.
Therapeutic Approaches to Diabetes Prevention and Treatment
The management of insulin resistance and T2D requires a multi-layered treatment plan targeting weight reduction, metabolic improvement, and long-term disease prevention. Based on evidence from large-scale trials, clinical guidelines, and recent studies, interventions can be organized into the following categories.
Lifestyle Modification
Lifestyle modification remains the first-line approach for prevention and management of insulin resistance. Weight loss achieved by restricting calories, reducing high–glycemic index carbohydrate intake, and lowering saturated fat and sodium intake has consistently been shown to delay or prevent T2D onset. These interventions work by reducing visceral body fat, alleviating chronic low-grade inflammation, improving mitochondrial efficiency, and restoring insulin signaling pathways. The U.S. National Diabetes Prevention Program (NDPP) found that prediabetic individuals across all age groups who achieved a 5–7% weight loss reduced their T2D risk by 58%, with an even greater 71% risk reduction in adults aged 60 years or older59. Long-term benefits can also persist for up to 20 years, even without major weight loss, as long as lifestyle changes are maintained72. Long-term weight maintenance is equally important, as evidenced by the reduced T2D incidence in individuals who transitioned from an obese BMI to a non-obese BMI55. Public health measures, such as improving access to nutritious foods and implementing community-level obesity prevention programs, are critical for sustaining these effects at the population level.
Based on this evidence, clinical guidelines highlight weight loss as one of the most effective modifiable risk factors for diabetes. In clinical practice, an initial weight loss of 5–10% of body weight is associated with significant glycemic and cardiometabolic improvements, including a 0.6–1.0% drop in A1C, improved blood pressure and lipid levels, and less need for medication. Even modest loss can lower fasting glucose by ~20 mg/dL, while sustained losses of 10% or more are linked to reduced cardiovascular disease and death73. Effective programs typically deliver high-intensity behavioral counseling (≈14–16 contacts over 6 months), encourage a daily 500–750 kcal deficit, and promote healthier eating habits (e.g., low-carbohydrate, low-fat, or Mediterranean), and incorporate physical activity, which is essential for long-term weight maintenance73. Similarly, a synthesis of landmark studies in high-risk adults—including the Da Qing Diabetes Prevention Study, the Finnish Diabetes Prevention Study, and the United States Diabetes Prevention Program—demonstrates risk reductions of 31–58% during the intervention phase with durability lasting up to ~20 years, suggesting that lifestyle change can prevent or postpone T2D even when weight loss is modest72. Contemporary evidence likewise suggests that lifestyle modifications (dietary habits, increased physical activity to  150 minutes/week, and 5–10% weight loss) can greatly improve metabolism and even lead to diabetes remission in some patients, particularly when utilizing approaches such as the Mediterranean or DASH diet, higher protein intake, greater dietary fiber intake, and adjusted carbohydrate timing tailored to the individual8.
150 minutes/week, and 5–10% weight loss) can greatly improve metabolism and even lead to diabetes remission in some patients, particularly when utilizing approaches such as the Mediterranean or DASH diet, higher protein intake, greater dietary fiber intake, and adjusted carbohydrate timing tailored to the individual8.
Across both prevention and treatment contexts, moderate weight loss consistently provides substantial clinical benefit and serves as the foundation for adding medications or procedures. Intensive, skills-based behavioral support can also be added to reach and maintain these results.
Pharmacological Therapy
When lifestyle change alone does not result in adequate blood glucose and weight control, or when instances of cardiovascular or renal comorbidities, medications can be added to treatment plans74. Metformin is widely used as initial therapy for T2D in the United States: it lowers the liver’s overproduction of glucose and helps the body use insulin more effectively, resulting in modest weight loss. GLP-1 receptor agonists and the dual GLP-1/GIP agonist tirzepatide can be used to reduce appetite, increase fullness, and produce more substantial weight loss. In fact, in adults with obesity but without diabetes, tirzepatide cut future T2D risk by 27%, and in patients with active T2D it lowered major cardiovascular events by 46%75. SGLT2 inhibitors lower glucose levels by promoting glucose excretion in the urine and have clear heart–kidney benefits as well. DPP-4 inhibitors gently increase post-meal insulin release with little effect on weight, while thiazolidinediones (TZDs) improve the body’s response to insulin but must be used selectively due to side effects.
Clinically, medications are started either when lifestyle changes alone do not reach targets, or when symptoms of hyperglycemia (high glucose levels) are present. If a person has, or is at high risk for, ASCVD, heart failure, or CKD, an SGLT2 inhibitor or GLP-1 agent is often prioritized even regardless of baseline A1C. When weight loss is a key goal, clinicians favor glucose-lowering drugs that support weight loss (metformin, GLP-1 agents, SGLT2 inhibitors) and, when appropriate, add stronger weight-management medications (e.g., phentermine–topiramate, naltrexone–bupropion, orlistat). Guidance highlights starting with a 5–10% weight-loss goal supported by intensive counseling, with pharmacological interventions added if needed to help maintain the loss and reach glucose and organ-protection goals73. Recent reviews highlight GLP-1/dual agonists (liraglutide, semaglutide, tirzepatide) and SGLT2 inhibitors as key treatments for obesity-related T2D, with anti-obesity medications used on a case-by-case basis8. Mechanistic reviews show that metformin, pioglitazone, SGLT2 inhibitors, and GLP-1 drugs remain key treatments for insulin resistance, while new therapies aimed at targeting inflammation, lipid metabolism, and the gut microbiome are under development16.
However, there are some important considerations. GLP-1/dual agonists commonly cause nausea or GI upset and can be inaccessible; SGLT2 inhibitors can increase urinary or genital infections and at times can cause euglycemic ketoacidosis; TZDs can lead to edema and weight gain (and rarely fractures); sulfonylureas and insulin may raise risks of hypoglycemia and weight gain. Injectable forms may reduce adherence, and stopping some medications abruptly can lead to rebound weight gain or even worse glucose control. Overall, medications complement rather than replace lifestyle change. The most effective care matches the appropriate pharmacological intervention to each person’s cardiometabolic profile, weight goals, preferences, and access, and effectiveness is best maintained with regular follow-up and education.
