
Abstract
Pancreatic cancer remains one of the most aggressive and lethal malignancies, with a five-year survival rate that continues to be lower than most other cancers. Traditional chemotherapy offers limited benefits, particularly in patients with underlying genetic mutations such as BRCA1 or BRCA2 mutations that impair DNA repair pathways. In recent years, PARP (poly ADP-ribose polymerase) inhibitors have emerged as targeted therapies for pancreatic cancer, especially in patients harboring BRCA1 or BRCA2 mutations. This paper reviews multiple clinical trials that assess the efficacy of PARP inhibitors, as either maintenance therapy or active treatment in advanced pancreatic cancer. Several studies demonstrated improved progression-free survival among BRCA-mutated patients receiving PARP inhibitors compared to placebo. However, the improvements in overall survival were less consistent. Treatment responses were influenced by factors such as disease stage, prior platinum-based chemotherapy, and specific mutation subtypes. Common side effects included fatigue, anemia, nausea, and hematologic toxicities, which may impact long-term use. Overall, this review highlights the therapeutic potential of PARP inhibitors in a genetically defined subgroup of pancreatic cancer patients, while acknowledging their limitations and the need for continued research to optimize their use.
Keywords: PARP inhibitors; pancreatic cancer; rucaparib; olaparib; niraparib; veliparib; BRCA mutation
Introduction
Pancreatic cancer is a highly aggressive tumor exhibiting a high rate of genomic alterations, frequent activation of oncogene and tumor suppressor gene mutations1. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer and has become the third leading cause of cancer-related death, only behind lung cancer and colorectal cancer in the United States, and is predicted to rise to the second by 20302. In 2025, an estimated 67,440 new diagnoses and 51,980 deaths from pancreatic cancer occurred in the United States3. The prognosis of pancreatic cancer remains dismal; 5-year survival is only 13% in 2025 in pancreatic cancer patients of all stages3.
Due to a combination of late symptom onset and aggressive pathophysiology, less than 20% of pancreatic cancer patients are eligible for surgery, which means that most patients are in an advanced stage at the time of diagnosis3. For patients with nonmetastatic or locally advanced pancreatic cancer, surgical resection of the pancreas is still the preferred treatment, but more than 50% of patients still experience recurrence within 12 months after surgery4. For patients with inoperable metastatic pancreatic cancer, cytotoxic chemotherapies were the only available systemic options to delay tumor progression over a long period of time. Due to the low sensitivity of pancreatic cancer to chemotherapy, treatment efficacy and prognosis are still unsatisfactory.
Targeted therapies are designed to target specific oncogenes and pathways at the cellular and molecular levels to interfere with and block specific target molecules involved in cancer growth and progression. They have emerged as promising strategies for pancreatic cancer treatment, as they offer greater targeting precision and improved therapeutic efficacy with fewer side effects compared to conventional chemotherapy5.
This study provides an overview of DNA repair and the concept of synthetic lethality, emphasizing the roles of poly (ADP-ribose) polymerase (PARP) and BRCA mutations. It highlights PARP inhibitors as targeted therapy based on the principle of synthetic lethality. It further explores the molecular mechanisms of PARP inhibitors and presents case studies illustrating their application in pancreatic cancer treatment, especially in patients with BRCA variants.
DNA Repair and Synthetic Lethality
Maintaining genome integrity is fundamental to cellular survival and proper function in organisms. Normal cells possess complex mechanisms to preserve the accuracy of DNA replication, preventing mutations that could lead to cellular dysfunction, transformation, or death. The genetic code must be accurately replicated during cell division and protected from various endogenous and exogenous sources of damage, including reactive oxygen species, UV radiation, chemical mutagens, and replication errors6. Even minor alterations in DNA sequence can disrupt protein function, change gene expression patterns, and disturb cellular homeostasis. Cells have evolved complex DNA damage response (DDR) mechanisms to detect, signal, and repair various types of DNA lesions7. The DDR serves as a surveillance system that monitors DNA accuracy and coordinates appropriate responses to maintain genomic stability7. When DNA damage is detected, DDR pathways activate cell cycle checkpoints to allow time for repair, induce DNA repair mechanisms, or trigger apoptosis if the damage is too extensive to repair8. If DDR activation fails or is overwhelmed, cells may undergo malignant transformation due to accumulated mutations, enter senescence, or die through apoptotic pathways7. The failure of DDR mechanisms is an indicator of cancer development, as it allows cells to tolerate high levels of genomic instability while continuing to proliferate9.
