
Abstract
Alzheimer’s Disease (AD) is a devastating neurodegenerative condition that primarily affects the elderly. It is characterized by memory impairment, synaptic dysfunction, and progressive cognitive decline. The hippocampus, pivotal to memory and learning processes, is a central locus of pathological alterations in AD. Examining the intrinsic properties of neurons affected by this disease provides insight into the cellular processes underlying hippocampal dysfunction. While much research has examined synaptic dysfunction in AD, far fewer studies have explored how changes in the intrinsic properties of individual neurons may influence disease progression, despite their potential as early biomarkers and therapeutic targets. The findings presented here are based on original experimental data collected by the Daou Lab and analyzed in this study. The aim is to identify key differences in intrinsic properties, such as amplitude, threshold, and spike width, of pyramidal neurons in the hippocampus of mice with AD compared with controls. Whole-cell current-clamp recordings were obtained from hippocampal slices of both AD model mice and control mice to quantify these electrophysiological parameters. Analysis of these recordings revealed that neurons from AD-affected mice were significantly more excitable than those from controls, and several intrinsic properties were altered in the AD case. These findings suggest that further pharmacological investigations should be applied to identify the specific ion channels responsible for the altered excitability observed in AD neurons, potentially offering new targets for therapeutic intervention.
Keywords: Alzheimer’s disease, hippocampus, pyramidal neurons, intrinsic excitability, spike amplitude, spike threshold, spike width, afterhyperpolarization (AHP), ion channel dynamics, electrophysiology, neurodegeneration
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that advances through distinct stages, with late-stage disease causing severe cognitive and physical decline, including loss of movement, communication, and awareness, and most commonly affecting individuals over 65, particularly women who are at higher risk due to longer lifespans1,2.
AD is a growing global health concern, with prevalence increasing as the world’s population ages. Currently, over 55 million people worldwide live with dementia, projected to rise to 139 million by 2050. AD accounts for 60–70% of cases, with nearly 10 million new diagnoses annually3,4. As the most common cause of dementia, AD contributes significantly to disability and dependency among older adults worldwide5. This rising prevalence underscores the urgent need to understand the disease’s underlying mechanisms to develop effective treatments and interventions. The increasing prevalence of AD poses major challenges to healthcare systems and societies, highlighting the urgent need for improved understanding of its underlying mechanisms4,6.
First, we discuss the pathology of AD (neurofibrillary tangles and amyloid-beta plaques), the concept of intrinsic plasticity, and the impact AD has on the hippocampus. Next, this study assesses differences in intrinsic properties of hippocampal pyramidal neurons in mice with and without AD. We assessed several key electrophysiological parameters, including spike threshold, which is the minimum membrane voltage needed to trigger an action potential; time to peak afterhyperpolarization (ttAHP), the time it takes for the neuron’s membrane potential to reach its maximum negative value following an action potential; afterhyperpolarization (AHP), the period when the membrane potential becomes more negative than its resting state and helps regulate neuron firing; and spike width and amplitude, which describe the duration and size of the action potential, respectively. When assessing the pathology of AD, attention typically centers on synaptic connections, the communication between neurons, and how these are affected by neurofibrillary tangles and amyloid plaques. However, far less consideration is given to how the intrinsic properties of neurons themselves contribute to disease progression. These intrinsic properties encompass the internal mechanisms that govern neuronal excitability and signalling. By studying differences in these intrinsic properties, we aim to better understand changes in fundamental cellular processes caused by AD within the hippocampus and provide insight into cellular mechanisms that may precede or accompany synaptic dysfunction.
Pathology of AD
Two of the most well-known biomarkers for AD are neurofibrillary tangles and amyloid-beta plaques. Neurofibrillary tangles consist of abnormal accumulations of abnormally phosphorylated microtubule protein tau within the perikaryal cytoplasm of pyramidal neurons7. The plaques consist of a peptide known as beta-amyloid, surrounded by abnormally configured neuronal processes or neurites8.
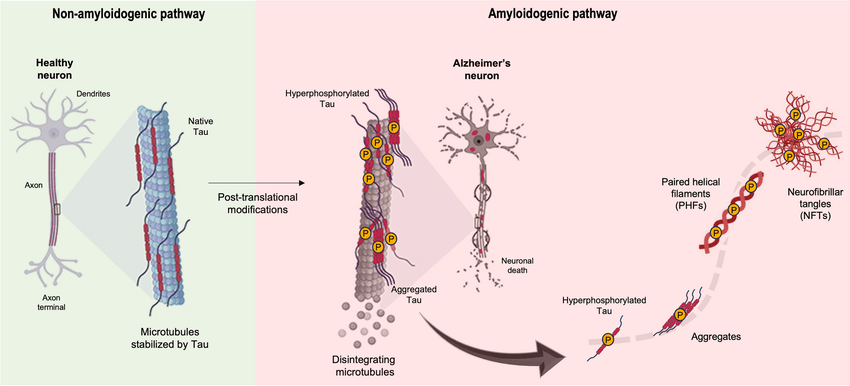
Neurofibrillary tangles (NFTs) are composed of abnormal fibrils about 10 nm in diameter, occurring in pairs and bound in a helical structure10. They can have many different protein components associated with it, namely, ubiquitin, cholinesterase and beta-amyloid 411. However, the most important element is the tau protein, whose abnormal phosphorylation leads to these tangles7. Tau proteins regulate the assembly of microtubules, playing an integral role in neuronal development and axonal growth12. The phosphorylation and dephosphorylation of tau are maintained by a group of tau phosphates and microtubule-associated kinases13. In certain pathological conditions, the downregulation of these phosphates causes hyperphosphorylation of tau, resulting in insoluble double helicase filaments14. These filaments clump to form protein complexes, creating Neurofibrillary tangles (Figure 1). The tau proteins in AD lose their usual abilities to bind and stabilize microtubules in the axon15. These structures lead to the eventual depredation of neurons and survive even after the death of affected neurons. After neuronal death, they are released extracellularly and interact with astrocytes and microglia16. Areas affected by neurofibrillary tangles include the layer II neurons of the entorhinal cortex, the CA1 and subicular regions of the hippocampus, the amygdala, the dorsal raphae and the nucleus basalis of Meynert17.
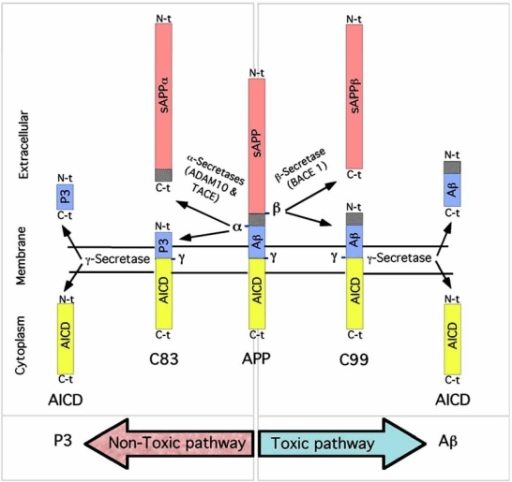
Another critical pathological lesion of AD is senile or neuritic plaques, otherwise known as amyloid plaques18. These plaques are extracellular accumulations of βA4, a peptide derived from the abnormal processing of amyloid precursor protein (APP)19. APP is a membrane protein in the endoplasmic reticulum and is transported to the Golgi complex to be cleaved by 3 secretases α, β, or γ20. Most notably, β-secretase cleaves the amino terminal while γ-secretase cleaves the carboxy-terminal, releasing βA4 (Figure 2)21. Diffused plaques are the initial deposition of βA4 in the cerebral cortex, lacking dystrophic neurites and are commonly associated with non-cognitive dysfunction18. Neuritic plaques have a central βA4 core and are surrounded by dystrophic neurites, they are pathognomonic of AD and are linked to cognitive decline18. Together these mechanisms ultimately disrupt synaptic function, trigger neuroinflammatory responses and contribute to neuronal death8.