Surgical Intervention
Bariatric/metabolic surgery causes significant, long-lasting weight loss, which lowers fat-driven inflammation, improves liver and muscle insulin sensitivity, and helps restore β-cell function. The Swedish Obese Subjects (SOS) study reported a 16.1% weight reduction at 10 years, accompanied by reduced T2D incidence and improved remission rates for T2D, hypertriglyceridemia, and low HDL cholesterol. Consistent with these findings, the landmark SOS cohort analysis showed that in patients with severe obesity, bariatric surgery produced sustained weight loss over a 10 year period and significantly improved or prevented major metabolic diseases, including T2D76. Surgery can also cause remission in 30–60% of T2D cases, although relapse is still possible72.
Precision Medicine Approaches
Emerging approaches classify obesity into phenotypes such as abnormal satiation (hungry brain), abnormal postprandial satiety (hungry gut), low energy expenditure (slow burn), and emotional hunger, with the goal of aligning treatment to the underlying pathophysiology77. For example, “slow burn” phenotypes may result in reduced brown adipose tissue thermogenesis, while “hungry brain” types involve hypothalamic leptin/insulin resistance. Phenotype-guided therapy has achieved greater weight loss than standard care, and tools like multi-omics profiling are being developed to better define phenotypes, predict treatment responses, and personalize interventions.
Gut Microbiome and Metabolomic Modulation
Gut dysbiosis and altered metabolomic profiles can cause chronic inflammation, increase intestinal permeability, and impair short-chain fatty acid production—all mechanisms that are closely linked to insulin resistance78’79. Recent studies show that higher microbial diversity correlates with better blood glucose control, and approximately two-thirds of diabetes-related plasma metabolites can be modified via diet or exercise79. Probiotic supplementation, prebiotic-rich diets, and microbiome-targeted therapies are also being studied as adjunct treatments to help restore metabolic balance.
Clinical studies have also been looking at microbiome-directed approaches as adjuncts rather than stand-alone therapies. In some studies, probiotics (e.g., Lactobacillus, Bifidobacterium, Akkermansia) and prebiotics (e.g., inulin-type fructans) have shown to improve insulin sensitivity or lipid profiles, but weight-loss effects have been inconsistent80. Fecal microbiota transplantation (FMT) from donors have shown early improvements in insulin sensitivity and function, yet more recent controlled studies have reported inconsistent results as well as safety and logistical constraints, which necessitates further study50’80. Large studies done on multi-omic cohorts indicate that the gut–metabolism connection is modifiable. Short-term dietary and exercise changes can alter over a hundred glucose-related markers, supporting the development of personalized treatment plans79. Overall, microbiome-targeted interventions are promising complementary interventions that can be used in addition to lifestyle changes and pharmacotherapy. However, more comprehensive, standardized human trials would be beneficial to more clearly define responders, durability, and safety profiles.
Public Health & Policy Interventions
Beyond individual-level strategies, population-wide public health initiatives and policy measures are essential for reducing the incidence of obesity, insulin resistance, and T2D at the population-level. Programs such as the U.S. National Diabetes Prevention Program (NDPP) have been implemented at a nation-wide level to provide structured lifestyle coaching, nutrition education, and physical activity promotion to at-risk populations59. Additionally, school-based interventions targeting childhood obesity such as nutrition education and increased opportunities for physical activity have shown promise in improving BMI trajectories and metabolic profiles. Policy-level measures, such as sugar-sweetened beverage taxation, mandatory front-of-package nutrition labeling, and regulations of trans fats, have been implemented in several countries and have successfully reduced the consumption of unhealthy foods and beverages. Urban design strategies that enhance walkability, expand public transportation, and increase access to recreational spaces target environmental factors that contribute to physical inactivity. Interventions such as subsidized access to healthy foods in low-income communities, culturally tailored educational resources, and expanded coverage of preventive healthcare services are intended to reduce disparities in diabetes burden. Mass media campaigns and community-based programs further promote public awareness, improve health literacy, and facilitate the early detection of high-risk cases. These interventions target upstream determinants of obesity and insulin resistance by shaping both diet and physical activity environment, reinforcing sustained lifestyle changes, and supporting long-term disease prevention.
Brown Adipose Tissue Activation
Adipose depots in mammals are made up of energy‐storing white adipose tissue (WAT) and heat‐producing brown adipose tissue (BAT); under stimuli such as exercise or colder temperatures, WAT can acquire brown-like features (“browning”), forming beige adipocytes and beige cells that arise de novo from resident progenitors81. In adults, 18F-FDG PET/CT has revealed metabolically active BAT/beige fat in the cervical–supraclavicular region that vigorously consumes glucose and fatty acids to serve as fuel for thermogenesis, aiding whole-body glucose–lipid homeostasis. Therefore, activating BAT/beige fat and inducing WAT browning are both promising ways to lower insulin demand and improve insulin sensitivity and glucose tolerance81’82.
Mechanistically, BAT can increase glucose uptake and oxidation via UCP1-mediated non-shivering thermogenesis and sympathetic β3-adrenergic→cAMP/PKA→PPAR/PGC-1α signaling with central circuits (CNS→IML→SNS), allowing upstream control81’82. AMPK is a primary regulator of BAT/beige development, mitochondrial quality, and thermogenic capacity: adipocyte AMPK loss in mice causes cold intolerance, hindered thermogenesis, and decreased NAFLD/insulin resistance. On the other hand, AMPK activation promotes BAT activation/browning; in humans, adipose AMPK activity is reduced in insulin-resistant states48.
Currently, however, the majority of evidence remains preclinical: in mice, cold, β3-agonism, and BAT transplantation improve energy expenditure, insulin sensitivity, glucose tolerance, and lipid handling (partly via IL-6/adiponectin). In humans, on the other hand, functional BAT/beige depots and increased BAT glucose uptake with cold or hormonal/β3 stimuli suggest translational likelihood. Still, translation is limited by interspecies differences in adult BAT amount/distribution, thermoneutral living, FDG-PET’s restrictions as a thermogenesis proxy, catecholamine resistance in obesity, and cardiovascular safety concerns with β3 agonists; intensive randomized trials are required to establish effect size, durability, standardized stimulation methods, and safety81’82.
