
Abstract
Neuronal migration (NM) is a regulated process essential for neurodevelopment and circuit formation. This systematic review aims to examine how Traumatic Brain Injury (TBI) disrupts interneuron migration, leading to enhanced risk of epilepsy. NM was previously thought to occur only during gestation; however, it’s recently been established migration continues postnatally throughout infancy. Continued migration after birth suggests a critical window of interneuron disruption that is susceptible to environmental factors, thereby increasing the risk of epilepsy in infants. Traumatic Brain Injury (TBI), an established epilepsy risk factor, may further increase the risk of disorders including epilepsy, by disrupting inhibitory neuronal migration through radial glial damage and scaffolding disorganization. Studies in pediatric populations have established that TBI is associated with an increased risk of epilepsy. Experimental evidence has established that TBI disrupts glial scaffolds, which are crucial for proper NM and disruption possibly contributes to migration abnormalities. Inhibitory neurons have also been shown to migrate along astrocytic sheaths and vascular scaffolds while relying on radial glial cells and cytoskeleton remodeling for proper directional movement. Trauma-induced disruptions of mechanical structures during interneuron migration in early development have been proposed to impair proper laminar integration of GABAergic interneurons, thereby impairing inhibitory signaling and causing an excitatory-inhibitory imbalance associated with epileptogenesis. To investigate therapeutic models, rodent studies indicated that iPSC-derived interneuron progenitors reduce seizures by restoring GABAergic-inhibition. However, translation to human systems remains limited due to increased migratory distance and ethical constraints. While these findings underscore their therapeutic potential, transition to human models remains challenging.
Keywords: neuronal migration, traumatic brain injury, epilepsy, inhibitory interneurons, stem cell therapy
Introduction
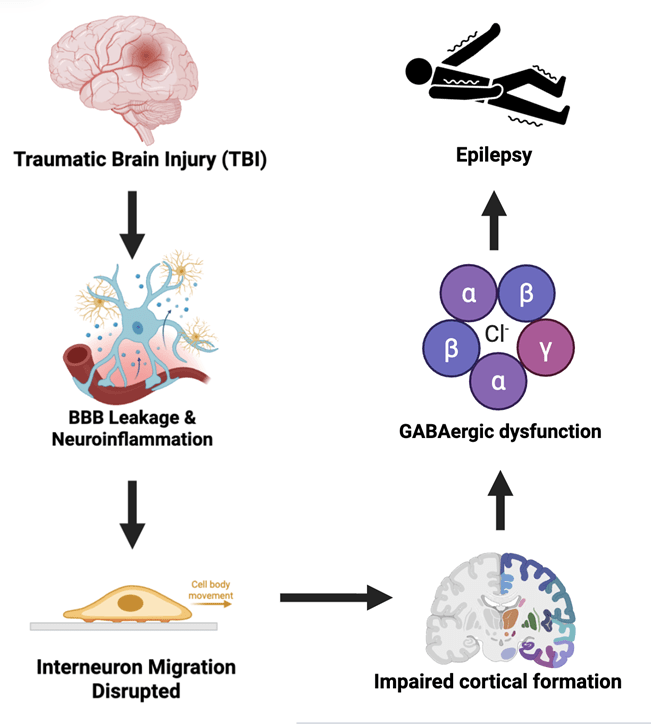
Neuronal migration is the fundamental process by which neurons travel from germinal zones (Ventricular and Sub-Ventricular Zones) to final positions in the brain after maturing. Proper positioning of these neurons is imperative, as it ensures proper cortical layering and circuit formation and provides a balance of excitatory and inhibitory signaling. It’s been established that the disruption of migratory processes was associated with abnormal cortical architecture and neurodevelopmental disorders, such as epilepsy, because of an imbalance in excitatory-inhibitory signaling1. Traumatic Brain Injury (TBI) is a well-established risk factor for epilepsy and is particularly prominent in pediatric populations2. Unlike other neurodevelopmental disorders, epilepsy arises from an excitatory-inhibitory imbalance caused by trauma induced scaffolding misalignment, showing the imperative for why interneurons mispositioned during development are critical factors for post-traumatic seizure susceptibility3. Population studies have shown a several-fold increase in lifetime risk of post-traumatic epilepsy following moderate to severe childhood TBI2. Despite substantial progress in characterizing TBI with epileptogenesis in adolescents, we know relatively little about the mechanical and