Intrinsic Plasticity
Amyloid plaques and Tau tangles are widely recognized as the primary pathological markers of AD, playing a critical role in disrupting normal neuronal function. Inflammation, as well as the progressive loss of synapses, forms the crux of neurological damage seen in AD22. However, crucial factors that have been overlooked are how the intrinsic features and changes of neurons affect this disease. These intrinsic properties (IPs) are critical because they determine how neurons respond to inputs, regulate firing patterns, and maintain network stability23. Changes in intrinsic excitability can alter information processing and disrupt signal propagation, even without synaptic modifications. In AD, such alterations may contribute to network dysfunction by creating states of excessive or reduced excitability, which can impair learning and memory24. Investigating these intrinsic changes is therefore important, as it provides insight into cellular-level mechanisms of the disease and may reveal targets for interventions beyond synaptic pathways.
Intrinsic changes are distinct from synaptic changes. While synaptic changes involve alterations at the synapse, intrinsic plasticity refers to activity-dependent modifications in a neuron’s own electrophysiological properties25. These changes are mediated by ion channels and affect processes such as synaptic integration, subthreshold signal propagation, spike generation, and meta-plasticity26. By adjusting these intrinsic properties, neurons can regulate their excitability and responsiveness independently of synaptic modifications.
A neuron’s ability to generate an action potential based on a stimulus is regulated by the number, type, and distribution of its voltage- and calcium-gated ion channels27. Furthermore, intrinsic plasticity is related to homeostatic regulation of neuronal firing. For example, depriving the neocortical or hippocampal neurons of electrical or synaptic stimulation for an extended period increased neuronal excitability, contrary to the results of synaptic plasticity28. Conversely, increasing neuronal input above the basal level was seen to decrease neuronal excitability. The changes and regulation of intrinsic excitability (IE) are attributed to the modification of ion channels in the neuron29.
The Hippocampus and AD
Structure of the Hippocampus
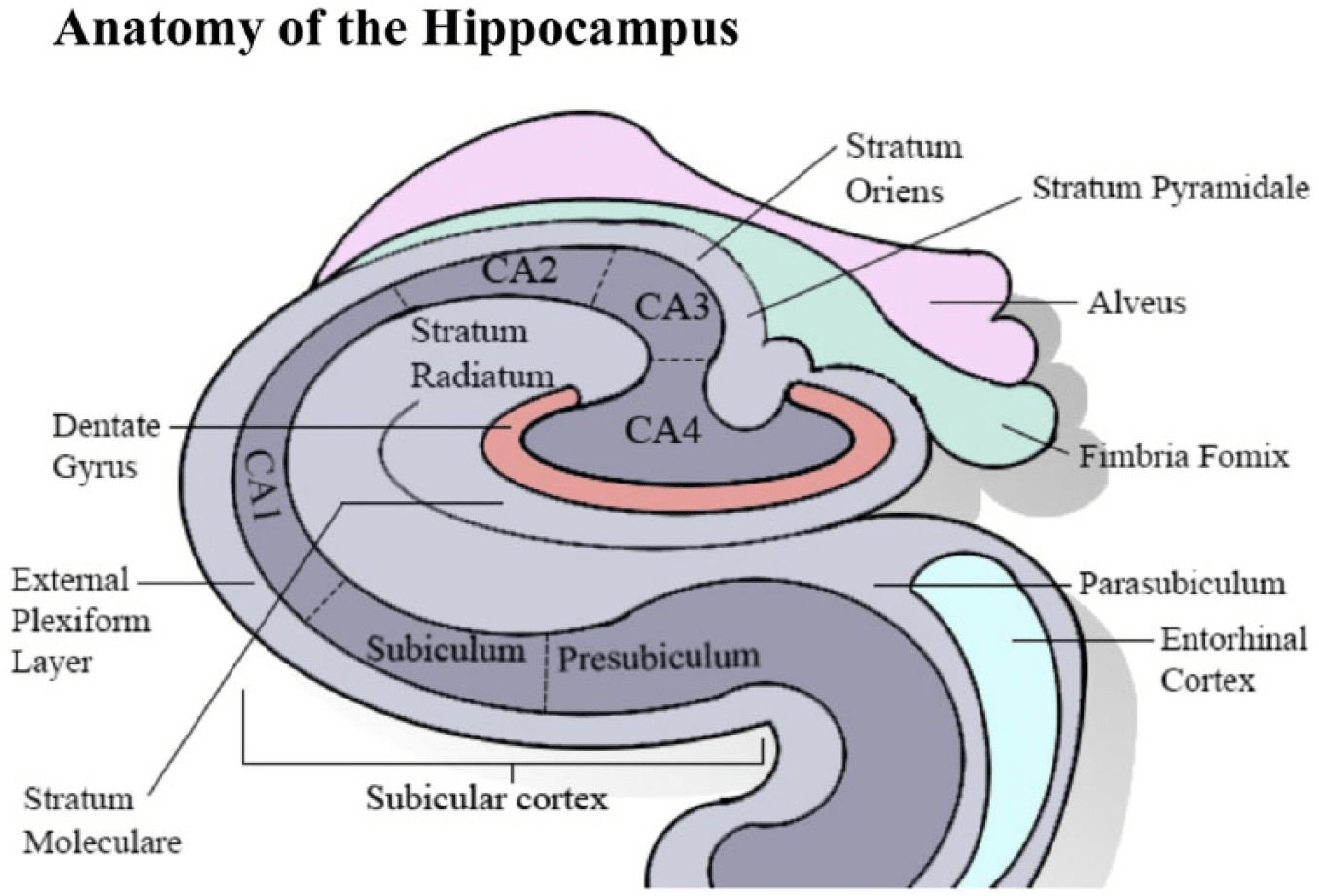
The hippocampus, a key structure for memory and decision-making, is particularly vulnerable in Alzheimer’s disease, where neuronal loss and synaptic dysfunction contribute to the characteristic memory deficits (refer to Figure 3 for the anatomical overview)31,32.