Candidate activation routes include cold exposure, exercise/myokines (irisin, Metrnl), hormones/cytokines (FGF21, thyroid hormones, IL-6, cardiac natriuretic peptides), microbiota/dietary interventions (intermittent fasting, caloric restriction, SIRT pathways), miRNAs/BMPs, and BAT transplantation, while selective recruitment of BAT/beige via defined CNS nodes may improve effectiveness without generalized sympathoactivation81’82. Comprehensively, BAT/beige-targeted “energy-expenditure” therapies could aid glucose-lowering drugs to address insulin resistance, dyslipidemia, and NAFLD, with adipose signaling hubs such as AMPK representing promising targets.
Underexplored Pathways & Further Research Priorities
Although existing management strategies target established pathways such as caloric excess, poor diet quality, and physical inactivity, several determinants of insulin resistance and T2D remain underexplored. For instance, therapeutic approaches targeting immune cell polarization within adipose tissue (a central driver of obesity-related chronic inflammation) remain in experimental phases.
Mitochondrial dysfunction and redox imbalance in insulin-sensitive tissues are key contributors to impaired glucose utilization, yet remain difficult to address directly. Emerging therapies that enhance mitochondrial biogenesis or mitigate oxidative stress may prove to be beneficial. Impaired branched-chain amino acid (BCAA) metabolism, which has been linked to the development of insulin resistance, has not yet been routinely assessed or targeted in clinical practice. However, dietary strategies and microbiome modulation could help.
Additionally, emerging research has suggested that impaired GLUT4-mediated glucose uptake and de novo lipogenesis in subcutaneous adipose tissue contribute to systemic insulin resistance. Decreased GLUT4 expression restricts glucose uptake in adipocytes, which reduces the synthesis of branched fatty acid esters of hydroxy fatty acids (FAHFAs), a newly identified group of fats that help improve insulin sensitivity, reduce inflammation, and boost incretin release. FAHFAs enhance insulin-stimulated glucose transport, promote glucose-stimulated GLP-1 and insulin secretion, and suppress inflammation, but their levels are markedly reduced in insulin-resistant individuals83. Despite their therapeutic potential, FAHFA-targeted interventions are still at the early stages of research, and haven’t reached clinical integration yet.
Research on the gut microbiome is rapidly advancing, but most interventions still rely on general probiotics or dietary changes as opposed to directly targeted manipulations of specific microbial metabolites that affect glucose control78’79’80. Multi-omics integration that combines genomics, metabolomics, proteomics, and microbiome data may allow for more precise phenotyping in order to match patients with appropriate treatment plans77. Future research should focus on developing interventions that address these underexplored mechanisms, integrating them into existing lifestyle, pharmacological, and policy approaches. By expanding therapeutic targets beyond traditional risk factors, more durable prevention and remission of T2D can be achieved.
Discussion
Limits to Generalizability and Heterogeneity among studies
The link between obesity and T2D is not a single linear relationship but a heterogeneous set of relationships with differing magnitudes and directions, impacted by both population and context. Although obesity is commonly measured using BMI and higher BMI is generally associated with higher diabetes incidence, studies in East Asian populations indicate elevated T2D risk at comparatively lower BMI, potentially due to more pronounced β-cell dysfunction and greater central adiposity. These findings illustrate why broad generalizations do not hold universally across populations.
Inconsistencies across studies point to underlying contextual heterogeneity. U.S. surveillance from 2012 to 2022 shows rising diabetes prevalence overall, with men slightly higher than women. In contrast, a global synthesis reports that, on average worldwide and in most regions, women have higher obesity prevalence than men. Thus, opposing results are present across these studies.
Overall, variation in ancestry, socioeconomic status, environment, and measurement practices influences how obesity relates to T2D. Because these influences are multilayered and contextual, the generalizability of definitions, thresholds, and effect estimates can be limited. Therefore, accurate identification of effect modifiers and contextual factors is critical for interpreting and comparing study results.
Socioeconomic Factors and Contextual Heterogeneity
Boutari and Mantzoros58 identify a reversal in the socioeconomic distribution of obesity. In many high income countries, obesity prevalence is higher in lower socioeconomic groups, whereas in many low and middle income countries it is higher in higher socioeconomic groups. Differences in economic development, inequality, diet and work settings, urbanization, social expectations, and policy systems help determine how socioeconomic status is associated, both in strength and in direction. Since the pathways linking socioeconomic status to obesity and diabetes risk differ by context, it cannot be generalized that lower or higher income consistently corresponds to greater prevalence. This context dependence limits the consistency of findings across settings.
Challenges in Causal Interpretation
Bowen et al.9 attribute the higher prevalence of obesity and T2D in lower income populations in the United States to structural conditions such as food insecurity, energy insecurity, and limited access to safe spaces for physical activity, which is further exacerbated by lower participation in prevention and management programs such as Diabetes Self Management Education and Support and the National Diabetes Prevention Program. This is a plausible explanation for the observed disparities. However, not all studies can deliver equally strong causal accounts due to limitations in data, measurement, and study design.
Large surveillance analyses can describe patterns with precision yet often fall short of clarifying the causal mechanisms behind them. For example, Neupane et al.56 analyzed 5.31 million U.S. adults from 2012 to 2022 and documented rising diabetes prevalence overall, with particularly high levels among non-Hispanic Black adults (15.8%), adults aged 65 years or older (23.86%), and adults with obesity (19.23%). Regionally, increases were greatest in the South and Midwest, rising from 9.2 percent to 12.8 percent, with Arkansas, Kentucky, and Nebraska particularly affected. However, while these results support clear patterns in the data, determining why these patterns emerged is considerably more difficult. Moreover, interpretations can vary across researchers depending on analytic choices and assumptions, which is an additional limitation.
Limitations in Measurement and Operational Definitions
Many studies define obesity and its severity solely by BMI. When exposure is operationalized only with BMI and then related to other factors, the resulting estimates are vulnerable to misclassification and bias. For example, research in East Asian populations reports elevated T2D risk at comparatively lower BMI, partly due to greater β-cell dysfunction and a more central pattern of adiposity. If one assumes that diabetes risk rises only at higher BMI, such findings can appear inconsistent.
Accordingly, recent work argues that studies should supplement BMI with distributional and qualitative indicators, including waist circumference, waist to height ratio, visceral adipose tissue, and organ specific fat such as liver fat in nonalcoholic fatty liver disease. Nevertheless, many published analyses still rely on BMI alone. Viewed in light of current evidence, those estimates may embed error and have limited transportability across populations and settings.
