
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline and neuronal loss, affecting millions of older adults worldwide. Cigarette smoking is increasingly recognized as a significant, modifiable risk factor for AD. This narrative review examines the mechanistic links between cigarette smoke exposure and AD, focusing on two mechanistic pathways: oxidative stress and blood-brain barrier (BBB) dysfunction. Smoking introduces neurotoxic compounds such as nicotine, nitric oxide (NO), and lead (Pb), which impair vascular and brain homeostasis. Oxidative stress, fueled by excess reactive oxygen species (ROS) from smoke, promotes lipid, protein, and DNA damage, contributing to neuronal degeneration. BBB breakdown, caused by inflammatory processes and oxidative stress, allows toxins to enter the brain, triggering neuroinflammation. This paper summarizes data from human and animal research, including studies that show increased oxidative damage, disrupted tight junction proteins, impaired signaling pathways, and increased BBB permeability following smoking exposure. The therapeutic implications highlight prevention and early intervention, as growing evidence suggests quitting smoking can lower AD risk and reverse some vascular and neural damage. However, the heavy reliance on non-human models limits the direct therapeutic relevance of these results, emphasizing the importance of more continuous and human-based research. Understanding these pathways is important for developing targeted neuroprotective medications and designing effective public health interventions against smoking to reduce AD risk. This review synthesizes mechanistic evidence to clarify how smoking related oxidative stress and BBB disruption may interact with established AD pathways.
Keywords: Alzheimer’s disease, smoking, blood-brain barrier, oxidative stress
Introduction
Alzheimer’s disease (AD) is a progressive neurological disorder, most commonly affecting individuals over the age of 65. It is characterized by memory loss, impaired thinking, and the inability to perform daily routines, and is driven by the accumulation of amyloid plaques and tau tangles in the brain. In 2024, an estimated 6.9 million Americans ages 65 and older had AD. This number is expected to rise to 13.8 million Americans by 2060, assuming there are no new medical advances to cure AD1. As the number of people affected by AD grows, it becomes increasingly necessary to identify the risk factors for developing the condition. Substantial evidence indicates that one factor may be cigarette smoking, which remains prevalent in millions of Americans. 11.5%, or about 28.3 million U.S. adults, smoked cigarettes in 20212. However, epidemiological evidence on smoking and AD remains controversial, with cohort studies showing mixed results due to age-dependent effects and methodological challenges like survival bias: some report smokers having a 72% increased risk of developing AD compared to nonsmokers, while others find null associations3. One explanation for these mixed results is that if heavy smokers die earlier from cardiovascular or pulmonary disease, then surviving smokers in late-life cohorts are a selectively resilient group, reducing measured AD risk (survivor bias), whereas midlife cohorts may capture the true harmful effect before this selective survival occurs. To further understand this relationship, it is necessary to examine the underlying pathological characteristics that define AD.
The most widely recognized pathological markers of AD are amyloid-beta (Aβ) plaques and neurofibrillary tangles composed of tau protein4. Aβ plaques are protein clusters that accumulate between nerve cells, disrupting communication and activating immune responses, while tau protein forms toxic neurofibrillary tangles inside neurons that lead to cell death and impaired signaling. This immune response causes neuroinflammation, a prolonged activation of the brain’s immune system that, while initially beneficial, can become harmful by supporting chronic inflammation, neuronal damage, and AD advancement. Aβ plaques and tau neurofibrillary tangles, together with genetic factors like the APOEε4 allele—the strongest known risk factor for AD—are associated with molecular changes that contribute to neuronal damage, and progression of AD. Beyond amyloid and tau pathologies, two prominent but underexplored pathways in smoking-AD research are oxidative stress and blood-brain barrier (BBB) dysfunction. Oxidative stress occurs when there is an imbalance between the excessive production of free radicals– specifically reactive oxygen species (ROS), which are normal byproducts of cellular metabolism–and insufficient antioxidant defenses. This imbalance leads to cellular damage and contributes to the progression of diseases like Alzheimer’s by damaging lipids, proteins, and DNA, and allowing the buildup of toxic proteins, ultimately impairing neuronal function. The BBB is composed of tightly packed endothelial cells that serve as a protective boundary for the brain from harmful substances that may be in the bloodstream. Damage to the BBB would result in increased permeability, allowing harmful toxins, including toxins from cigarette smoke, to enter the brain. This results in neuronal damage and neuroinflammation, both of which contribute to AD development.
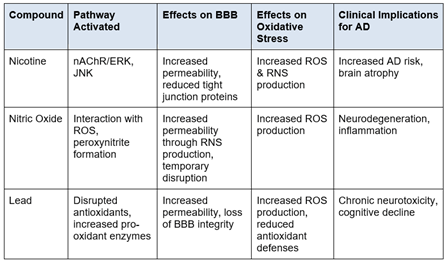
Researchers have identified modifiable lifestyle risk factors that may increase the risk of AD, such as smoking. Smoking exposes the body to harmful toxins and heavy metals such as nicotine, nitric oxide (NO), and lead (Pb), whose effects may be enhanced by genetic vulnerability like the APOEε4 allele. Nicotine is an alkaloid compound naturally found in tobacco plants. It is highly addictive and acts on nicotinic acetylcholine receptors in the brain, influencing neurotransmitter release and neural signaling5. NO is a small, gaseous molecule produced naturally in the body. It is an important signaling molecule in the nervous system, regulating blood flow, neurotransmission, and immune responses. However, excess NO can react with other molecules to form reactive nitrogen species (RNS), contributing to oxidative stress and neuronal damage6. Pb is a dense, toxic heavy metal that can easily accumulate in tissues and disrupt several biological processes in the nervous system. Pb exposure, particularly in early life, is associated with reduced IQ, poorer cognitive performance, and increased behavioral problems as it may disrupt neurotransmitter signaling and damage vulnerable brain regions during development7. These smoke-derived toxins connect to AD as they converge on core disease mechanisms: nicotine compromises BBB integrity to enable toxicant entry, excess NO amplifies oxidative damage, and Pb induces chronic neuroinflammation. Collectively, these compounds accelerate amyloid and tau pathology, neuronal loss, and cognitive decline.
There is substantial evidence that smoking increases AD risk, particularly since smoking is officially recognized as a modifiable risk factor for dementia8. Despite mixed epidemiology, smoking’s role in dementia risk is apparent, but the mechanistic understanding is insufficient. No prior reviews synthesize how specific cigarette smoke toxins (nicotine, NO, Pb) simultaneously target oxidative stress and BBB dysfunction—two interconnected AD accelerators—or integrate dose, duration, and sex effects with human relevance limitations. With AD prevalence doubling by 2060, this paper clarifies why smoking cessation timing matters and identifies gaps for neuroprotective interventions regardless of survivor bias1. This paper begins by outlining the basic roles of oxidative stress and the blood-brain barrier in AD pathology, followed by an examination of how neurotoxic compounds found in cigarette smoke, including nicotine, NO, and lead, influence each of these processes. When evaluating this evidence, human longitudinal cohort studies and biomarker-confirmed AD research are the most relevant, followed by mammalian in vivo models with smoker-relevant exposure patterns. In vitro, non-mammalian, and isolated toxin studies largely illustrate mechanisms rather than determine human smoking-related AD risk. By clarifying these mechanisms, this paper seeks to increase awareness of the negative impacts of smoking on public health and brain disease.
Microglia are the CNS’s natural immune cells, and they play an important role in brain homeostasis, injury response, and neuroinflammation9. In the context of AD, accumulating data suggest that microglial activation occurs early and persists throughout the disease process. Additionally, microglial findings imply that smoking alters neuroimmune responses, either decreasing preventive regulation or sustaining maladaptive activation in ways that may reduce the threshold for AD-related pathology.
Alzheimer’s Disease & Smoking
The increasing amount of evidence from human and animal studies suggests that cigarette smoke is a significant modifiable risk factor for AD. As previously mentioned, a meta-analysis that accounted for the tobacco industry connection indicated that smokers had a 72% greater chance of developing AD compared to nonsmokers3, confirming the association between smoking and neurodegenerative diseases. Moreover, the Rotterdam Study, which examined smoking patterns in a large, community-based cohort, adds to the evidence for this association10. The results found that current smokers at the time had a significantly higher risk of both dementia and AD, whereas former smokers had no increased risk. This shows that smoking’s negative impacts on cognitive function can be minimized by quitting, highlighting an important issue for public health: quitting smoking may be able to reverse, or at least reduce, the increased risk of dementia. This large human cohort provides the strongest direct evidence that quitting smoking had a stronger protective impact in people without the APOEε4 allele compared to smaller animal models of acute exposure. The relationship between smoking and dementia risk was weaker in APOEε4 carriers compared to noncarriers.
Several studies have been conducted to better understand the mechanisms underlying smoking-induced neurodegeneration. For instance, the link between smoking in humans and the risk of dementia may involve a reduction in brain volume, as seen in the results of a large-scale brain imaging study conducted by Chang et al., 2024. This study included over 32,000 people, examining smoking’s impact on brain health and dementia11. The results found that cigarette smoking is closely associated with a decrease in global brain volume, which in turn raises the risk for AD. Notably, this large-scale human imaging study offers strong clinical evidence that this impact was dose-dependent and persisted long after people stopped smoking. This demonstrates greater relevance than rodent models examining intermediate oxidative markers alone, implying that smoking-induced brain atrophy may be permanent. This decrease in brain volume, specifically in critical regions for memory and executive functions such as the hippocampus and the frontal cortex, has been linked to approximately 14% of Alzheimer’s cases globally, further emphasizing its clinical significance.