Function and Impact of AD of the Hippocampus
The hippocampus is involved in various cognitive functions, particularly memory, learning, and spatial navigation33. It is one of the few regions in the brain where neurogenesis—mainly in the dentate gyrus—continues into adulthood34. Though initially thought to have little impact, this ongoing neurogenesis is now understood to contribute to memory processing, as new neurons integrate into existing neural networks and assist in memory consolidation35. The hippocampus plays a crucial role in encoding and retrieving spatial, episodic, and semantic memories and communicates with the neocortex—particularly during memory encoding (hippocampus) and retrieval (neocortex)36. Memory is processed through two primary pathways: the polysynaptic and direct intra-hippocampal pathways. The polysynaptic pathway involves afferent connections from the parietal, temporal, and occipital lobes via the entorhinal cortex, traveling through the dentate gyrus, CA3, CA1, and subiculum, before reaching the anterior thalamus and cingulate cortex37. The direct pathway carries inputs from the temporal association cortex through entorhinal areas directly to CA1, impacting episodic and spatial memory36. The hippocampus also influences decision-making and memory consolidation, particularly in transforming short-term into long-term memory, which supports future decision-making38.
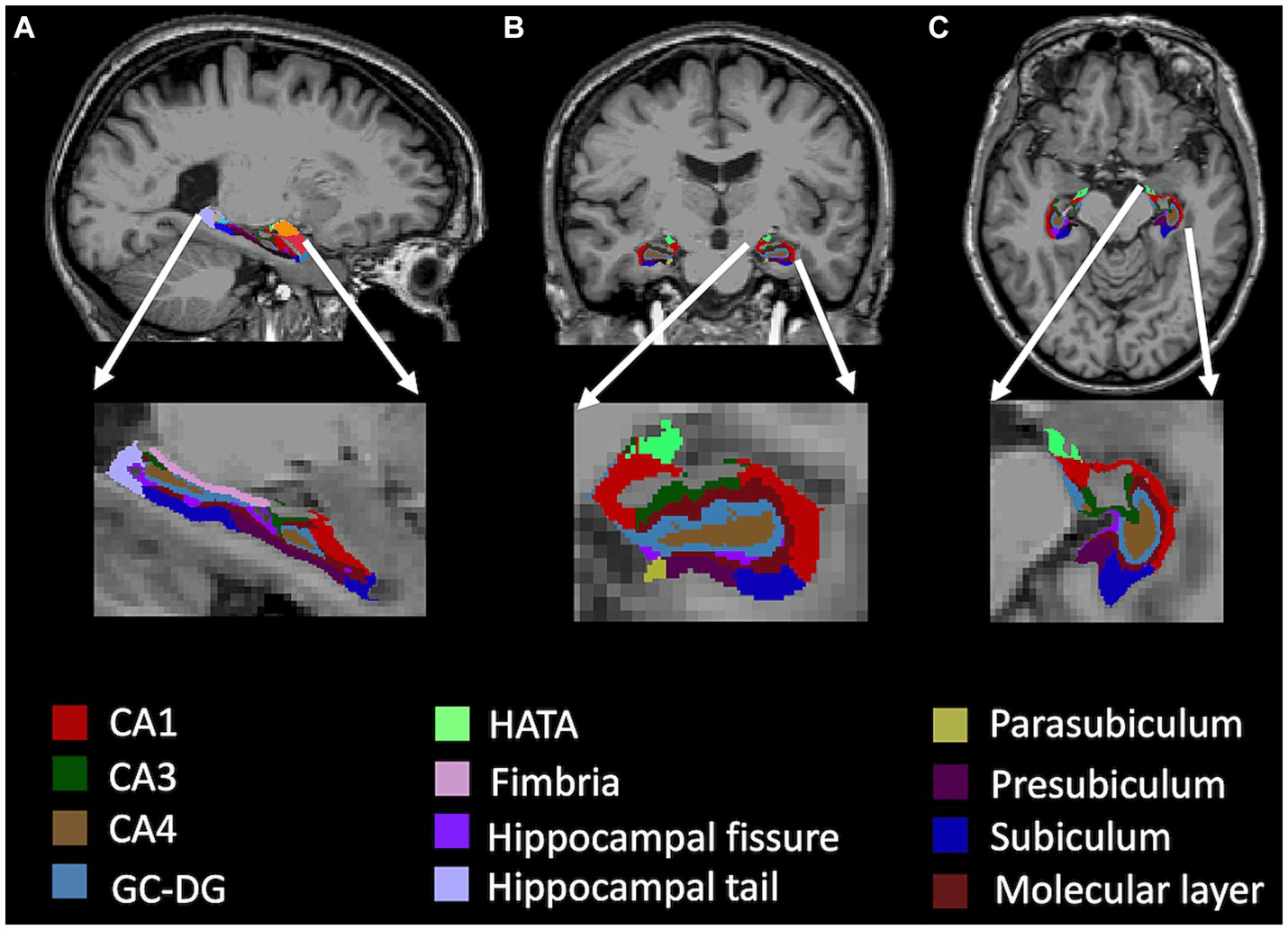
Specific to AD, neuronal loss and gliosis in the hippocampal region is a prevalent pathological feature18. Early dysfunction and synaptic loss that primarily affects excitatory transmission in the hippocampus and cerebral cortex, are key contributors to memory loss in AD40. Vulnerable regions include glutamatergic neurons in the entorhinal cortex and pyramidal neurons in CA141. Histopathologically, AD is characterized by neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein, first appearing in the CA1 region before spreading to the subiculum, CA2, CA3, and dentate gyrus (DG)18 (Figure 4). Concurrently, amyloid-beta (Aβ) plaques accumulate in the entorhinal cortex and later invade the hippocampus, disrupting synaptic communication18. These degenerative changes are exacerbated by neuroinflammation, oxidative stress, and gliosis, resulting in hippocampal atrophy, reduced cortical connectivity, and cognitive dysfunction, as described by the “hippocampo-cortical dissociation” hypothesis42. Imaging studies show bilateral atrophy in hippocampal layers such as the stratum radiatum, stratum lacunosum, and subiculum’s stratum pyramidale, all detectable through MRI43. Disruptions in glutamatergic, serotonergic, and noradrenergic neurotransmitter systems correlate with neuronal loss. Experimental models also link tau and Aβ pathology with disrupted DG connectivity, paralleling the progressive degeneration observed in human AD12. Clinically, hippocampal damage manifests as memory deficits, including both anterograde and retrograde amnesia, with advanced AD stages marked by severe cognitive decline and confabulations44.
Methods
This experiment focuses on extracting and comparing the intrinsic features of hippocampal pyramidal neurons in both Alzheimer’s and Control groups. The data was provided by the Daou Lab.
Alzheimer’s disease model mice used in the current study were generated using the APP transgenic mice (PDAPP), which causes robust amyloid-beta plaque deposition. Control animals, referred to as the “intact” group, were not genetically modified and served as the control. All the animals were maintained under controlled settings and manipulated in line with the animal research ethics guidelines. All the procedures were subject to approval by the University of Chicago Institutional Animal Care and Use Committee (IACUC).
