
Abstract
Neurotransmitter regulation is essential for proper thinking, feeling, and movement. Three important enzymes, acetylcholinesterase (AChE), butyrylcholinesterase (BChE), and monoamine oxidase (MAO), work in coordination to control brain signaling. Although many studies have examined each enzyme individually, fewer have explored how they interact, particularly in the context of genetic mutations affecting BChE. This review investigates the roles of these enzymes in maintaining brain chemistry and examines the consequences of their disruption due to mutations. Variants in the BChE gene, such as the atypical (dibucaine resistant), K variant, and silent forms, can significantly reduce enzymatic efficiency. For example, the BChE K variant has been linked to a more rapid decline in cognitive function among individuals with mild memory impairment who are treated with donepezil. Genetic testing for such variants may improve treatment selection and clinical outcomes. The review was conducted using a structured literature-based methodology to analyze the physiological and pathological roles of AChE, BChE, and MAO in neurotransmitter regulation. The search also examined how genetic variability in these enzymes contributes to neurological and psychiatric disorders. A comprehensive literature search was performed using PubMed, Google Scholar, ScienceDirect, and the NCBI Gene database. Relevant keywords were combined with Boolean operators to identify pertinent studies. Search terms included acetylcholinesterase, butyrylcholinesterase, monoamine oxidase, cholinesterase polymorphisms, BChE K variant, neurotransmitter regulation, oxidative stress, donepezil response, and multi target therapy in neurodegeneration. Filters were applied to prioritize peer-reviewed articles published between 2000 and 2024, with an emphasis on studies from the last decade. Studies were selected according to predefined inclusion and exclusion criteria. Included articles consisted of peer-reviewed primary research and reviews that discussed enzyme function, genetic variants (especially involving BChE), and their physiological or pathological roles in the central nervous system. Additional selection criteria focused on therapeutic implications, including the effectiveness of enzyme-targeting drugs in treating Alzheimer’s disease, Parkinson’s disease, and mood disorders. Articles were excluded if they were not published in English, lacked experimental or clinical data, or focused only on peripheral cholinesterase activity unrelated to brain function. Editorials, opinion pieces, and conference abstracts without sufficient methodology or results were also excluded. All selected articles were reviewed in full, and relevant data were extracted to identify major themes and findings. These included the molecular functions of AChE, BChE, and MAO; the clinical relevance of known BChE mutations such as the K variant, atypical forms, and silent alleles; compensatory interactions between enzymes; and therapeutic outcomes related to drugs such as donepezil and MAO inhibitors. Special attention was given to gene–environment interactions, modulation of enzyme activity, and the potential of multi target therapeutic strategies. This review did not involve original experimental work or research involving human or animal subjects. All data were obtained from publicly available, peer-reviewed sources, and full citations were provided to ensure academic transparency and reproducibility.
Keywords: Neurotransmitter regulation; Acetylcholinesterase (AChE); Butyrylcholinesterase (BChE); Monoamine oxidase (MAO); BChE-K variant; Cholinesterase polymorphisms; Donepezil response; Brain disorders; Multi-target therapy; Gene-environment interaction; AChE Mutations; BuChE Mutations; Enzyme Polymorphisms.
Introduction
Neurotransmitter regulation is a cornerstone of cognitive function, emotional stability, and neurological health. Precise modulation of neurotransmitter levels relies on the activity of enzymes that control their synthesis, release, and degradation. Among these, acetylcholinesterase (AChE), butyrylcholinesterase (BuChE), and monoamine oxidase (MAO) are central to maintaining neuronal communication and function. Dysregulation of these enzymes disrupts neurotransmitter balance and has been associated with neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease, as well as psychiatric disorders like depression. Understanding how these enzymes function in both healthy and pathological states is essential for uncovering the molecular mechanisms underlying these conditions.
AChE is a critical enzyme responsible for hydrolyzing acetylcholine, terminating cholinergic signaling at synapses. This precise termination ensures that neurotransmitter levels do not overwhelm receptors, maintaining normal synaptic function1. BuChE, a related enzyme, provides a backup mechanism for acetylcholine hydrolysis, becoming increasingly active in the later stages of Alzheimer’s disease when AChE levels decline.
The functional overlap and interdependence of AChE, BuChE, and MAO reveal a complex regulatory network1. Disruptions in this network have far-reaching implications for synaptic activity, oxidative balance, and neuronal survival. Advances in pharmacology have demonstrated the potential of AChE and BuChE inhibitors in improving cognitive outcomes in Alzheimer’s disease, while MAO inhibitors have shown promise in mitigating oxidative stress and improving motor symptoms in Parkinson’s disease2’3’1. Despite these successes, there remain critical gaps in understanding how these enzymes interact within larger neurological systems.
Recent findings emphasize the need to explore the interconnected roles of AChE, BuChE, and MAO in regulating neurotransmitter balance under healthy and pathological conditions1’4. Their collective influence on oxidative stress, synaptic function, and disease progression underscores the importance of developing more targeted and effective treatments. Insights into these mechanisms may pave the way for innovative therapeutic approaches that address the underlying molecular dysfunctions of neurodegenerative and psychiatric disorders3.
This research fits into the current understanding of AChE, BuChE, and MAO by taking a more holistic approach to their roles in neurotransmitter regulation across a variety of neurodegenerative diseases1’3). While previous studies have mainly focused on Alzheimer’s and Parkinson’s disease, this review explores how these enzymes interact and contribute to additional conditions, including Huntington’s disease and depression1’4. The review delves deeper into the molecular mechanisms behind these enzymes, particularly how they work together to influence neurotransmitter balance1’4. This broader perspective could help identify new therapeutic strategies, especially those targeting both AChE and BuChE simultaneously, offering more precise treatment options5. By connecting the roles of these enzymes and their impact on neurodegenerative diseases, this research contributes valuable insights into potential approaches for improving treatment of these debilitating disorders1’4.
Materials and Methods
A comprehensive literature review was conducted using the PubMed and Scopus databases, covering publications from January 2000 to June 2025. Search terms included acetylcholinesterase, butyrylcholinesterase, monoamine oxidase, neurodegenerative diseases, oxidative stress, and enzyme inhibitors. Titles and abstracts were screened for relevance to enzymatic mechanisms, physiological and pathological functions, and therapeutic interventions. Studies published prior to 2000 were included only if cited within otherwise eligible articles. Following full-text review, 26 studies met inclusion criteria.
These included three experimental studies: one examining organophosphate-induced cholinesterase inhibition in murine models6, another investigating drug responses across butyrylcholinesterase genotypes7, and a third evaluating novel butyrylcholinesterase inhibitors8.
Four correlation studies explored associations between enzymatic activity and clinical or biomarker outcomes. Remaining studies provided mechanistic, structural, or therapeutic insights into enzyme behavior.
Data extracted from each study included sample size, model type, intervention or exposure, primary enzymatic and behavioral outcomes, and any reported biomarker or clinical trial findings. Due to variability in study design, populations, and outcome measures, a structured narrative synthesis approach was applied rather than a formal meta-analysis. Results were categorized by enzyme-specific physiological roles, the influence of genetic and biochemical markers on disease progression, and emerging strategies for combination therapy development.
Enzymes
Introduction to Acetylcholinesterase
Acetylcholinesterase (AChE), a member of the carboxylesterase family (EC 3.1.1.7), is a serine protease critical for maintaining acetylcholine levels in the nervous system. AChE catalyzes the hydrolysis of acetylcholine into choline and acetic acid, ensuring proper neurotransmission at neuromuscular junctions and cholinergic synapses (Figure 1)9. This rapid reaction, occurring within microseconds, prevents the accumulation of acetylcholine in the synaptic cleft, thereby maintaining the precision of synaptic signaling. AChE is an α/β protein with a 12-stranded central β-sheet surrounded by 14 α-helices, featuring a deep, narrow gorge lined with 14 conserved aromatic residues that lead to the active site (Figure 2)10.
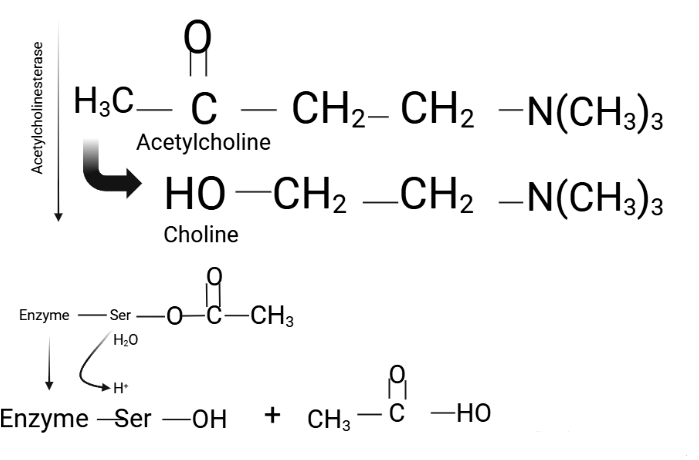

The enzyme features two active subsites: an anionic subsite, which binds acetylcholine, and an esteratic subsite, which facilitates hydrolysis. During neurotransmission, acetylcholine is released into the synaptic cleft and interacts with postsynaptic acetylcholine receptors to propagate signals responsible for muscle contraction9. Simultaneously, AChE hydrolyzes acetylcholine, terminating the signal and preventing excessive stimulation. The resulting choline and acetic acid are recycled to replenish neurotransmitter reserves9.
Normal Physiology of AChE
According to the International Union of Biochemistry and Molecular Biology (IUBMB), acetylcholinesterase (AChE) is classified as a hydrolase enzyme that acts on ester bonds, specifically targeting esters of carboxylic acids. While its primary role is the breakdown of acetylcholine in cholinergic synapses, AChE also exhibits a range of non-classical functions essential to neural development and maintenance. It plays a critical role in forming neuromuscular junctions, guiding thalamocortical projections, and supporting axonal growth during the development of the nervous system. Additionally, AChE contributes to neurogenesis by influencing cell structure and participates in neuroplastic changes within the neocortex. Due to its structural similarity to cell adhesion molecules, AChE is also believed to assist in cellular adhesion. Experimental studies have shown that increased expression of AChE correlates with stronger substrate adhesion, and this adhesion can be inhibited by anti-AChE antibodies or specific inhibitors such as BW284c51. Similar patterns have been observed in fibroblasts and astrocytes, further supporting its adhesive function. Moreover, AChE is involved in the regulation of apoptosis. Cells with higher AChE levels are more prone to undergo programmed cell death, while suppression of AChE activity has been shown to delay or prevent apoptosis. This effect is linked to AChE’s interactions with molecules like caveolin and cytochrome c, facilitating apoptosome assembly. Notably, silencing the AChE gene interferes with caspase-9 activation and disrupts the interaction between cytochrome c and apoptotic protease activating factor-1, ultimately impeding the apoptotic cascade11.
Post-Translational Modifications (PTMs) of AChE
After translation, acetylcholinesterase (AChE) undergoes a series of post-translational modifications (PTMs) that are essential for proper folding, assembly, and targeting to the synapse. One key PTM is N-linked glycosylation, which occurs in the endoplasmic reticulum and ensures correct protein folding and progression through the secretory pathway. Improper glycosylation causes retention and degradation in the ER, whereas properly modified AChE becomes resistant to endoglycosidase H and is efficiently trafficked to the plasma membrane12.
Disulfide bond formation is equally critical, as it stabilizes the tertiary structure and enables AChE to form dimers and tetramers—the functional multimeric forms required for synaptic efficacy. AChE tetramers form through interactions between a tryptophan amphiphilic tetramerization (WAT) domain and proline-rich attachment domains (PRADs) on anchoring proteins. In the nervous system, AChE anchors to PRiMA to form tetrameric membrane-bound G4 structures; in muscle, it binds to ColQ, generating asymmetric A12 complexes localized in the basal lamina12’13.
The four-helix bundle (FHB) domain also plays a key role in guiding subunit specificity during oligomerization. It promotes selective AChE-AChE interactions while minimizing cross-assembly with butyrylcholinesterase (BChE). Although hybrid AChE/BChE oligomers have been observed under engineered conditions, AChE normally assembles into homomeric structures in vivo12.
