
Abstract
Huntington’s disease (HD) is a hereditary neurodegenerative disorder. There are no FDA-approved therapies that can modify the disease. Caspase-6 plays a role in HD by creating harmful mutant huntingtin fragments and promoting axonal degeneration. This makes caspase-6 an important target for treatment. Computational molecular docking offers an effective way to screen small molecules for potential interactions with the protein caspase-6 before testing them experimentally. In this study, in-silico docking was used to evaluate a range of FDA-approved drugs for neurological disorders and previously reported caspase inhibitors. The goal was to predict their binding to the active site of human caspase-6 using the SwissDock platform. This study mainly generates hypotheses through a docking-based ranking analysis. It aims to identify small molecules that could be repurposed against caspase-6 in Huntington’s disease. The analysis found several compounds with predicted binding energies and interactions within the caspase-6 catalytic pocket, with Levacetylleucine showing the highest predicted affinity. While docking alone cannot confirm biological inhibition, these findings show caspase-6 as an important therapeutic target in HD. They also identify several small molecules that should be prioritized for future experimental validation.
Keywords: Huntington’s disease; Caspase-6; molecular docking; in silico screening; neurodegeneration; mutant huntingtin; small-molecule inhibitors; drug repurposing; SwissDock; therapeutic target
Introduction
Huntington’s disease (HD) is a genetically inherited neurodegenerative disorder with a CAG trinucleotide repeat expansion encoding a glutamine-rich repeat region and an abnormally long poly glutamine sequence. The mutant huntingtin protein (mHTT) that arises from this mutation disrupts many aspects of neuronal function, including cell signaling, the transport system in the neuronal environment, and synaptic structure, culminating in progressive neuronal degeneration in the striatum and cortex. HD affects 3-7 per 100,000 people globally, onset occurs in mid-adulthood, and death follows over 15-20 years of steady decline1.
The symptoms include involuntary choreiform movements, poor coordination, dystonia, mental decline, and behavioral disorders such as obsessive and compulsive disorders, bipolar disorders, and apathy1,2. Typically, there is heterogeneous symptom presentation in HD where some symptoms seem worse and have a greater effect on function, which change during the disease1,2.
The underlying molecular causes of HD involve both a gain-of-function toxicity of mHTT, and a partial loss-of-function of the normal huntingtin (HTT) gene. Although the mutation is genetically simple, the molecular effects are widespread, encompassing protein aggregation, altered proteolysis, abnormal gene regulation, epigenetic modifications, dysfunctional neuronal transport, and aberrant synaptic signaling2.
Expansion of the polyQ sequence is known to induce folding of HTT into the aggregation prone β-sheet-rich structures and subsequent polymerization into amyloid aggregates. Aggregates can be observed in both the nucleus and the cytoplasm of neurons and in neuronal processes and are composed not only of mHTT, but also ubiquitin, components of the proteasome, molecular chaperones, transcription factors, and at times wild type HTT3,4. It was once assumed that aggregates themselves were toxic, although increasingly it appears that they may act as a safe-storage system to trap less toxic soluble oligomers. Current thinking includes that soluble forms of mHTT (especially oligomers and N-terminal species) may be a toxic entity5. Recent evidence also suggests that perinuclear aggregates are particularly neurotoxic due to the triggering of the abnormal cell cycle while intranuclear aggregates appear much less toxic5,6.
mHTT is cleaved by caspases and calpains to form, in part, N-terminal fragments which readily polymerize into very aggregated structures. These fragments show marked accumulation in vulnerable populations of cells and a correlation to cell death. Alternative pathways such as the splicing of exon 1 HTT have also been shown to produce N-terminal fragments5. Inhibiting the formation of such fragments by both direct inhibition of these proteases and by altering the phosphorylation status of HTT to reduce cleavage offers another therapeutic target5.
mHTT appears to alter gene regulation by binding to regions of the glutamine-rich transactivation domains of transcription factors. Such transcription factors include p53, CREB, CBP, Sp1/TAFII130 and PGC-1. An important result of these interactions is a decrease in production and axonal transport of BDNF which is essential for survival of the striatal medium spiny neurons6.
This allows the wild-type HTT protein to function as a scaffold that helps facilitate the microtubule-associated movement of trafficking vesicles, organelles and trophic factors as illustrated in Figure 17,8. BDNF and its corresponding receptor TrkB fail to reach distal neuronal locations. Furthermore, impairment in the proteasomal and autophagic mechanisms of clearance result in increased cellular stress and protein accumulation. The presence of ubiquitin-proteasome machinery dysfunction, failure of autophagy, and sequestration of important proteins into inclusions compromise neuronal stability.