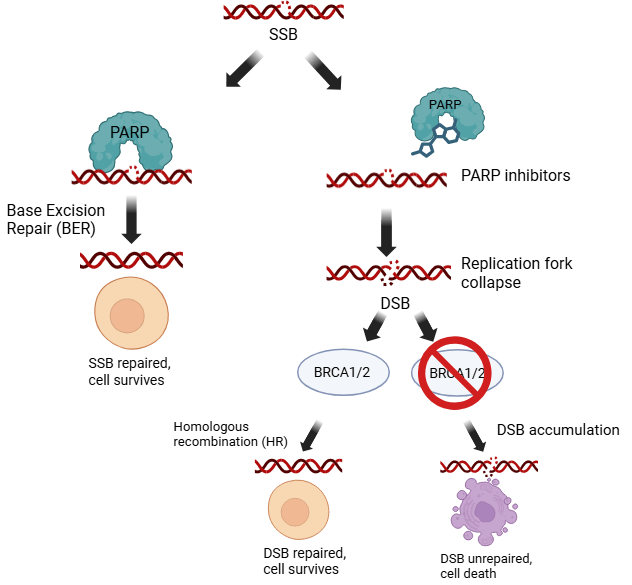
The cellular response to DNA damage involves three major repair pathways, and each is specialized for different types of lesions7. Base excision repair (BER) primarily handles single-strand breaks (SSBs) and small base modifications caused by oxidative damage or spontaneous base loss; when BER fails, SSBs can convert to more dangerous double-strand breaks during replication7. Failure of SSB repair mechanisms can result in the accumulation of unrepaired breaks, leading to replication fork stalling, collapse, and ultimately the formation of double-stranded DNA breaks (DSBs). When SSB repair is impaired, DSBs can form, triggering the activation of two alternative pathways to compensate for the deficiency. The first is homologous recombination repair (HRR), a high-fidelity process that uses the sister chromatid as a template to accurately restore the DNA sequence, particularly during S and G2 phases of the cell cycle10. Non-homologous end joining (NHEJ) also repairs DSBs but through direct ligation of broken ends, which is faster but more error-prone than HRR and operates throughout the cell cycle10.
The concept of synthetic lethality was first described in classical genetics in 1946 through studies on Drosophila12. He discovered that while mutations in two separate genes were individually compatible with survival, their combination within the same organism proved lethal13. This finding highlighted how genetic interactions can establish conditionally essential relationships, where the loss of one gene could be compensated by the normal function of another, but the simultaneous loss of both would result in death8. This foundational concept demonstrated that synthetic lethal interactions reveal functional relationships between genes and pathways that might not be apparent from studying single gene mutations alone9.
The application of synthetic lethality concepts to cancer research has revolutionized targeted therapy by exploiting the genetic vulnerabilities inherent in cancer cells9. Cancer cells often harbor mutations in tumor suppressor genes or DNA repair pathways, creating dependencies on alternative survival mechanisms that normal cells do not require9. By identifying and targeting these cancer-specific dependencies, synthetic lethality offers a means to selectively eliminate malignant cells while sparing healthy tissue9. This approach has been particularly effective in cancers with DNA repair defects, where inhibiting complementary repair pathways can disrupt cancer cell survival without harming normal cells.
Role of PARP and BRCA Mutation
Pancreatic cancer is the third most common cancer related to early-onset gene mutation in breast cancer as well as ovarian cancer. Family history of pancreatic cancer is an essential risk factor, and germline BRCA mutations significantly increase pancreatic cancer risk. Germline BRCA1 and BRCA2 mutations are found in approximately 4-7% of pancreatic cancer patients, representing one of the most significant genetic risk factors for this malignancy14. The hereditary component of pancreatic cancer is increasingly recognized, with BRCA1/2 mutations being the most common genetic alterations in familial pancreatic cancer cases. The molecular mechanisms underlying pancreatic cancer development in BRCA mutation carriers involve defective homologous recombination repair, leading to genomic instability that drives the accumulation of oncogenic mutations and chromosomal aberrations. The pancreatic tissue may be particularly susceptible to BRCA-related tumorigenesis due to its high proliferative capacity and exposure to various DNA-damaging agents, including reactive oxygen species generated during normal metabolism and exogenous carcinogens.