To investigate the underlying foundations of these impacts, animal models have provided intriguing results. While limited by non-human translation, a study conducted by Shan Ho et al. 2012 examined how chronic second-hand exposure to cigarette smoke affects the brain and contributes to pre-Alzheimer-like neuropathology in rats12. They investigated this by exposing male rats to 4% cigarette smoke for 1 hour per day over 8 weeks to simulate second-hand smoking. The study discovered higher markers of oxidative damage in the hippocampus of rats continuously exposed to cigarette smoke, including raised 8-OHG and 8-OHdG, which are biomarkers for oxidative stress and indicators of RNA and DNA damage, respectively. The changes were followed by lower levels of presynaptic proteins such as synaptophysin and synapsin-1, indicating early synaptic degeneration. Furthermore, tau protein was discovered to be hyperphosphorylated at numerous Alzheimer-relevant locations (Thr231, Thr205, and Ser 404), which was most likely caused by oxidative stress-induced Erk1/2 and JNK activation. Normally, the ERK pathway helps regulate healthy cell growth and survival, but when abnormally activated, such as by oxidative stress, it can contribute to diseases like cancer or AD by promoting harmful processes like tau hyperphosphorylation13. The JNK pathway is typically activated in response to cellular stress and, when overactivated, can lead to inflammation, cell damage, and cell death14. These rats also accumulated Aβ in the hippocampus, mirroring several key aspects of AD pathology, which provides mechanistic support rather than direct human risk evidence.
Additional research has found molecular mediators that may link cigarette use to AD in the brain. One study conducted by Sun et al., 2023 examined how chronic exposure to cigarette smoke causes neurotoxicity by exposing mice to various concentrations of cigarette smoke for 6 months, and exposing human neural cells to cigarette smoke extract15. The study found that cigarette smoke exposure in mice resulted in hippocampal injury, substantial neuronal loss, and memory issues. These effects were facilitated in part by the microRNA miR-153-3p, which inhibits the production of PIK3R1— a gene that helps make the PI3K protein involved in insulin signaling. However, when PIK3R1 does not work correctly or is suppressed, it disrupts normal cell communication. This disruption can result in tau becoming excessively phosphorylated, as well as synaptic toxicity, both of which are well-known hallmarks of AD.
Together, these studies illustrate the complex relationship between cigarette smoking and AD, revealing how smoking contributes to oxidative damage, synaptic dysfunction, altered brain signaling, and reduced brain volume. This emphasizes how quitting smoking is crucial not just for cardiovascular and respiratory health but also for long-term brain health and dementia prevention.
General Mechanisms of Oxidative Stress and the Blood-Brain Barrier in Alzheimer’s Disease and Smoking
Oxidative Stress
Oxidative stress is a physiological imbalance between the creation of ROS, or free radicals, and the body’s ability to counteract their damaging effects via antioxidant defenses. Cigarette smoke is a significant external source of oxidative stress since it includes over 4,000 harmful compounds, including a substantial number of free radicals16. Additionally, each puff of cigarette smoke generates around 10 trillion free radicals, significantly overwhelming cellular antioxidant mechanisms. This constant input of free radicals affects both micromolecules and macromolecules throughout the body, causing damage that is most noticeable in tissues with high oxygen demands, such as the lungs and brain. This section demonstrates how cigarette smoke-induced oxidative stress at the cerebral level can convert a typically adaptive oxidative system into a chronic cause of neuronal and barrier damage related to AD.
A study conducted by Lee et al., 2017 confirms that cigarette smoke directly promotes ROS production inside brain tissue17. The results showed that when exposed to cigarette smoke residue in vitro, neurons and astrocytes showed enhanced oxidative stress and ROS generation. This impact became more significant with increased exposure, reaching the highest ROS levels at 16 mg/ml of cigarette smoke residue. This shows that increased exposure to toxic cigarette compounds may drastically increase the production of oxidative stress. However, it lacks direct relevance to non-cancerous AD neuronal loss.
This process is driven not only by direct ROS intake from smoke but also by the activation of endogenous pro-oxidant enzymes such as inducible nitric oxide synthase18. These results come from a study by Khanna et al., 2013 that examined neuroinflammatory markers in rats after being exposed to cigarette smoke for 6 weeks. These pro-oxidant enzymes normally play a key role in generating ROS and RNS during cellular stress and inflammation. The enzymes were overactivated in response to cigarette smoke exposure, which worsened the consequences of oxidative damage that contribute to neurodegeneration. This study shows cigarette smoke exposure is associated with oxidative stress responses in animals, consistent with oxidative mechanisms observed in human smoking research.
Cells have adaptive methods for reducing oxidative damage, including the activation of antioxidant pathways such as the Nrf2 pathway18,19. Under increased oxidative stress, cells such as astrocytes and BBB endothelial cells activate Nrf2, a transcription factor that controls the production of several antioxidant proteins. In a study conducted by Prasad et al., 2015, researchers exposed human blood-brain barrier endothelial cells (hCMEC/D3) to high glucose or cigarette smoke extract to assess their impact on barrier integrity, cytokine release, protein expression, and drug efflux activity using cellular and molecular techniques used to analyze features such as barrier strength (TEER), protein levels (ELISA, western blot), and drug transport (rhodamine 123 assays)19. The researchers found that following cigarette smoke exposure, Nrf2 activation rises, which may function as a protective antioxidant reaction. Nevertheless, the high ROS load from chronic cigarette smoking exposure may override these protective systems, leading to long-term oxidative damage to neuronal cells and the BBB. As a result, it leads to the development of neurodegenerative and other oxidative stress-related diseases.
A study by Ewees et al., 2025 examined how chronic cigarette smoke exposure affects vascular, blood-brain barrier, oxidative, inflammatory, neurodegenerative, and cognitive outcomes in mice20. This was done by exposing male mice to either cigarette smoke or air (control) for 2 hours per day over five days a week for up to 60 weeks to mimic typical human smoking patterns. The cigarette smoke, consisting of various toxins, also includes nicotine, NO, and Pb. The mice’s brain tissues were assessed using histological, biochemical, and immunohistochemical analysis to evaluate various mechanisms such as oxidative stress and neuroinflammation. The findings suggest exposure to cigarette smoke generates high levels of ROS in the cerebral microvasculature, leading to oxidative stress, endothelial cell damage, and disruption of capillary homeostasis. Figure 1 combines these mechanisms into a single diagram, demonstrating how converging oxidative insults at the microvascular level can trigger or accelerate neurodegenerative pathways in AD. These studies do not support a universal ROS-driven pathology. Instead, activation of Nrf2 suggests that smoking overwhelms endogenous adaptive defenses rather than simply producing nonspecific oxidative damage.
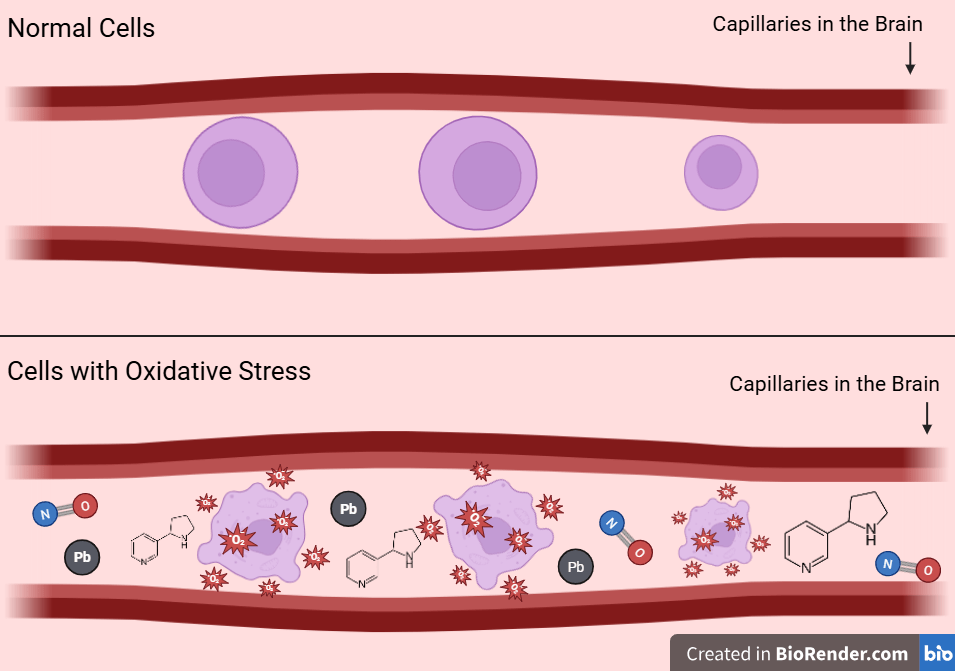
Blood Brain Barrier Permeability
Establishing how BBB permeability impacts the brain is critical for understanding AD pathology, particularly in relation to oxidative stress and environmental risk factors such as smoking. In the context of Alzheimer’s disease, BBB dysfunction has been suggested to allow toxins, inflammatory mediators, and immune cells to enter the brain, leading to neurodegeneration and cognitive impairment. Evidence suggests that smoking does not simply open up the BBB, but rather gradually reduces its strength, allowing aging, amyloid, and vascular risk factors to easily reflect in neuroinflammation.
A magnetic resonance imaging (MRI) study conducted by Starr et al., 2009 directly examined BBB permeability in AD by comparing a group of 15 AD patients to 15 healthy older adults, neither of whom smoke21. The MRI scan was performed repeatedly, nine times both before and after a 20 ml Gd-DTPA bolus injection, which helps the MRI highlight blood vessels and leaks across the BBB. Researchers found that there was no significant variation in BBB permeability between the two groups; however, the speed and behavior of the leakage differed from healthy controls. The healthy group showed the predicted progressive rise in MRI signal 15 minutes after contrast. AD patients, on the other hand, had an abnormal pattern of an initial quick spike in gray matter signal, followed by another increase at a later time, rather than the gradual progression seen in healthy individuals. This altered timing of spiking, indicating leakage, shows a more subtle disturbance in BBB function in AD, stressing that permeability abnormalities may not necessarily result in leaking but may be associated with unusual physiological responses to vascular stresses.