Also found in HD is synaptic dysfunction and structural change as Illustrated in Figure 16. Both the lack of wild-type HTT’s scaffold and transcriptional function result in loss of normal corticostriatal synapses. These structural and functional synaptic changes might occur early on in the course of HD and are related to deficits in glutamatergic signaling, changes in NMDAR function and lack of normal postsynaptic density development. In fact, many of the structural changes occur prior to significant neuronal loss, which indicates that synaptic dysfunction is a key factor in causing early HD phenotypes.
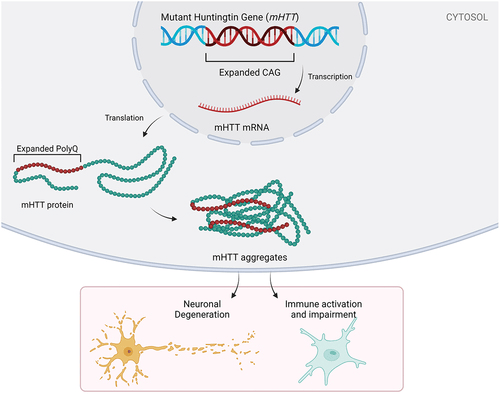
The most relevant protease to HD biology is caspase-6. Amongst all the proteases capable of cleaving mutant HTT, caspase-6 cleaves at a unique site which produces an N-terminal fragment with very high toxicity8. This fragment can easily enter the nucleus and can therefore repress gene expression. This is thought to contribute to the widespread pattern of gene repression at an early stage in HD, as the fragment blocks transcription and alters the normal chromatin structure8. Interestingly, animals lacking either the caspase-6 cleavage site or depleted of the enzyme’s activity display significantly delayed behavioral deficits and neurodegeneration.
Caspase-6 plays a role in several pathways deregulated in HD in addition to its role in generating toxic fragments. Caspase-6 is a protease strongly activated by neuronal stress, and is implicated in the axonal degeneration that is thought to be one of the earliest events in HD. Caspase-6 is known to be capable of cleaving cytoskeletal and transport related proteins. Thus this protease can weaken axonal integrity, slowing down long-range trafficking of vital cargoes (such as BDNF-carrying vesicles). This is especially relevant given that fragile striatal neurons rely heavily on the constant supply of cortical BDNF. This explains how this protease exacerbates the most significant vulnerability in the corticostriatal circuitry10.
Caspase-6 also interacts with apoptotic pathways. While mHTT may not directly initiate classical apoptosis in some situations, mHTT-expressing cells are far more susceptible to stress-induced caspase activation. Positioning above caspase-3 in some pathways, caspase-6 may therefore act as a ‘gatekeeper’ preparing the neurons for additional neurodegeneration. The convergence of these actions clearly places caspase-6 as a central component and its inhibition as a therapy target.
Although our understanding of HD biology continues to improve, current treatment is exclusively symptomatic and has no impact on the disease course10. None of the existing FDA-approved drugs are capable of preventing neuronal death by directly addressing the specific events which induce neuronal dysfunction, including production of toxic mHTT fragments and initial axon damage11. This lack of targeted therapy has generated increased interest in upstream interventions in the disease pathway13. One such intervention, inhibition of caspase-6 activity (a critical mediator of N-terminal fragment production as well as early degeneration), represents a highly attractive candidate for slowing or preventing HD progression11,8.
In the search for treatments that can do more than manage HD symptoms, computational techniques have begun to play a more significant role in the drug discovery pipeline. Instead of designing new compounds from scratch in the lab, these approaches leverage artificial intelligence and in silico modeling to prioritize the compounds which are most likely to bind successfully after screening large, diverse chemical libraries against the target protein. In this manner, these techniques produce data regarding binding affinity, orientation, and other molecular properties which are impractical to acquire experimentally on a large scale12.
Computational molecular docking offers one readily available example of an AI-driven method used to examine a target protein’s interaction with small molecules. In modeling potential binding positions and calculating approximate binding affinity, molecular docking makes it feasible to computationally screen thousands of small molecules against an enzyme targeted for inhibition (e.g., an enzyme mediating a pathological effect in a neurodegenerative disease) far more quickly and inexpensively than is possible experimentally12.
The objective of this study is to perform in silico docking using the platform Swiss Dock to identify small molecules that can act as caspase-6 inhibitors and potential drug candidates for treating Huntington’s disease. This study focuses only on ranking small candidate molecules based on docking results where emphasis is given on the repurposing of drugs, not to compare how good inhibitor each compound is or validating use in any experiment. It is hypothesized that compounds that have high predicted binding affinity to caspase-6 active site can be identified through docking that can be a potential candidate drug for targeting caspase-6.