Hippocampal tissue slices were prepared from the CA1 region of the hippocampus of a total of 11 adult male mice (5 control and 6 Alzheimer’s disease model), with each animal contributing one neuron to the dataset. The CA1 subfield was targeted given its selective vulnerability to neurofibrillary tangle accumulation and synaptic loss in AD. After deep anesthesia with isoflurane, animals were decapitated, and brains were rapidly removed and placed in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 119 NaCl, 2.5 KCl, 1.3 MgCl₂, 2.5 CaCl₂, 1.0 NaH₂PO₄, 26.2 NaHCO₃, and 22 glucose (pH 7.2–7.3; 285–295 mOsm), aerated with a 95% O₂ / 5% CO₂ gas mix. Slices were cut at 300–400 μm thickness using a vibratome and placed directly into an oxygenated ACSF-filled incubation chamber at 37°C. Slices were incubated at 37°C for at least 1 hour and then allowed to equilibrate to room temperature (~24–25°C) prior to recordings. Whole-cell current-clamp recordings were obtained using borosilicate glass pipettes with a resistance of 3–8 MΩ, filled with intracellular solution containing (in mM): 135 K-gluconate, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, 0.2 EGTA, pH adjusted to 7.2–7.3 with KOH. Series resistance (Rs) was monitored throughout each recording and compensated online when necessary. Cells were included in the analysis only if they met the following criteria: resting membrane potential ≤ −65 mV at baseline and stable throughout the recording, series resistance between 3.5–10 MΩ, and consistent spiking across the recording period. Input resistance was measured from small hyperpolarizing current steps (5–25 pA, 200–300 ms), and leak currents were monitored to ensure recording stability. All recordings were conducted blind to experimental group. Tight-seal recordings (>1 GΩ) were obtained, and neurons were allowed to dialyze for several minutes before stimulation. Recordings were obtained under visual control using a Multiclamp 700B amplifier, and data were sampled at 50 kHz.
Pharmacological blockers were utilized to suppress synaptic input throughout electrophysiological recordings, thereby isolating intrinsic neuronal excitability from synaptic contributions. All drug treatments were administered via a gravity-fed perfusion system that facilitated continuous and accurate delivery to the tissue slices. Synaptic transmission was blocked in all standard recordings using NMDA, AMPA/kainate, GABAₐ, and GABA₂ receptor antagonists (CNQX, CPP, Gabazine, and CGP 35348), ensuring that the action potential properties measured reflect intrinsic excitability rather than network-driven activity. Additional voltage-gated channel blockers—including cadmium chloride, TEA, cesium chloride, and TTX—were applied selectively in mechanistic sub-experiments designed to isolate specific ionic conductances, and were not used during the standard action potential recordings reported here.
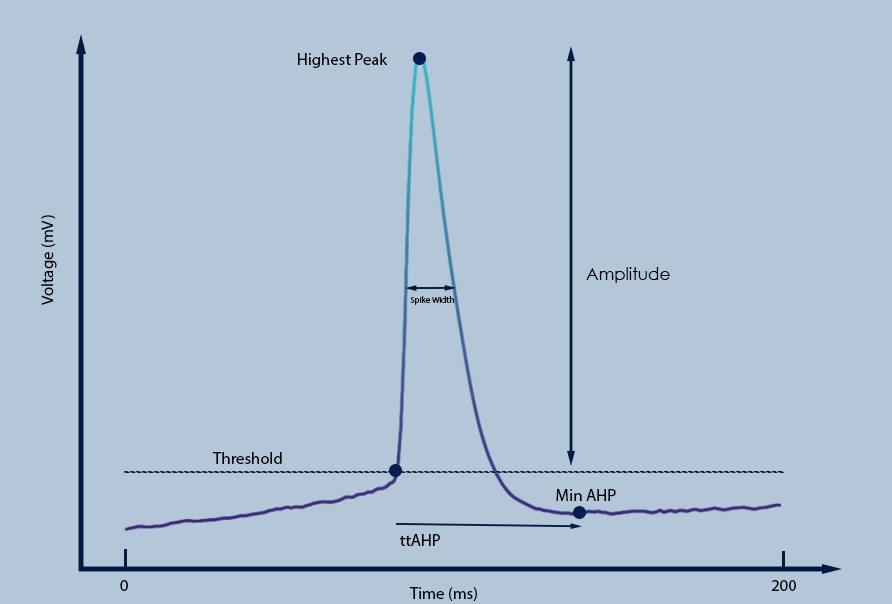
For the purposes of this paper, the primary coding language used to extract the features of the spikes is Python. Using Python allowed for leverage of the versatile and rich libraries that were available for scientific computing and data visualization. In addition, the Spyder software was used to run the code as well as curate graphs and visualizations for the given data. For each detected spike, a local window around the peak was extracted to isolate individual action potential (AP) events. Within this window, three principal components of the AP were quantified: the threshold, defined as the first point where the membrane potential’s rate of change (dV/dt) exceeded 0.5 mV per sample—equivalent to 25 mV/ms at the 50 kHz sampling rate—consistent with the derivative-based approach described in Daou and Margoliash (2020)45. Prior to computing dV/dt, voltage traces were smoothed using a Gaussian kernel with a standard deviation of 1 sample (~0.02 ms) to reduce high-frequency noise while preserving the rapid depolarization of genuine action potentials; the peak, corresponding to the maximum voltage within the spike; and the afterhyperpolarization (AHP), defined as the difference between the threshold and the minimum voltage following the peak within the same window. Spike amplitude was calculated as the difference between the peak and the threshold voltage (Figure 5). The width of the spike was estimated as the duration over which the membrane potential exceeded half the spike amplitude. The time to peak and time to AHP were computed as the respective latencies from the threshold point. Spike frequency was calculated as the total number of spikes divided by the duration of the detected stimuli (in seconds), yielding a measure of firing rate in Hertz. For all pairwise comparisons between AD and control neurons, independent samples t-tests were performed using Python’s SciPy library. A threshold of p < 0.01 was used to define statistical significance.