The importance of vascular risk factors, specifically cigarette smoking, was emphasized by a large-scale, biomarker-confirmed study of 368 AD patients conducted by Bonomi et al., 202422. This study specifically assessed the effects of smoking on BBB permeability correlating to cognitive decline and neurodegeneration through mediation analysis and multivariate backward regressions. The study concluded that higher vascular risk, with smoking being a key factor, was related to increased BBB permeability, an effect that was more prevalent in late-onset AD than in early-onset forms. Furthermore, in early and very late-onset AD, smoking and increased vascular risk factors connected more directly with neurodegeneration than with measured BBB permeability. The findings indicate that, while increased BBB permeability in the presence of smoking appears to play a role in cognitive impairment, its effects may be secondary or interacting with other neurodegeneration mechanisms, highlighting BBB disruption as one component of the broader pathology of AD.
Insights from experimental models further support how cigarette smoke mechanistically impairs BBB integrity. In a study conducted by Prasad et al., 2015, when cultured human endothelial cells were exposed to cigarette smoke extract and elevated glucose (to stimulate coexisting metabolic risk), the tight junction protein ZO-1, critical for BBB selectivity, was reduced19. This loss of tight junction structure resulted in significant increases in permeability, particularly when both stressors were present concurrently. Moreover, exposure to cigarette smoke extract or high glucose did not induce cell death, but rather triggered a strong inflammatory response with significant increases in cytokines such as IL-6, IL-8, PECAM-1, and ICAM-1. These changes reveal how the BBB’s physical function can be undermined not just by cell loss, but by reduced cohesion and inflammation, conditions that closely resemble those seen in human smokers.
Similar discoveries in more complex biological models help us understand the timing and resistance of BBB disruption under sustained stress. In the study conducted by Lee et al., 2017, researchers used human brain cells (T98G), astrocytes (U-373 MG), and brain endothelial cells (HBMEC) cultured individually and together to assess the effects of commercial cigarette smoke condensate (CCSC) at different concentrations and durations on cell viability, oxidative stress (ROS production), and cellular integrity17. The results showed that short-term exposure (24 hours) caused relatively modest barrier disruption, with most HBMECs retaining their protective function. However, extending exposure to 48-120 hours resulted in considerably greater weakening of the endothelial layer and decreased protective capacity. Furthermore, the T98G brain cells were more sensitive to this damage than U-373 MG astrocytes, but the co-culture with HBMEC offered protection. These observations emphasize the dependence of the BBB on the dose and duration of smoking, a factor specifically relevant to chronic smokers at risk for AD.
Animal studies further verify the impact of environmental exposures on the BBB dynamics at the molecular and organismal levels. The previously mentioned study by Khanna et al., 2013 shows that in rodent models, cigarette smoke exposure led to increased levels of proinflammatory cytokines (TNF-α, IL-1β, IL-6), all of which are well recognized for their ability to disrupt tight junctions and, as a result, increase BBB permeability18. This inflammatory process reflects more than just an acute response, as it signifies a chronic, self-amplifying process that links peripheral immune activation to progressive barrier dysfunction and ultimately, neurodegeneration.
Oxidative stress, induced by cigarette smoke, is another widely recognized driver of BBB dysfunction as cigarette smoke generates ROS, directly damaging endothelial cell proteins, lipids, and DNA23. This oxidative injury both weakens tight junctions and disrupts homeostasis, leading to BBB breakdown and dysregulation. Moreover, continuous ROS production encourages inflammation, resulting in the effects described above.
Additionally, Bernard et al., 2019 tested cigarette smoke extract on mouse brain endothelial cells and measured BBB permeability and oxidative stress by paracellular permeability assays24. Researchers found that cigarette smoke extract significantly worsens BBB permeability in cerebral endothelial cells via oxidative and inflammatory mechanisms. This supports the idea that smoking worsens BBB integrity and allows neurotoxicants to enter the brain, as exposure to substances like nicotine can damage endothelial tight junction proteins, such as occludin. While individual studies report changes in specific tight junction proteins or permeability tracers, their combined implications for BBB function can be difficult to visualize. Figure 2 synthesizes these results into a unified model, illustrating how cigarette smoke toxins may progressively worsen barrier integrity and increase brain exposure to circulating neurotoxins relevant to AD. In this review, BBB dysfunction is viewed as a dynamic interaction, where smoke-induced oxidative stress and inflammation weaken the barrier, leading to further oxidative and inflammatory harm in brain tissue. Collectively, these studies support a bidirectional model in which cigarette smoke-induced oxidative stress and inflammation progressively impair BBB integrity. The resulting barrier leak increases brain exposure to circulating toxins, increasing oxidative and inflammatory stress rather than functioning as a standalone pathology.
Finally, an integrative view is that if individuals already have age-related or amyloid-related BBB vulnerability, then smoking acts mainly as a factor that accelerates tight junction loss and inflammatory leak. In contrast, in younger or healthy brains, only sustained, high-dose smoking may be sufficient to initiate clinically significant BBB breakdown. This conditional framing helps explain why some AD cohorts show strong smoking-BBB associations while others show subtler permeability changes.
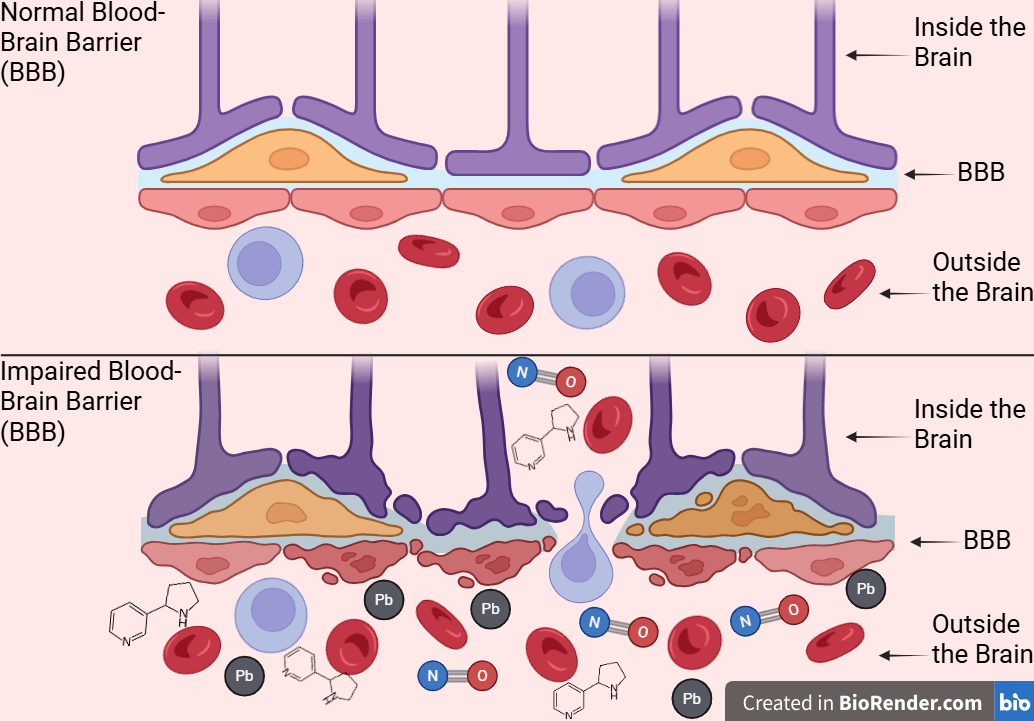
Nicotine
Oxidative Stress
Nicotine’s effects on oxidative stress are complex and context-dependent, with evidence indicating both harmful and potentially beneficial functions under various conditions. A study conducted by Naik et al., 2014 examined the toxic impact of smoke extract from different cigarette products (full flavor, ultra low nicotine, and “nicotine free”) on endothelial cells (hCMEC/D3) in a BBB model through immunofluorescence and western blotting analysis25. Results revealed that exposure to cigarette smoke extract, whether from regular, ultra-low nicotine, or even “nicotine free” cigarettes, led to significant increases in the production of ROS, RNS, and hydrogen peroxide. Notably, the level of oxidative stress was shown to be far more strongly correlated with the tar concentration of cigarettes than with the nicotine content. Chronic exposure to ultra-low nicotine and nicotine-free smoke extracts resulted in the greatest levels of oxidative stress and disruption of proteins that maintain the BBB’s tight connections. This shows that nicotine-free or reduced-nicotine cigarette products are not always safer in terms of oxidative and vascular harm, and may even have a higher oxidation potential due to additional components such as tar.
In order to investigate the direct effects of nicotine, Barr et al., 2007 examined rat mesencephalic (midbrain) nerve cells using techniques such as enzyme mobility shift assays, immunoblot analysis, and kinase assays26. Specifically, the kinase assays demonstrated activation of ROS-sensitive signaling pathways, supporting the conclusion that nicotine alone can rapidly dose-dependently elevate intracellular ROS. Furthermore, even low amounts of nicotine raised ROS by about 35%, with larger doses resulting in levels of up to 80%. The increase in ROS was noticeable within 30 minutes and continued to grow with ongoing exposure; however, these effects were shown to be reversible following nicotine elimination. Nicotine’s quick and powerful production of oxidative stress demonstrates its capacity to contribute to cellular damage, especially during prolonged exposure. This illustrates a mechanism that could contribute to AD progression if occurring chronically in human smokers as this production of oxidative stress also impairs neuronal functions vital to the body.