Methodology
Literature Mining Process
Candidate small molecules were first identified using an exhaustive search of the literature and prominent chemical databases. The search of the literature and reference sites (PubMed, Google Scholar, Cambridge Structural Database, etc.) was aimed at identifying molecules with previously reported effects on caspase-6, proteolysis related to HD, or general mechanisms of neurodegeneration. Searches were performed using keywords such as “caspase-6 inhibition”, “Huntington’s disease proteolysis”, “polyglutamine-mediated degeneration”, and “FDA-approved neurological therapeutics”. A list of molecules which were identified to interact with caspase at least to some extent or that have some degree of clinical significance to neurological diseases was developed. From this initial list, molecules with available structural data (SMILES, 3D structure) were prioritized. Those more clearly related to the nervous system were identified, and those which had been implicated in caspase action or proteolysis were highlighted. Non-small molecules (biologics, gene therapies) that did not have a defined small molecule structure were excluded from the docking studies but are included for discussion.
Use of Swiss Dock
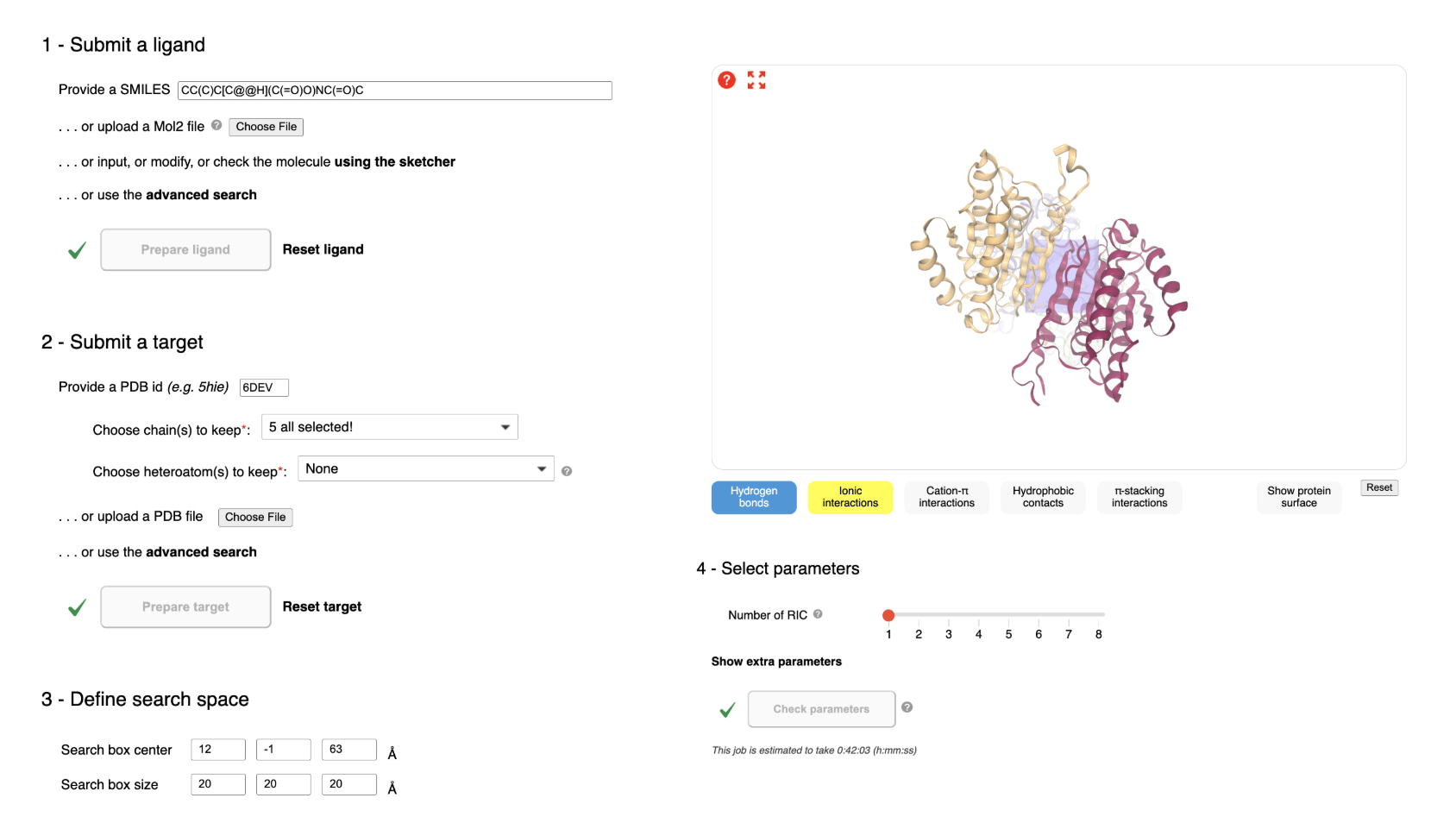
SwissDock (https://www.swissdock.ch/dock.php)13 was used to evaluate the docking behavior of a curated set of FDA-approved, investigational, and literature-reported molecules against the protease caspase-6. Identification of suitable inhibitors required a standardized workflow for ligand preparation, target setup, search-space definition, and sampling configuration within the SwissDock environment.