Results
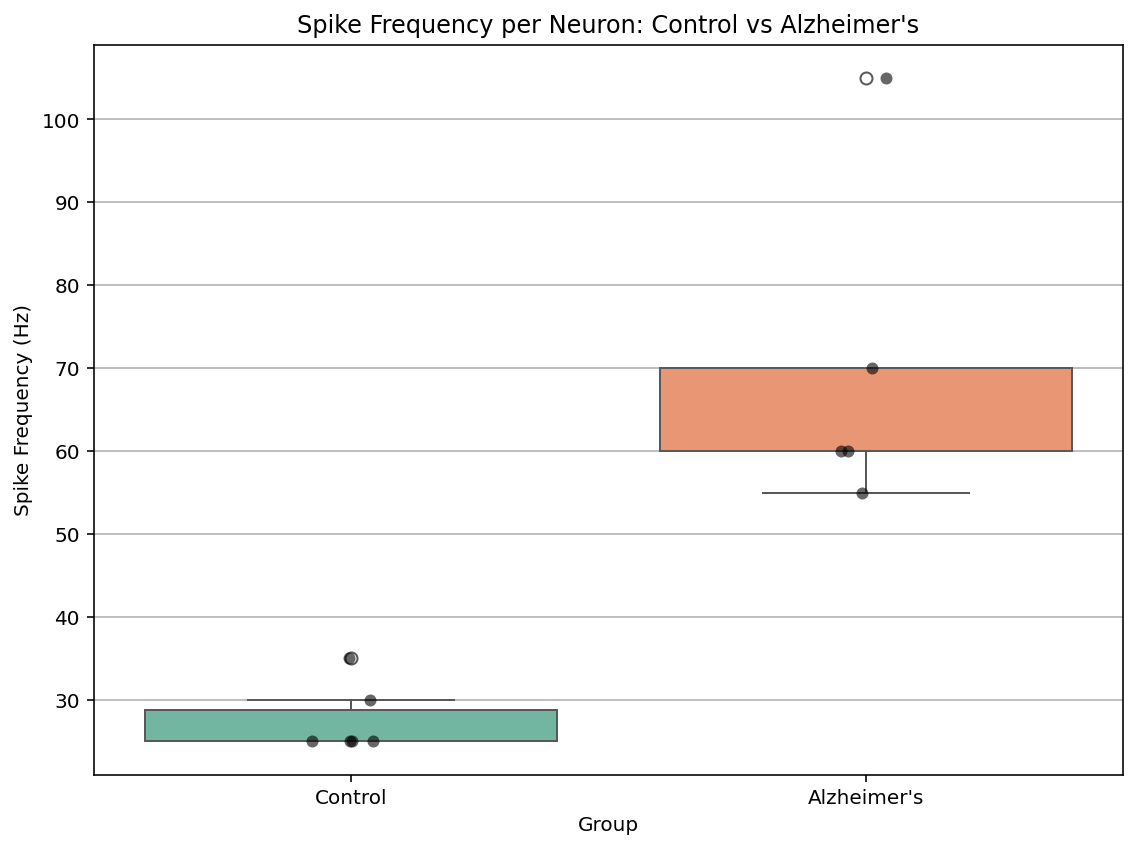
To investigate the IPs of hippocampal pyramidal neurons in Alzheimer’s disease (AD) mouse model, we firstly visualized the spikes trains of their elicited membrane potential traces in response to current stimuli and compared them with quantitative metrics of spiking dynamics between AD and control groups. Representative traces revealed that AD neurons (Figure 6) exhibited a dramatically markedly increased elevated spike frequency, narrower spike widths, and lower peaks in comparison with control neurons (Figure 6).
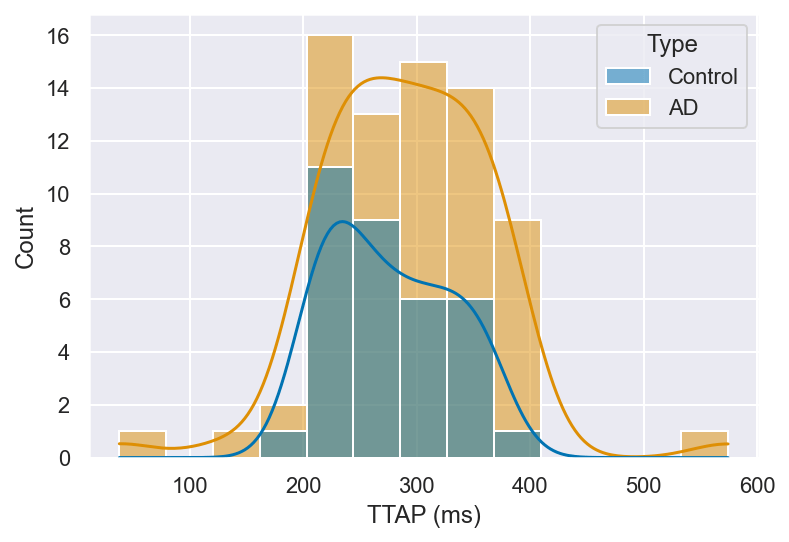
The spike amplitude of APs directly influences the strength of neuronal firing and the efficacy of subsequent synaptic transmission, while spike width and time to peak play crucial roles in determining the temporal precision of information encoding and the ability to sustain high-frequency firing. A detailed analysis of these features can thus provide important insights into the altered intrinsic properties of neurons in AD. With this consideration in mind, we quantified spike amplitude, spike width, and time to peak in both control and AD neurons, as illustrated in Figure 7. (t = 11.63), p < 0.01).
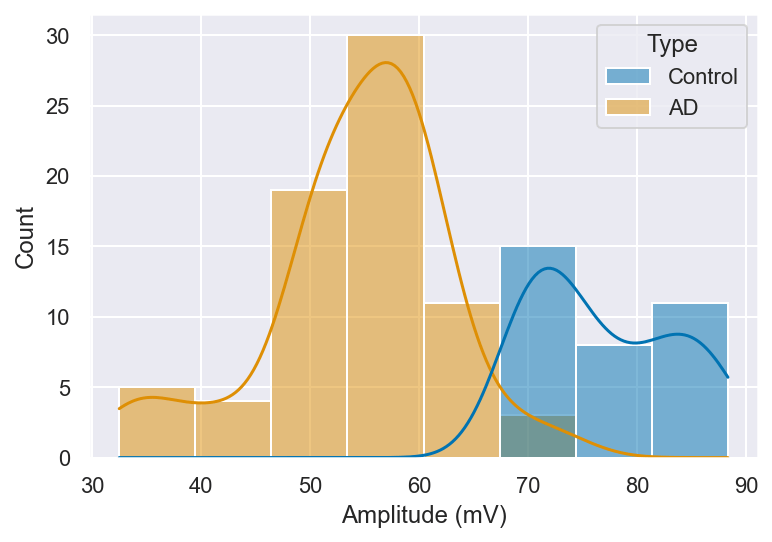
Possible explanations as to what lower spike amplitudes could signify include reduced voltage-gated sodium channel availability or partial channel inactivation, which would limit the inward Na⁺ current during the depolarization phase and thereby reduce peak voltage. Upregulation of potassium channel conductance could further truncate the amplitude by accelerating repolarization before the peak is reached. Furthermore, physiological changes to neurons such as synaptic loss and axonal degradation may result in weaker spike propagation and reduced signal strength. The changes seen in Figure 7 could represent the loss of functional homeostasis in the neural network, potentially contributing to cognitive deficits seen in AD.
The next feature measured was the spike threshold, which was seen to be significantly more depolarized in AD samples compared to controls (t = -4.99, p < 0.01). Examining the thresholds for each individual spike elicited in both control and AD pyramidal neurons showed significant changes among the two groups (Figure 8). Spike threshold is a critical determinant of IE and dictates the minimum voltage required to initiate an AP, it thereby influencing the neuron’s responsiveness to incoming stimuli.
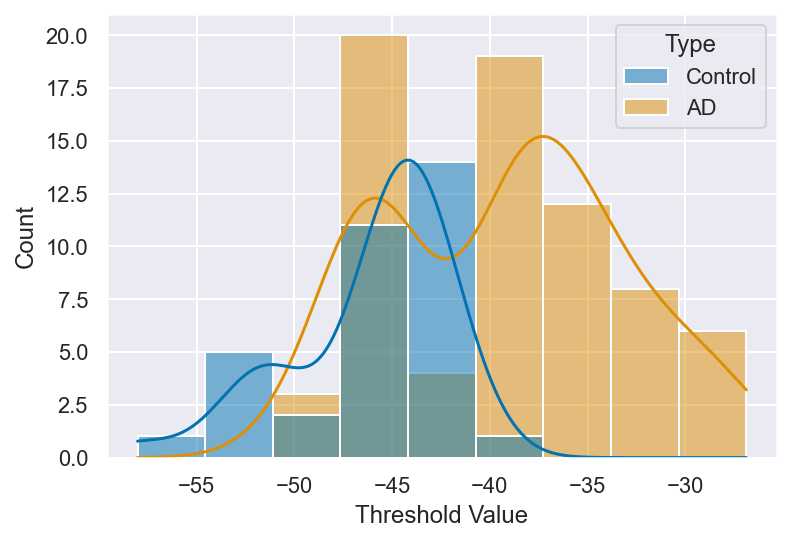
A spike’s threshold is largely determined by voltage-gated sodium channels, which open to allow the influx of sodium ions during the initiation of an action potential. The increased depolarization of the neurons in the AD group could suggest reduced sensitivity of sodium channels or altered gating mechanisms. With a higher spike threshold, neurons are less responsive to excitatory input and may fail to fire altogether during low-frequency stimulation.