Pathogenesis Involving AChE
Acetylcholinesterase (AChE) plays an important role in the pathogenesis of neurodegenerative diseases by influencing inflammation, apoptosis, oxidative stress, and the aggregation of pathological proteins. Neurodegenerative disorders are often linked to cholinergic dysfunction because acetylcholine, the main neurotransmitter in this system, is broken down by AChE. For this reason, AChE inhibitors or muscarinic and nicotinic receptor agonists are frequently used in treatment14. Beyond breaking down acetylcholine, AChE also affects cell adhesion, neural development, synapse formation, and programmed cell death, which are altered in both neurodegenerative and depressive disorders14.
In Alzheimer’s disease, overactivity of AChE lowers acetylcholine levels and leads to degeneration of the cholinergic system. Although AChE inhibitors can relieve symptoms, they do not stop disease progression14. AChE also contributes to the buildup of β‑amyloid (Aβ) plaques. Enzyme activity is found within amyloid cores, diffuse pre‑amyloid deposits, and cerebral blood vessels, and forms of AChE in Alzheimer’s disease respond differently to inhibitors such as indoleamine and bacitracin14.
Building on this, therapeutic research has shifted toward enhancing cholinergic tone more effectively. Moss highlights that current short‑acting inhibitors such as donepezil, rivastigmine, and galantamine achieve only modest central inhibition (about 25 to 35 percent) because of dose‑limiting gastrointestinal toxicity, which falls short of the roughly 50 percent threshold believed necessary for stronger neuroprotection. Moss also describes how irreversible AChE inhibitors like methanesulfonyl fluoride (MSF) form stable covalent complexes and exploit the slower turnover of central AChE compared to peripheral tissues. This pharmacokinetic difference allows for higher and more selective brain inhibition, reaching up to 65 to 80 percent in preclinical and early clinical studies without the peripheral side effects seen with reversible inhibitors14.
Alongside synthetic approaches, naturally occurring polyphenolic compounds—particularly flavonoids such as quercetin, apigenin, kaempferol, and naringenin—have emerged as promising AChE inhibitors with multifaceted neuroprotective actions15. These compounds not only inhibit AChE activity by binding to its active and peripheral anionic sites but also mitigate oxidative stress, reduce neuroinflammation, and disrupt Aβ aggregation. For example, quercetin exhibits high binding affinity to AChE’s catalytic site (Ser200 and His440), activates the Nrf2/ARE antioxidant pathway, and suppresses pro‑inflammatory cytokines like TNF‑α and IL‑6, collectively improving cognitive function in animal models of Alzheimer’s disease15. Similarly, apigenin and kaempferol demonstrate dual cholinergic and anti‑amyloidogenic effects, remodeling toxic Aβ oligomers into non‑toxic aggregates while enhancing brain‑derived neurotrophic factor (BDNF) expression to support synaptic plasticity15. Naringenin, another citrus‑derived flavonoid, increases synaptic acetylcholine levels while preserving mitochondrial integrity and reducing calcium dysregulation, underscoring its potential to address multiple pathological hallmarks simultaneously15.
Effects of Organophosphates in AChE inhibition
However, AChE can be inactivated by organophosphate (OP) compounds, which form covalent bonds with the serine residue at the enzyme’s active site16. This inhibition disrupts the enzyme’s function, leading to sustained muscle contraction and, in severe cases, paralysis. There are also neurotoxins such as Soman and sarin that inhibit acetylcholinesterase by binding to its active center, but Soman causes much faster aging, leaving the enzyme irreversibly inactivated17.
The reactions hold significant importance for both medicine and national security, as OP agents that block AChE act as acute neurotoxins. Recent occurrences highlight this issue: Iraq deployed tabun against Iranian forces during the Iran–Iraq war, terrorists have utilized sarin against the civilian population, authoritarian leaders have employed sarin to carry out mass killings of their own citizens, and a dictator sanctioned the execution of his own brother using VX18. OP compounds are also used in agriculture as pesticides, in industry as flame retardants, and in chemical warfare as nerve agents. When exposed to high doses or chronic levels of OPs, they can be extremely toxic or even lethal19. This is primarily due to the inhibition of acetylcholinesterase, which causes a buildup of acetylcholine and leads to severe consequences such as cholinergic crises and prolonged seizures, which can be fatal if not controlled19. Furthermore, even if death is avoided, improper seizure management may lead to long-term brain damage19. In addition to acute exposure, chronic, low-dose exposure to OPs can also result in brain damage, but through mechanisms such as calcium dyshomeostasis and oxidative stress, which occur independently of acetylcholinesterase inhibition and seizures19.
Study of Acetylcholinesterase by effects of physical aging
Inhibition of AChE activity by commonly used insecticides—such as organophosphates (OPs) and carbamates—as well as certain nerve agents, leads to the accumulation of acetylcholine, resulting in significant physiological alterations and neurotoxicity20. The acute toxicity of OP nerve agents and pesticides is based on the irreversible inhibition of the serine esterase acetylcholinesterase21. A study by Suarez-Lopez et al. measured AChE activity from a single finger-stick blood sample collected from 746 participants aged 4 to 26 years across 3,100 observations who resided in an agricultural county in Ecuador. The aim of the study was to measure the acetylcholinesterase levels of citizens who have been in contact with farmers20.
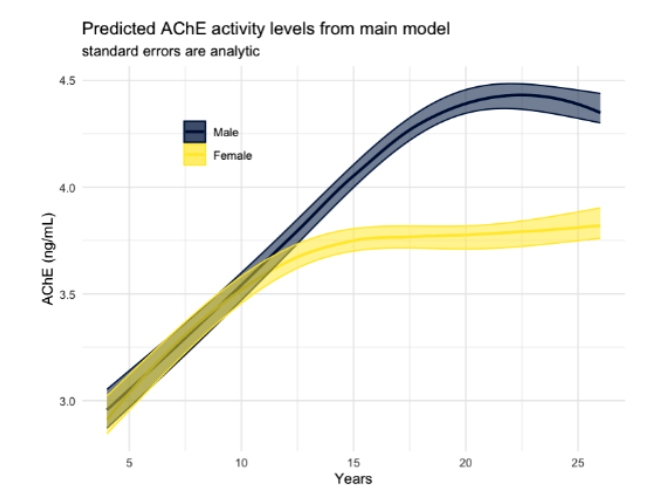
The study concluded that acetylcholinesterase (AChE) activity levels increase nonlinearly with age, with significant differences between males and females. Males typically show higher AChE activity, though these levels tend to appear later in life compared to females. A linear increase in AChE activity was observed between the ages of 5 and 12 or 18 years, after which further aging did not result in substantial changes. The primary factor influencing variability in AChE activity levels was aging, suggesting that developmental factors, rather than other external variables, play a significant role in the observed trends. These findings underline the importance of considering age-related changes in AChE activity, particularly in populations exposed to agricultural chemicals, during the design of relevant therapeutics when they are needed.
While the findings provide insight into normative developmental trends in AChE activity, they remain correlational rather than causal. The study did not directly link increased AChE activity to specific neurological outcomes or disease states, limiting its ability to confirm whether altered enzyme levels contribute to the etiology of neurodegenerative conditions such as Alzheimer’s disease. Additionally, other potential confounding variables, such as genetic polymorphisms, nutritional status, inflammation, or concurrent exposure to other neurotoxic agents, were not controlled or explored. These omissions leave open the possibility that observed AChE differences reflect broader systemic factors rather than targeted enzyme dysregulation.
Involvement of Acetylcholinesterase in Seizure Initiation via the Basolateral Amygdala After Organophosphate Exposure
Organophosphates (OPs) primarily target acetylcholinesterase and inhibit it via binding to its active site, forming a covalent bond with the enzyme’s critical serine residue22. This inhibition leads to the accumulation of acetylcholine, resulting in overstimulation of both nicotinic and muscarinic receptors22. The excessive activation of nicotinic receptors at the neuromuscular junction can cause muscle twitching, jerking, and eventually flaccid paralysis due to a depolarizing block22. Additionally, the overstimulation of muscarinic receptors can induce severe symptoms, including bradycardia, bronchospasm, and respiratory failure, highlighting the life-threatening consequences of OP exposure22. The persistent presence of acetylcholine in the central nervous system can lead to CNS depression, coma, and seizures22. These toxic effects are compounded by the presence of solvents and surfactants in many OP formulations, which can increase the overall toxicity and contribute to complications like aspiration pneumonitis and adult respiratory distress syndrome22.
The effects of organophosphates (OPs), such as nerve agents, on seizure initiation in the brain have been a subject of extensive research. McDonough and colleagues demonstrated that convulsions could be induced by microinjections of nerve agents into the basolateral nucleus of the rat amygdala, but not into other brain regions such as the hippocampus or piriform cortex23. This finding highlighted the unique role of the amygdala in the onset of seizures following OP exposure. Further studies revealed that inhibition of acetylcholinesterase (AChE) in the basolateral amygdala was crucial for the induction of status epilepticus (SE), a condition characterized by prolonged or repeated seizures without recovery of consciousness between episodes, often leading to brain damage or death if untreated19. Rats injected with soman, an OP nerve agent, but not exhibiting SE showed no significant AChE inhibition in the amygdala, while rats that developed SE exhibited marked AChE inhibition in this region23 (Figure 5). Additionally, rapid increases in extracellular glutamate in the amygdala after soman exposure were observed, occurring before similar increases in the hippocampus23. These findings suggest that the amygdala plays a pivotal role in seizure initiation, and highlight the importance of AChE inhibition in the development of OP-induced seizures.

Although the data suggest a correlation between local AChE inhibition and seizure activity, they do not fully establish a singular causal pathway. The role of the amygdala in seizure propagation is well-supported, but further studies are needed to clarify whether AChE inhibition is sufficient on its own to induce SE or whether it acts as one trigger within a multifactorial cascade. Future investigations should include multiregional brain analyses, real-time neurochemical monitoring, and assessment of additional neurotransmitter systems to better contextualize AChE’s contribution to seizure disorders following toxic exposures.
AChE Isoforms and Impacts
| AChE-S (Synaptic/Tailed) Variant | A high-activity tetramer anchored in neurons and muscles. It’s essential for breaking down acetylcholine at synapses. Reduced in Alzheimer’s and Parkinson’s, and increased in stress, where it may worsen neuronal damage. Also promotes amyloid-β aggregation24. |
| AChE-R (Read-Through) Variant | A low-activity soluble monomer upregulated by stress. It is neuroprotective, counteracts Aβ toxicity, and may help detoxify organophosphates. Regulated by miR-132, making it a target in Alzheimer’s and toxic exposure25. |
| AChE-E/H (Erythrocytic/Hydrophobic) Variant | A moderate-activity dimer found on red blood cells and placenta. Less involved in neurotransmission but may help regulate systemic acetylcholine. Its role in disease is less defined26. |
| N-Extended AChE Variants (e.g., N-AChE-S) | Isoforms with extended N-terminals, often linked to cell stress or cancer. May affect apoptosis and tumor growth. Still under investigation for clinical use27. |
Introduction to Butyrylcholinesterase
Butyrylcholinesterase (EC 3.1.1.8; BChE), also known as cholinesterase, pseudocholinesterase, or plasma cholinesterase, is a sister enzyme of acetylcholinesterase28\(V. Whittaker, 2010). BChE is essential for catalyzing the hydrolysis of butyrylcholine, breaking it down into choline and butyrate29. BChE is a tetrameric, 342 kDa glycoprotein synthesized in the liver, released into the bloodstream, and found in the brain, heart, muscles, lungs, small intestine, and adipose tissue1. BChE and acetylcholinesterase share a similar three-dimensional structure and are believed to have evolved from an ancestral AChE gene duplication29. Due to this, the structure of AChE and BChE are similar (Figure 6).