Limitation of the study
This review is a narrative synthesis that integrates epidemiological, clinical, and mechanistic evidence rather than a quantitative meta analysis, so effect sizes were not statistically pooled. The scope is restricted to peer-reviewed studies written in English, which may omit non-English evidence. Studies that focused exclusively on gestational diabetes or mainly on other diseases such as cardiovascular disease with only tangential effects leading to diabetes were excluded from the search, which can limit mechanistic depth and the breadth of perspectives. Variability in study designs and populations reduces comparability across studies and may contribute to inconsistencies in findings.
There is no unifying model or schematic that explicates how obesity duration, severity, and fat distribution interact mechanistically. Section 3.5 has been expanded to synthesize these interactions, but a unifying figure is not included in this revision, with the integrated framework articulated narratively. Additionally, this review explores studies conducted on mice and presents possible extrapolation to humans, but the review is not limited to specific human studies.
In regards to terminologies, broader descriptions regarding obesity duration such as “from young adults to midlife” is referenced, but specifically how many years constitutes a longer duration of obesity was not noted in any of the referenced studies and therefore omitted from this review.
Implications and future application
Given that implications for measurement and study design are clear, future studies should investigate deeper than BMI by adding distributional and organ specific indicators such as waist circumference, waist to height ratio, visceral adipose tissue, and liver fat. Analyses should explore the effects by age, sex and menopausal status, ancestry, socioeconomic conditions, comorbidities, and geography, and describe setting and policy environment. Duration and severity of obesity should be measured using integrated burden metrics. Microbiome and metabolomic endpoints need consistent functional markers with validation to improve comparability.
Implications for prevention are linked with cumulative adiposity as one of the main drivers of risk. Priorities are to delay the onset of obesity, shorten its duration, and reduce its severity, with structured lifestyle programs as the foundation, pharmacotherapy layered when indicated, and surgery for severe obesity with metabolic disease. At the community level, programs such as the National Diabetes Prevention Program and Diabetes Self Management Education and Support, combined with policies that reshape food and activity environments, address upstream risk. Future work should target adipose immune polarization, mitochondrial function and redox balance, branched chain amino acid pathways, adipose tissue glucose handling and FAHFA biology, and brown adipose tissue activation and browning, while implementing multi omics phenotyping and implementation approaches that take into consideration social determinants to match therapies to individual biology and local context.
Conclusion
This review synthesizes the biological and physiological mechanisms by which obesity initiates and accelerates T2D and considers how genetics, age, sex, ethnicity, socioeconomic status (SES), comorbidities, medication exposure, and the duration and severity of obesity shape risk. Obesity promotes adipose tissue inflammation, imbalances in adipokines, free fatty acids, and cytokines, and ectopic lipid deposition with disruption of insulin signaling, producing organ-specific insulin resistance in the liver, skeletal muscle, and adipose tissue, and β-cell dysfunction in the pancreas. Dysregulation of the gut microbiota–intestinal barrier axis and brown adipose tissue (BAT) dysfunction further amplify these disturbances, increasing insulin demand and driving β-cell overload and exhaustion, with consequent impairment of glucose homeostasis. Viewed in conjunction, the evidence indicates that these interlinked pathways induce and intensify insulin resistance and increase β-cell workload, reinforcing positive feedback loops rather than a single linear route to T2D onset and progression.
This cascade is influenced by systemic and population-level contextual factors (age, sex, ethnicity, SES, comorbidities, medication exposure) and by the cumulative adiposity burden caused by obesity severity and duration. As these factors interact, heterogeneity in effect size and direction emerges across individuals and groups, such that the same exposure can yield different outcomes. Accordingly, effect sizes and thresholds are context-sensitive, and broad generalization across populations does come with inherent limits.
Although treatment options are wide-ranging, the solution most consistently emphasized across studies is weight reduction. Weight loss lowers ectopic fat and free-fatty-acid flux, attenuates adipose inflammation, benefits insulin signaling, and decreases β-cell workload—making it a mechanism-aligned first-line strategy with consistent benefits for delaying onset and slowing illness progression, while being cost-effective and scalable. In practice, structured lifestyle intervention forms the foundation; when indicated, this is layered with weight- and organ-protective pharmacotherapies (e.g., metformin, GLP-1 receptor agonists/dual GLP-1–GIP agonists, SGLT2 inhibitors) and metabolic (bariatric) surgery in a tailored, stepwise combination. Notably, realistic and low-cost strategies such as dietary adjustment and postprandial walking alone can yield meaningful improvements, supporting both individual adherence and population-level impact.
Looking ahead, the clinical utility of mechanism-based emerging approaches—including microbiome/metabolome modulation and BAT activation of white adipose tissue—should be tested across a much more diverse group that considers real-world settings. Public health programs are already in place, yet not many people know about these programs and some individuals that are aware hesitate to participate; pairing these programs with policies that improve food and activity environments can reduce upstream risk and strengthen access and equity. Implemented together, such multidimensional strategies offer the most practical and cost-effective route to delay onset, slow progression, and reduce the long-term individual and societal (including economic) burden of T2D.