The AHP values for the action potentials were significantly higher in AD neurons compared to controls (Figure 9) (t = -7.18, p < 0.01). The AHP that follows each spike is an important determinant of intrinsic excitability. It sets the refractory window and modifies the pattern and accuracy of recurrent firing. The amplitude of the AHP and the time it takes to reach its peak (time to AHP peak) determine how effectively a neuron can manage incoming signals and how quickly it can get back to firing. Thus, changes in these properties could have great effects on how neuronal networks perform.
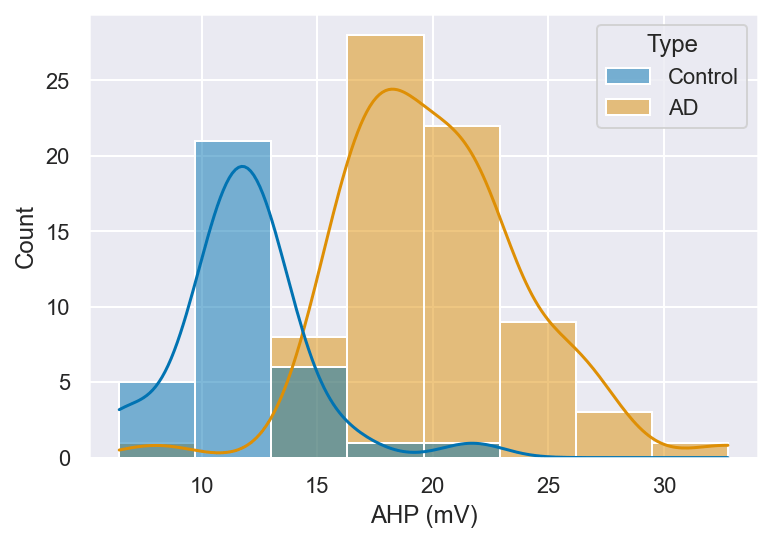
AHP is primarily mediated by calcium-activated potassium channels, particularly small-conductance (SK) and large-conductance (BK) channels, and contributes to the resetting of the membrane potential following each action potential. In this study, the AHP amplitude was significantly higher in AD neurons compared to controls. Importantly, this larger measured AHP amplitude does not contradict the observed increase in spike frequency. In AD, downregulation of SK channel expression and activity is well established, which functionally reduces the inhibitory SK-mediated K⁺ current that normally suppresses repetitive firing. When SK channel currents are diminished, the inhibitory brake on neuronal firing is weakened, allowing neurons to fire more rapidly despite the presence of a larger AHP—a dissociation that has been documented in models of hippocampal hyperexcitability. The enlarged AHP amplitude observed here may therefore reflect a compensatory upregulation of other potassium conductances, or altered calcium-activated channel kinetics, rather than a net increase in inhibitory tone. Critically, it is also important to note that a central feature of AD neurotoxicity is the disruption of intracellular calcium homeostasis. Elevated intracellular Ca²⁺ levels resulting from dysregulated endoplasmic reticulum (ER) Ca²⁺ release and impaired buffering can directly activate calcium-sensitive potassium channels including SK and BK channels, thereby modifying AHP amplitude and contributing to altered firing patterns. Resting membrane potential was not significantly different between groups, indicating that the increased excitability is primarily driven by altered ion channel dynamics rather than a shift in baseline membrane state. No significant difference was found in time to peak AHP between control and AD samples (Figure 10) (t = -0.75, p = 0.46). This suggests that the timing of the AHP remains largely consistent between the two groups. The preservation of time-to-peak AHP suggests that while the amplitude of potassium channel-mediated currents may be altered in AD neurons, the kinetics of channel activation and deactivation remain largely intact. This dissociation between amplitude and timing may indicate that the number or conductance of active channels is affected, rather than the fundamental gating properties of the channels themselves.
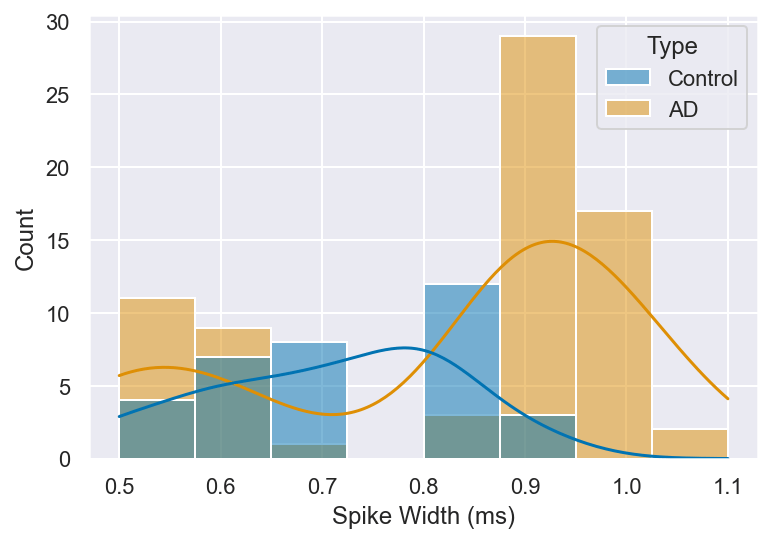
Finally, the spike widths in AD neurons tended to be narrower, suggesting that the duration of the action potential was reduced compared to the control group (Figure 11). This could imply that the depolarization phase of the action potential is shorter, possibly reflecting faster repolarization and altered ion channel dynamics. The narrowing of spike width may reflect enhanced potassium channel conductance and/or reduced sodium channel availability, both of which would accelerate repolarization. Upregulation of voltage-gated K⁺ channels such as Kv3 or Kv4.2 could drive faster repolarization, while reduced Nav channel density would limit the duration of the inward Na⁺ current sustaining the depolarization plateau. Statistically, the differences between the two groups did not reach the pre-defined significance threshold (t = 2.26, p = 0.03), and this trend warrants further investigation with larger sample sizes.
Conclusion
Our results reveal robust alterations in intrinsic properties of hippocampal pyramidal neurons in the AD mouse model compared to healthy controls, extending current understanding of AD-related hippocampal dysfunction. Specifically, we observed consistent changes across several features, including spike amplitude, spike width, time to peak, AHP amplitude, and time to AHP peak. These findings support our original hypothesis that hippocampal neurons in AD undergo intrinsic excitability changes that may reflect early biophysical correlates of cognitive dysfunction.