An in vivo study by Sukketsiri et al., 2019 supports the association between nicotine and oxidative damage, where young male mice were divided into control and nicotine groups27. Over 15 days, the mice’s blood markers were assessed along with the integrity of vascular tissue, injury by oxidative stress, and protein changes using histological and biochemical analyses. When mice were given nicotine, it was found that the nicotine increased vascular oxidative stress and an enzyme that promotes further deterioration of vascular tissue called matrix metalloproteinase-2 (MMP‑2), indicating early BBB-relevant vascular injury. These results are directly like nicotine-induced vascular oxidative stress and structural damage to the larger mechanisms by which cigarette smoke impairs BBB integrity and accelerates neurodegenerative processes.
In addition, nicotine is not always harmful in all circumstances or concentrations. According to research conducted by Dong et al., 2020, low nicotine concentrations can provide some protection for particular neurons during oxidative stress28. This was found by examining mouse hippocampal cells to assess the effects of various drugs on oxidative stress, apoptosis, and cell viability. When hippocampal neurons were exposed to oxidative insults, low-dose nicotine was shown to lower intracellular ROS, promote cell survival, preserve mitochondrial membrane potential, and avoid cell cycle arrest. The neuroprotective effects were partially caused by the α7-nicotinic acetylcholine receptor (α7-nAChR) and the Erk1/2 signaling pathway (see Table 1 for overview of key mechanistic pathways). Importantly, the protective effect of nicotine was lost at greater nicotine concentrations, indicating a limited therapeutic opportunity. This suggests that mechanistically, lower doses of nicotine may activate protective signalling pathways in the BBB, while higher doses could overwhelm these mechanisms and trigger oxidative stress and inflammation.
Overall, data suggest that nicotine is a strong inducer of oxidative stress in vascular and brain tissues, particularly when administered in the form of cigarette smoke, where additional cigarette elements, such as tar, can worsen oxidative and inflammatory harm. Across human, animal, and cell models, there is consistent convergence that the presence of nicotine in smoke increases ROS and oxidative damage, despite the small given number of cell studies reporting low-dose neuroprotection under highly controlled conditions. Although low-dose nicotine may provide some neuroprotection in certain instances, the overall effect of nicotine exposure in the setting of smoking is overwhelmingly damaging to both vascular and brain health. These findings highlight that nicotine-free cigarettes and low-nicotine formulations do not reduce, and may potentially increase, the oxidative risks associated with smoking. Importantly, these protective mechanistic findings occur at nicotine concentrations unlikely to be achieved through smoking alone, whereas the BBB-damaging effects occur at physiologically relevant smoker levels. These protective effects are model- and dose-dependent exceptions that do not appear in all animal or human exposures that more closely resemble smoking. Human imaging evidence has the most therapeutic relevance, whereas cell/animal models at smoker-relevant nicotine doses (100 ng/mL) show mechanistic support as low-dose neuroprotection findings from isolated cell culture remain uncertain for human smokers. Taken together, nicotine-driven oxidative stress accelerates neuronal injury, brain atrophy, and AD progression.
Blood-Brain Barrier Permeability
Nicotine regulates BBB permeability by a variety of well-defined mechanisms. Abbruscato et al., 2002 used in vitro models of the BBB with astrocyte co-culture, which was exposed to 100 ng/mL (concentration relevant to smokers) of nicotine for 12-24 hours29. To assess BBB permeability, the movement of molecular tracers-such as Evans blue dye, and sucrose were tracked to determine how easily molecules can cross from the bloodstream into the brain through the BBB. The study demonstrates that short-term exposure (12-24 hours) to nicotine at levels comparable to those reported in smokers (100 ng/mL) causes a considerable, time-dependent increase in paracellular permeability in an in vitro BBB co-culture model. This indicates an acute permeability enhancing effect, when nicotine, alone or in combination with its metabolite cotinine, promotes the flow of molecular tracers such as sucrose across the BBB. Antagonists of the α7-nAChR can inhibit the increase in permeability, indicating that the impact is specifically mediated by these receptors on brain endothelial cells. This demonstrates that nicotine directly impairs BBB integrity via receptor mediated pathways, supporting the idea that chronic nicotine exposure not only affects neural signaling but also weakens protective barriers in the brain, making it more vulnerable to toxins.
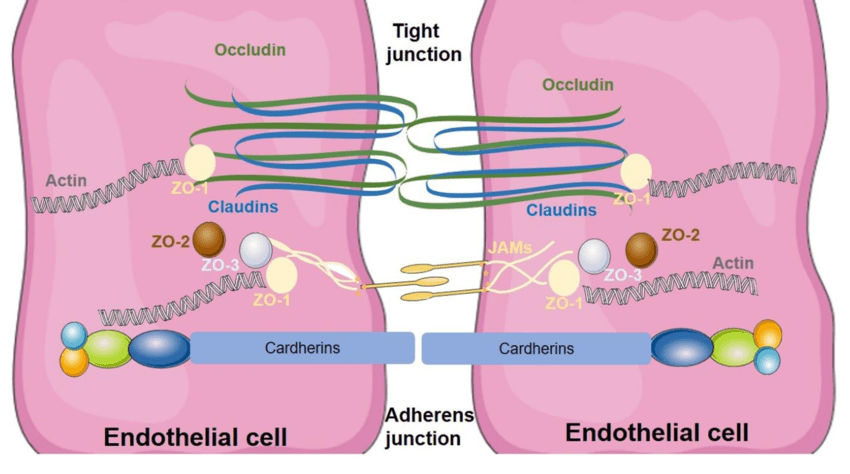
At the molecular level, nicotine exposure damages the tight junction complex, which is essential for BBB integrity23. Specifically, the expression of the tight junction ZO-1 protein (Figure 3) decreases when nicotine is paired with cotinine, resulting in an uneven distribution and altered cell shape rather than the typical border-localized pattern of ZO-1. These modifications indicate that nicotine not only inhibits tight junction proteins but also affects their cellular localization, making the BBB more permeable to circulating molecules. Taken together, Abbruscato et al. and Hawkins et al. indicate that acute nicotine exposure at smoker-relevant doses generally increases BBB permeability and disrupts tight junction organization in endothelial and rodent models. These findings are based on tracers, such as sucrose and Evans blue dye, and mislocalization of tight junction proteins, such as ZO‑1 and occludin. Therefore, these changes primarily reflect those in paracellular transport rather than all transport pathways. Since molecular details of tight junction organization can be abstract, Figure 3 provides a structural framework for interpreting how smoke-induced changes in ZO‑1, occludin, and claudins translate into functional barrier disruption. Moreover, Figure 3 helps connect specific protein-level findings to system-level BBB failure in AD.
Increased cigarette smoke exposure, including extract from both normal and nicotine-free cigarettes, worsens BBB functioning. According to research by Naik et al., 2014, cigarette smoke extract significantly reduces ZO-1 and occludin levels at endothelial cell junctions, resulting in demonstrable increases in permeability to dextran molecules (tracers used to test BBB permeability) of various sizes25. Occludin is a protein found in tight junctions where it helps maintain barrier function of structures like the BBB by regulating its permeability. Furthermore, nicotine-free and ultra-low nicotine cigarettes pose even larger dangers to the BBB because of greater levels of non-nicotine toxicants such as tar and NO. These observations highlight how cigarette smoke-induced disruption of tight junction proteins impairs BBB function, supporting that exposure to cigarette smoke, regardless of nicotine content, allows the pathological processes linked to AD.
An animal experiment conducted by Hawkins et al., 2004 confirms nicotine’s disrupting effects on the BBB31. The study examined how nicotine exposure affects BBB permeability and the organization of tight junction proteins in the brain using a rat model with continuous nicotine administration at levels comparable to heavy smokers. The results revealed that both acute and chronic nicotine treatment in rats increases BBB permeability in vivo, resulting in disturbed distribution of tight junction proteins such as ZO-1, although the total amount of these proteins may not decrease significantly. This suggests that altered protein organization is crucial in increased BBB permeability, supporting the link between nicotine exposure from cigarette smoke and AD progression.
Hawkins et al., 2005 further highlights the link between nicotine and BBB permeability through the activation of nAChRs on brain endothelial cells using a rat model32. The rats were administered nicotine systemically to achieve physiologically relevant concentrations. To assess changes in BBB permeability, researchers infused radiolabeled sucrose entry into brain tissue. The outcome of the study showed that nicotine administration led to a significant increase in the rate at which sucrose crossed the BBB, indicating increased permeability. This supports the hypothesis that nicotine compromises BBB function in vivo. Mechanistically, the effect was attributed to the activation of nAChRs located on the cerebral endothelial cells, as these receptors play a role in regulating tight junction dynamics and the movement of molecules across the barrier. This finding suggests that nicotine can directly affect the structural and functional integrity of the BBB, therefore increasing the brain’s exposure to circulating substances found in cigarette smoke. These acute effects can create a phase specific positive feedback loop in which nicotine induced BBB disruption allows greater entry of smoke derived toxins, which then further damage the barrier. As the BBB is damaged, it increases risks for AD by allowing various other toxins found in cigarette smoke to enter and harm the brain.
However, persistent nicotine exposure provides a more complicated picture as indicated in a study by Lockman et al., 2005, which investigated the effects of chronic nicotine exposure on BBB function in rats33. This was investigated by exposing rats to 4.5 mg of nicotine per kg of body weight, which was given daily for 28 days using osmotic minipumps to achieve plasma nicotine levels similar to those found in heavy human smokers. After the exposure period, the brain uptake rates of radiolabeled methyllycaconitine (3H-MLA)–a nAChR antagonist–and 14C-thiourea, a small polar molecule that served as a tracer for passive diffusion across the BBB, were measured. The findings showed that 28 days of nicotine exposure led to a 60% reduction in the brain uptake of 3H-MLA and reduced the passive permeability of 14C-thiourea by approximately 24% compared to control animals. Lockman’s findings suggest that 28 days of chronic nicotine exposure affects BBB permeability for specific small, passively diffusing tracers, indicating an adaptive ‘tightening’ of specific pathways rather than a widespread leak. Importantly, this endpoint-specific tightening does not restore normal barrier function or prevent the accumulation of bigger neurotoxic proteins like Aβ and tau, and it happens in a rodent model under continuous dosing conditions that differ from acute exposure settings. Therefore, nicotine’s effect on BBB permeability is time and endpoint dependent. Acute exposure increases paracellular permeability, but chronic exposure can reduce permeability for some small molecules, even if oxidative and inflammatory harm to the neurovascular system persists.