Target Structure Selection and Preparation
The first step was locating a high-quality structural template for human caspase-6. Using PubMed and PubChem queries, supplemented with reports in the protein-engineering literature, the crystal structure PDB ID: 6DEV was selected as the docking target. The 6DEV structure was selected due to its high-resolution representation of human caspase-6 and availability within the SwissDock-compatible PDB database. Although this structure may include engineered or stabilized features, it provides a practical and commonly used model for active-site docking studies.
The structure was imported into SwissDock by entering the PDB ID directly, prompting the server to retrieve the coordinates from PDB. Once loaded, the interface displayed all protein chains and associated molecules. These components were inspected to determine which should be retained for docking; the core caspase-6 chains were selected. Upon finalizing this selection, the target-preparation command is activated. The process of using SwissDock can be seen in Figure 2.
Prior to docking, the protein structure was used as provided by SwissDock without additional manual energy minimization. Non-essential heteroatoms and solvent molecules were excluded during target preparation where applicable. Default protonation states and charge assignments provided by the SwissDock pipeline were used for both protein and ligands.
Ligand Collection, SMILES Retrieval, and Preparation
The experimental molecule set consisted of three broad categories: FDA-approved neurological drugs, clinically investigated drugs, or non-approved molecules previously reported in caspase-related or protease-inhibitor literature12. Only small-molecule compounds with defined chemical structures were included in docking simulations; biologics and gene-based therapies are listed for context but were not computationally evaluated.
| Compound name | Drug/Molecule Type |
| Z-VEID-FMK peptidic inhibitor | Previously validated caspase-6 inhibitor |
| Arimoclomol, Levacetylleucine | FDA approved Child Neurology drugs |
| Atidarsagene Autotemcel, Givinostat, Generic Deflazacort, Delandistrogene Moxeparvovec-rokl | FDA approved Neuro-Muscular Disorder drugs |
| Valbenazine, Deutetrabenazine, Cerliponase Alfa, Carbidopa, Levodopa, Eladocagene Exuparvovec-tneq | FDA approved Muscular Disorder drugs |
| Vorasidenib | FDA approved Neuro-Oncology drug |
| Ravulizumab-cwvz, Edgar-tigimod Alfa & Hyaluronidase-qvfc | FDA approved Multiple Sclerosis and Immune Disorders drugs |
| Ocrelizumab & Hyaluronidase-ocsq | FDA approved Muscular-Skeletal Disorder drugs |
| Vinyl sulfone and Isatin sulfonamide | From published caspase-inhibitor literature, non-FDA approved drugs |
For every molecule with a known structure, SMILES strings were obtained from PubChem or derived from supporting literature. Molecules lacking any structural information (including several gene-therapy vectors and large biologics) could not be docked and were excluded from computational evaluation.
Each available SMILES string was imported into SwissDock either by direct pasting into the input box or by uploading the corresponding structure file. The system displayed the molecule in the text field for visual inspection. Each ligand was checked to ensure proper charge state and valence representation prior to preparation. After confirmation, the ligand-preparation button became active. A check mark indicated successful processing; molecules that failed preparation were associated with specific error messages pinpointing structural issues requiring correction.
Ligands were processed using default SwissDock preparation protocols, including automated geometry optimization and charge assignment.
Defining the Docking Search Region
After ligand and target preparation, the docking search space was established. Using the integrated molecular viewer, the search-box center was positioned directly over the caspase-6 active site, identified through visual inspection of catalytic residues and the substrate-binding cleft.