While AChE plays a critical role in the hydrolysis of acetylcholine at the neuromuscular junction, the roles of BChE remain less defined, though it is involved in hydrolyzing succinylcholine and bambuterol, which are used as muscle relaxants in anesthesiology29 (Figure 7).
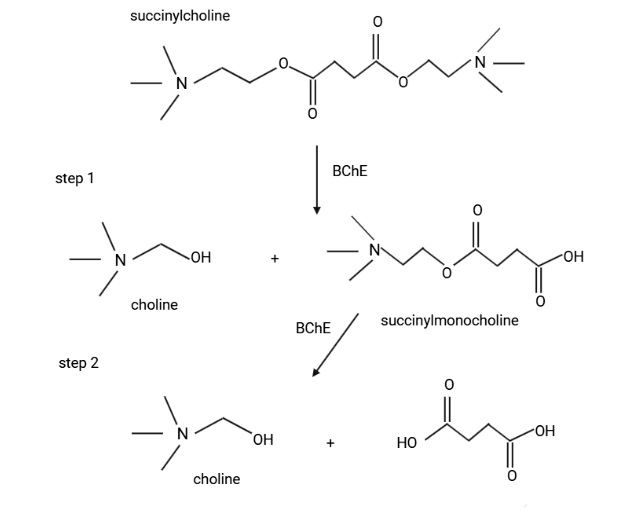
Normal Physiology and pathology of BuChE
Butyrylcholinesterase (BChE, EC 3.1.1.8), also known as serum cholinesterase, is primarily synthesized in the liver and plays an essential role in regulating biological metabolism12. Approximately one‑third of BChE circulates in the serum, reflecting its systemic presence and involvement in physiological processes beyond hepatic synthesis. Because its levels fluctuate in response to various insults, BChE serves as a sensitive biomarker: changes in BChE activity can indicate the occurrence of disease and thus provide an important measure of overall health status. Numerous studies have linked BChE activity to a spectrum of pathologies. In the liver, its activity is associated with acute hepatitis, chronic liver disease, liver cancer, and cirrhosis while alterations in BChE have also been observed in diabetes, cardiovascular disease, trait anxiety, Parkinson’s disease, and Alzheimer’s disease. Indeed, the level of BChE in living organisms is closely tied to disease occurrence. Using the fluorescent probe LAN‑bche, Xu and colleagues confirmed that BChE expression is downregulated in liver cancer and liver injury models, likely reflecting impaired hepatic capacity to generate BChE due to cell damage and malignant transformation, whereas BChE expression is upregulated in diabetes.
Beyond chronic diseases, BChE has gained prominence as a prognostic marker in acute inflammatory conditions such as COVID‑19. A large‑scale study of 462 PCR‑confirmed COVID‑19 patients demonstrated that BChE activity declines significantly with increasing disease severity, particularly in critically ill individuals, and closely correlates with 30‑day mortality30. Notably, patients with severe respiratory symptoms (pneumonia, dyspnea) exhibited markedly reduced BChE levels, whereas those with neurological symptoms (anosmia, ageusia, headache) showed comparatively higher activity, highlighting a symptom‑specific pattern of cholinergic dysregulation. BChE levels also decreased with age and were lower among vaccinated individuals, suggesting complex interactions between immune activation, systemic inflammation, and cholinergic pathways during viral infection. These findings extend earlier observations of BChE’s prognostic value in sepsis and cardiovascular disease, reinforcing its potential as a multifaceted biomarker bridging neurodegenerative, metabolic, and acute infectious pathologies.
Beyond its role as a biomarker, BChE has emerging functions in lipid metabolism that extend beyond classic cholinergic pathways. Muslum Gok, Cigdem Cicek, and Ebru Bodur demonstrated that BChE can hydrolyze lipogenic substrates such as ghrelin, 4‑methylumbelliferyl (4‑mu) palmitate, and arachidonoylcholine, suggesting it serves as a back‑up enzyme for lipolysis when traditional lipases are less effective. This activity is particularly relevant in tissues with active lipid turnover—liver, plasma, intestine, and adipose tissue—where BChE levels are abundant. Moreover, polyunsaturated fatty acids like linoleic and alpha‑linolenic acids not only regulate BChE activity but also increase its expression in hepatic cells, hinting at a feedback loop between lipid mediators and cholinesterase activity. These findings propose a novel link between BChE and lipid homeostasis, with implications for metabolic disorders and neurodegenerative diseases where altered lipid metabolism and elevated BChE levels are observed31.
Recent clinical studies further highlight BChE’s utility as a predictive biomarker in surgical settings. In a prospective single‑center study of 402 colorectal surgery patients, low BChE levels on the first and third postoperative days were independently associated with a two‑ to threefold increased risk of developing surgical site infections (SSIs), even after adjusting for confounders such as operative approach and comorbidities32. Notably, patients with SSI exhibited significantly lower mean BChE levels (3.90 KU/L vs. 4.54 KU/L) by the third postoperative day compared to uncomplicated cases, and ROC analysis demonstrated high predictive accuracy (AUC 0.981)33. These results suggest that early postoperative BChE monitoring could serve as a cost‑effective adjunct to standard infection surveillance, enabling earlier intervention in high‑risk patients.
Butyrylcholinesterase mutations
BuChE does not have multiple functional isoforms in the way that AChE does (e.g., AChE‑S, AChE‑R, AChE‑E) through alternative splicing. However, BuChE does exhibit significant polymorphic variation at the genetic level, which leads to differences in enzyme activity, stability, and clinical behavior.
It is important to know that mutations of BuChE can alter its enzymatic activity. A study conducted by Zhu and colleagues focused on classifying pseudocholinesterase (BChE) deficiency based on genetic variants and their clinical implications for succinylcholine metabolism34. The researchers identified various genotypes associated with BChE activity, ranging from normal to severe deficiency. They determined that individuals with no genetic variants showed normal enzyme activity and typical responses to succinylcholine. Those with mild BChE deficiency carried one copy of the K variant, resulting in slightly reduced enzyme activity. Moderate BChE deficiency was associated with one copy of variants A, F1, F2, or S1, or two copies of the K variant. These individuals may experience rare but significant neuromuscular blockade prolongation after succinylcholine use. Severe BChE deficiency, identified in homozygotes of A, F1, F2, or S1, or those with two or more of these variants, predicted prolonged neuromuscular blockade and required careful management. The study also analyzed the frequency of BChE genotypes across different ethnic groups in the United States, highlighting the diversity in genetic distribution34.
| Mutation | Effect on Enzymatic Activity |
| Atypical (Dibucaine‐Resistant) Variant | A point mutation alters the enzyme’s active site so that it becomes resistant to inhibition by dibucaine. Homozygous carriers have a very low “dibucaine number” (typically around 20–30 compared to ~80 in normal individuals). This leads to significantly reduced BChE activity, resulting in prolonged hydrolysis times for drugs such as succinylcholine. Consequently, patients may experience extended neuromuscular blockade and apnea during or after anesthesia. |
| K Variant | A common mutation (often an A539T substitution) that causes moderately reduced enzyme activity. Carriers (whether heterozygous or homozygous) may show a somewhat prolonged duration of action for muscle relaxants like succinylcholine. The effect is usually less dramatic than that seen with the atypical variant, but under stressful conditions (e.g., during surgery), there may still be an increased risk for extended paralysis. |
| Silent (Null) Variant | Mutations that result in little to no production of functional BChE protein. Individuals with a “silent” allele (or those who are homozygous for such mutations) have nearly undetectable BChE activity. This places them at very high risk for severe, prolonged paralysis and apnea following the administration of ester drugs (such as succinylcholine) and may complicate anesthesia management. |
| Homozygous BChE Deficiency (Compound Heterozygotes) | Inheriting two defective alleles (for example, one atypical and one silent variant, or two copies of a severely dysfunctional allele) leads to markedly low overall enzyme activity. Severe BChE deficiency dramatically increases the risk of adverse reactions to drugs that rely on BChE for metabolism (e.g., prolonged neuromuscular blockade, respiratory depression). Anesthesiologists must be aware of such deficiencies to avoid complications during surgery. |
| Fluoride-Resistant Variant (Less Common) | A rarer mutation that alters the enzyme’s sensitivity to fluoride inhibition. Although the full clinical implications are not as well defined as for the atypical or silent variants, this variant may affect the metabolism of certain anesthetic agents. Altered drug interactions could necessitate dosage adjustments during anesthesia, with potential implications for patient safety. |
Genetic variants of BChE affect its enzymatic activity and drug metabolism, with the wild-type protein functioning normally while atypical variants such as Asp70Gly and Asp70His reduce activity. Fluoride-resistant variants including Thr243Met, Gly390Val, and Leu330Ile alter inhibitor sensitivity, and the H variant (Val142Met) demonstrates significantly diminished function35. Moderate reductions in activity occur in the New York (Ala184Val), K (Ala539Thr), and J (Glu497Val) variants, while neutral mutations such as Glu255Asp, Gly75Arg, and Ala162Ala have little effect. Silent mutations like Ile4 deletion, Gly117fs, and Ser198Gly can severely impair or eliminate activity35. Some variants, including Thr250Pro and Trp471Arg, retain minimal function, while high-activity variants such as Cynthiana, Johannesburg, and C5+ enhance enzymatic performance, influencing drug response and toxicity risk35.
Sokolow and colleagues investigated the effect of the butyrylcholinesterase (BChE) K variant on treatment outcomes in individuals aged 55 to 90 diagnosed with mild cognitive impairment (MCI), a condition marked by primary memory impairment with relative preservation of other cognitive domains7. Participants met diagnostic criteria for amnestic MCI of degenerative etiology, defined by impaired memory performance with a Logical Memory delayed-recall score 1.5 to 2 standard deviations below education-adjusted norms, a Clinical Dementia Rating (CDR) of 0.5 (indicating very mild dementia), and a Mini-Mental State Examination (MMSE) score of 24 to 30, signifying impairment without full dementia. The treatment group received donepezil, beginning at 5 mg daily and increasing to 10 mg after six weeks, while the control group comprised individuals from placebo and 2000 IU vitamin E arms, as vitamin E demonstrated no therapeutic benefit7.
The study assessed cognitive function using the MMSE and CDR-SB, with lower MMSE scores and higher CDR-SB scores indicating greater impairment. Genotyping was performed on DNA extracted from blood, analyzed using the Illumina 610Quad array, with quality control via PLINK. The impact of the BChE-K polymorphism (rs1803274) on cognition was examined using linear regression models, adjusting for age, gender, and APOE4 status. Mixed models analyzed interactions between treatment, genotype, and therapy duration. Kaplan-Meier curves estimated Alzheimer’s disease progression, while Cox-proportional hazard models and z-tests compared survival rates between treatment and control groups over time.
The study found that approximately 33% of participants carried the rs1803274 K-variant of BChE, with similar frequencies in the control (31%) and treatment (35%) groups. No significant differences in baseline cognitive function were observed between these groups. In the control group, the BChE-K variant did not significantly affect MMSE decline or CDR-SB increase over 36 months. However, among those treated with donepezil, the K-variant was associated with a greater cognitive decline. Specifically, homozygous K-carriers experienced a more pronounced drop in MMSE scores (−7.2 ± 3.4) compared to heterozygous carriers (−2.2 ± 0.5) and non-carriers (−0.9 ± 0.4; P = 0.0004)(Figure 8). A similar pattern was observed in CDR-SB scores, with homozygous K-carriers showing a greater increase (4.1 ± 1.9) compared to 2full cohort, including placebo, vitamin E, and donepezil groups, no overall association between BChE-K genotype and cognitive decline was observed, suggesting that the impact of the genotype was specific to the donepezil-treated group.