Altogether, nicotine reduces tight junction protein expression and disrupts their structural organization in many acute models, primarily through nAChR signaling pathways on endothelial cells. However, chronic exposure may also induce adaptive tightening for some small compounds. In vitro and animal models converge on nicotine-induced disruption of tight junctions and increased permeability, whereas the partial “tightening” observed with some chronic dosages appears model-specific and limited to specific tracers rather than global barrier repair. The overall theme is a heterogeneous, dose and duration dependent modulation of BBB permeability, in which even partial tightening does not prevent oxidative and inflammatory damage or the accumulation of AD relevant proteins. This framework also helps explain why some cell studies report neuroprotection while epidemiologic and in vivo smoking models consistently show harm. This emphasizes nicotine’s role in AD risk through chronic neurovascular damage, despite adaptive responses. Among these studies, human AD cohort data (primarily from Bonomi et al.) linking smoking to increased BBB permeability in clinically diagnosed patients carries the highest clinical relevance, while cell/animal models using smoke extract demonstrate reasonable mechanisms at smoker-relevant doses.
Nitric Oxide
Oxidative Stress
Nitric Oxide may trigger oxidative stress in various ways, depending on the environment. In certain circumstances, NO increases the production of ROS, which harms cells, while in others, it helps reduce oxidative stress. The overall impact of NO is determined by its source, concentration, and the surrounding biological environment.
Endogenous NO levels, specifically when elevated due to inflammation or exposure to air pollution, were shown by Lelieveld et al., 2024 to induce increased oxidative stress34. The study used an extended kinetic multilayer model (KM-SUB-ELF) to simulate the chemical interactions between inhaled pollutants and endogenous molecules within the three compartments of the epithelial lining fluid. The results showed that NO quickly interacts with superoxide (O2-) to produce peroxynitrite (ONOO-), a highly reactive molecule. Peroxynitrite then decomposes to form hydroxyl radicals (OH), which significantly contribute to oxidative stress and eventually cellular damage. This mechanism explains why individuals who have inflammatory airway disorders or who are exposed to pollutants (like smokers) are more vulnerable to oxidative damage, neurodegeneration, and BBB dysfunction. This study provides a mechanistic explanation for the higher sensitivity reported in people with persistently elevated airway NO, thus connecting inflammation, NO production, and injury pathways caused by radicals. This mechanistic pathway highlights how elevated NO-induced oxidative stress in vulnerable individuals can promote neurodegeneration and BBB dysfunction relevant to AD progression.
Further evidence gathered from Wei et al., 2000 suggests that NO donors increase ROS levels inside cells, specifically the mitochondria35. This finding was found by using primary cultures of rat cerebellar granule cells exposed to the S-Nitroso-N-acetyl-DL-penicillamine (SNAP)—an NO donor that slowly releases NO under physiological conditions—to assess NO-induced oxidative stress and apoptosis. DNA fragmentation, indicated by apoptosis, was assessed with ELISA detecting cytoplasmic nucleosomes released during apoptotic DNA. Cytosolic and mitochondrial ROS were quantified by flow cytometry with dyes that fluoresce upon ROS oxidation. Flow cytometry measures increases in fluorescence intensity in individual cells, providing quantitative analysis of intracellular ROS levels. Elevated amounts of hydrogen peroxide and lipid peroxidation products were found, both of which are indications of oxidative stress. When NO donors increase ROS levels in nerve cells, it triggers apoptosis. This is indicated by a significant increase in DNA breakage markers, specifically mono- and oligonucleosomes. Importantly, when cells receive antioxidants such as superoxide dismutase before NO exposure, it can lower both oxidative stress and cell death, showing that ROS production plays a key role in NO-related nerve damage. In short, an increase in NO causes excessive ROS production, which not only leads to oxidative stress and neuronal death, but also increases AD risks.
However, NO’s effect on oxidative stress is not always negative; under some situations, it might even be beneficial. Kaur et al., 2015 used a plant model where wheat seedlings grown without soil were exposed to Pb stress with or without the NO donor sodium nitroprusside (SNP), and then analyzed for ROS, oxidative damage, and antioxidant enzymes using biochemical assays and histochemical staining36. They found that in plant models, exogenous NO supplementation lowers oxidative stress indicators, ROS accumulation, membrane damage, and ion leakage in wheat roots subjected to Pb poisoning. While NO reduced oxidative stress and membrane damage, it is important to note that NO did not fully restore growth or reverse all enzyme activity changes, especially with prolonged or high-dose Pb exposure. Although plant systems differ significantly from human neurons, this demonstrates a common basic biochemical principle: NO may influence ROS homeostasis and protect against heavy metal toxicity by regulating antioxidant enzymes. These findings point to a possible comparable pathway in animal cells where NO may prevent oxidative damage from smoke-derived toxins, but human neuroprotective benefits require direct evidence in neuronal and BBB models.
In summary, NO regulates oxidative stress through both direct chemical interactions, creating highly reactive species when present in excess, and indirect processes, such as triggering antioxidant response and mitochondrial adaptation. The balance of these functions is considerably determined by the physiological environment, NO supply and concentration, and the presence of oxidative or antioxidant components. The findings across species show that NO exposure increases oxidative stress, with the majority of damage occurring due to dose and exposure context differences (acute vs chronic), implying a strong model dependence. Excessive NO, especially in inflammatory conditions or after pollutant exposure, tends to aggravate oxidative damage, whereas regulated NO levels may activate protective cellular pathways. Moreover, in cigarette smoke, NO regularly circulates with other pro-oxidant toxins, causing inflammation, overwhelming endogenous responses, and promoting neuronal injury. A potential integrative model is that in a low-oxidative stress environment, low levels of NO may stimulate antioxidant enzymes and reduce heavy metal toxicity. However, in the presence of ROS produced through cigarette smoke, excess NO can form RNS that promote oxidative damage. Therefore, depending on the initial oxidative activity and dosage, NO’s role shifts from protective to harmful.
Blood-Brain Barrier Permeability
Numerous studies show that NO and NO-releasing compounds modulate BBB integrity in animal models. Shukla et al., 1996 examined how NO and NO-releasing compounds affect BBB permeability in rat brains, as well as administered the NO donor sodium nitroprusside, the SIN-1 peroxynitrite donor, and the NO precursor L-arginine through the carotid artery37. BBB permeability was then assessed by measuring the entry of tracers such as sodium fluorescein, and Evans blue into brain tissue. Their findings highlighted that the NO-releasing compounds caused a dose-dependent increase in BBB permeability. This effect was observed across tracer molecules of different sizes and characteristics. Specifically, SIN-1 was found to increase BBB permeability in rat brains, which was widespread and dose-dependent. This suggests that NO’s impact is not restricted to specific types of solutes. Mechanistically, some of this disruption may be due to the generation of RNS as a result of NO interaction with superoxide radicals, which increases oxidative stress and contributes to endothelial dysfunction. High doses of L-arginine–the natural substrate for nitric oxide synthase–also resulted in considerable increases in BBB permeability, supporting the hypothesis that both endogenous and exogenous NO may produce similar effects. In particular, the SIN-1 findings imply that permeability changes may be caused by a combination of NO and free radical activity, rather than solely NO. Additionally, these results suggest that both endogenous and exogenous NO can weaken the BBB and provide mechanistic support, rather than direct proof, for NO-related contributions to AD pathology.
In pathological contexts, Weyerbrock et al., 2011 examined how NO delivered via NO donors affect BBB permeability and cerebral blood flow in a rat model of glioma38.They administered the NO donor PROLI/NO to male rats with intracerebral C6 gliomas, and measured permeability to radiolabeled tracers and regional blood flow. The results showed that in glioma tumor models (not neurodegeneration) NO donor delivery in intracerebral C6 gliomas led to a temporary increase in permeability at the blood-tumor barrier. The increased permeability was mainly noticeable in tumor tissue and surrounding brain areas, while normal cortex and white matter were unaffected. This specificity suggests potential therapeutic utility in tailored drug delivery techniques, in which brief bursts of NO may be employed to temporarily increase chemotherapeutic absorption into brain tumors without damaging the entire BBB. This temporary tumor barrier effect does not directly model chronic AD BBB breakdown in normal aging brains and serves mainly as evidence for the concept of NO’s permeability role. Moreover, this emphasizes that NO’s effect on the BBB are context dependent and could either facilitate targeted drug delivery or influence the neurovascular processes relevant to AD.
As a non-mammalian model far from human AD context, evidence from Kovacic et al., 2015 suggests that non-mammalian models may support NO’s function in BBB integrity39. The study used healthy 2-year-old common carp and administered the vasodilator glyceryl trinitrate (1 mg/kg) or saline via intraperitoneal injection to assess NO-related changes in BBB integrity and brain pathology. Serum nitrate/nitrite levels were measured and BBB permeability was evaluated using Evans blue dye. The findings reveal that NO donor treatment in common carp resulted in a significant but reversible increase in BBB permeability, which peaked at about 6 hours after administration. This was seen by considerable blue dye leakage into the brain tissue. Importantly, BBB function reverted to baseline within 12-25 hours, demonstrating that NO-induced alterations are both temporary and recoverable in this species. These findings highlight the importance of timing, dosage, and physiological context (including disease status) in determining the amount and duration of NO’s effects on BBB permeability. Furthermore, these results suggest that NO’s impact on BBB integrity can be reversible, implying that short term NO exposure may not always contribute to lasting neurodegenerative changes. However, this evidence cannot support direct inferences about smoker-related BBB dysfunction in AD as it is from a fish model.