To refine search-space placement, SwissDock visualization tools were used to feature potential ligand-protein interactions, including hydrogen bonds, hydrophobic contacts, aromatic stacking, and ionic interactions, as seen to the right in Figure 2. Protein surface rendering helped assess cavity depth and accessibility, ensuring that the search region accurately captured the physiologically relevant binding environment for caspase-6 inhibitors.
The final selected docking region corresponds to the known catalytic pocket of caspase-6, which includes the active-site cysteine and substrate-recognition residues. The search space was defined to encompass this region based on visual alignment with the substrate-binding cleft rather than exhaustive blind docking.
Sampling Configuration and Parameter Validation
Before computations could proceed, all parameters were verified using the “Check parameters” function. This internal validation ensured that the search box contained at least one detectable cavity (for AC), the estimated runtime remained within allowable server limits (under 1 hour for AC), and the configuration did not conflict with SwissDock constraints.
Docking Execution and Monitoring
Once all validation rules for the ligand, target, search space and sampling were fulfilled, the START DOCKING command was activated. All ligands were run independently and SwissDock pushed them onto its queue. Status, time remaining and progress bar appeared on the jobs monitoring page once calculation was running:
Once calculation finished, the user was redirected to the results page. On that page, a 3D representation of the protein-ligand complex is given for each molecule, a 3D representation of the ranking docking position, interaction displays (showing the contacts of the molecule with caspase-6) and links to other SwissDrugDesign functionalities such as position clustering or score analysis.
From these results, candidate molecules were then compared in terms of docking performance, most efficient caspase-6 inhibitors were selected and molecules were also classified by the probability to bind to the active site of caspase-6.
Data Collection
Protein-Ligand Interaction Analysis
The docking workflow provides a close look at where each candidate molecule resides within the caspase-6 binding site. Interaction maps, generated by SwissDock, display which areas of the protein bind most favorably to the ligand. These views were inspected to identify if a molecule is located in the active site cleft, interacting with the important amino acid residues responsible for recognition of the substrate, or is nested in a nearby allosteric site that might affect its activity. Care is taken to evaluate the interaction between active-site cysteine and nearby amino acids, because these interactions often play a significant role in effective caspase inhibition. Typical patterns of hydrogen bonding, hydrophobic interaction, and stacking are monitored for comparison across various chemical scaffolds in the context of the caspase-6 enzyme14.
Binding Affinity Predictions
SwissDock gives numerical prediction of binding energy by predicted free energy (AC) scores (ΔG). The lower the value of the ΔG, the better and stable is the protein-ligand complex1,6. The predicted energy values help in identifying the list of possible inhibitors that bind strongly enough to the caspase-6 molecule and affect its enzymatic activity5. When all candidates are compared together, the compound with favorable ΔG values at many possible positions indicates overall good binding with the enzyme’s structural characteristics. The clustering of predicted energy minima indicates if the candidate binds in a single favored conformation or a group of conformations5.
Results
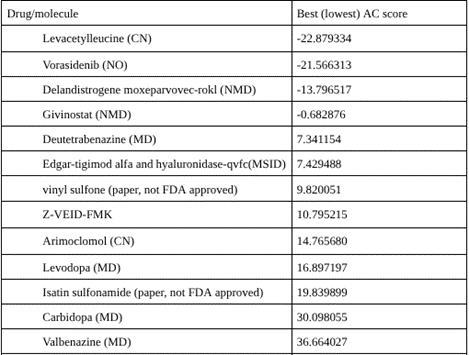
SwissDock returned multiple AC scores for each molecule, corresponding to different predicted binding positions. To assign a single representative value per compound, the most favorable (lowest, most negative) AC score was selected. These representative scores were then used to rank all molecules, with more negative values indicating stronger predicted binding.
Although multiple docking poses were generated for each ligand, only the most favorable (lowest) AC score was selected for initial comparison. However, this approach does not capture pose consistency or clustering behavior, which may influence binding reliability.
Discussion
Among all molecules docked to SwissDock, Levacetylleucine, commercially known as Aqneursa (Figure 3) had the most favorable predicted binding affinity to caspase-6 with an AC score of -22.8793. AC scores are predictions of binding free energy, therefore the more negative a value, the more spontaneous the ligand protein binding. In more practical terms, it takes relatively less energy (or less energy input from other molecules) for Levacetylleucine to assume the optimal pose within the active site and the residues within the catalytic pocket and have high complementary interactions with this molecule. It should be mentioned that there can be discrepancies in the sign (positive vs negative values) of reported AC scores, and it is possible that these vary between outputs from SwissDock or transcription. Here we rank the molecules based on relative AC scores rather than the magnitude of the AC score.