Further analysis revealed a significant interaction between treatment duration and BChE-K genotype in the donepezil group, with K-homozygous individuals experiencing the fastest decline in MMSE (P < 0.0038) and the most substantial rise in CDR-SB (P < 0.0017). Compared to untreated controls, K-homozygous participants treated with donepezil exhibited significantly greater cognitive decline (P = 0.0096 for MMSE, P = 0.008 for CDR-SB), but this difference was no longer apparent at the 36-month follow-up. Stratification by APOE4 status showed that in APOE4 carriers receiving donepezil, BChE-K homozygosity was significantly associated with an accelerated cognitive decline (P = 0.003 for MMSE, P < 0.0001 for CDR-SB). Similar associations were observed when comparing treated and untreated APOE4 carriers (P = 0.011 for MMSE, P = 0.0007 for CDR-SB). However, no significant genotype-related differences were found among APOE4 non-carriers. Adjustments for baseline MMSE and CDR-SB scores and the exclusion of the vitamin E control group did not alter these findings.
BChE-K polymorphisms are associated with accelerated cognitive decline in patients with mild cognitive impairment (MCI) undergoing three years of donepezil therapy. This finding suggests that BChE-K genotyping should be performed to identify subsets of patients at higher risk of adverse outcomes during cholinesterase inhibitor treatment, particularly those who also carry the APOE4 allele. In such cases, prescribing donepezil off-label for MCI management should be avoided due to the potential for worsened clinical outcomes7.
Butyrylcholinesterase Inhibitors
In the search for effective treatments for Alzheimer’s disease (AD), the current approach focuses on developing drugs that target multiple pathways simultaneously. One strategy is to inhibit both acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), two enzymes implicated in AD. AChE plays a critical role in cognitive functions, particularly memory, and its inhibition has been a standard treatment approach for AD36. Recent research has shifted some focus to BChE, as its inhibition may provide additional therapeutic benefits and can produce fewer side effects than AChE inhibition36.
Thiazole, a class of compounds known for their biological activity, has emerged as a potential source for novel BChE inhibitors. Thiazole derivatives, including talipiprazole and riluzole, have shown diverse biological activities, such as cholinesterase inhibition36. In efforts to identify more effective BChE inhibitors, Fazal Rahim and colleagues designed and synthesized new thiazole derivatives (Figure 9). These compounds were synthesized using inexpensive, widely available reagents under mild conditions, making the process efficient and scalable36. Activity testing revealed that most compounds displayed stronger inhibition of BChE than AChE, with IC₅₀ values ranging from 1.59 μM to 389.25 μM. Among these, compound 17 stood out with the highest selectivity for BChE, demonstrating a selectivity index of 232.9, suggesting its potential as a promising candidate for further development36.

Selective butyrylcholinesterase (BChE) inhibitors, studied by Greig et al., including cymserine, phenyl ethyl carbamate (PEC), and butyl norcymserine (BNC), demonstrate high potency and specificity in targeting BChE over acetylcholinesterase (AChE)37.
PEC exhibited exceptional selectivity, showing over 5,000-fold preference for human BChE, indicating its potential therapeutic value. These compounds also demonstrated excellent brain penetration, with cymserine achieving a 40:1 brain-to-plasma concentration ratio. Notably, PEC significantly elevated acetylcholine (ACh) levels in the parietal cortex, increasing them by up to 290% in a dose-dependent manner without inhibiting AChE. This cholinergic enhancement was accompanied by improved long-term potentiation (LTP) in hippocampal slices and enhanced cognitive performance in aged rats, demonstrated by reduced errors and faster maze completion times. Beyond cholinergic effects, PEC reduced levels of amyloid precursor protein (APP) and amyloid-beta peptides (Aβ40 and Aβ42) in human neuroblastoma cells and Alzheimer’s disease mouse models, with reductions of up to 54% for Aβ40 and 47% for Aβ4237.
Introduction to Mono-Amine Oxidase (MAO) type A and B
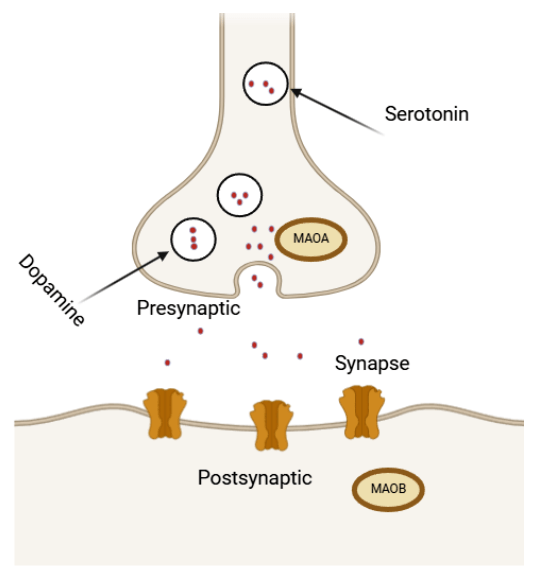
Monoamine oxidase (MAO) is an essential flavin enzyme that catalyzes the oxidation of monoamine neurotransmitters in the brain, such as dopamine and serotonin. This process is vital for maintaining neurotransmitter balance, but it also generates toxic by-products, including aldehydes and hydrogen peroxide, which can induce oxidative stress and neuronal cell death38. There are two types of Monoamine-Oxidase isoforms, MAO-A and MAO-B. Monoamine oxidase (MAO) is a critical enzyme that catalyzes the oxidative deamination of biogenic amines, including neurotransmitters like serotonin (5-HT), norepinephrine (NE), dopamine (DA), and the neuromodulator phenylethylamine (PEA)39. Two forms of MAO, MAO A and MAO B, have been identified based on their biochemical properties and gene structures.

MAO A exhibits a higher affinity for serotonin and NE and is inhibited by clorgyline, whereas MAO B prefers PEA and benzylamine and is inhibited by deprenyl. These enzymes are central to neurological research due to their role in neurotransmitter metabolism, with studies linking MAO B activity to psychiatric conditions such as bipolar disorder, suicidal behavior, and poor impulse control. Smoking has been shown to inhibit both MAO A and MAO B activity, which renders it a risk factor towards psychiatric disorders. The genes for MAOA and MAOB are located on human Xp11.2-11.4, alongside the Norrie disease (ND) gene, and their deletion can cause congenital blindness through neuroretinal degeneration39.
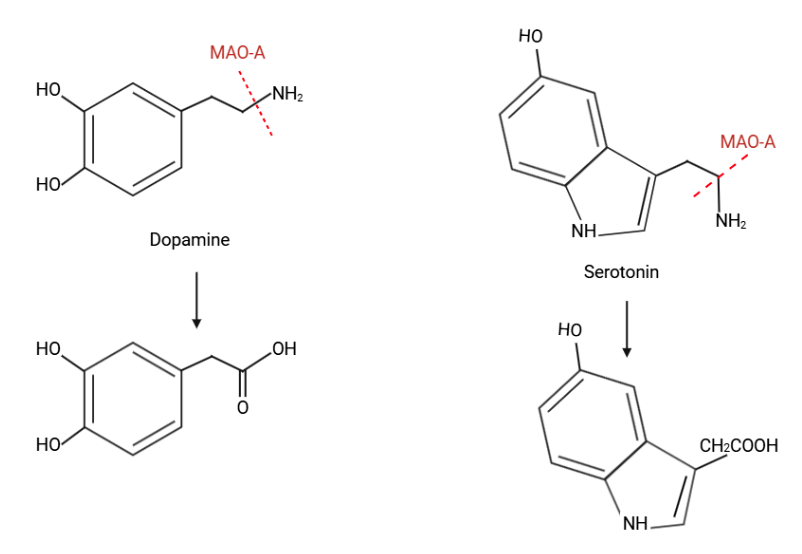
The MAO-A is the key enzyme for the degradation of serotonin and catecholamines which has been associated with a higher risk for antisocial behavior (ASB) and violence, particularly in males with a history of child maltreatment40. Chemical degradation of these organic compounds can be found in Figure 10.
The MAO-B isoform, in particular, has been implicated in the pathology of neurodegenerative diseases like Parkinson’s disease (PD) and Alzheimer’s disease (AD), as its overexpression is associated with insufficient dopamine concentrations, a hallmark of these conditions38. Targeting MAO-B through selective inhibition has emerged as a promising strategy for mitigating neurodegeneration by reducing oxidative damage and preserving dopaminergic function.
Natural products have become valuable in the search for effective MAO-B inhibitors, as they provide diverse and structurally unique scaffolds. Compounds like chromone, coumarin, chalcone, caffeine, and aurone have shown potential in drug discovery due to their ability to inhibit MAO-B activity selectively. Despite significant research efforts, only a few reversible and selective MAO-B inhibitors have been approved for clinical use, highlighting the need for continued exploration of these natural scaffolds. This approach holds promise for developing innovative treatments that leverage the therapeutic potential of nature while addressing the limitations of current drug development strategies38.
The genetic evidence for MAOs’ role in human behavior is exemplified by Brunner et al.’s study of a Dutch family, where a C→T mutation in the MAOA gene led to a null allele, resulting in impulsive aggression and borderline mental retardation in eight males. Behavioral traits associated with MAOA and MAOB polymorphisms include aggression, substance abuse, and bipolar disorder. Research in MAO A–deficient mice reveals that high central nervous system (CNS) levels of monoamine neurotransmitters promote aggressive behavior, which can be mitigated by drugs like ketanserin and tetrabenazine in a dose-dependent manner. Moreover, untreated wild-type males exhibit less aggression compared to MAO A-knockout mice, emphasizing MAO A’s significant role in modulating aggression39.
Isoforms and Polymorphisms of MAO-A
Monoamine oxidase A (MAO A) is unique among neuroenzymes in that it is expressed as a single protein with no alternative splicing isoforms identified in humans. Functional diversity in MAO A activity therefore arises almost entirely from genetic polymorphisms and epigenetic regulation rather than isoform heterogeneity. This characteristic makes MAO A an ideal system for studying how specific variants in the gene directly influence enzymatic activity, monoamine metabolism, and behavioral outcomes.
Several promoter and coding polymorphisms in MAO A have been extensively characterized. These variants can alter transcriptional activity, mRNA processing, or catalytic efficiency, leading to measurable differences in serotonin, dopamine, and norepinephrine catabolism. Their clinical significance spans a range of psychiatric and neurodevelopmental disorders including impulsivity, aggression, anxiety, and depression, and they are frequently studied in the context of interactions between genes and the environment such as early life stress or trauma.
Epigenetic factors add another layer of complexity. Promoter methylation can dynamically suppress MAO A transcription independent of genotype, and altered methylation patterns have been observed in conditions like PTSD and major depressive disorder. This integration of polymorphic and epigenetic regulation provides a nuanced framework for understanding how MAO A influences behavior and psychiatric vulnerability.
Table 3 below summarizes the principal polymorphisms of MAO A, detailing their genomic location, effect on enzymatic activity, and known associations with behavioral or clinical outcomes.
| Polymorphism | Effect on Enzymatic Activity and Clinical Implications |
| MAOA-uVNTR (Variable Number Tandem Repeat) | A variable number of 30 base-pair repeats in the MAOA promoter region affects gene transcription levels. The 3.5 and 4 repeat alleles cause high MAO-A expression, while the 2, 3, and 5 repeat alleles lead to low expression. High-expression variants are associated with increased breakdown of monoamines (serotonin, norepinephrine, dopamine), influencing mood and behavior. Low-expression variants have been linked to impulsivity and aggression, especially when combined with environmental stressors. |
| rs6323 (T941G) SNP | A synonymous single nucleotide polymorphism in exon 8, changing a thymine (T) to guanine (G), which does not alter the amino acid but may influence enzyme folding or mRNA translation efficiency. The G allele has been correlated with altered MAO-A activity and has associations with psychiatric conditions such as depression and anxiety. |
| rs3027399 (Intronic variant) | Located in an intronic region of MAOA, this polymorphism may affect mRNA splicing or stability, though exact mechanisms are unclear. It has been studied for links to autism spectrum disorder and mood regulation, but causality remains uncertain. |
| rs1137070 (Exonic synonymous variant) | A silent mutation in the coding region which does not change the amino acid sequence but may affect mRNA processing. Some studies suggest associations with aggression, impulsivity, and behavioral phenotypes in specific populations, though findings are inconsistent. |
| MAOA-LPR (Linked to VNTR) | A combination of promoter polymorphisms including the VNTR that together modulate transcriptional activity more precisely. These haplotypes have been used to better predict behavioral risk factors, particularly in studies on antisocial behavior and response to environmental stress. |
| Epigenetic Modification: Promoter Methylation | Hypermethylation of the MAOA promoter reduces gene expression and consequently MAO-A enzyme levels. Altered methylation patterns have been observed in psychiatric disorders like PTSD, anxiety, and depression, suggesting an epigenetic regulation layer affecting MAO-A activity beyond DNA sequence variants. |
Isoforms and Polymorphisms of MAO-B
Monoamine oxidase B (MAO‑B), like MAO‑A, is expressed as a single protein in humans with no known alternative splicing isoforms. Functional variation in MAO‑B activity is therefore driven primarily by single nucleotide polymorphisms (SNPs) and epigenetic modifications rather than isoform heterogeneity. These polymorphisms influence dopamine and phenylethylamine metabolism and have been implicated in neurodegenerative conditions such as Parkinson’s and Alzheimer’s disease, as well as in mood and behavioral disorders.