Collectively, these studies identify NO as a possible regulator of pathological barrier breakdown as well as having controlled influence of BBB characteristics in therapeutic applications. In cigarette smoke, NO works with other toxins to break down tight junctions and increase permeability, allowing more neurotoxic substances to enter the brain. In cigarette smoke, NO allows neurotoxic substances to enter the brain due to BBB breakdown contributing to elevated AD risk for smokers. This positions smoke-derived NO at a beginning stage of the pathway from environmental exposure to BBB dysfunction and, ultimately, increased AD risk in smokers.
Lead
Oxidative Stress
Lead exposure is a well-recognized environmental hazard that disrupts the body’s oxidative balance by increasing ROS production and impairing antioxidant defenses, leading to cellular and tissue damage in a variety of organisms. Among humans, this oxidative stress occurs across diverse demographic groups, as shown in the Obeng-Gyasi, 2018 life course study of U.S. adults40. The study analyzed data from the 2007–2010 NHANES, a nationally representative U.S. adult survey, to examine the relationship between blood lead levels (BLLs) and the oxidative stress biomarker gamma-glutamyl transferase (GGT). BLL were divided into tertiles and analyzed using log-transformed linear regression models. Results revealed that higher BLLs were strongly associated with increased GGT, an indicator of oxidative stress. This implies that as Pb exposure increases, so does oxidative stress. This association is especially evident among older adults, men, ethnic minorities, those with lower incomes and education levels, and those working in industries such as construction and agriculture, which have historically had higher Pb exposure. While women are more likely to develop AD, men’s higher likelihood of working in occupations with high Pb exposure, such as construction, may increase their risk through environmental processes. Furthermore, these disadvantaged socioeconomic and minority groups interact with established AD risk factors, as lower education is associated with reduced cognition, and ethnic minorities have a significantly higher AD prevalence.
Moreover, Pb generates oxidative stress by increasing the production of ROS such as superoxide and hydrogen peroxide, increasing the human vascular endothelial and smooth muscle cells. A human study by Ni et al., 2004 examined how Pb exposure affects the production of specific ROS in human coronary artery endothelial cells and vascular smooth muscle cells, and investigated the changes in antioxidant enzyme expression and activity41. Human coronary endothelial cells and vascular smooth muscle cells were exposed to either 1 parts per million (ppm) or 10 ppm Pb acetate, or sodium acetate as a control, for short-term (1-30 minutes) or long-term (60 hours) periods in culture. ROS production (superoxide and hydrogen peroxide) was quantified by flow cytometry, and antioxidant/oxidant enzyme levels (Cu/Zn SOD, catalase, glutathione peroxidase, and NAD(P)H oxidase) were measured. The results indicated that short-term Pb exposure immediately increased ROS generation, whereas longer exposure caused permanent increases. These increases were particularly found in hydrogen peroxide, due to changes in antioxidant enzyme activity. This oxidative imbalance harms critical cellular structures and alters essential signaling pathways, contributing to cardiovascular and neurological conditions over time.
Animal studies further support the harmful effects of Pb on oxidative stress. A meta-analysis of 108 rat and mice studies conducted by Fan et al., 2020 examined the extent of oxidative damage induced by Pb42. A comprehensive literature search was conducted in Embase, PubMed, Web of Science, Medline, China National Knowledge Infrastructure, and Chinese Biological Medicine databases to identify all relevant English or Chinese language studies published from the earliest available date in each database through July 22, 2018. The results demonstrated that Pb exposure dramatically increases levels of oxidant markers–malondialdehyde, glutathione disulfide, ROS, and H₂O₂–while simultaneously decreasing antioxidant defenses, including catalase, glutathione peroxidase, and superoxide dismutase. Higher Pb doses and longer exposure periods caused more severe oxidative damage, especially in the kidney, brain, and blood. Brain tissue, in particular, showed decreased activity of antioxidant enzymes and higher amounts of oxidative markers, indicating potential neurotoxicity. This is significant because the neurotoxicity produced may accelerate AD pathology.
While quite distinct from human AD pathology, the previously mentioned study by Kaur et al., 2015 investigations in wheat roots highlight the impact of Pb-induced oxidative stress as a general oxidative stress principle, but provide little significance in predicting the smoking-AD risk in humans36. Their findings highlight that Pb exposure in wheat roots caused higher ROS levels, membrane degradation, and alterations in antioxidant enzyme activity, which is a stress response indicating basic physiological disturbance. Furthermore, exogenous NO supplementation reduced Pb-induced oxidative stress by detoxifying ROS. This indicates potential preventive methods against Pb poisoning, but with insufficient restoration of normal cellular function after extended exposure. These results highlight the role of oxidative stress in Pb toxicity and suggest that limiting ROS accumulation may help reduce neurodegenerative damage relevant to AD progression.
Overall, these studies suggest that Pb exposure triggers oxidative stress in people, animals, and plants by increasing oxidant production while decreasing antioxidants. The biological effects of Pb exposure explain much of the pathology associated with Pb toxicity, including cardiovascular disease, neurodegeneration, and impaired organ function. The findings across species show that Pb exposure increases oxidative stress, with conflicts largely related to dose and duration, indicating strong model dependence. Furthermore, this highlights the necessity for preventative interventions, especially for vulnerable groups who are more severely impacted by Pb exposure. When Pb exposure occurs through cigarette smoke, it adds a chronic, low-level oxidative damage that interacts with other smoke-derived toxins to further accelerate the AD-related mechanisms outlined in this review.
Blood-Brain Barrier Permeability
Chronic exposure to Pb, even at environmentally relevant or moderate concentrations, has a significant influence on the structure and function of the BBB in both adult and developing animal models. Research by Struzynska et al,. 1997 examined whether chronic, environmentally relevant lead exposure alters the structure and function of the BBB in adult rats43. This was done by chronically exposing adult rats to lead acetate in drinking water, and assessing BBB integrity using horseradish peroxidase tracer experiments along with light and electron microscopy to detect structural and permeability changes. Lead amounts in brain capillaries and nerve endings, along with changes in cell membrane fats, were measured to link chemical changes with observed damage to the BBB. Results showed that adult rats exposed to prolonged Pb treatment had a significant accumulation of Pb in both their brain capillaries and nerve endings, suggesting Pb was collecting in nerve cells. During tests with continuously given horseradish peroxidase, significant BBB failure was observed as the barrier became “leaky”, allowing chemicals normally rejected from brain tissue to pass through. The effect is characterized by the opening of interendothelial tight junctions, which are necessary for maintaining BBB integrity. Electron microscopy showed that endothelial cells had increased fluid uptake, pericytes were engulfing particles, and the fat composition in capillary walls had changed, all of which made the membrane unusually permeable. Overall, these findings demonstrate that chronic Pb exposure by cigarette smoke compromises BBB integrity in ways that may allow the acceleration of processes associated with AD pathology.
Furthermore, a study by Gu et al., 2020 using dynamic imaging with AD mouse models demonstrated that both toxic and subtoxic chronic oral Pb doses dramatically increased the BBB’s permeability44. This was found by administering mice varying doses of Pb-acetate or Na-acetate (control), with Pb levels in the brain measured by atomic absorption spectrophotometry. Contrast-enhanced computed tomography imaging was used to analyze brain volumes and time courses, and group differences were assessed with t-tests. The results revealed higher BBB permeability, indicated by real-time imaging. Additionally, the impact was not entirely dose-dependent as even low, “safe” lead concentrations showed a trend towards greater permeability. This implies that prolonged Pb exposure, regardless of dosage, can directly disrupt the BBB, affecting its role as a neuronal protective barrier and perhaps leading to neurodegenerative disorders such as AD.
In an early developmental study conducted by Moorhouse et al., 2002, researchers exposed newborn rats to 0.1% Pb acetate via their mothers’ milk for three weeks, after which the BLLs were measured by atomic absorption spectrophotometry45. BBB integrity and transport function were assessed using programmed intravenous infusion and radiolabeled solutes. The results showed that when newborn rats received low doses of Pb through maternal milk, there was neither widespread nor severe BBB structural breakdown. Cerebrovascular permeability for most indicators remained constant, but there were selective, region-specific increases in the transfers of specific nutrients such as thiamine. This suggests subtle abnormalities in BBB function, likely due to altered brain metabolism or delayed development, rather than apparent barrier collapse. Therefore, while persistent low-level lead exposure in developing brains may not result in visible BBB leakage, it can selectively change nutrition availability, which may impact brain development, potentially conditioning the brain for later-life neurodegeneration rather than directly modeling AD.
All together, these studies from various models and approaches support that persistent Pb exposure, even at levels considered subtoxic, can affect BBB function by increasing permeability and altering substrate transport. Among the Pb studies, adult rat BBB disruption (Struzynska et al.) and Pb-induced permeability increases in AD mouse models (Gu et al.) are most directly relevant to AD, because they demonstrate that chronic, even subtoxic, Pb can weaken the barrier in brains already on an AD trajectory. Developmental and non-AD models therefore serve largely to demonstrate Pb’s ability to disrupt barrier function across the life course, whereas adult human and AD-transgenic data give the strongest support for Pb as a moderator of late-life AD neurodegeneration. Pb-induced BBB weakening is consistent in both in vitro and animal models, though distribution changes vary by model. The BBB’s role as a selective barrier is impaired, which may allow for increased Pb buildup in the brain as well as the admission of other neurotoxic compounds, raising the risk of neurodevelopmental and degenerative illnesses. Additionally, in the context of cigarette smoking, low-dose, chronic Pb carried in smoke likely acts as a vascular co-stressor that increases oxidative stress and BBB leakage established by nicotine and NO, rather than as an isolated cause of AD.