The increased spike frequency in AD neurons suggests enhanced intrinsic excitability, likely driven by altered ion channel function. Downregulation of SK channels reduces the afterhyperpolarization-mediated inhibitory brake on repetitive firing, while downregulation of Kv1 channels shortens interspike intervals and weakens spike-frequency adaptation. Possible downregulation of Kv3 channels, which support fast spiking, may further contribute to this phenotype. In parallel, reduced Nav1.6 availability or a depolarizing shift in its inactivation curve may lower the spike threshold and accelerate depolarization, facilitating earlier firing despite reduced peak amplitude. Together, these intrinsic mechanisms likely underlie the heightened excitability observed here, and this increased excitability is consistent with previous studies linking AD to excitotoxicity in the hippocampus46. It is important to note that while the recordings in this study were performed under synaptic blockade—isolating intrinsic excitability from network-driven input—in the intact AD brain, synaptic changes would act in concert with the intrinsic alterations described here. In particular, AD-related downregulation of inhibitory GABAₐ currents and upregulation of excitatory AMPA receptor activity would further promote neuronal hyperexcitability beyond what is observed from intrinsic mechanisms alone. The results presented here therefore represent a lower bound on the degree of excitability change in the living AD brain.
One important interpretive framework for these findings is homeostatic plasticity. The observed changes—including elevated AHP amplitude, higher spike threshold, and reduced spike amplitude—may not represent purely pathological deterioration, but rather a compensatory response by which neurons attempt to counteract an initial state of hyperexcitability driven by amyloid-beta toxicity, impaired calcium buffering, and disrupted inhibitory tone. Under this framework, the neuron upregulates inhibitory conductances in an attempt to restore firing to a physiological set point. However, if SK channel downregulation simultaneously reduces the effectiveness of the AHP as an inhibitory brake, the net result is paradoxical: compensatory changes fail to suppress firing, and neurons remain hyperexcitable despite the presence of a larger AHP—a dissociation that highlights the complexity of excitability changes in AD and underscores the need for mechanistic follow-up studies to disentangle adaptive from maladaptive alterations.
These findings highlight the importance of intrinsic neuronal properties in the pathophysiology of AD, emphasizing that alterations in the ionic mechanisms governing action potential generation and repolarization can lead to impaired neuronal communication, network dysfunction, and ultimately cognitive decline. In doing so, our work contributes to filling a gap in the literature by quantitatively linking specific biophysical changes in hippocampal neurons to potential early drivers of AD pathology, providing a cellular-level perspective that complements existing synaptic and molecular studies. Understanding these changes in neuronal excitability provides crucial insights into the underlying cellular mechanisms of AD, with potential implications for the development of targeted therapeutic strategies aimed at restoring normal neuronal function and mitigating the cognitive impairments associated with the disease. For example, future research could employ pharmacological modulation of specific potassium and sodium channels in vivo to determine whether normalizing excitability can rescue memory performance in AD models or use high-resolution imaging to track how these excitability changes progress during disease stages.
One limitation of this study is the relatively modest sample size, which may affect the statistical power and generalizability of certain findings. In conclusion, the observed alterations in neuronal excitability may represent an early and targetable component of AD pathology, suggesting that therapies aimed at stabilizing intrinsic firing properties could complement approaches focused on reducing amyloid or tau pathology. By advancing our understanding of how the disease alters the fundamental electrical behavior of hippocampal neurons, this study underscores the importance of integrating electrophysiological biomarkers into both basic and translational AD research, helping to build a clearer connection between cellular-level changes and the progression of cognitive symptoms in Alzheimer’s disease.
References
- B. Reisberg, S. H. Ferris, M. J. de Leon, T. Crook. Functional staging of dementia of the Alzheimer type. Annals of the New York Academy of Sciences, 435, 481–483 (1984). https://doi.org/10.1111/j.1749-6632.1984.tb13859.x [↩]
- M. T. Ferretti, M. F. Iulita, E. Cavedo, P. A. Chiesa, A. S. Dimech, A. S. Chadha, H. Hampel. Sex and gender differences in Alzheimer’s disease: current challenges and implications for clinical practice. European Journal of Neurology, 27, 928–943 (2020). https://doi.org/10.1111/ene.14174 [↩]
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to dementia. London: ADI (2019). [↩]
- World Health Organization. Dementia (2021). https://www.who.int/news-room/fact-sheets/detail/dementia [↩] [↩]
- M. Prince, G. C. Ali, M. Guerchet, A. M. Prina, E. Albanese, Y. T. Wu. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimer’s Research & Therapy, 8, 23 (2016). https://doi.org/10.1186/s13195-016-0188-8 [↩]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted trends through 2050: an analysis for the Global Burden of Disease Study 2019. The Lancet Public Health, 7, e105–e125 (2022). https://doi.org/10.1016/S2468-2667(21)00249-8 [↩]
- K. Iqbal, I. Grundke-Iqbal. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. Journal of Cellular and Molecular Medicine, 12, 38–55 (2008). https://doi.org/10.1111/j.1582-4934.2008.00225.x [↩] [↩]
- R. H. Takahashi, T. Nagao, G. K. Gouras. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathology International, 67, 185–193 (2017). https://doi.org/10.1111/pin.12520 [↩] [↩]
- Z. L. Almeida, D. C. Vaz, R. M. Brito. Morphological and molecular profiling of amyloid-β species in Alzheimer’s pathogenesis. Molecular Neurobiology, 62, 4391–4419 (2024). https://doi.org/10.1007/s12035-024-04543-4 [↩]
- T. F. Gendron, L. Petrucelli. The role of tau in neurodegeneration. Molecular Neurodegeneration, 4, 13 (2009). https://doi.org/10.1186/1750-1326-4-13 [↩]
- D. P. Perl. Neuropathology of Alzheimer’s disease. Mount Sinai Journal of Medicine: A Journal of Translational and Personalized Medicine, 77, 32–42 (2010). https://doi.org/10.1002/msj.20157 [↩]
- T. F. Gendron, L. Petrucelli. The role of tau in neurodegeneration. Molecular Neurodegeneration, 4, 13 (2009). https://doi.org/10.1186/1750-1326-4-13 [↩] [↩]