References
- World Health Organization. (2024a, November 14). Diabetes. World Health Organization. https://www.who.int/news-room/fact-sheets/detail/diabetes [↩]
- International Diabetes Federation. (2025). IDF Diabetes Atlas (11th ed.). https://diabetesatlas.org/ [↩]
- Centers for Disease Control and Prevention. (n.d.). Diabetes data & research. Retrieved July 10, 2025, from https://www.cdc.gov/diabetes/php/data-research/index.html [↩]
- American Diabetes Association. (2013). Diagnosis and classification of diabetes mellitus. Diabetes Care, 36(Suppl. 1), S67–S74. https://doi.org/10.2337/dc13-S067 [↩]
- Centers for Disease Control and Prevention. (n.d.). Diabetes and vision loss. https://www.cdc.gov/diabetes/diabetes-complications/diabetes-and-vision-loss.html [↩]
- Centers for Disease Control and Prevention. (n.d.). Preventing diabetes-related amputations. https://www.cdc.gov/diabetes/diabetes-complications/preventing-diabetes-related-amputations.html [↩]
- Chandrasekaran, P., & Weiskirchen, R. (2024). The role of obesity in type 2 diabetes mellitus—An overview. International Journal of Molecular Sciences, 25(3), 1882. https://doi.org/10.3390/ijms25031882 [↩] [↩]
- Garg, C., & Daley, S. F. (2025). Obesity and type 2 diabetes. In StatPearls. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/sites/books/NBK592412/ [↩] [↩] [↩]
- Bowen, S., Alamian, A., & Onufrak, S. (2025). Public health research and program strategies for diabetes prevention and management. Preventing Chronic Disease, 22, E50. https://doi.org/10.5888/pcd22.240501 [↩] [↩] [↩] [↩]
- World Health Organization. (2024b, March 1). Obesity and overweight. World Health Organization. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight [↩]
- James, D. E., Stöckli, J., & Birnbaum, M. J. (2021). The aetiology and molecular landscape of insulin resistance. Nature Reviews Molecular Cell Biology, 22, 751–771. https://doi.org/10.1038/s41580-021-00390-6 [↩] [↩]
- Lee, S.-H., Park, S.-Y., & Choi, C. S. (2022). Insulin resistance: From mechanisms to therapeutic strategies. Diabetes & Metabolism Journal, 46(1), 15–37. https://doi.org/10.4093/dmj.2021.0280 [↩]
- Cui, Y., Tang, T.-Y., Lu, C.-Q., & Ju, S. (2022). Insulin resistance and cognitive impairment: Evidence from neuroimaging. Journal of Magnetic Resonance Imaging, 56(6), 1621–1649. https://doi.org/10.1002/jmri.28358 [↩]
- Gutt, M., Davis, C. L., Spitzer, S. B., Llabre, M. M., Kumar, M., Czarnecki, E. M., Schneiderman, N., Skyler, J. S., & Marks, J. B. (2000). Validation of the insulin sensitivity index (ISI0,120): Comparison with other measures. Diabetes Research and Clinical Practice, 47(3), 177–184. https://doi.org/10.1016/S0168-8227(99)00116-3 [↩]
- Sears, B. & Perry, M. (2015). The role of fatty acids in insulin resistance. Lipids in Health and Disease, 14, 121. https://doi.org/10.1186/s12944-015-0123-1 [↩]
- Zhao, X., An, X., Yang, C., Sun, W., Ji, H., & Lian, F. (2023). The crucial role and mechanism of insulin resistance in metabolic disease. Frontiers in Endocrinology, 14, 1149239. https://doi.org/10.3389/fendo.2023.1149239 [↩] [↩] [↩] [↩] [↩]
- Polonsky, W. H., Arsenault, J., Fisher, L., Kushner, P., Miller, E. M., Pearson, T. L., Tracz, M., Harris, S., Hermanns, N., Scholz, B-M., Pollom, R. K., Perez-Nieves, M., Pollom, R. D., & Hadjiyianni, I. (2017). Initiating insulin: How to help people with type 2 diabetes start and continue insulin successfully. International Journal of Clinical Practice, 71(8), e12973. https://doi.org/10.1111/ijcp.12973 [↩]
- Lebovitz, H. E. (2011). Insulin: Potential negative consequences of early routine use in patients with type 2 diabetes. Diabetes Care, 34(Suppl 2), S225-S230. https://doi.org/10.2337/dc11-s225 [↩]
- Kwon, H., & Pessin, J. E. (2013). Adipokines mediate inflammation and insulin resistance. Frontiers in Endocrinology, 4, 71. https://doi.org/10.3389/fendo.2013.00071 [↩] [↩] [↩] [↩]
- Sethi, J. K. & Hotamisligil, G. S. (2021). Metabolic messengers: Tumour necrosis factor. Nature Metabolism, 3(10), 1302–1312. https://doi.org/10.1038/s42255-021-00470-z [↩] [↩] [↩]
- Orliaguet, L., Ejlalmanesh, T., & Alzaid, F. (2020). Metabolic and molecular mechanisms of macrophage polarisation and adipose tissue insulin resistance. International Journal of Molecular Sciences, 21(16), 5731. https://doi.org/10.3390/ijms21165731 [↩]
- Arneth, B. (2024). Mechanisms of insulin resistance in patients with obesity. Endocrines, 5(2), 153–165. https://doi.org/10.3390/endocrines5020011 [↩] [↩] [↩]
- Klein, S., Gastaldelli, A., Yki-Järvinen, H., & Scherer, P. E. (2022). Why does obesity cause diabetes? Cell Metabolism, 34(1), 11–20. https://doi.org/10.1016/j.cmet.2021.12.012 [↩]
- Clemente-Suárez, V. J., Redondo-Flórez, L., Beltrán-Velasco, A. I., Martín-Rodríguez, A., Martínez-Guardado, I., Navarro-Jiménez, E., Laborde-Cárdenas, C. C., & Tornero-Aguilera, J. F. (2023). The role of adipokines in health and disease. Biomedicines, 11(5), 1290. https://doi.org/10.3390/biomedicines11051290 [↩] [↩] [↩]
- Yamauchi, T., Kamon, J., Waki, H., Terauchi, Y., Kubota, N., Hara, K., Mori, Y., Ide, T., Murakami, K., Tsuboyama-Kasaoka, N., Ezaki, O., Akanuma, Y., Gavrilova, O., Vinson, C., Reitman, M. L., Kagechika, H., Shudo, K., Yoda, M., Nakano, Y., … Kadowaki, T. (2001). The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nature Medicine, 7(8), 941-946. https://doi.org/10.1038/90984 [↩] [↩]