Several MAO‑B variants are located in regulatory regions such as introns, promoters, and the 3′ untranslated region (3′ UTR), where they may affect transcription factor binding, mRNA stability, or splicing efficiency. While some variants, like rs1799836 in intron 13, show strong associations with enzyme activity and dopamine metabolism, others, including promoter or 3′ UTR variants, have more subtle or uncertain clinical effects. Epigenetic modifications, particularly promoter methylation, further modulate MAO‑B activity and are increasingly recognized for their role in neuropsychiatric and neurodegenerative disease risk.
Table 4 summarizes key MAO‑B polymorphisms, highlighting their genomic locations, functional effects, and relevance to brain disorders and behavioral phenotypes41’42’43.
| Polymorphism | Effect on Enzymatic Activity and Clinical Implications |
| rs1799836 (Intron 13, A/G) | A common SNP influencing enzyme activity; the G allele is linked with increased MAO-B activity, affecting dopamine metabolism and neuropsychiatric traits. |
| rs6651806 (Promoter region) | Potentially modifies gene expression by affecting transcription factor binding, though clinical implications remain unclear. |
| rs9140 (3’ UTR) | May impact mRNA stability and regulation; associations with neurodegenerative disease risk are under study but not definitive. |
| Exon 14 coding variants | Rare mutations causing amino acid changes; possible minor effects on enzyme function but limited data. |
| Epigenetic regulation (methylation) | Changes in promoter methylation can modulate MAO-B expression, linked to altered enzyme levels in brain diseases such as Parkinson’s and Alzheimer’s. |
Normal Physiology and Catalytic Mechanism
Monoamine oxidases A and B are flavin-dependent enzymes located on the outer membrane of mitochondria. They are responsible for the oxidative deamination of dietary amines and monoamine neurotransmitters, thereby helping to regulate synaptic neurotransmitter levels44. During this reaction, amines are converted into aldehydes, with ammonia and hydrogen peroxide released as byproducts. These aldehydes are then further processed by enzymes such as aldehyde dehydrogenase or aldehyde reductase44.
The efficient turnover of neurotransmitters like serotonin, norepinephrine, and dopamine is essential for maintaining emotional stability, motor coordination, sensory processing, and cognitive function44. MAO A has a higher affinity for serotonin and norepinephrine, while MAO B preferentially acts on phenylethylamine; both contribute to dopamine metabolism, though the dominant isoform involved differs by species and brain region—MAO A being more active in rodents and MAO B in humans and other primates44.
Structurally, MAO A and B share about 70% sequence identity, each containing a conserved FAD-binding motif and being anchored to the mitochondrial membrane through their C-terminal domain44. Their distribution within the brain further defines their roles: MAO A is primarily found in catecholaminergic neurons, whereas MAO B is located in serotonergic and histaminergic neurons as well as in astrocytes44. These biochemical and anatomical distinctions enable precise regulation of neurotransmitter levels across different brain regions.
Pathogenesis and Oxidative Stress
Although MAO is essential for neurotransmitter degradation, its enzymatic activity produces byproducts that may be harmful to neurons. These include hydrogen peroxide, ammonia, and various aldehydes, all of which possess neurotoxic potential44. Hydrogen peroxide, in particular, can promote the formation of reactive oxygen species, which in turn can damage mitochondria and induce neuronal cell death. This oxidative stress is believed to play a role in the development of neurodegenerative diseases such as Parkinson’s disease44.
When MAO activity is excessive or poorly regulated, it may lead to pathological conditions through the accumulation of reactive oxygen species and aldehyde toxicity. Findings from genetic studies further support this: mice lacking MAO exhibit notable changes in behavior and neural function, demonstrating the importance of MAO in maintaining neurochemical balance. These models not only underscore the enzyme’s physiological significance but also point to potential therapeutic strategies targeting MAO function44.
Role of Monoamine Oxidase-A in Aggression and Violence
Aggression, a multifaceted behavior aimed at causing harm, can serve adaptive purposes such as enhancing social status and defensive reactions45. However, pathological aggression often leads to criminal activities and interpersonal dysfunction, with reactive and proactive forms presenting distinct neural correlates45. Reactive aggression, characterized by impulsive responses to provocation, is linked to a hypoactive prefrontal cortex (PFC) and hyperactive limbic regions like the amygdala45. In contrast, proactive aggression, which involves premeditated actions for personal gain, correlates with increased grey matter density in the dorsomedial PFC and altered functional connectivity between the left precuneus and PFC40. Notably, low activity of the MAO‑A gene, which regulates serotonin degradation (Figure 11), has been strongly associated with aggression, particularly in males who experience early-life adversity40. This gene–environment interaction underscores the complex neurobiological underpinnings of aggressive behavior, as demonstrated in both human studies and animal models40. MAO‑A’s function affects the epinephrine/norepinephrine system, which facilitates fight‑or‑flight reactions and autonomic nervous system activity46. The MAO‑A gene, located on the X chromosome, is also known as the warrior gene, since abnormal versions of the gene often result in aggressive behaviors46.
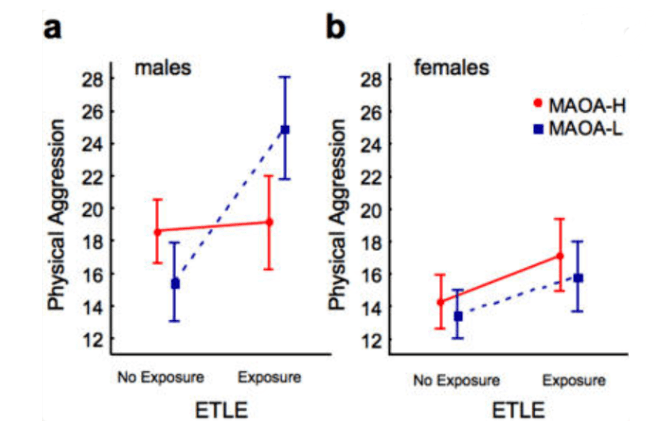
Frazzetto and colleagues conducted a study to investigate the interaction between MAOA genotypes, early traumatic life events (ETLE), gender, and physical aggression using an ANOVA model. The study found that 43% of males carried low‑activity MAOA alleles, while 57% had high‑activity alleles, and female genotypes were grouped as low activity for heterozygotes. Notably, 34% of participants reported at least one ETLE before age 15, with the most common events being marital problems (26.16%), family violence (13.50%), and parental alcohol addiction (9.28%). Gender (F = 36.77, P < 0.0001) and ETLE (F = 27.50, P < 0.0001) significantly impacted aggression levels, with males and ETLE‑exposed individuals showing higher scores. A significant gene–environment interaction (F = 8.20, P = 0.005) revealed that males with low‑activity MAO‑A alleles and ETLE exposure had the highest aggression scores (mean = 25.69). In contrast, females showed no significant differences in aggression across genotypes and ETLE exposure. The study concluded that low‑activity MAO‑A alleles amplify the effects of ETLE on aggression, particularly in males, explaining 6.6% of the variance47.
Genetic Variances in Monoamine Oxidase and its Impact
Genetic variations in the Monoamine Oxidase A (MAOA) gene, particularly in the uVNTR polymorphism, are associated with differing levels of enzymatic activity and have been implicated in aggressive and impulsive behaviors. According to Stetler et al., low-activity MAOA variants (2- and 3-repeat alleles) were significantly more common among violent offenders (61.22%) compared to non-violent offenders (20%), suggesting a robust link between these alleles and violent behavior48. This association was especially significant in the Caucasian subgroup (P<0.01), while a marginal difference was observed in African-American convicts (P=0.08). Furthermore, individuals with low-activity MAOA genotypes exhibited higher impulsivity, particularly in BIS-11 total and attentional-impulsiveness scores (P<0.05), although no significant interaction was found between genotype and ethnicity or violent crime status. These findings suggest that MAOA genetic polymorphisms may contribute to behavioral dysregulation, particularly in populations with environmental or psychosocial stressors48. Result graphs are seen in Figure 13.

In-depth review of Monoamine Oxidase B in Alzheimer’s disease
Monoamine oxidase B (MAO-B) is an enzyme primarily located in cerebral glial cells, platelets, and hepatic cells, where it plays a crucial role in neurotransmitter metabolism. Within the blood-brain barrier (BBB), MAO-B acts as a metabolic barricade, breaking down exogenous amines and preserving neuronal function. This enzymatic activity is essential for maintaining neural homeostasis, but alterations in MAO-B expression have been linked to neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). In AD, immunostaining studies have shown that MAO-B activity is significantly elevated in the cortical and hippocampal regions, areas crucial for memory and cognition. This increase in MAO-B expression is associated with oxidative stress and neurotransmitter impairment, particularly affecting the adrenergic and cholinergic systems, both of which play a vital role in cognitive function. Additionally, MAO-B preferentially metabolizes benzylamine and 2-phenylethylamine, further influencing neurotransmitter levels. Its activation has been shown to negatively impact cholinergic transmission, which is essential for processes related to memory and emotion. The overexpression of MAO-B in AD brains is thought to contribute to neurodegeneration through enhanced oxidative stress and gliosis, leading to progressive cognitive decline. Given its significant role in neurotransmitter regulation and neurodegenerative processes, targeting MAO-B has become a critical area of research for developing therapeutic interventions in AD and other neuropsychiatric disorders.
Study of Major Depressive Episodes and Monoamine Oxidase B
Monoamine‑oxidase activity in the brain correlates with the length of major depressive episodes. Moriguchi et al. conducted a case‑control study from April 2014 to August 2018 to explore brain chemistry differences in people with major depressive episodes (MDEs) compared to healthy controls. The study involved 20 patients with unmedicated MDEs from Toronto in a specialized psychiatric hospital and 20 healthy age‑matched controls. Participants were rigorously screened to rule out other psychiatric or neurological disorders, and all refrained from alcohol, grapefruit juice, and caffeine on the day of the scan. Using advanced imaging techniques, including a high‑resolution 3‑dimensional PET scanner combined with MRI, the researchers measured the total distribution volume (VT) of monoamine oxidase B (MAO‑B), an enzyme that helps break down brain chemicals and plays a key role in mood regulation S. Moriguchi et al., 2019. They found that patients with MDEs had, on average, a 26% higher MAO‑B VT in the prefrontal cortex compared to healthy controls (Figure 14)—a robust difference that persisted even after excluding an outlier value S. Moriguchi et al., 2019. Moreover, this elevated enzyme activity was not uniform across the brain; it was especially pronounced in regions such as the ventrolateral, dorsolateral, and orbitofrontal prefrontal cortices, the thalamus, and the inferior parietal cortex S. Moriguchi et al., 2019. Importantly, among the depressed patients, a longer duration of illness was strongly linked to even higher MAO‑B levels, suggesting that chronic depression may be associated with progressive changes in brain chemistry S. Moriguchi et al., 2019. Graph of results are shown in figure 14.