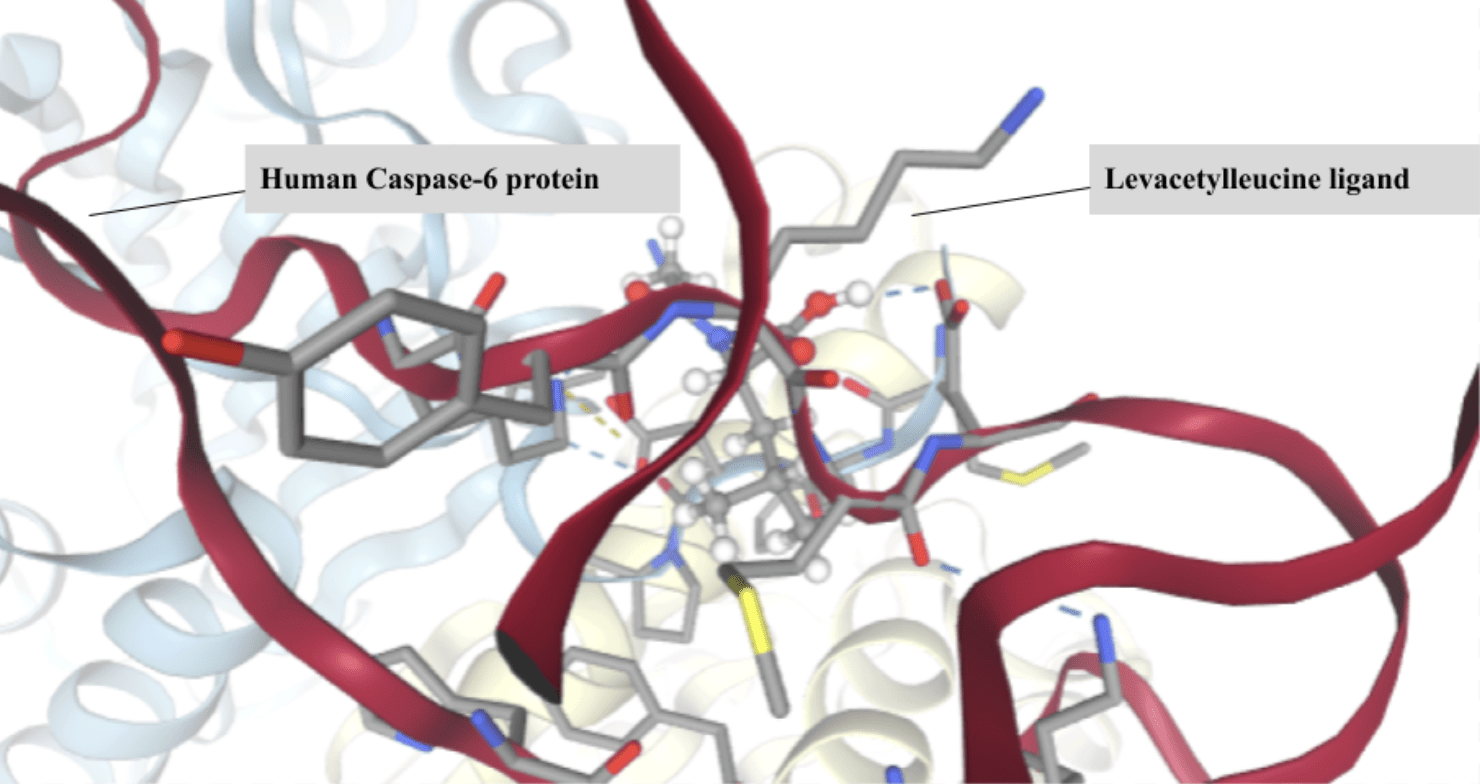
It should be pointed out that the greatest weakness that impacts on the entire ranking is that covalent, and mechanism-based, inhibition is ignored by most standard docking protocols. The majority of well-established caspase inhibitors bind through a covalent link to the catalytic cysteine residue which Swiss Dock scoring would ignore. It is possible that these inhibitors will be artificially ranked less favorably, hence not representative of their true biological inhibitory capacity. The high predicted affinity is attributed to Levacetylleucine’s amino acid backbone and side chain, giving rise to many of the interactions that protease active sites generally prefer5. Caspase-6 recognizes peptide substrates and hence the backbone structure and side chain features present in Levacetylleucine make it readily adopt similar geometry, giving it a good shape complementarity within the enzyme’s substrate binding cleft15,8. Its functional groups allow it to establish hydrogen bonds, polar interactions, and hydrophobic interactions, thus allowing the ligand to readily interact with residues surrounding the catalytic species. The geometry, polarity, and size of the molecule allows Levacetylleucine to slot nicely into the pocket, introducing no steric clashes, and hence reducing the binding energy. Although known primarily for its application in child neurology, this data implies that Levacetylleucine does contain favorable properties for interaction with Caspase-613. Levacetylleucine is indicated for the treatment of neurological symptoms associated with Niemann-Pick type C (NPC) in pediatric patients15. NPC is a rare, progressive genetic disorder in which lipids accumulate within cells and lead to accumulation in multiple organs including the brain, which causes severe progressive neurological symptoms15,16. Computational docking alone cannot determine inhibitory potential but this high predicted affinity identifies Levacetylleucine as a viable candidate for experimental confirmation as it may be useful in the treatment of neurodegenerative diseases, where Caspase-6 is known to contribute to pathology.