- Y. Xia, S. Prokop, B. I. Giasson. “Don’t phos over tau”: recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Molecular Neurodegeneration, 16, (2021). https://doi.org/10.1186/s13024-021-00460-5 [↩]
- A. Schneider, J. Biernat, M. von Bergen, E. Mandelkow, E. M. Mandelkow. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry, 38, 3549–3558 (1999). https://doi.org/10.1021/bi981874p [↩]
- Y. Chen, Y. Yu. Tau and neuroinflammation in Alzheimer’s disease: interplay mechanisms and clinical translation. Journal of Neuroinflammation, 20, (2023). https://doi.org/10.1186/s12974-023-02853-3 [↩]
- H. S. Kwon, S. Koh. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Translational Neurodegeneration, 9, (2020). https://doi.org/10.1186/s40035-020-00221-2 [↩]
- C. M. Moloney, V. J. Lowe, M. E. Murray. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: a clinicopathologic perspective for biomarker research. Alzheimer’s & Dementia, 17, 1554–1574 (2021). https://doi.org/10.1002/alz.12321 [↩] [↩]
- M. A. DeTure, D. W. Dickson. The neuropathological diagnosis of Alzheimer’s disease. Molecular Neurodegeneration, 14, (2019). https://doi.org/10.1186/s13024-019-0333-5 [↩] [↩] [↩] [↩] [↩] [↩]
- M. M. Rahman, C. Lendel. Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology. Molecular Neurodegeneration, 16, (2021). https://doi.org/10.1186/s13024-021-00465-0 [↩]
- Y. W. Zhang, R. Thompson, H. Zhang, H. Xu. App processing in Alzheimer’s disease. Molecular Brain, 4, 3 (2011). https://doi.org/10.1186/1756-6606-4-3 [↩]
- S. L. Cole, R. Vassar. The Alzheimer’s disease beta-secretase enzyme, BACE1. Molecular Neurodegeneration, 2, 22 (2007). https://doi.org/10.1186/1750-1326-2-22 [↩]
- M. Colom-Cadena, T. Spires-Jones, H. Zetterberg, K. Blennow, A. Caggiano, S. T. DeKosky, J. E. Harrison, L. S. Schneider, P. Scheltens, W. de Haan, M. Grundman, C. H. van Dyck, N. J. Izzo, S. M. Catalano. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimer’s Research & Therapy, 12, 21 (2020). https://doi.org/10.1186/s13195-020-00588-4 [↩]
- J. Gjorgjieva, R. A. Mease, W. J. Moody, A. L. Fairhall. Intrinsic neuronal properties switch the mode of information transmission in networks. PLoS Computational Biology, 10, e1003962 (2014). https://doi.org/10.1371/journal.pcbi.1003962 [↩]
- T. Guo, D. Zhang, Y. Zeng, T. Y. Huang, H. Xu, Y. Zhao. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Molecular Neurodegeneration, 15, (2020). https://doi.org/10.1186/s13024-020-00391-7 [↩]
- G. Daoudal, D. Debanne. Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learning & Memory, 10, 456–465 (2003). https://doi.org/10.1101/lm.64103 [↩]
- A. X. Patel, D. Burdakov. Mechanisms of gain control by voltage-gated channels in intrinsically-firing neurons. PLOS ONE, 10, e0115431 (2015). https://doi.org/10.1371/journal.pone.0115431 [↩]
- A. Daou, D. Margoliash. Intrinsic plasticity and birdsong learning. Neurobiology of Learning and Memory, 180, 107407 (2021). https://doi.org/10.1016/j.nlm.2021.107407 [↩]
- J. Echegoyen, A. Neu, K. D. Graber, I. Soltesz. Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLOS ONE, 2, e700 (2007). https://doi.org/10.1371/journal.pone.0000700 [↩]
- T. O’Leary, M. C. W. van Rossum, D. J. A. Wyllie. Homeostasis of intrinsic excitability in hippocampal neurones: dynamics and mechanism of the response to chronic depolarization. The Journal of Physiology, 588, 157–170 (2010). https://doi.org/10.1113/jphysiol.2009.181024 [↩]
- C. C. Pang, C.Kiecker, J. T.O’Brien, W.Noble and R. C. Chang, Ammon’s horn 2 (CA2) of the hippocampus: a long-known region with a new potential role in neurodegeneration. The Neuroscientist, 25, 167–180 (2018). https://doi.org/10.1177/1073858418778747 [↩]
- M. A. Meyer. The hippocampus. In Neurologic Disease (pp. 127–141). Springer, Cham (2016). https://doi.org/10.1007/978-3-319-39581-4_6 [↩]
- L. A. Fogwe, V. Reddy, F. B. Mesfin. Neuroanatomy, hippocampus. StatPearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK482171/ (2023). [↩]
- H. Eichenbaum. The role of the hippocampus in navigation is memory. Journal of Neurophysiology, 117, 1785–1796 (2017). https://doi.org/10.1152/jn.00005.2017 [↩]
- R. Knoth, I. Singec, M. Ditter, G. Pantazis, P. Capetian, R. P. Meyer, V. Horvat, B. Volk, G. Kempermann. Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLOS ONE, 5, e8809 (2010). https://doi.org/10.1371/journal.pone.0008809 [↩]
- A. Wu, J. Zhang. Neuroinflammation, memory, and depression: new approaches to hippocampal neurogenesis. Journal of Neuroinflammation, 20, (2023). https://doi.org/10.1186/s12974-023-02964-x [↩]
- K. T. Sumadevi. The hippocampus: anatomy, function and clinical correlation. Sri Lanka Anatomy Journal, 8, 6–20 (2024). https://doi.org/10.4038/slaj.v8i1.220 [↩] [↩]
- S. Ohara, S. Sato, K. Tsutsui, M. P. Witter, T. Iijima. Organization of multisynaptic inputs to the dorsal and ventral dentate gyrus: retrograde trans-synaptic tracing with rabies virus vector in the rat. PLOS ONE, 8, e78928 (2013). https://doi.org/10.1371/journal.pone.0078928 [↩]
- L. R. Squire, L. Genzel, J. T. Wixted, R. G. Morris. Memory consolidation. Cold Spring Harbor Perspectives in Biology, 7, a021766 (2015). https://doi.org/10.1101/cshperspect.a021766 [↩]
- Y. Xiao, Y. Hu, K. Huang. Atrophy of hippocampal subfields relates to memory decline during the pathological progression of Alzheimer’s disease. Frontiers in Aging Neuroscience, 15, (2023). https://doi.org/10.3389/fnagi.2023.1287122 [↩]
- L. Crews, E. Masliah. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Human Molecular Genetics, 19, R12–R20 (2010). https://doi.org/10.1093/hmg/ddq160 [↩]
- G. W. van Hoesen, B. T. Hyman, A. R. Damasio. Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus, 1, 1–8 (1991). https://doi.org/10.1002/hipo.450010102 [↩]
- B. T. Hyman, G. W. Van Hoesen, A. R. Damasio, C. L. Barnes. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science, 225, 1168–1170 (1984). https://doi.org/10.1126/science.6474172 [↩]
- E. Frankó, O. Joly, Alzheimer’s Disease Neuroimaging Initiative. Evaluating Alzheimer’s disease progression using rate of regional hippocampal atrophy. PLOS ONE, 8, e71354 (2013). https://doi.org/10.1371/journal.pone.0071354 [↩]
- W. B. Scoville, B. Milner. Loss of recent memory after bilateral hippocampal lesions. The Journal of Neurology, Neurosurgery, and Psychiatry, 20, 11–21 (1957). https://doi.org/10.1136/jnnp.20.1.11 [↩]
- A. Daou, D. Margoliash. Intrinsic neuronal properties represent song and error in zebra finch vocal learning. Nature Communications, 11, (2020). https://doi.org/10.1038/s41467-020-14738-7 [↩]
- Q. Deng, X. Yu, Y. Qin, S. Chen, J. Zhao, H. Zhang. Microglia and astrocytes in Alzheimer’s disease: significance and summary of recent advances. Aging and Disease, 14, (2023). https://doi.org/10.14336/ad.2023.0907 [↩]



{kind=link}