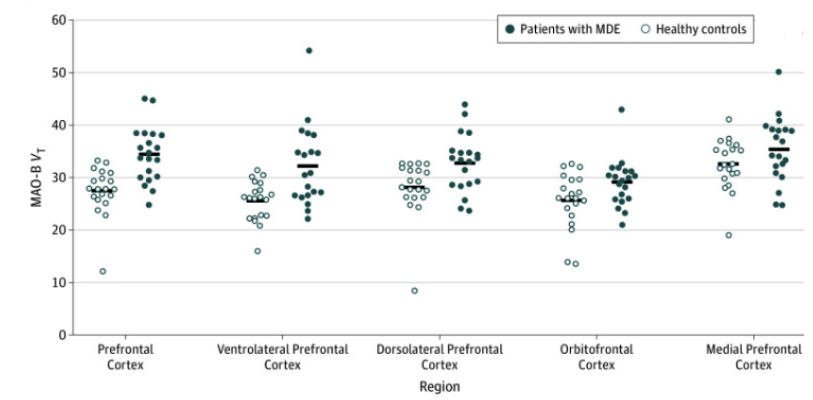
A similar pattern arose when other regions of the brain were measured (Figure 15). Figure 15 illustrates significantly elevated monoamine oxidase B (MAO-B) total distribution volume (VT) in individuals with major depressive episodes (MDEs) compared to healthy controls across key brain regions, analyzed using analysis of variance (ANOVA). The F value is the ratio of the two different estimates of variance. The prefrontal cortex showed the most pronounced increase (F(1,38) = 19.6, P < .001), highlighting its role in mood regulation and executive function impairments. The thalamus (F(1,38) = 8.8, P = .005) and inferior parietal cortex (F(1,38) = 9.0, P = .005) also exhibited significantly higher MAO-B VT, suggesting disruptions in sensory processing, attention, and cognitive function. These findings reinforce the hypothesis that increased MAO-B activity is a neurochemical hallmark of depression, potentially serving as a biomarker for diagnosis and treatment monitoring. Given MAO-B’s role in breaking down neurotransmitters like dopamine and serotonin, these results provide a foundation for exploring MAO-B inhibitors as a therapeutic strategy to restore neurochemical balance in depression.

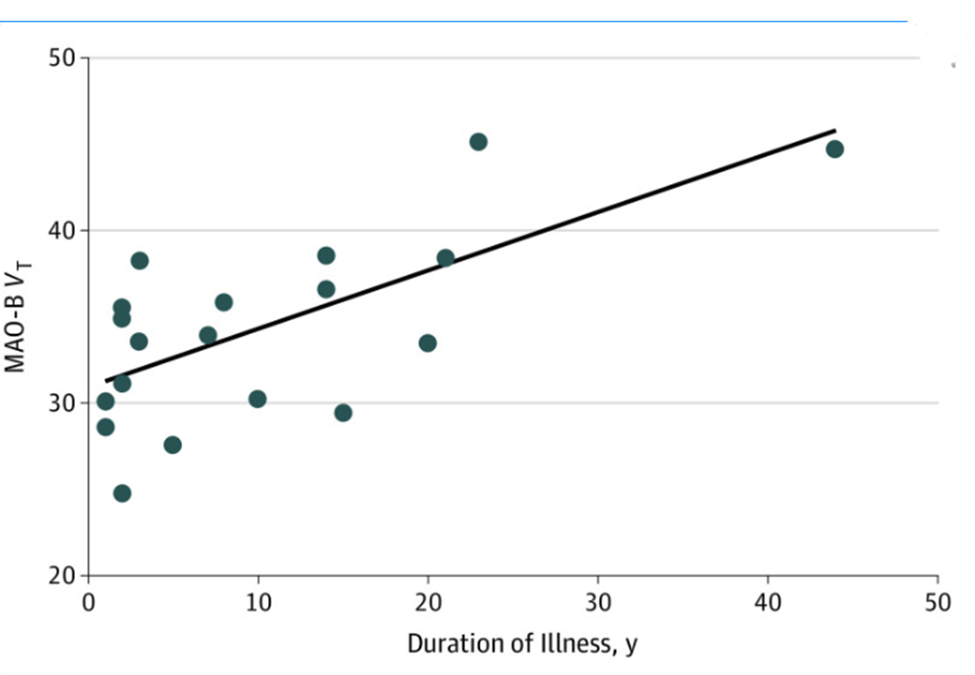
In individuals with MDEs, a longer illness duration correlated with higher MAO-B VT across multiple brain regions. A repeated-measures ANCOVA revealed a significant overall association between illness duration and MAO-B VT (F(1,18) = 18.1, P < .001), along with a notable interaction between duration and specific brain regions (F(7,12) = 9.1, P = .02). Increased MAO-B VT was particularly evident in the ventrolateral and dorsolateral prefrontal cortices, orbitofrontal cortex, anterior cingulate cortex (ACC), thalamus, inferior parietal cortex, temporal cortex, and occipital cortex, suggesting a widespread impact of prolonged depression on brain chemistry. There is a correlation between MAO-B VT and Duration of Illness with r = 0.65 (Figure 16).
MAOs Inhibitors
Monoamine oxidase inhibitors (MAOIs) are a class of drugs that block the activity of monoamine oxidase (MAO), an enzyme responsible for breaking down neurotransmitters like serotonin, dopamine, and norepinephrine. These inhibitors are divided into two main types: irreversible and reversible. Irreversible MAOIs, such as phenelzine, tranylcypromine, and isocarboxazid, permanently deactivate the enzyme, meaning the body must create new MAO enzymes for normal function to return. Some, like selegiline and rasagiline, specifically target MAO‑B, which is involved in dopamine breakdown and is commonly treated in Parkinson’s disease. In contrast, reversible inhibitors (RIMAs) like moclobemide and toloxatone temporarily block MAO‑A, allowing neurotransmitter levels to increase without permanently affecting enzyme function. Because reversible inhibitors do not completely stop MAO activity, they have fewer side effects and dietary restrictions compared to irreversible MAOIs. Additionally, safinamide acts as a selective MAO‑B inhibitor with additional effects on dopamine regulation. Understanding these inhibitors is essential in treating psychiatric and neurological disorders, as they help manage conditions such as depression and Parkinson’s disease by increasing neurotransmitter availability in the brain.
Irreversible MAO inhibitors such as phenelzine, tranylcypromine, and isocarboxazid raise serotonin, norepinephrine, and dopamine levels for extended periods but require strict dietary restrictions and carry significant risk of hypertensive crises, which limits patient adherence and complicates trial enrollment39. Reversible MAO‑A inhibitors including moclobemide and toloxatone allow rapid restoration of MAO‑A activity and have a better safety profile, yet clinical evidence for sustained benefit in major depressive episodes is limited and focused primarily on short‑term symptom relief49. Selective MAO‑B inhibitors such as selegiline, rasagiline, and safinamide have provided transient improvements in motor function for Parkinson’s disease but do not slow neurodegeneration, and natural scaffolds like38. Although genetic variants and PET‑measured MAO‑B total distribution volume clearly influence drug response and disease progression, no major trial has yet stratified participants by MAO genotype or incorporated PET and CSF biomarkers of enzyme inhibition to confirm target engagement39. Future development of MAO‑targeted therapies must focus on reversible, isoform‑selective compounds, integrate validated PET and CSF biomarkers to demonstrate durable enzyme inhibition, and apply genotype‑guided trial designs in order to translate biochemical modulation into meaningful clinical outcomes.
The Interplay between Enzymes in Alzheimer’s Disease
In Alzheimer’s disease, the enzymes acetylcholinesterase (AChE), butyrylcholinesterase (BuChE), and monoamine oxidase B (MAO-B) operate within a complex and mutually reinforcing biochemical network that contributes significantly to synaptic dysfunction, oxidative stress, and neurodegeneration. AChE is primarily responsible for the rapid hydrolysis of acetylcholine (ACh) at synaptic junctions, a process essential for maintaining cholinergic tone and ensuring the timely termination of neurotransmission50. In the early stages of Alzheimer’s disease, degeneration of basal forebrain cholinergic neurons results in a marked decline in AChE levels, impairing the breakdown of acetylcholine and disrupting cholinergic signaling51.
In regions where AChE expression diminishes, BuChE expression is often found to increase, particularly within astrocytes and glial cells surrounding amyloid-β plaques52’53. Although BuChE can hydrolyze acetylcholine, its lower substrate specificity and reduced regulatory control result in erratic neurotransmitter clearance and further impair synaptic precision54’55. The upregulation of BuChE may initially appear compensatory, but its altered kinetics exacerbate cholinergic imbalance and may contribute to plaque-associated neurotoxicity52,56.
Simultaneously, MAO-B catalyzes the oxidative deamination of monoamines such as dopamine and phenylethylamine, producing hydrogen peroxide and aldehydes as byproducts57’58. These reactive species increase oxidative stress, damage mitochondrial membranes, and suppress AChE gene transcription through redox-sensitive pathways18. Hydrogen peroxide can also directly modify AChE’s structure, reducing its enzymatic efficiency and destabilizing cholinergic homeostasis18.
The neuroinflammatory milieu of Alzheimer’s disease further modulates this enzymatic network. Proinflammatory cytokines such as interleukin-1β and tumor necrosis factor-α, released in response to amyloid deposition, have been shown to downregulate AChE transcription while enhancing BuChE activity59’60. These changes are further reinforced by epigenetic modifications such as DNA methylation and histone deacetylation at the ACHE gene locus, contributing to long-term suppression of AChE expression61.
Together, the decline in AChE, the compensatory upregulation of BuChE, and the elevation of MAO-B form a pathological triad that drives progressive neurochemical imbalance. This triad facilitates neurotransmitter dysregulation, impairs synaptic signaling, and promotes oxidative and inflammatory damage in the Alzheimer’s brain. As such, it represents a compelling therapeutic target. Interventions that address this network collectively—rather than focusing on a single enzyme—may provide more durable clinical benefits. Multi-target strategies that combine cholinesterase and MAO-B inhibition, especially those based on selective or natural scaffolds, have the potential to restore synaptic integrity and attenuate disease progression56’57.

The Interplay between Enzymes in Parkinson’s Disease
Parkinson’s disease (PD) emerges from a progressive depletion of dopamine-producing neurons in the substantia nigra, disrupting the equilibrium between dopaminergic and cholinergic signaling within the basal ganglia. This neurochemical imbalance contributes directly to hallmark motor symptoms such as rigidity, tremor, and bradykinesia, and implicates multiple enzymatic systems in its pathogenesis.
Monoamine oxidase B (MAO-B), expressed primarily in glial cells, plays a central role in the oxidative metabolism of dopamine. Its catalytic activity generates hydrogen peroxide and other reactive oxygen species as byproducts, which exacerbate oxidative stress and potentiate neuronal injury. Increased MAO-B expression in the brains of PD patients further accelerates dopaminergic loss, creating a self-reinforcing loop of degeneration. Pharmacologically, selective MAO-B inhibitors such as selegiline, rasagiline, and safinamide are used to preserve dopamine levels and mitigate oxidative damage, offering symptomatic relief and potential neuroprotection62.
On the cholinergic side, acetylcholinesterase (AChE) is responsible for terminating synaptic acetylcholine signaling by hydrolyzing the neurotransmitter in both central and peripheral synapses. In PD, the decline in dopamine levels disinhibits cholinergic interneurons in the striatum, leading to excess acetylcholine activity. While AChE remains active, especially in early stages, cognitive decline in later disease may reflect progressive loss of cholinergic neurons and subsequent reduction in AChE expression. Enhancing acetylcholine through AChE inhibition, particularly in Parkinson’s disease dementia, has shown modest cognitive benefit.
Butyrylcholinesterase (BuChE), which becomes increasingly expressed in glial cells during cholinergic system failure, acts as a secondary acetylcholine regulator. Its enzymatic activity typically rises as AChE declines, but it lacks the same precision in synaptic clearance. Elevated BuChE levels have been associated with both cognitive impairment and disease severity in PD patients, suggesting its potential as a biomarker for diagnosis and progression monitoring63. Dual inhibitors like rivastigmine, which target both AChE and BuChE, have demonstrated improved efficacy in treating PD-related cognitive symptoms compared to AChE-selective drugs alone.