inflammatory injury mechanisms that drive the imbalance in inhibitory interneuron signaling2. Understanding this process is imperative, as positional disruption of interneurons permanently predisposes chronic seizure disorders.2. Traditionally, neuronal migration was considered a dominant prenatal process; however, recent studies have indicated that specific interneurons continue to migrate into early infancy. Recent studies have identified migratory streams of immature neurons that targeted frontal cortical regions during postnatal brain development4 vulnerability to environmental factors that can disrupt interneuron migratory processes, including TBI. Thus, this extended postnatal window contributes to a sensitive developmental phase in which disturbances of the excitatory-inhibitory balance may occur, resulting from disrupted postnatal migration, and may precipitate long-term network instability and an increased risk of epileptogenesis. Despite growing evidence of the TBI linkage to epilepsy, the potential of postnatal interneuron migration in post-traumatic epileptogenesis has received limited attention. This systematic review aims to review current evidence on how TBI may disrupt inhibitory interneuron migration via vascular and glial scaffolding, thereby causing an imbalance in excitatory-inhibitory signaling, and to evaluate the implications of these disturbances for epileptogenesis, while highlighting the possible implications of future human stem-cell-based models.
Methods
This systematic review was conducted in accordance with the PRISMA 2020 guidelines to ensure reproducibility and transparency. Literature searches were conducted between January 2018 and December 2024 using three databases: PubMed, Paper Guide, and Google Scholar. Search strategies included free-text keywords and Medical Subject Headings (MeSH) terms, including “Neuronal Migration Disorders” [MeSH], “Traumatic Brain Injury” [MeSH], “inhibitory interneurons”, and “epileptogenesis”. A total of 82 studies were identified across all databases, with 45 articles screened by abstract and 23 meeting the inclusion criteria for full review. Duplicates were removed prior to abstract screening. The full-text articles were excluded if they lacked relevance to neuronal migration, traumatic brain injury, or insufficient detail. The author screened titles, abstracts, and all full texts, and inclusion details were based on the predefined eligibility criteria. The study selection process was summarized using a PRISMA 2020 flow diagram. The inclusion criteria required studies to be (1) peer-reviewed, (2) published between 2018 and 2024, (3) written in English, (4) focus on TBI, epileptogenesis, neuronal migration, or inhibitory interneurons, and (5) available in full-text. To preserve theoretical context and background information, many foundational studies (1997-2007) that described mechanisms of neuronal migration and cortical malformation were also included. Exclusion criteria eliminated conference abstracts, non-peer-reviewed material, duplicates, and articles unrelated to neuronal migration or neurotrauma. The study quality was assessed qualitatively based on the experimental design, the relevance of inhibitory interneuron migration, and the strength of the evidence in the review. Many studies with indirect or speculative links to migratory disruption were downplayed in priority during the literature synthesis.