This paper’s synthesis proposes a clear “toxin-barrier-stress” model in which cigarette smoke toxins, such as nicotine, NO, and Pb first damage the BBB by disrupting proteins such as ZO-1 and occludin, allowing excess ROS and RNS production and fuel oxidative stress in an ongoing process that drives amyloid and tau damage. This positions cigarette smoking as a key cause of early-onset AD, explaining mixed study results such as survivor bias and APOEε4 effects through critical dose and duration thresholds, where barrier failure results in permanent degeneration. The review’s primary contribution is this integrated perspective, which emphasizes new treatment approaches centered around the barrier-stress link rather than isolated fixes such as antioxidants alone.
Therapeutic Implications
Cigarette smoke exposure supports the pathological processes of AD through mechanisms such as oxidative stress, BBB dysfunction, and neuroinflammation, as outlined in this review. Importantly, some of these negative effects appear to be reversible, which suggests promise for the development of targeted preventative and therapeutic interventions.
Multiple studies indicate that eliminating cigarette smoke exposure can reduce the risk of developing dementia and cognitive impairment, specifically in individuals who do not carry the APOEε4 allele10. This highlights the potential for effective public health interventions that focus on quitting smoking as a main method of reducing the risk of AD. In laboratory models, the structural and functional damage caused by cigarette smoke–such as damaged tight junction proteins and increased BBB permeability–may be reversed through eliminating the source of oxidative stress and stimulating the endogenous antioxidant response (such as the activation of the Nrf2 pathway). These adaptive processes indicate that the brain does have a repairing ability and may restore BBB integrity and cellular homeostasis after the harmful exposure is removed. Moreover, targeting the BBB without addressing oxidative and inflammatory causes is unlikely to be effective, given smoking-related insults affect both the barrier and the inflammatory conditions collectively (as previously indicated in the bidirectional model).
However, considerations should be taken when interpreting research on low-dose exposures. While some research has suggested that very low concentrations of nicotine or NO may activate protective signalling pathways and therefore reduce cellular oxidative damage, other evidence, such as the finding that ultra-low nicotine and nicotine-free cigarettes cause even greater oxidative stress, contradicts this idea and suggests a limited therapeutic window. For instance, Prasad et al., 2015 and Naik et al., 2014 reveal that non-nicotine components (such as tar) may contribute as much or more harm as nicotine itself, demonstrating that switching to low-nicotine products does not mitigate the danger19,46.
Additionally, recent findings by Kumar et al., 2024 examined how chronic nicotine exposure and subsequent withdrawal affect BBB permeability and the role of overactivated microglia in exacerbating these processes47. Kumar et al., 2024 chronically treated mice with nicotine (12 mg/kg/day), then induced 48 hour withdrawal, to measure BBB permeability, tight junction proteins expression, and neuroinflammation. Microglial depletion was completed prior to the 46 hour withdrawal with PLX5622 chow (mouse food mixed with a drug called PLX5622, which causes microglia to temporarily disappear), and its effects on these outcomes were compared to the control group. Microglia depletion was shown to selectively prevent withdrawal-induced increases in BBB permeability and inflammation in female mice. These sex-specific results suggest microglia-targeted therapies could address withdrawal-induced BBB dysfunction, providing specific smoking cessation for women at AD risk.
Overall, these findings highlight the necessity of prevention, emphasizing that the greatest therapeutic effect comes from reducing or eliminating all exposure to tobacco smoke and associated toxins. Advances in understanding neuroprotective techniques, such as antioxidants, anti-inflammatory drugs, and BBB protectants, may provide further benefits, especially for individuals unable to completely avoid environmental risks. However, the reliance on animal or cell models limits the therapeutic application of these results, since such research may not fully represent the complexities of human disease or the impact of chronic smoking in diverse populations. To further clarify the therapeutic importance of these findings, future research should address these gaps through more extensive, continuous, and human-centered investigations. Ultimately, the reversibility of some smoke-induced brain damage emphasizes the need for early intervention, active risk factor management, and ongoing research into medicines that address both the vascular and neuroinflammatory concerns that drive AD development in smokers.
Future Directions
Although significant progress has been made in determining the function of cigarette smoke in oxidative stress, BBB permeability, and AD progression, significant gaps remain. The most urgent gap is the comprehensive dose-response studies across species, brain regions, exposure durations, and human cohorts, as Schilling et al., 1992 showed nicotine enhances BBB permeability only at toxic levels in cats but failed to address physiological doses or human relevance48. Sex-specific human imaging studies using dynamic contrast MRI evaluating nicotine withdrawal effects are crucial for expanding on Kumar et al., 2024 discovery of female-specific prefrontal BBB breakdown reversed by microglial reduction47. Finally, longitudinal cohort models that incorporate smoke components such as nicotine, NO, and Pb, APOEε4 allele status, genetic variability, and realistic exposure patterns are also required to identify modifiable risk variables contributing to AD risk.
Conclusion
In conclusion, despite mixed epidemiological evidence, cigarette smoke emerges as a plausible modifiable contributor to AD risk through interconnected oxidative stress, BBB disruption, and neuroinflammation. Nicotine, NO, and Pb are among the neurotoxic substances found in cigarette smoke. They seem to influence vascular and brain homeostasis and regulate BBB permeability in a manner dependent on dosage, duration, and model, with acute exposures generally increasing permeability and chronic exposures resulting in more complex, often endpoint-specific adaptations, while overall worsening neurodegenerative changes in preclinical models. Some effects show model-specific reversibility upon cessation, suggesting potential benefits from early intervention, though the direct relevance for humans remains uncertain. Finally, a thorough knowledge of these pathways will inform the development of effective neuroprotective therapies and public health initiatives aimed at improving outcomes for individuals at risk of neurodegenerative diseases worldwide. This review reframes smoking’s role in AD from a vascular risk to a multi-pathway accelerator via oxidative and BBB damage. These processes increase amyloid and tau pathology as well as neuroinflammation, helping explain why the timing of smoking cessation and the use of targeted antioxidant and BBB-protective interventions can significantly alter AD progression.
References
- Alzheimer’s Association. 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. 20, 3708–3821 (2024). [↩] [↩]
- M.E. Cornelius, C.G. Loretan, A. Jamal, B.C. Davis Lynn, M. Mayer, I.C. Alcantara, L. J. Neff. Tobacco Product Use Among Adults – United States, 2021. Morb Mortal Wkly Rep. 72, 475–483 (2023). [↩]
- J.K. Cataldo, J.J. Prochaska, S.A. Glantz. Cigarette smoking is a risk factor for Alzheimer’s Disease: an analysis controlling for tobacco industry affiliation. J Alzheimers Dis. 19, 465–480 (2010). [↩] [↩]
- I. Grundke-Iqbal, K. Iqbal, Y.C. Tung, M. Quinlan, H.M. Wisniewski, L.I. Binder. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 83, 4913–4917 (1986). [↩]
- R.E. Wittenberg, S.L. Wolfman, M. De Biasi, J.A. Dani. Nicotinic Acetylcholine Receptors and Nicotine Addiction: A Brief Introduction. Neuropharmacology. 177, 108256 (2020). [↩]
- V.L. Dawson, T.M. Dawson, E.D. London, D.S. Bredt, S.H. Snyder. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 88, 6368–6371 (1991). [↩]
- A. Reuben, A. Caspi, D.W. Belsky, J. Broadbent, H. Harrington, K. Sugden, R.M. Houts, S. Ramrakha, R. Poulton, T.E. Moffitt. Association of Childhood Blood Lead Levels With Cognitive Function and Socioeconomic Status at Age 38 Years and With IQ Change and Socioeconomic Mobility Between Childhood and Adulthood. JAMA. 317, 1244–1251 (2017). [↩]