Taken together, these enzymes—MAO-B, AChE, and BuChE—form an interdependent network whose dysregulation fuels both motor and cognitive deterioration in Parkinson’s disease. Targeting this enzymatic triad through selective or combined inhibition strategies remains a cornerstone of current therapeutic efforts and a promising avenue for future drug development. This interaction can be seen in Figure 18.
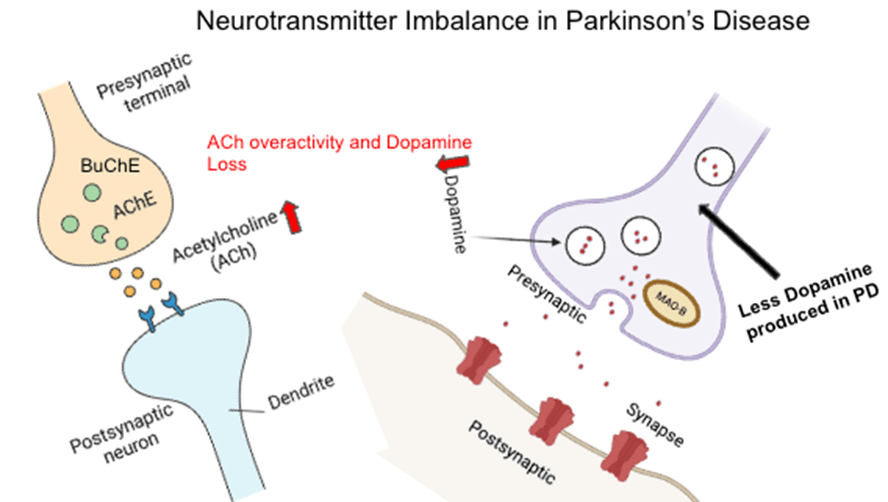
This diagram shows how dopamine loss and acetylcholine overactivity contribute to motor symptoms in Parkinson’s disease. Reduced dopamine release and MAO-B activity are shown on the right, while increased acetylcholine signaling with AChE and BuChE activity is shown on the left. The imbalance between these two systems underlies key features of the disease and informs targeted treatments.
Discussion
The findings from this review highlight the critical interplay between acetylcholinesterase (AChE), butyrylcholinesterase (BuChE), and monoamine oxidase (MAO) in maintaining neurotransmitter balance and their collective impact on neurological health. AChE’s role in terminating acetylcholine signaling is essential for synaptic precision, while BuChE compensates during pathological states like late-stage Alzheimer’s disease. MAO’s regulation of monoamines, though vital, can lead to oxidative stress when dysregulated, exacerbating neurodegeneration.
Although donepezil, rivastigmine, and galantamine yield modest 2–4-point improvements on the ADAS-Cog over 24 weeks, their symptomatic benefit plateaus after 12–18 months and fails to slow neurodegeneration, while gastrointestinal adverse events affect up to 30 % of patients and cause 10–15 % to discontinue therapy. Tacrine’s transient cognitive relief was overshadowed by hepatotoxicity in roughly one-third of users, underscoring the narrow safety window of active-site blockade. These clinical outcomes expose a clear therapeutic gap that reversible, competitive inhibitors cannot fill. Non-competitive allosteric or peripheral-site modulators (e.g., an investigational compound engaging the peripheral anionic site) hold promise for reducing on-target toxicity, but no candidate has advanced beyond early-phase trials. Compounding this, past studies have lacked robust target-engagement biomarkers—such as CSF AChE activity assays and amyloid-PET or synaptic-density tracers—to confirm durable enzyme inhibition and correlate it with clinical outcomes64. Future research must therefore pivot toward non-competitive binding modes and integrate biomarker-guided trial designs to move AChE targeting from transient symptom relief to genuine disease modification.
Although butyrylcholinesterase (BChE) is a sensitive systemic biomarker—its serum activity fluctuating with liver injury, diabetes, and neurodegeneration65—efforts to target BChE therapeutically remain at the preclinical stage. Thiazole-based inhibitors synthesized by Rahim et al. showed IC₅₀ values from 1.59 to 389.25 μM, with compound 17 achieving a selectivity index of 232.9, yet no pharmacokinetic or safety data in humans have been reported66. Similarly, carbamate-derived molecules such as cymserine, PEC, and BNC exhibit >5,000-fold specificity for BChE, excellent brain penetration, dose-dependent acetylcholine elevation, enhanced long-term potentiation, cognitive improvement in aged rats, and up to 54 % reductions in Aβ40/42 in cell and mouse models, but none have advanced to clinical trials67. Compounding these translational gaps, common BChE polymorphisms—including the K, atypical, and silent variants—profoundly affect enzyme activity and drug metabolism: for example, Sokolow et al. demonstrated that BChE-K homozygotes treated with donepezil experience accelerated cognitive decline, indicating the necessity of genotype-guided therapy7. To bridge the gap between bench and bedside, future development must prioritize early-phase human studies that integrate validated BChE activity biomarkers, stratify participants by BChE genotype, and assess longitudinal cognitive outcomes.
Irreversible MAO inhibitors such as phenelzine, tranylcypromine, and isocarboxazid raise serotonin, norepinephrine, and dopamine levels for extended periods but require strict dietary restrictions and carry significant risk of hypertensive crises, which limits patient adherence and complicates trial enrollment39. Reversible MAO-A inhibitors including moclobemide and toloxatone allow rapid restoration of MAO-A activity and have a better safety profile, yet clinical evidence for sustained benefit in major depressive episodes is limited and focused primarily on short-term symptom relief49. Selective MAO-B inhibitors such as selegiline, rasagiline, and safinamide have provided transient improvements in motor function for Parkinson’s disease but do not slow neurodegeneration, and natural scaffolds like chromone, coumarin, chalcone, caffeine, and aurone remain at the preclinical stage despite showing promising in vitro MAO-B selectivity68. Although genetic variants and PET-measured MAO-B total distribution volume clearly influence drug response and disease progression, no major trial has yet stratified participants by MAO genotype or incorporated PET and CSF biomarkers of enzyme inhibition to confirm target engagement69’49. Future development of MAO-targeted therapies must focus on reversible, isoform-selective compounds, integrate validated PET and CSF biomarkers to demonstrate durable enzyme inhibition, and apply genotype-guided trial designs in order to translate biochemical modulation into meaningful clinical outcomes.
Conclusions
This review underscores the importance of AChE, BuChE, and MAO in neurotransmitter regulation and their involvement in neurodegenerative and psychiatric disorders. The works of Dhull et al. elucidated AChE’s critical role in synaptic precision, while Zhou and Huang highlighted the therapeutic potential of targeting BuChE for Alzheimer’s disease. Lv et al. contributed valuable insights into MAO’s link to oxidative stress and its implications for conditions like Parkinson’s and Alzheimer’s disease.
In Alzheimer’s disease, acetylcholinesterase (AChE), butyrylcholinesterase (BuChE), and monoamine oxidase B (MAO-B) intersect through their combined effects on neurotransmitter breakdown, oxidative stress, and synaptic dysfunction. AChE is responsible for degrading acetylcholine (ACh) in the synapse, and its early decline contributes to cognitive deficits. As the disease progresses, BuChE activity increases—particularly in the hippocampus and glial cells—further depleting ACh levels and intensifying cholinergic dysfunction. MAO-B, which breaks down monoamines like dopamine, is overexpressed in Alzheimer’s-affected brain regions such as the cortex and hippocampus. This enzymatic activity generates hydrogen peroxide and aldehydes, promoting oxidative stress and neuronal damage. While AChE inhibitors such as donepezil offer early symptom relief, they lose effectiveness over time; BuChE-selective or dual inhibitors like rivastigmine may be more beneficial in later stages. MAO-B inhibitors have shown potential to reduce oxidative damage, but current treatments remain limited to symptomatic management. The shared roles of these enzymes in neurotransmitter metabolism and neurodegeneration suggest that multi-target strategies addressing AChE, BuChE, and MAO-B simultaneously could offer improved outcomes by restoring chemical balance and mitigating neuronal loss.
While current therapeutic strategies have shown success in managing symptoms, they often fail to address the interconnected effects of these enzymes. Advancing research into multi-target approaches, including selective inhibitors and natural product-based therapies, may lead to innovative treatments. By focusing on the shared roles of these enzymes in oxidative stress and neurotransmitter balance, future therapies have the potential to improve outcomes for patients with neurodegenerative and psychiatric illnesses.