- Petersen, M. C., & Shulman, G. I. (2018) Mechanisms of insulin action and insulin resistance. Physiological Reviews, 98(4), 2133–2223. https://doi.org/10.1152/physrev.00063.2017 [↩]
- Wondmkun, Y. T. (2020). Obesity, insulin resistance, and type 2 diabetes: Associations and therapeutic implications. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy, 13, 3611–3616. https://doi.org/10.2147/DMSO.S275898 [↩]
- Schmitz-Peiffer, C. (2020). Deconstructing the role of PKC epsilon in glucose homeostasis. Trends in Endocrinology & Metabolism. 31(5). 344-356. https://doi.org/10.1016/j.tem.2020.01.016 [↩]
- Boden, G. (2011). Obesity, insulin resistance and free fatty acids. Current Opinion in Endocrinology, Diabetes, and Obesity, 18(2), 139–143. https://doi.org/10.1097/MED.0b013e3283444b09 [↩]
- Xu, C., Zhou, L., Wu, K., Li, Y., Xu, J., Jiang, D. & Gao, L. (2019). Abnormal glucose metabolism and insulin resistance are induced via the IRE1α/XBP-1 pathway in subclinical hypothyroidism. Frontiers in Endocrinology. 10(303). 1-9. https://doi.org/10.3389/fendo.2019.00303 [↩] [↩]
- Luukkonen, P. K., Qadri, S., Ahlholm, N., Porthan, K., Männistö, V., Sammalkorpi, H., Penttilä, A. K., Hakkarainen, A., Lehtimäki, T. E., Gaggini, M., Gastaldelli, A., Ala-Korpela, M., Orho-Melander, M., Arola, J., Juuti, A., Pihlajamäki, J., Hodson, L. & Yki-Järvinen, H. (2022). Distinct contributions of metabolic dysfunction and genetic risk factors in the pathogenesis of non-alcoholic fatty liver disease. Journal of Hepatology, 76(3), 526-535. https://doi.org/10.1016/j.jhep.2021.10.013 [↩] [↩] [↩] [↩]
- Gastaldelli, A. (2011). Role of beta-cell dysfunction, ectopic fat accumulation and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Diabetes Research and Clinical Practice, 93(Suppl. 1), S60–S65. https://doi.org/10.1016/S0168-8227(11)70015-8 [↩] [↩] [↩] [↩]
- Yki-Järvinen, H. (2002). Ectopic fat accumulation: An important cause of insulin resistance in humans. Journal of the Royal Society of Medicine, 95(Suppl 42), 39–45. https://pmc.ncbi.nlm.nih.gov/articles/PMC1308944/ [↩] [↩] [↩]
- Trouwborst, I., Bowser, S. M., Goossens, G. H., & Blaak, E. E. (2018). Ectopic fat accumulation in distinct insulin resistant phenotypes; Targets for personalized nutritional interventions. Frontiers in Nutrition, 5, 77. https://doi.org/10.3389/fnut.2018.00077 [↩] [↩] [↩] [↩] [↩]
- Snel, M., Jonker, J. T., Schoones, J., Lamb, H., de Roos, A., Pijl, H., Smit, J. W. A., Meinders, A. E., & Jazet, I. M. (2012). Ectopic fat and insulin resistance: Pathophysiology and effect of diet and lifestyle interventions. Journal of Obesity, 2012, 983814. https://doi.org/10.1155/2012/983814 [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- Byrne, C. D. (2013). Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proceedings of the Nutrition Society, 72(3), 412–419. https://doi.org/10.1017/S0029665113001249 [↩] [↩] [↩] [↩]
- Iatcu, C. O., Steen, A., & Covasa, M. (2022). Gut microbiota and complications of type-2 diabetes. Nutrients, 14(1), 166. https://doi.org/10.3390/nu14010166 [↩]
- Takeuchi, T., Kubota, T., Nakanishi, Y., Tsugawa, H., Suda, W., Kwon, A. T.-J., Yazaki, J., Ikeda, K., Nemoto, S., Mochizuki, Y., Kitami, T., Yugi, K., Mizuno, Y., Yamamichi, N., Yamazaki, T., Takamoto, I., Kubota, N., Kadowaki, T., Arner, E., … Ohno, H. (2023). Gut microbial carbohydrate metabolism contributes to insulin resistance. Nature, 621(7978), 389–395. https://doi.org/10.1038/s41586-023-06466-x [↩] [↩]
- Zhang, L., Chu, J., Hao, W., Zhang, J., Li, H., Yang, C., Yang, J., Chen, X., & Wang, H. (2021). Gut microbiota and type 2 diabetes mellitus: Association, mechanism, and translational applications. Journal of Diabetes Research, 2021, 5110276. https://doi.org/10.1155/2021/5110276 [↩] [↩] [↩]
- Cunningham, A. L., Stephens, J. W., & Harris, D. A. (2021). Gut microbiota influence in type 2 diabetes mellitus (T2DM). Gut Pathogens, 13, 50. https://doi.org/10.1186/s13099-021-00446-0 [↩] [↩]
- Crudele, L., Gadaleta, R. M., Cariello, M., & Moschetta, A. (2023). Gut microbiota in the pathogenesis and therapeutic approaches of diabetes. eBioMedicine, 97, 104821. https://doi.org/10.1016/j.ebiom.2023.104821 [↩] [↩] [↩]
- Salamone, D., Rivellese, A. A., & Vetrani, C. (2021). The relationship between gut microbiota, short-chain fatty acids and type 2 diabetes mellitus: The possible role of dietary fibre. Acta Diabetologica, 58(9), 1131–1138. https://doi.org/10.1007/s00592-021-01727-5 [↩] [↩] [↩]
- Bielka, W., Przezak, A., & Pawlik, A. (2022). The role of the gut microbiota in the pathogenesis of diabetes. International Journal of Molecular Sciences, 23(1), 480. https://doi.org/10.3390/ijms23010480 [↩] [↩] [↩]
- Iatcu, C. O., Steen, A., & Covasa, M. (2022). Gut microbiota and complications of type-2 diabetes. Nutrients, 14(1), 166. https://doi.org/10.3390/nu14010166 [↩]
- Cunningham, A. L., Stephens, J. W., & Harris, D. A. (2021). Gut microbiota influence in type 2 diabetes mellitus (T2DM). Gut Pathogens, 13, 50. https://doi.org/10.1186/s13099-021-00446-0 [↩]