Limitations
All of the reported docking results for this paper are for the static 3D structures of caspase-6 and the selected ligands. While the static approximation derived from SwissDock may be a close representation of interactions at a molecular level, such representations lack the physiological dynamics in which caspase-6 typically operates17. In the body, protein conformation changes dynamically, with changes in pH and ion concentration and interactions with a multitude of other cellular components. None of these factors are simulated during the docking environment and as a consequence, expected binding location cannot fully represent that found in biology13.
There was no formal docking protocol validation undertaken during this study (i.e., re-docking of a known ligand or benchmarking against experimental structures). This leads to caution when examining absolute rank order in the binding analysis, as the result is not necessarily a confirmation of binding in the active site.
This computational screening, however, has proven useful by significantly reducing the number of compounds to a manageable list of possible active agents that are in a favorable position to bind caspase-618,19. The trends across the FDA and non-FDA approved compounds show that certain candidates, and more significantly Levacetylleucine, might inhibit caspase-620. From the information gained by docking these molecules, there is promising binding compatibility with the enzyme21,22 but requires validation through wet lab investigation including enzyme activity and cell based modeling/testing for toxicity to verify the results.
Conclusions
When all selected/screened compounds were docked using SwissDock, many docked into the active site of caspase-6, an enzyme that has been found to be correlated with Huntington’s Disease. Of all the compounds tested, Levacetylleucine docked the best with regards to both energy as well as positioning in the active site, hinting that it may bind better according to its docking score. The potential that molecules binding stably to the caspase-6 enzyme may be viable drugs needs to be considered23. Few reports have investigated small molecule inhibitors to caspase-6 in a comprehensive manner, nor have many FDA approved neurologically targeting drugs been explored to see if they may be useful inhibitors. Combining literature mining with in silico docking analysis allows for several interesting compounds to be studied in more detail. The interaction predicted by Levacetylleucine is strong and may point to an unexplored direction for existing drugs against caspase-6 mediated neurodegeneration. These findings may only be seen as predictions for a drug, and need to be confirmed by further experimentation.
Acknowledgements
Thank you for the guidance of Dr. Ilkem Sevgili, postdoctoral research fellow at Harvard Medical School and Mass Eye and Ear, in the development of this research paper.
References
- M. J. Leyva et al., “Identification and evaluation of small molecule pan-caspase inhibitors in Huntington’s disease models,” Chem Biol, vol. 17, no. 11, 2010, doi: 10.1016/j.chembiol.2010.08.014 [↩] [↩] [↩] [↩]
- L. Zhou, J. Flores, A. Noël, O. Beauchet, P. J. Sjöström, and A. C. Leblanc, “Methylene blue inhibits Caspase-6 activity, and reverses Caspase-6-induced cognitive impairment and neuroinflammation in aged mice,” Acta Neuropathol Commun, vol. 7, no. 1, 2019, doi: 10.1186/s40478-019-0856-6 [↩] [↩] [↩]
- S. K. Custer et al., “Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport,” Nat Neurosci, vol. 9, no. 10, 2006, doi: 10.1038/nn1750 [↩]
- J. Donaldson, S. Powell, N. Rickards, P. Holmans, and L. Jones, “What is the Pathogenic CAG Expansion Length in Huntington’s Disease,” 2021. doi: 10.3233/JHD-200445 [↩]