| Study | Model & Age | TBI Model | Migration Evidence | Interneuron Type | Epilepsy Outcome | Key Finding |
| Zhu et al., 2019 | Mouse, Juvenile | CCI | Reduced interneuron density (IHC) | PV+, SST+ | Spontaneous seizures | Hippocampal interneuron loss linked to seizure phenotype |
| Hunt et al., 2013 | Rodent, Juvenile | Experimental TBI | Dentate gyrus interneuron loss | Hilar interneurons | Seizure susceptibility | Reduced inhibitory populations in epileptogenic hippocampus |
| Brizuela et al., 2017 | Mouse, Postnatal | CCI | Altered neurite orientation | Calretinin+ | Not primary | Injury-induced remodeling near lesion site |
| Raghupathi, 2004 | Rodent, Developing | Experimental TBI | Radial glial fiber disruption | Not specified | Not measured | Scaffold misalignment after injury |
| Super et al., 2000 | Rodent, Developmental | Experimental manipulation | Radial glial mapping | Migrating interneurons | Not measured | Proper scaffold alignment required for laminar positioning |
| Bressan & Saghatelyan, 2021 | Human, Postnatal | N/A | Migratory stream tracing | Immature interneurons | Not measured | Postnatal interneuron migration persists into infancy |
| Gressens, 2000 | Developmental model | N/A | Cortical malformation analysis | GABAergic interneurons | Epilepsy association | Migration disorders linked to seizure risk |
| Powell et al., 2003 | Rodent | Migration disorder model | Interneuron mislocalization | GABAergic interneurons | Epileptiform activity | Improper positioning weakens synaptic inhibition |
| Olsen & Avoli, 1997 | Experimental epilepsy model | N/A | Electrophysiology (GABA_A) | GABAergic interneurons | Network hyperexcitability | Reduced GABA_A inhibition increases excitability |
| Fritschy, 2008 | Epileptic tissue | N/A | GABA receptor mapping | GABAergic interneurons | Epileptogenic tissue | Altered GABA_A distribution in epilepsy |
Results
Traumatic Brain Injury Disrupts Radial Glial and Vascular Migratory Scaffolds Through DAMP-Mediated Neuroinflammation
Across multiple studies, Traumatic Brain Injury (TBI) was consistently associated with the disruption of the vascular and glial scaffolds, which were required for postnatal inhibitory interneuron migration. Multiple experimental TBI models in mice have reported damage to radial glial fibers following traumatic insult, resulting in loss of scaffold alignment and thus reduced guidance capacity for migrating neuroblasts5. The mechanical deformation and secondary inflammatory responses after injury have been shown to misalign the radial glial scaffolds, which are essential for directional neuronal migration during early developmental windows5. Recent studies have demonstrated that following minutes of traumatic injury, an immune response develops under sterile conditions initiated by the release of damage-associated molecular patterns (DAMPs) from dying cells, which, once released to the extracellular space, activate toll-like receptors and initiate reactive gliosis that rapidly propagates injury to distal tissues6. Following injury, key DAMPs such as High-Mobility Group Box 1 (HMGB1), ATP, mitochondrial DNA (mtDNA), and Heat-Shock Proteins (HSP’s) are rapidly released from damaged neurons and glia, where they activate purinergic receptors to initiate sterile inflammation7. As DAMP-mediated signaling activates distant microglia and astrocytes, the glial cells that react release pro-inflammatory cytokines that further amplify neuroinflammation and worsen disruptions to local migratory scaffolding environments. Pro-neuroinflammatory responses, such as cytokines IL-6 and TNF, have been shown to create an altered cellular environment in regions where migratory scaffolding structures are disrupted5 This cytokine-rich inflammatory environment destabilizes the radial glial scaffolds by activating JAK/STAT signaling pathways, which increase GFAP and vimentin expression, thereby driving cytoskeletal reorganization, loss of scaffold alignment, and removal of laminar orientation cues necessary for interneuron migration guidance8. In addition to radial glial disruption, Astrocytic sheaths and blood-vessel-associated substrates which normally support chain migration and long-distance movement of inhibitory interneurons, undergo reactive gliosis9. These components were found to be structurally reorganized following brain trauma4 Following TBI, inflammatory signaling has been shown to induce a shift in astrocytes towards a neurotoxic A1 phenotype, which upregulates and releases a complement component C3, which can tag neurons and oligodendrocytes for degeneration and further disrupt the local migratory environment10 Collectively, the studies show that TBI induces misalignment of radial glial fibers, upregulates astrocytic processes, and further disrupts vascular scaffolding pathways, thereby undermining the directional migration required for postnatal interneuron integration.