References
- İ. Yapici, E. İzol, Effects of acetylcholinesterase and butyrylcholinesterase enzymes on human memory. 2023. [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- S. C. Godar, et al., The role of monoamine oxidase A in aggression: current translational developments and future challenges. Prog Neuropsychopharmacol Biol Psychiatry. 69, 90–100 (2016). [↩]
- D. Garcia-Arocena, The genetics of violent behavior. https://www.jax.org/news-and-insights/jax-blog/2015/december/the-genetics-of-violent-behavior (2015). [↩] [↩] [↩]
- D. Garcia-Arocena, The genetics of violent behavior. https://www.jax.org/news-and-insights/jax-blog/2015/december/the-genetics-of-violent-behavior (2015).) [↩] [↩] [↩] [↩]
- S. Brimijoin, V. P. Chen, Y. P. Pang, L. Geng, Y. Gao, Physiological roles for butyrylcholinesterase: a BChE-ghrelin axis. Chem Biol Interact. 259, 271–275 (2016). [↩]
- E. L. Robb, M. B. Baker, Organophosphate toxicity. https://www.ncbi.nlm.nih.gov/books/NBK470430/ (2019). [↩]
- S. Sokolow, X. Li, L. Chen, K. D. Taylor, J. I. Rotter, R. A. Rissman, P. S. Aisen, L. G. Apostolova. Deleterious effect of butyrylcholinesterase K‑variant in donepezil treatment of mild cognitive impairment. Journal of Alzheimer’s Disease 56, 229‑237 (2017). [↩] [↩] [↩] [↩] [↩] [↩]
- S. Zhou, G. Huang, The biological activities of butyrylcholinesterase inhibitors. Biomedicine & Pharmacotherapy. https://www.sciencedirect.com/science/article/pii/S0753332221013433 (2021). [↩]
- V. Dhull, A. Gahlaut, N. Dilbaghi, V. Hooda, Acetylcholinesterase biosensors for electrochemical detection of organophosphorus compounds: a review. Biochem Res Int. 2013, 731501 (2013). [↩] [↩] [↩]
- H. Dvir, A. Silman, J. L. Sussman, et al., Acetylcholinesterase: from 3D structure to function. Chem Biol Interact. 187, 10–22 (2010). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2894301/, https://doi.org/10.1016/j.cbi.2010.01.042. [↩]
- Ł. J. Walczak-Nowicka, M. Herbet, Acetylcholinesterase inhibitors in the treatment of neurodegenerative diseases and the role of acetylcholinesterase in their pathogenesis. Int. J. Mol. Sci. 22, 9290 (2021). [↩]
- V. P. Chen, W. K. W. Luk, W. K. B. Chan, K. W. Leung, A. J. Y. Guo, G. K. L. Chan, S. L. Xu, R. C. Y. Choi, K. W. K. Tsim. Molecular assembly and biosynthesis of acetylcholinesterase in brain and muscle: the roles of t‑peptide, FHB domain, and N‑linked glycosylation. Front Mol Neurosci. 4, 36 (2011). https://doi.org/10.3389/fnmol.2011.00036 [↩] [↩] [↩] [↩]
- M. A. Benner, L. L. Rachinsky, C. G. Bon, S. J. Camp, M. J. Massoulie. Acetylcholinesterase tetramers are stabilized by the WAT domain: relevance to PRAD binding and collagen anchoring. J Biol Chem. 273, 24502–24509 (1998). [↩]
- H. Hampel, M. M. Mesulam, A. C. Cuello, M. R. Farlow, E. Giacobini, G. T. Grossberg, A. S. Khachaturian, A. Vergallo, E. Cavedo, P. J. Snyder, Z. S. Khachaturian. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 141, 1917–1933 (2018). [↩] [↩] [↩] [↩] [↩]
- N. Cichon, W. Grabowska, L. Gorniak, M. Stela, P. Harmata, M. Ceremuga, M. Bijak. Mechanistic and therapeutic insights into flavonoid-based inhibition of acetylcholinesterase: implications for neurodegenerative diseases. Nutrients. 17, 78 (2024). [↩] [↩] [↩] [↩]
- V. Dhull, A. Gahlaut, N. Dilbaghi, V. Hooda. Acetylcholinesterase biosensors for electrochemical detection of organophosphorus compounds: a review. Biochem Res Int. 2013, 731501 (2013). [↩]
- B. Hoskins, et al. Relationship between the neurotoxicities of Soman, Sarin and Tabun, and acetylcholinesterase inhibition. Toxicology Letters. 30, 121–129 (2002). [↩]
- D. Quinn, et al. Why is aged acetylcholinesterase so difficult to reactivate? Molecules. 22, 1464 (2017). [↩] [↩] [↩]
- V. Aroniadou-Anderjaska, T. H. Figueiredo, M. de Araujo Furtado, V. I. Pidoplichko, M. F. M. Braga. Mechanisms of organophosphate toxicity and the role of acetylcholinesterase inhibition. Toxics. 11, 866 (2023). [↩] [↩] [↩] [↩] [↩] [↩]
- J. Suarez-Lopez, et al. Change in acetylcholinesterase activity from childhood to young adulthood. bioRxiv. (2024). https://doi.org/10.1101/2024.10.21.24315881. [↩] [↩]
- F. Worek, et al. On-site analysis of acetylcholinesterase and butyrylcholinesterase activity with the ChE Check Mobile test kit—determination of reference values and their relevance for diagnosis of exposure to organophosphorus compounds. Toxicology Letters. 249, 22–28 (2016). [↩]
- E. L. Robb, M. B. Baker, Organophosphate Toxicity. StatPearls Publishing, 2019. www.ncbi.nlm.nih.gov/books/NBK470430/ [↩] [↩] [↩] [↩] [↩] [↩]
- B. J. McDonough, et al., Mechanisms of Organophosphate-Induced Seizures. Toxicology, 1987 [↩] [↩] [↩] [↩]
- G. F. Chen, T. H. Xu, Y. Yan, Y. R. Zhou, Y. Jiang, K. Melcher, H. E. Xu, Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacologica Sinica. 38, 1205–1235 (2017). https://doi.org/10.1038/aps.2017.28 [↩]
- R. El Fatimy, et al., MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathologica. 136, 537–555 (2018). https://doi.org/10.1007/s00401-018-1880-5 [↩]
- D. Li, J. Wu, X. Xiong, The role of the acetylcholine system in common respiratory diseases and COVID-19. Molecules. 28(3), 1139 (2023). https://doi.org/10.3390/molecules28031139 [↩]
- X. J. Zhang, D. S. Greenberg, Acetylcholinesterase involvement in apoptosis. Front Mol Neurosci. 5, 40 (2012). https://doi.org/10.3389/fnmol.2012.00040 [↩]
- B. Johnson, S. W. Moore, Why Has Butyrylcholinesterase Been Retained? Structural and Functional Diversification in a Duplicated Gene. Neurochemistry International. 61, 783–797 (2012). https://doi.org/10.1016/j.neuint.2012.06.016 [↩]
- G. R. Sridhar, L. Gumpeny, Emerging significance of butyrylcholinesterase. World J Exp Med. 14(1), 87202 (2024). https://doi.org/10.5493/wjem.v14.i1.87202 [↩] [↩] [↩]
- T. Fazekaš, L. Kováčik, M. M. Rad, X. G. Barrios, S. M. Iftimie, J. Camps, A. Hrabovská. Serum butyrylcholinesterase activity as a predictor of severity and mortality in COVID‑19 patients. Sci Rep. 15, 23437 (2025). https://doi.org/10.1038/s41598-025-07017-2 [↩]
- M. Gok, C. Cicek, E. Bodur, et al., Journal of Neurochemistry. 168(4), 381‑385 (2023) [↩]
- G. I. Verras, F. Mulita. Butyrylcholinesterase levels correlate with surgical site infection risk and severity after colorectal surgery: a prospective single‑center study. Front Surg. 11, 1379410 (2024). https://doi.org/10.3389/fsurg.2024.1379410 [↩]
- G. I. Verras, F. Mulita, Front Surg. 11, 1379410 (2024 [↩]
- Y. Zhu, H. Li, J. Wang, L. Patel, R. S. Smith. Genetic classification of pseudocholinesterase deficiency: implications for succinylcholine metabolism and perioperative management. Clinical Genetics 104, 214‑226 (2023). [↩] [↩] [↩]
- O. Lockridge, R. B. Norgren Jr., R. C. Johnson, T. A. Blake. Naturally occurring genetic variants of human acetylcholinesterase and butyrylcholinesterase and their potential impact on the risk of toxicity from cholinesterase inhibitors. Chemical Research in Toxicology 29, 1381‑1392 (2016). [↩] [↩] [↩]
- S. Zhou, G. Huang. The biological activities of butyrylcholinesterase inhibitors. Biomedicine & Pharmacotherapy 144, 112260 (2021). www.sciencedirect.com/science/article/pii/S0753332221013433 [↩] [↩] [↩] [↩] [↩]
- N. H. Greig et al., Proceedings of the National Academy of Sciences USA 102, 17213–17218 (2005). doi:10.1073/pnas.0508575102 [↩] [↩]
- Y. Lv, Z. Li, M. Liu, W. Sun, X. Zhang, X. Zhang. Monoamine oxidase B inhibitors based on natural privileged scaffolds: A review of systematically structural modification. International Journal of Biological Macromolecules 251, 126158 (2023). https://doi.org/10.1016/j.ijbiomac.2023.126158 [↩] [↩] [↩] [↩]
- J. C. Shih, R. F. Thompson. Monoamine oxidase in neuropsychiatry and behavior. The American Journal of Human Genetics 65, 593‑598 (1999). https://doi.org/10.1086/302562 [↩] [↩] [↩] [↩] [↩] [↩]
- N. J. Kolla, M. Bortolato. The role of monoamine oxidase A in the neurobiology of aggressive, antisocial, and violent behavior: A tale of mice and men. Progress in Neurobiology 194, 101875 (2020). https://doi.org/10.1016/j.pneurobio.2020.101875 [↩] [↩] [↩] [↩]
- Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet. 103(3), 273‑279 (1998). https://doi.org/10.1007/s004390050816 [↩]
- Zhu QS, Grimsby J, Chen K, Shih JC. Promoter organization and activity of human monoamine oxidase (MAO) A and B genes. J Neurosci. 12(11), 4437‑4446 (1992). https://doi.org/10.1523/JNEUROSCI.12-11-04437.1992 [↩]
- Parsian A, Racette B, Zhang ZH, Rundle M, Perlmutter JS. Association of variations in monoamine oxidases A and B with Parkinson’s disease subgroups. Genomics. 83(3), 454‑460 (2004). https://doi.org/10.1016/j.ygeno.2003.09.002 [↩]
- M. Bortolato, K. Chen, J. C. Shih. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Advanced Drug Delivery Reviews 60, 1527‑1533 (2008). https://doi.org/10.1016/j.addr.2008.06.002 [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- N. J. Kolla, M. Bortolato. The role of monoamine oxidase A in the neurobiology of aggressive, antisocial, and violent behavior: a tale of mice and men. Progress in Neurobiology 194, 101875 (2020). https://doi.org/10.1016/j.pneurobio.2020.101875 [↩] [↩] [↩]
- D. Garcia-Arocena. The genetics of violent behavior. The Jackson Laboratory (2015). www.jax.org/news-and-insights/jax-blog/2015/december/the-genetics-of-violent-behavior [↩] [↩]
- G. Frazzetto, et al. Early trauma and increased risk for physical aggression during adulthood: the moderating role of MAOA genotype. PLoS One 2, e486 (2007). https://doi.org/10.1371/journal.pone.0000486 [↩] [↩]
- D. A. Stetler, et al. Association of low-activity MAOA allelic variants with violent crime in incarcerated offenders. Journal of Psychiatric Research 58, 69–75 (2014). https://doi.org/10.1016/j.jpsychires.2014.07.006 [↩] [↩] [↩]
- S. Moriguchi, A. A. Wilson, L. Miler, P. M. Rusjan, N. Vasdev, S. J. Kish, G. Rajkowska, J. Wang, M. Bagby, R. Mizrahi, B. Varughese, S. Houle, J. H. Meyer. Monoamine oxidase B total distribution volume in the prefrontal cortex of major depressive disorder: An [11C]SL25.1188 positron emission tomography study. JAMA Psychiatry 76, 634–641 (2019). https://doi.org/10.1001/jamapsychiatry.2019.0044 [↩] [↩] [↩] [↩] [↩] [↩]
- V. Dhull et al., Biochem Res Int., 2013:731501 (2013). [↩]
- M. B. Colović et al., Curr Neuropharmacol., vol. 11, no. 3, pp. 315–335 (2013). [↩]
- S. Brimijoin et al., Chem Biol Interact., vol. 259, pp. 271–275 (2016). [↩] [↩]
- G. Johnson and S. W. Moore, Neurochem. Int., vol. 61, no. 5, pp. 783–797 (2012). [↩]
- İ. Yapici and E. İzol, Effects of Acetylcholinesterase and Butyrylcholinesterase Enzymes on Human Memory (2023). [↩]
- G. R. Sridhar and L. Gumpeny, World J Exp Med., vol. 14, no. 1, p. 87202 (2024). [↩]
- S. Zhou and G. Huang, Biomedicine & Pharmacotherapy (2021). [↩] [↩]
- Y. Lv et al., Int. J. Biol. Macromol., vol. 251, p. 126158 (2023). [↩] [↩]
- M. Bortolato et al., Adv. Drug Deliv. Rev., vol. 60, pp. 1527–1533 (2008). [↩]
- V. P. Chen et al., Front. Mol. Neurosci., vol. 4, p. 36 (2011). [↩]
- D. Garcia-Arocena, The Jackson Laboratory (2015). [↩]
- X. J. Zhang and D. S. Greenberg, Front. Mol. Neurosci., vol. 5, p. 40 (2012). [↩]
- Y.-Y. Tan et al., Journal of Parkinson’s Disease, vol. 12, no. 2, pp. 477–493 (2022). [↩]
- M.-X. Dong et al., BioMed Research International, vol. 2017, pp. 1–9 (2017). [↩]
- Colović, M. B., Krstić, D. Z., Lazarević-Pašti, T. D., Bondžić, A. M., Vasić, V. M. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol. 11, 315–335 (2013). https://doi.org/10.2174/1570159X11311030006 [↩]
- Yapici, İ., & İzol, E. Effects of acetylcholinesterase and butyrylcholinesterase enzymes on human memory. 2023 [↩]
- Zhou, S., & Huang, G. The biological activities of butyrylcholinesterase inhibitors. Biomedicine & Pharmacotherapy. Elsevier Masson (2021). www.sciencedirect.com/science/article/pii/S0753332221013433 [↩]
- Brimijoin, S., Chen, V. P., Pang, Y. P., Geng, L., Gao, Y. Physiological roles for butyrylcholinesterase: A BChE-ghrelin axis. Chem Biol Interact. 259, 271–275 (2016). https://doi.org/10.1016/j.cbi.2016.02.013 [↩]
- Lv, Y., et al. Monoamine oxidase B inhibitors based on natural privileged scaffolds: A review of systematically structural modification. Int J Biol Macromol. 251, 126158 (2023). https://doi.org/10.1016/j.ijbiomac.2023.126158 [↩]
- Shih, J. C., and Thompson, R. F. Monoamine oxidase in neuropsychiatry and behavior. Am J Hum Genet. 65, 593–598 (1999). https://doi.org/10.1086/302562 [↩]



{kind=link}