- T. Ratovitski et al., “Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis,” Cell Cycle, vol. 11, no. 10, 2012, doi: 10.4161/cc.20423 [↩] [↩] [↩] [↩] [↩] [↩]
- M. Jimenez-Sanchez, F. Licitra, B. R. Underwood, and D. C. Rubinsztein, “Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies,” Cold Spring Harb Perspect Med, vol. 7, no. 7, pp. 1–22, Jul. 2017, doi: 10.1101/CSHPERSPECT.A024240 [↩] [↩] [↩] [↩]
- L. Qi, L. Wang, M. Jin, M. Jiang, L. Li, and Y. Li, “Caspase‐6 is a key regulator of cross‐talk signal pathway in cancer,” Immunology, vol. 169, no. 3, pp. 245–259, Jul. 2023, doi: 10.1111/imm.13633 [↩]
- N. S. Caron et al., “Mutant huntingtin is cleared from the brain via active mechanisms in Huntington disease,” Journal of Neuroscience, vol. 41, no. 4, 2021, doi: 10.1523/JNEUROSCI.1865-20.2020 [↩] [↩] [↩] [↩] [↩]
- S. E. Field, A. J. Curle, and R. A. Barker, “Inflammation and Huntington’s disease – a neglected therapeutic target?,” Expert Opin. Investig. Drugs, vol. 33, no. 5, pp. 451-467, 2024, doi: 10.1080/13543784.2024.2348738 [↩]
- M. Zheng, R. Karki, P. Vogel, and T. D. Kanneganti, “Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense,” Cell, vol. 181, no. 3, 2020, doi: 10.1016/j.cell.2020.03.040 [↩] [↩]
- P. McColgan and S. J. Tabrizi, “Huntington’s disease: a clinical review,” 2018. doi: 10.1111/ene.13413 [↩] [↩]
- O. E. Eje, C. V. Ogbonna, C. S. Onoyima, and F. O. Nduka, “Huntington Disease: Mechanism of Pathogenesis and Recent Developments in Its Therapeutic Strategies-A Short Review,” 2023. doi: 10.22034/jcr.2023.362508.1194 [↩] [↩] [↩]
- X.-Y. Meng, H.-X. Zhang, M. Mezei, and M. Cui, “Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery,” Current Computer Aided-Drug Design, vol. 7, no. 2, 2012, doi: 10.2174/157340911795677602 [↩] [↩] [↩]
- M. Jimenez-Sanchez, F. Licitra, B. R. Underwood, and D. C. Rubinsztein, “Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies,” Cold Spring Harb Perspect Med, vol. 7, no. 7, 2017, doi: 10.1101/cshperspect.a024240 [↩]
- D. E. Ehrnhoefer et al., “Partial reduction of huntingtin expression by antisense oligonucleotides is sufficient to restore function in a mouse model of Huntington’s disease,” Chem Biol, vol. 26, no. 7, 2019, doi: 10.1016/j.chembiol.2019.07.001 [↩] [↩] [↩]
- R. van Gool et al., “Levacetylleucine (N-acetyl-l-leucine) for Niemann-Pick disease type C,” Trends Pharmacol Sci. 2025 Apr;46(4):386-387, doi: 10.1016/j.tips.2025.02.003 [↩]
- D. E. Ehrnhoefer et al., “A quantitative method for the specific assessment of caspase-6 activity in cell culture,” PLoS One. 2011;6(11):e27680, doi: 10.1371/journal.pone.0027680 [↩]
- A. Tubeleviciute-Aydin et al., “Identification of Allosteric Inhibitors against Active Caspase-6,” Sci Rep. 2019 Apr 2;9(1):5504, doi: 10.1038/s41598-019-41930-7 [↩]
- Z. Ma et al., “Docking strategies for predicting protein-ligand interactions and their application to structure-based drug design,” Commun Inf Syst. 2024;24(3):199-230, doi: 10.4310/cis.241021221101 [↩]
- S. Feleus et al., “Medication Use and Treatment Indications in Huntington’s Disease; Analyses from a Large Cohort,” Mov Disord Clin Pract. 2024 Dec;11(12):1530-1541, doi: 10.1002/mdc3.14230 [↩]
- S. J. Tabrizi et al., “Potential disease-modifying therapies for Huntington’s disease: lessons learned and future opportunities,” Lancet Neurol. 2022 Jul;21(7):645-658, doi: 10.1016/S1474-4422(22)00121-1 [↩]
- A. Grosdidier et al., “SwissDock, a protein-small molecule docking web service based on EADock DSS,” Nucleic Acids Res. 2011 Jul;39(Web Server issue):W270-7, doi: 10.1093/nar/gkr366 [↩]
- M. R. Hossain et al., “Identification of molecular targets and small drug candidates for Huntington’s disease via bioinformatics and a network-based screening approach,” J Cell Mol Med. 2024 Aug;28(16):e18588, doi: 10.1111/jcmm.18588 [↩]



{kind=link}