Traumatic Brain Injury-Associated Neuronal Development Dysfunction
Evidence from developmental and pediatric injury studies has hinted towards TBI during early postnatal periods being associated with altered migration patterns of inhibitory interneurons, which include focal migration arrest, mislocalization, and impaired laminar integration1 The abnormalities of migration are shown to be region and population specific, consistent with localized migratory streams which persist during postnatal development4. We know that during postnatal development, migrating inhibitory interneurons are highly dependent on intact radial glial and astrocytic scaffolds for directional guidance and laminar integration. We can see that disruption of these structures following TBI is associated with improper positioning of GABAergic interneurons, suggesting that injury during this critical developmental window interferes with migration trajectories rather than causing widespread interneuron loss5. Using a controlled cortical impact (CCI) model of moderate TBI in mice, researchers induced a localized cortical lesion while preserving hippocampal structural integrity. Histological analysis showed substantial tissue loss at the cortical impact site but little to no detectable hippocampal tissue loss11. Further, it has been shown that TBI induces neural stem cells (NPCs) to shift their fate toward neurogenesis rather than astrocyte formation11. RNA velocity analysis in CCI rodent models predicted “backflow transitions” among cells in the astrocytic lineage, increasing their transition toward early neuronal states while decreasing their transition toward astrocytic states, suggesting that injury reshapes progenitor output in a manner that may destabilize the glial support environment required for proper interneuron migration and laminar polishing.11. According to the UMAP representation from scRNA-seq analysis of CCI rodent tissue models, TBI also produced no change in the developmental lineages of NSC-derived neurons and astrocytes.11.
Association Between Migration Disruption and Excitation-Inhibition Imbalance
Across reviewed studies, disruption of inhibitory interneuron migration is frequently shown to alter the excitation-inhibition balance within affected neural circuits. Studies examining neuronal migration disorders have reported that improper positioning of GABAergic interneurons has corresponded with reduced inhibitory signaling rather than with loss of excitatory neurons12. This impaired inhibitory signaling increased network excitability, making it more susceptible to synchronous firing patterns, as observed in epileptogenic tissue. Migration-related mislocalization of inhibitory interneurons is shown to weaken synaptic inhibition by reducing GABAergic control over local circuits, thereby shifting the excitation-inhibition balance toward hyperexcitability12. We can see how fast inhibitory transmission in cortical networks is primarily mediated by y-aminobutyric acid (GABA), acting on ionotropic GABA_A receptors, which function as ligand-gated chloride (Cl⁻) channels whose activation ultimately generates inhibitory postsynaptic currents that reduce action potential firing and synchronize network activity13. Thus, reductions in the strength and distribution of GABA_A receptor-mediated inhibition are associated with increased neuronal excitability and a higher tendency toward synchronous discharge patterns in epileptogenic tissue14. Knowing this, studies of neuronal migration disorders report that circuit dysfunction results from impaired inhibitory signaling and interneuron integration rather than from the death or loss of excitatory neurons, supporting the idea that improper interneuron positioning can cause persistent hyperexcitability12. TBI is also the occurrence of spreading depolarization (SD). SDs are characterized by a massive wave of neuronal and glial depolarization that travels at 205 mm/min and is followed by electrical silence as neurons become temporarily refractory15. Moreover, Glutamatergic mossy cells located in the dentate hilus are extremely vulnerable to injury following TBI and seizures. In the LFPI (Lateral Fluid Percussion Injury) model, it’s observed that mossy cells, which survive the insult, are hyperexcitable15.