- G. Livingston, J. Huntley, K.Y. Liu, S.G. Costafreda, G. Selbæk, S. Alladi, D. Ames, S. Banerjee, A. Burns, C. Brayne, N.C. Fox, C.P. Ferri, L.N. Gitlin, R. Howard, H. C. Kales, M. Kivimäki, E. B. Larson, N. Nakasujja, K. Rockwood, Q. Samus, K. Shirai, A. Singh-Manoux, L.S. Schneider, S. Walsh, Y. Yao, A. Sommerland, N. Mukadam. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. The Lancet. 404, 572–628 (2024). [↩]
- A. Nimmerjahn, F. Kirchhoff, F. Helmchen. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 308, 1314–1318 (2005). [↩]
- C. Reitz, T. den Heijer, C. van Duijn, A. Hofman, M.M.B. Breteler. Relation between smoking and risk of dementia and Alzheimer disease: the Rotterdam Study. Neurology. 69, 998–1005 (2007). [↩] [↩]
- Y. Chang, V. Thornton, A. Chaloemtoem, A.P. Anokhin, J. Bijsterbosch, R. Bogdan, D.B. Hancock, E.O. Johnson, L.J. Bierut. Investigating the Relationship Between Smoking Behavior and Global Brain Volume. Biol Psychiatry Glob Open Sci. 4, 74–82 (2024). [↩]
- Y.S. Ho, X. Yang, S.C. Yeung, K. Chiu, C.F. Lau, A.W.T. Tsang, J.C.W. Mack, R.C.C. Chang. Cigarette Smoking Accelerated Brain Aging and Induced Pre-Alzheimer-Like Neuropathology in Rats. PLoS ONE. 7, e36752 (2012). [↩]
- G. Drewes, B. Lichtenberg-Kraag, F. Döring, E.M. Mandelkow, J. Biernat, E. Mandelkow. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 11, 2131–2138 (1992). [↩]
- Z. Xia, M. Dickens, J. Raingeaud, R.J. Davis, M.E. Greenberg. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 270, 1326–1331 (1995). [↩]
- Q. Sun, H. Wang, M. Yang, H. Xia, Y. Wu, Q. Liu, H. Tang. miR-153-3p via PIK3R1 Is Involved in Cigarette Smoke-Induced Neurotoxicity in the Brain. Toxics. 11, 969 (2023). [↩]
- D.F. Church, W.A. Pryor. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 64, 111–126 (1985). [↩]
- S.B. Lee, J.H. Kim, M.H. Cho, E.S. Choe, K.S. Kim, S.M. Shim. Impact of commercial cigarette smoke condensate on brain tissue co-cultured with astrocytes and blood-brain barrier endothelial cells. J Toxicol Environ Health A. 80, 533–541 (2017). [↩] [↩]
- A. Khanna, M. Guo, M. Mehra, W. Royal. Inflammation and Oxidative Stress Induced by Cigarette Smoke in Lewis Rat Brains. J Neuroimmunol. 254, 69–75 (2013). [↩] [↩] [↩]
- S. Prasad, R.K. Sajja, J.H. Park, P. Naik, M.A. Kaisar, L. Cucullo. Impact of cigarette smoke extract and hyperglycemic conditions on blood-brain barrier endothelial cells. Fluids Barriers CNS. 12, 18 (2015). [↩] [↩] [↩] [↩]
- M.G. Ewees, M.A. El-Mahdy, Y. Hannawi, J.L. Zweier. Tobacco cigarette smoking induces cerebrovascular dysfunction followed by oxidative neuronal injury with the onset of cognitive impairment. J Cereb Blood Flow Metab. 45, 48–65 (2025). [↩]
- J.M. Starr, A.J. Farrall, P. Armitage, B. McGurn, J. Wardlaw. Blood–brain barrier permeability in Alzheimer’s disease: a case–control MRI study. Psychiatry Res Neuroimaging. 171, 232–241 (2009). [↩]
- C.G. Bonomi, C. Motta, M.G. Di Donna, M. Poli, M. Nuccetelli, S. Bernardini, N.B. Mercuri, G. Koch, A. Martorana. Age of onset moderates the effects of Vascular Risk Factors on Neurodegeneration, Blood-Brain-Barrier permeability, and cognitive decline in Alzheimer’s Disease. Alzheimers Res Ther. 16, 248 (2024). [↩]
- H. Kadry, B. Noorani, U. Bickel, T.J. Abbruscato, L. Cucullo. Comparative assessment of in vitro BBB tight junction integrity following exposure to cigarette smoke and e-cigarette vapor: a quantitative evaluation of the protective effects of metformin using small-molecular-weight paracellular markers. Fluids Barriers CNS. 18, 28 (2021). [↩] [↩]
- A. Bernard, J.M. Ku, R. Vlahos, A.A. Miller. Cigarette smoke extract exacerbates hyperpermeability of cerebral endothelial cells after oxygen glucose deprivation and reoxygenation. Sci Rep. 9, 15573 (2019). [↩]
- P. Naik, N. Fofaria, S. Prasad, R.K. Sajja, B. Weksler, P.O. Couraud, I.A. Romero, L. Cucullo. Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: is smoking reduced or nicotine-free products really safe? BMC Neurosci. 15, 51 (2014). [↩] [↩]
- J. Barr, C.S. Sharma, S. Sarkar, K. Wise, L. Dong, A. Periyakaruppan, G.T. Ramesh. Nicotine induces oxidative stress and activates nuclear transcription factor kappa B in rat mesencephalic cells. Mol Cell Biochem. 297, 93–99 (2007). [↩]
- W. Sukketsiri, K. Hoshino, H. Kugo, T. Nakamura, T. Sasoh, T. Moriyama, N. Zaima. Isoflavone Ameliorated Oxidative Stress and Vascular Damages in Nicotine-Administrated Mice. J Oleo Sci. 68, 1241–1249 (2019). [↩]
- Y. Dong, W. Bi, K. Zheng, E. Zhu, S. Wang, Y. Xiong, J. Chang, J. Jiang, B. Liu, Z. Lu, Y. Cheng. Nicotine Prevents Oxidative Stress-Induced Hippocampal Neuronal Injury Through α7-nAChR/Erk1/2 Signaling Pathway. Front Mol Neurosci. 13, 557647 (2020). [↩]
- T.J. Abbruscato, S.P. Lopez, K.S. Mark, B.T. Hawkins, T.P. Davis. Nicotine and Cotinine Modulate Cerebral Microvascular Permeability and Protein Expression of ZO-1 through Nicotinic Acetylcholine Receptors Expressed on Brain Endothelial Cells. J Pharm Sci. 91, 2525–2538 (2002). [↩]
- Q.Q. Tao, R.R. Lin, Y.H. Chen, Z.Y. Wu. Discerning the role of blood-brain barrier dysfunction in Alzheimer’s disease. Aging Dis. 13, 1391–1404 (2022). [↩]
- B.T. Hawkins, T.J. Abbruscato, R.D. Egleton, R.C. Brown, J.D. Huber, C.R. Campos, T.P. Davis. Nicotine increases in vivo blood–brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 1027, 48–58 (2004). [↩]
- B.T. Hawkins, R.D. Egleton, T.P. Davis. Modulation of cerebral microvascular permeability by endothelial nicotinic acetylcholine receptors. American Journal of Physiology-Heart and Circulatory Physiology. 289, 212–219 (2005). [↩]
- P.R. Lockman, C.J. Van der Schyf, T.J. Abbruscato, D.D. Allen. Chronic nicotine exposure alters blood–brain barrier permeability and diminishes brain uptake of methyllycaconitine. Journal of Neurochemistry. 94, 37–44 (2005). [↩]
- S. Lelieveld, J. Lelieveld, A. Mishra, A. Daiber, A. Pozzer, U. Pöschl, T. Berkemeier. Endogenous Nitric Oxide Can Enhance Oxidative Stress Caused by Air Pollutants and Explain Higher Susceptibility of Individuals with Inflammatory Disorders. Environmental Sci Technol. 58, 1823–1831 (2024). [↩]
- T. Wei, C. Chen, J. Hou, W. Xin, A. Mori. Nitric oxide induces oxidative stress and apoptosis in neuronal cells. Biochim Biophys Acta BBA – Mol Cell Res. 1498, 72–79 (2000). [↩]
- G. Kaur, H.P. Singh, D.R. Batish, P. Mahajan, R.K. Kohli, V. Rishi. Exogenous Nitric Oxide (NO) Interferes with Lead (Pb)-Induced Toxicity by Detoxifying Reactive Oxygen Species in Hydroponically Grown Wheat (Triticum aestivum) Roots. PLoS ONE. 10, e0138713 (2015). [↩] [↩]
- A. Shukla, M. Dikshit, R.C. Srimal. Nitric oxide-dependent blood-brain barrier permeability alteration in the rat brain. Experientia. 52, 136–140 (1996). [↩]
- A. Weyerbrock, S. Walbridge, J.E. Saavedra, L.K. Keefer, E.H. Oldfield. Differential effects of nitric oxide on blood-brain barrier integrity and cerebral blood flow in intracerebral C6 gliomas. Neuro-Oncol. 13, 203–211 (2011). [↩]
- S. Kovacić, L. Rumora, E. Gjurcevic, M.Š. Klaric, G. Ivkic. Effects of nitric oxide on blood-brain barrier permeability in common carp (Cyprinus carpio L.). Am J Vet Res. 76, 615-624 (2015). [↩]
- E. Obeng-Gyasi. Lead Exposure and Oxidative Stress—A Life Course Approach in U.S. Adults. Toxics. 6, 42 (2018). [↩]
- Z. Ni, S. Hou, C.H. Barton, N.D. Vaziri. Lead exposure raises superoxide and hydrogen peroxide in human endothelial and vascular smooth muscle cells. Kidney Int. 66, 2329–2336 (2004). [↩]
- Y. Fan, X. Zhao, J. Yu, J. Xie, C. Li, D. Liu, C. Tang, C. Wang. Lead-induced oxidative damage in rats/mice: A meta-analysis. J Trace Elem Med Biol. 58, 126443 (2020). [↩]
- L. Struzyńska, M. Walski, R. Gadamski, B. Dabrowska-Bouta,U. Rafałowska. Lead-induced abnormalities in blood-brain barrier permeability in experimental chronic toxicity. Mol Chem Neuropathol. 31, 207–224 (1997). [↩]
- H. Gu, P.R. Territo, S.A. Persohn, A.A. Bedwell, K. Eldridge, R. Speedy R, Z. Chen, W. Zheng, Y. Du. Evaluation of chronic lead effects in the Blood Brain Barrier System by DCE-CT. J Trace Elem Med Biol Organ Soc Miner Trace Elem GMS. 62, 126648 (2020). [↩]
- S.R. Moorhouse, S. Carden, P.N. Drewitt, B.P. Eley, R.J. Hargreaves, D. Pelling. The effect of chronic low level lead exposure on blood-brain barrier function in the developing rat. Biochem Pharmacol. 37, 4539–4547 (1988). [↩]
- P. Naik, N. Fofaria, S. Prasad, R.K. Sajja, B. Weksler, P.O. Couraud, I.A. Romero, L. Cucullo. Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: is smoking reduced or nicotine-free products really safe? BMC Neurosci. 15, 51 (2014). [↩]
- M. Kumar, J. Keady, S.P. Aryal, M. Hessing, C.I. Richards, J.R. Turner. The Role of Microglia in Sex- and Region-Specific Blood-Brain Barrier Integrity During Nicotine Withdrawal. Biol Psychiatry Glob Open Sci. 4, 182–193 (2024). [↩] [↩]
- L. Schilling, A. Bultmann, M. Wahl. Lack of effect of topically applied nicotine on pial arteriole diameter and blood-brain barrier integrity in the cat. Clin Investig. 70, 210–217 (1992). [↩]



{kind=link}