- Blondin, D. P., Labbé, S. M., Noll, C., Kunach, M., Phoenix, S., Guérin, B., Turcotte, É. E., Haman, F., Richard, D., & Carpentier, A. C. (2015). Selective impairment of glucose but not fatty acid or oxidative metabolism in brown adipose tissue of subjects with type 2 diabetes. Diabetes, 64(7), 2388–2397. https://doi.org/10.2337/db14-1651 [↩] [↩]
- Hanssen, M. J. W., Wierts, R., Hoeks, J., Gemmink, A., Brans, B., Mottaghy, F. M., Schrauwen, P., & van Marken Lichtenbelt, W. D. (2015). Glucose uptake in human brown adipose tissue is impaired upon fasting-induced insulin resistance. Diabetologia, 58(3), 586–595. https://doi.org/10.1007/s00125-014-3465-8 [↩] [↩]
- Desjardins, E. M., & Steinberg, G. R. (2018). Emerging role of AMPK in brown and beige adipose tissue (BAT): Implications for obesity, insulin resistance, and type 2 diabetes. Current Diabetes Reports, 18(8), 80. https://doi.org/10.1007/s11892-018-1049-6 [↩] [↩]
- Xu, L., & Ota, T. (2018). Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: Focus on fat browning and macrophage polarization. Adipocyte, 7(2), 121–128. https://doi.org/10.1080/21623945.2017.1413516 [↩]
- Cerf, M. E. (2013). Beta cell dysfunction and insulin resistance. Frontiers in Endocrinology, 4, 37. https://doi.org/10.3389/fendo.2013.00037 [↩] [↩] [↩]
- LeRoith, D. (2002). β-cell dysfunction and insulin resistance in type 2 diabetes: Role of metabolic and genetic abnormalities. The American Journal of Medicine, 113(6, Suppl), 3–11. https://doi.org/10.1016/S0002-9343(02)01276-7 [↩] [↩] [↩]
- Khin, P. P., Lee, J. H., & Jun, H.-S. (2023). Pancreatic beta-cell dysfunction in type 2 diabetes. European Journal of Inflammation, 21, 1721727X231154152. https://doi.org/10.1177/1721727X231154152 [↩] [↩] [↩] [↩]
- Eguchi, N., Vaziri, N. D., Dafoe, D. C., & Ichii, H. (2021). The role of oxidative stress in pancreatic β cell dysfunction in diabetes. International Journal of Molecular Sciences, 22(4), 1509. https://doi.org/10.3390/ijms22041509 [↩]
- Chung, S. T., Galvan-De La Cruz, M., Aldana, P. C., Mabundo, L. S., DuBose, C. W., Onuzuruike, A. U., Walter, M., Gharib, A. M., Courville, A. B., Sherman, A. S., & Sumner, A. E. (2019). Postprandial insulin response and clearance among Black and White women: The Federal Women’s Study. The Journal of Clinical Endocrinology & Metabolism, 104(1), 181–192. https://doi.org/10.1210/jc.2018-01032 [↩]
- Stokes, A., Collins, J. M., Grant, B. F., Scamuffa, R. F., Hsiao, C.-W., Johnston, S. S., Ammann, E. M., Manson, J. E., & Preston, S. H. (2018). Obesity progression between young adulthood and midlife and incident diabetes: A retrospective cohort study of U.S. adults. Diabetes Care, 41(5), 1025–1031. https://doi.org/10.2337/dc17-2336 [↩] [↩]
- Neupane, S., Florkowski, W. J., Dhakal, U., & Dhakal, C. (2024). Regional disparities in type 2 diabetes prevalence and associated risk factors in the United States. Diabetes, Obesity and Metabolism, 26(10), 4776–4782. https://doi.org/10.1111/dom.15797 [↩] [↩] [↩] [↩] [↩] [↩]
- Bays, H. E., Gonsahn-Bollie, S., Younglove, C., & Wharton, S. (2022). Obesity Pillars roundtable: Body mass index and body composition in Black and female individuals. Race-relevant or racist? Sex-relevant or sexist? Obesity Pillars, 4, 100044. https://doi.org/10.1016/j.obpill.2022.100044 [↩]
- Boutari, C., & Mantzoros, C. S. (2022). A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism, 133, 155217. https://doi.org/10.1016/j.metabol.2022.155217 [↩] [↩] [↩] [↩]
- Cameron, N. A., Petito, L. C., McCabe, M., Allen, N. B., O’Brien, M. J., Carnethon, M. R., & Khan, S. S. (2021). Quantifying the sex–race/ethnicity–specific burden of obesity on incident diabetes mellitus in the United States, 2001 to 2016: MESA and NHANES. Journal of the American Heart Association, 10(4), e018799. https://doi.org/10.1161/JAHA.120.018799 [↩] [↩] [↩]
- Blüher, M. (2020). Metabolically healthy obesity. Endocrine Reviews, 41(3), bnaa004. https://doi.org/10.1210/endrev/bnaa004 [↩]
- Sun, H., Saeedi, P., Karuranga, S., Pinkepank, M., Ogurtsova, K., Duncan, B. B., Stein, C., Basit, A., Chan, J. C. N., Mbanya, J. C., Pavkov, M. E., Ramachandran, A., Wild, S. H., James, S., Herman, W. H., Zhang, P., Bommer, C., Kuo, S., Boyko, E. J., & Magliano, D. J. (2022). IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Research and Clinical Practice, 183, 109119. https://doi.org/10.1016/j.diabres.2021.109119 [↩] [↩]
- Benyshek, D. C., Martin, J. F., & Johnston, C. S. (2001). A reconsideration of the origins of the type 2 diabetes epidemic among Native Americans and the implications for intervention policy. Medical Anthropology, 20(1), 25–64. https://doi.org/10.1080/01459740.2001.9966186 [↩]
- Yabe, D., Seino, Y., Fukushima, M., & Seino, S. (2015). β cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes in East Asians. Current Diabetes Reports, 15, Article 36. https://doi.org/10.1007/s11892-015-0602-9 [↩]
- Zhang, X., Liu, J., Ni, Y., Yi, C., Fang, Y., Ning, Q., Shen, B., Zhang, K., Liu, Y., Yang, L., Li, K., Liu, Y., Huang, R., & Li, Z. (2024). Global prevalence of overweight and obesity in children and adolescents: A systematic review and meta-analysis. JAMA Pediatrics, 178(8), 800–813. https://doi.org/10.1001/jamapediatrics.2024.1576 [↩]
- Pofi, R., Caratti, G., Ray, D. W., & Tomlinson, J. W. (2023). Treating the Side Effects of Exogenous Glucocorticoids; Can We Separate the Good From the Bad? Endocrine Reviews, 44(6), 975–1011. https://doi.org/10.1210/endrev/bnad016 [↩]