Postnatal interneuron migration creates developmental vulnerability
Development of the central nervous system (CNS) is prolonged after birth, with up to 8 weeks of postnatal development and neurogenesis. Neural tube formation during neurulation occurs from Gestational Days (GD) 26 to GD 28 in humans; the anterior neuropore closes first, while the posterior neuropore closes later16. This prolonged period of neural tube formation makes the brain susceptible to environmental insults, such as TBI, which disrupts the natural developmental process16. Furthermore, after initial neuronal circuits are established, extensive neuronal migration ceases, but migration continues postnatally in specific niches to fine-tune the network in response to external stimuli17. Moreover, neuronal precursors are continuously integrated into the adult olfactory bulb (OB), with most of these precursor cells arising from intricate pathways that depend on both cell-autonomous factors and extrinsic regulation provided by the local environment18. Studies also identified that disruption of neuroblast migration not only produces neurons that fail to reach their target circuits but also imperfectly recapitulate the original program of maturation, and, as a result, neurons synaptically integrate into inappropriate neural networks18. GABAergic interneurons originate mainly from subcortical structures and migrate long distances to reach specific cortical layers, where their precise positioning ensures targeted inhibition of excitatory networks19. This spatial diversity directly ties to their regional vulnerability, since improper placement during postnatal neurogenesis can alter circuit balance and synaptic efficiency, leading to epileptogenesis19. Postnatal neurons migrating from the subventricular zone (SVZ) to sites such as the OB depend on astrocytes and the ECM for directional cues. Thus, disruptions of these components cause polarity defects and increased excitability18.
Therapeutic Strategies Targeting Migration Related to Circuit Dysfunction
Current treatments aim to target migration to restore proper circuitry function after interneuron displacement during neurogenesis. Transplanting interneuron precursors from the medial ganglionic eminence (MGE) into neonatal brains reduces behavioral disorders by strengthening GABAergic inhibition, rebalancing signaling, and promoting the migration of interneurons to the proper cortical layers20. Furthermore, human Induced Pluripotent Stem Cells (iPSCs) can be differentiated into medial ganglionic eminence-like progenitors using SHH, Wnt, and FGF signaling modulators, creating GABAergic interneurons that express markers such as Lhx6 and fire inhibitory action potentials21. Transplanted iPSC cells have been shown in rodent models to migrate into host tissue, integrate into pre-existing neural circuits, and provide inhibitory synaptic signaling that rebalances network activity imbalanced by neurogenesis malformations22. Recent studies used Kainic acid-induced status epilepticus rat models, in which intrahippocampal transplantation of neural stem cells preserved normal neurogenesis while promoting differentiation into GABAergic interneuron lineages, thereby reducing epileptogenic processes22.
Discussion
Collectively, the reviewed literature suggests that TBI may disrupt postnatal inhibitory interneuron development through not only tissue damage but also neuroinflammatory remodeling of migratory scaffolds, altered allocation of progenitor fates, and impaired integration of GABAergic interneurons into developing neural circuits11. Taken together, findings support the claim that post-traumatic epileptogenesis dysfunction arises not just from neuronal loss, but from miswiring in neurogenesis development caused by disrupted migration of interneurons during vulnerable postnatal periods in the developing CNS.
These findings are significant because they provide an understanding that post-traumatic dysfunction may emerge even after tissue preservation is relatively intact. Further, postnatal brain development occurs as interneurons continue to migrate along intrinsic niche pathways, increasing vulnerability to environmental trauma, as many brain regions are still undergoing development17. This increased vulnerability provides a critical window in which TBI may disrupt interneuron positioning, or inhibitory-circuitry assembly, without necessitating widespread neuronal ablation 11. As a result, long-term damage and disorders may arise from structural damage, and neurogenesis miswiring that alters the excitation-inhibition balance and increases susceptibility to hyperexcitability and epileptogenesis.
Future Directions
Future research regarding interneuron dysfunctionality following TBI should focus on direct experimental tracking of postnatal interneuron migration and integration into neural circuitry. Further, a comparison of interneuron development tracing between TBI and healthy control models should be performed to map changes in inhibitory interneuron development. Additional work should also aim to determine whether post-injury neurogenesis is functionally restorative and whether intrinsic neuronal injury and tissue disruption are the dominant drivers of circuit dysfunction that cause epileptogenesis. Moreover, human-derived iPSC-based models should be transplanted into more human-relevant models, where inhibitory interneurons must travel greater distances and integrate into more complex brain architecture. Additional studies to improve optimization could include improving graft survival and minimizing the risk of uncontrolled proliferation and ectopic integration. Further gene-editing approaches to refine inhibitory interneurons for transplantation could be explored, potentially lowering the risk of rejection and enabling greater precision in transplantation into epileptogenic regions.
Limitations
Despite the findings of this review, many limitations in the literature remain. Much of the evidence illustrated is derived from experiments conducted in rodent models of TBI, which may not fully capture the timing of development, migratory distances, or the complexity of the human brain16. Furthermore, many studies of various injuries suggest interneuronal dysfunction through vascular scaffolding and chemical markers; however, limited tracing of the direct migration and integration of postnatal inhibitory interneurons has been conducted 16. Similar translational limitations apply to emerging stem-cell therapeutic models, as successful integration of interneuron progenitors in rodent brains may not be accurately replicated in the human brain, given longer migratory distances and stricter safety barriers, which pose additional challenges for iPSC therapy22. Additionally, hyperexcitability following TBI is likely multifactorial and cannot be attributed exclusively to interneuron migration dysfunction.
Conclusion
Overall, current evidence supports the idea that TBI may impair postnatal circuit development not only through direct tissue damage but also through impaired interneuron development and integration into pre-existing neural circuitry. By synthesizing how neuroinflammatory scaffold remodeling, developmental lineage changes, and excitation-inhibitory imbalance arise from interneuron migration dysfunction, we can highlight a mechanism by which TBI may cause epileptogenesis. Recognizing new developmental mechanisms through which post-traumatic dysfunction occurs, as a problem of interneuron miswiring rather than structural injury, may help guide future therapies towards restoration of circuitry to control epilepsy rather than tissue-damage control alone.
References
- Gressens P. Mechanisms and disturbances of neuronal migration. Pediatric Research. 2000;48(6):725–730. https://doi.org/10.1203/00006450-200012000-00004. [↩] [↩]
- Gleeson JG. Neuronal migration disorders. Mental Retardation and Developmental Disabilities Research Reviews. 2001;7(3):167–171. https://doi.org/10.1002/mrdd.1024. [↩] [↩] [↩] [↩]
- Trevelyan AJ, Schevon CA. How inhibition influences seizure propagation. Neuropharmacology. 2013;69:45–54. https://doi.org/10.1016/j.neuropharm.2012.06.015. [↩]
- Bressan C, Saghatelyan A. Intrinsic mechanisms regulating neuronal migration in the postnatal brain. Frontiers in Cellular Neuroscience. 2021;14:620379. https://doi.org/10.3389/fncel.2020.620379. [↩] [↩] [↩]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathology. 2004;14(2):215–222. https://doi.org/10.1111/j.1750-3639.2004.tb00056.x. [↩] [↩] [↩] [↩]