- Chen, K., Shen, Z., Gu, W., Lyu, Z., Qi, X., Mu, Y., & Ning, Y., for the Meinian Investigator Group. (2023). Prevalence of obesity and associated complications in China: A cross-sectional, real-world study in 15.8 million adults. Diabetes, Obesity and Metabolism, 25(11), 3390–3399. https://doi.org/10.1111/dom.15238 [↩]
- Mongraw-Chaffin, M., Foster, M. C., Kalyani, R. R., Vaidya, D., Burke, G. L., Woodward, M., & Anderson, C. A. M. (2016). Obesity severity and duration are associated with incident metabolic syndrome: Evidence against metabolically healthy obesity from the Multi-Ethnic Study of Atherosclerosis. The Journal of Clinical Endocrinology & Metabolism, 101(11), 4117–4124. https://doi.org/10.1210/jc.2016-2460 [↩]
- Norris, T., Cole, T. J., Bann, D., Hamer, M., Hardy, R., Li, L., Ong, K. K., Ploubidis, G. B., Viner, R., & Johnson, W. (2020). Duration of obesity exposure between ages 10 and 40 years and its relationship with cardiometabolic disease risk factors: A cohort study. PLOS Medicine, 17(12), e1003387. https://doi.org/10.1371/journal.pmed.1003387 [↩]
- Abdullah, A., Amin, F. A., Hanum, F., Stoelwinder, J., Tanamas, S., Wolf, R., Wong, E., & Peeters, A. (2016). Estimating the risk of type-2 diabetes using obese-years in a contemporary population of the Framingham Study. Global Health Action, 9(1), 30421. https://doi.org/10.3402/gha.v9.30421 [↩]
- Hu, Y., Bhupathiraju, S. N., de Koning, L., & Hu, F. B. (2014). Duration of obesity and overweight and risk of type 2 diabetes among US women. Obesity (Silver Spring), 22(10), 2267–2273. https://doi.org/10.1002/oby.20851 [↩]
- Abdullah, A., Stoelwinder, J., Shortreed, S., Wolfe, R., Stevenson, C., Walls, H., de Courten, M., & Peeters, A. (2011). The duration of obesity and the risk of type 2 diabetes. Public Health Nutrition, 14(1), 119–126. https://doi.org/10.1017/S1368980010001813 [↩]
- Tuomilehto, J., Schwarz, P., & Lindström, J. (2011). Long-term benefits from lifestyle interventions for type 2 diabetes prevention: Time to expand the efforts. Diabetes Care, 34(Suppl 2), S210–S214. https://doi.org/10.2337/dc11-S222 [↩] [↩] [↩]
- Bramante, C. T., Lee, C. J., & Gudzune, K. A. (2017). Treatment of obesity in patients with diabetes. Diabetes Spectrum, 30(4), 237–243. https://doi.org/10.2337/ds17-0030 [↩] [↩] [↩]
- Freeman, A. M., Acevedo, L. A., & Pennings, N. (2023). Insulin resistance. In StatPearls. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK507839/ [↩]
- Anson, M., Henney, A. E., Broadwell, N., Zhao, S. S., Ibarburu, G. H., Lip, G. Y. H., Wilding, J. P. H., Cuthbertson, D. J., & Alam, U. (2024). Incidence of new-onset type 2 diabetes in adults living with obesity treated with tirzepatide or semaglutide: Real-world evidence from an international retrospective cohort study. EClinicalMedicine, 75, 102777. https://doi.org/10.1016/j.eclinm.2024.102777 [↩]
- Sjöström, L., Lindroos, A.-K., Peltonen, M., Torgerson, J., Bouchard, C., Carlsson, B., Dahlgren, S., Larsson, B., Narbro, K., Sjöström, C. D., Sullivan, M., & Wedel, H. [for the Swedish Obese Subjects Study Scientific Group]. (2004). Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. The New England Journal of Medicine, 351(26), 2683–2693. https://doi.org/10.1056/NEJMoa035622 [↩]
- Espinosa, M. A., de J Rivera Gutierrez, R., Villamarin, J., & Acosta, A. (2025). Precision medicine for obesity treatment. Journal of the Endocrine Society, 9(9), bvaf102. https://doi.org/10.1210/jendso/bvaf102 [↩] [↩]
- Carletti, M., Pandit, J., Gadaleta, M., Chiang, D., Delgado, F., Quartuccio, K., Fernandez, B., Raygoza Garay, J. A., Torkamani, A., Miotto, R., Rossman, H., Berk, B., Baca-Motes, K., Kheterpal, V., Segal, E., Topol, E. J., Ramos, E., & Quer, G. (2025). Multimodal AI correlates of glucose spikes in people with normal glucose regulation, pre-diabetes and type 2 diabetes. Nature Medicine. Advance online publication. https://doi.org/10.1038/s41591-025-03849-7 [↩] [↩]
- Wu, H., Lv, B., Zhi, L., Shao, Y., Liu, X., Mitteregger, M., Chakaroun, R., Tremaroli, V., Hazen, S. L., Wang, R., Bergström, G., & Bäckhed, F. (2025). Microbiome–metabolome dynamics associated with impaired glucose control and responses to lifestyle changes. Nature Medicine, 31, 2222–2231. https://doi.org/10.1038/s41591-025-03642-6 [↩] [↩] [↩] [↩]
- Cheng, Z., Zhang, L., Yang, L., & Chu, H. (2022). The critical role of gut microbiota in obesity. Frontiers in Endocrinology, 13, 1025706. https://doi.org/10.3389/fendo.2022.1025706 [↩] [↩] [↩]
- Cheng, L., Wang, J., Dai, H., Duan, Y., An, Y., Shi, L., Lv, Y., Li, H., Wang, C., Ma, Q., Li, Y., Li, P., Du, H., & Zhao, B. (2021). Brown and beige adipose tissue: A novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte, 10(1), 48–65. https://doi.org/10.1080/21623945.2020.1870060 [↩] [↩] [↩] [↩] [↩]
- Hankir, M. K., Cowley, M. A., & Fenske, W. K. (2016). A BAT-centric approach to the treatment of diabetes: Turn on the brain. Cell Metabolism, 24(1), 31–40. https://doi.org/10.1016/j.cmet.2016.05.003 [↩] [↩] [↩] [↩]
- Smith, U., & Kahn, B. B. (2016). Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. Journal of Internal Medicine, 280(5), 465–475. https://doi.org/10.1111/joim.12540 [↩]



{kind=link}