- Cieri MB, Ramos AJ. Astrocytes, reactive astrogliosis, and glial scar formation in traumatic brain injury. Neural Regeneration Research. 2024;20(4):973–989. https://doi.org/10.4103/nrr.nrr-d-23-02091. [↩]
- Sun B, Zhang J, Li Z, Wang J, Zhao C, Xu X. Role of damage-associated molecular patterns in the pathogenesis and therapeutics of traumatic brain injury. Burns & Trauma. 2025;13. https://doi.org/10.1093/burnst/tkaf043. [↩]
- Molofsky AV, Krenick R, Ullian E, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH. Astrocytes and disease: A neurodevelopmental perspective. Genes & Development. 2012;26(9):891–907. https://doi.org/10.1101/gad.188326.112. [↩]
- Amlerova Z, Chmelova M, Anderova M, Vargova L. Reactive gliosis in traumatic brain injury: A comprehensive review. Frontiers in Cellular Neuroscience. 2024;18. https://doi.org/10.3389/fncel.2024.1335849. [↩]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. https://doi.org/10.1038/nature21029. [↩]
- Bielefeld P, Martirosyan A, Martín-Suárez S, Apresyan A, Meerhoff GF, Pestana F, Poovathingal S, Reijner N, Koning W, Clement RA, Van der Veen I, Toledo EM, Polzer O, Durá I, Hovhannisyan S, Nilges BS, Bogdoll A, Kashikar ND, Lucassen PJ, Belgard TG. Traumatic brain injury promotes neurogenesis at the cost of astrogliogenesis in the adult hippocampus of male mice. Nature Communications. 2024;15(1). https://doi.org/10.1038/s41467-024-49299-6. [↩] [↩] [↩] [↩] [↩] [↩]
- Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. The Journal of Neuroscience. 2003;23(2):622–631. https://doi.org/10.1523/JNEUROSCI.23-02-00622.2003. [↩] [↩] [↩]
- Olsen RW, Avoli M. GABA and epileptogenesis. Epilepsia. 1997;38(4):399–407. https://doi.org/10.1111/j.1528-1157.1997.tb01728.x. [↩]
- Fritschy JM. E/I balance and GABAA receptor plasticity. Frontiers in Molecular Neuroscience. 2008;1. https://doi.org/10.3389/neuro.02.005.2008. [↩]
- Ngwenya LB, Danzer SC. Impact of traumatic brain injury on neurogenesis. Frontiers in Neuroscience. 2019;12. https://doi.org/10.3389/fnins.2018.01014. [↩] [↩]
- Rice D, Barone S. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environmental Health Perspectives. 2000;108(Suppl 3):511–533. https://doi.org/10.1289/ehp.00108s3511. [↩] [↩] [↩] [↩]
- Nakajima C, Sawada M, Sawamoto K. Postnatal neuronal migration in health and disease. Current Opinion in Neurobiology. 2020;66:1–9. https://doi.org/10.1016/j.conb.2020.06.001. [↩] [↩]
- Belvindrah R, Nissant A, Lledo PM. Abnormal neuronal migration changes the fate of developing neurons in the postnatal olfactory bulb. The Journal of Neuroscience. 2011;31(20):7551–7562. https://doi.org/10.1523/JNEUROSCI.6716-10.2011. [↩] [↩] [↩]
- Wonders C, Anderson SA. Cortical interneurons and their origins. The Neuroscientist. 2005;11(3):199–205. https://doi.org/10.1177/1073858404270968. [↩] [↩]
- Valente MF, Romariz S, Calcagnotto ME, Ruiz L, Mello LE, Frussa-Filho R, Longo BM. Postnatal transplantation of interneuronal precursor cells decreases anxiety-like behavior in adult mice. Cell Transplantation. 2013;22(7):1237–1247. https://doi.org/10.3727/096368912X657422. [↩]
- Kim TG, Yao R, Monnell T, Cho JH, Vasudevan A, Koh A, Peeyush KT, Moon M, Datta D, Bolshakov VY, Kim KS, Chung S. Efficient specification of interneurons from human pluripotent stem cells by dorsoventral and rostrocaudal modulation. Stem Cells. 2014;32(7):1789–1804. https://doi.org/10.1002/stem.1704. [↩]
- Alayli A, Lockard G, Gordon J, Connolly J, Monsour M, Schimmel S, Dela Peña I, Borlongan CV. Stem cells: Recent developments redefining epilepsy therapy. Cell Transplantation. 2023;32:09636897231158967. https://doi.org/10.1177/09636897231158967. [↩] [↩] [↩]

 and Family-Integrated Care (FIC): Global Trends and Local Provider Awareness in Fresno County, California")

{kind=link}