Clinically, BRCA-mutated pancreatic cancers exhibit distinct characteristics, including improved response to DNA-damaging chemotherapy such as platinum-based agents. Patients with germline BRCA mutations have received increased attention due to advances in management, with some studies showing a favorable prognosis compared to sporadic pancreatic cancer, likely due to additional therapeutic options and enhanced chemosensitivity15. The HRD phenotype renders these tumors more sensitive to chemotherapies that induce DNA damage, providing a therapeutic window that can be exploited to improve treatment outcomes in this traditionally treatment-resistant malignancy.
BRCA1 and BRCA2 are tumor suppressor genes that encode proteins essential for maintaining genomic stability through homologous recombination (HR) repair of DNA double-strand breaks16. BRCA1 functions as a scaffold protein that coordinates the DNA damage response by recruiting various repair proteins and regulatory factors, playing roles in DNA damage signaling, checkpoint activation, and repair pathway choice17. BRCA2 has a more direct role in HR by facilitating RAD51, known as DNA repair protein RAD51 homolog 1, loading onto single-strand DNA, a critical step in homologous recombination16. Both proteins are indispensable for maintaining genomic stability, particularly in rapidly dividing cells during S and G2 phases of the cell cycle.
BRCA genes are critically important in DNA damage response pathways because their loss of function leads to defective HR repair, resulting in the accumulation of chromosomal aberrations and genomic instability. This deficiency creates a state known as homologous recombination deficiency (HRD), where cells become particularly vulnerable to DNA-damaging agents (Figure 1). The HRD phenotype makes these cells dependent on alternative repair pathways for survival, creating therapeutic opportunities that can be exploited18.
The Poly(ADP-ribose) polymerase (PARP) family in humans comprises 17 members that catalyze protein ADP-ribosylation, the addition of ADP-ribose molecules to proteins, a critical post-translational modification involved in DNA repair and cellular homeostasis19. These enzymes are categorized into five distinct subgroups based on their domain structure and cellular functions (Table 1)20. DNA Damage-Dependent PARPs include PARP1 (ARTD1), PARP2 (ARTD2), and PARP3 (ARTD3), which are primarily nuclear enzymes that respond to DNA damage through single-strand and double-strand break repair mechanisms20. Tankyrases consist of PARP5a (TNKS1) and PARP5b (TNKS2), which regulate telomere maintenance and Wnt signaling pathways19. Cys-Cys-Cys-His Zinc finger (CCCH)-type PARPs contain PARP7 (TIPARP), PARP12 (ZC3HDC1), and PARP13 (ZAP), which are involved in stress responses and antiviral immunity. Macro-PARPs, which contain macro domains for ADP-ribose binding and regulate transcriptional and immune responses, include PARP9 (BAL1), PARP14 (BAL2), and PARP15 (BAL3)19. Other PARPs encompass PARP4 (vPARP), PARP6 (ARTD17), PARP8 (ARTD16), PARP10 (ARTD10), PARP11 (ARTD11), and PARP16 (ARTD15), which have diverse cellular functions including drug resistance, cell cycle regulation, and ER stress responses21.
| Name | Other Aliases | Subgroup | Main Functions |
| PARP1 | ADPRT, ARTD1 | DNA-dependent PARPs | DNA damage detection and repair coordination (base excision repair) Chromatin remodeling and transcriptional regulation Cell death pathway regulation |
| PARP2 | ARTD2 | DNA-dependent PARPs | DNA damage response and repair (base excision repair) Chromatin structure regulation Transcriptional regulation |
| PARP3 | ARTD3 | DNA-dependent PARPs | DNA damage response and signaling Chromatin remodeling and nucleosome modification Mono-ADP-ribosylation activity (attachment of a single ADP-ribose to a target molecule, such as proteins, DNA, RNA)22 |
| PARP4 | ARTD4, vPARP | Other PARPs | Vault particle regulation and cellular transport (Manages vault complexes for transport)23 Mono-ADP-ribosylation activity Cellular structure and potentially immune functions |
| PARP5a | ARTD5, TNKS1 | Tankyrases | Telomere maintenance and regulation Wnt signaling pathway activation Mitosis regulation and cell division control |
| PARP5b | ARTD6, TNKS2 | Tankyrases | Wnt/β-catenin signaling regulation Telomere maintenance and cellular processes Protein stability regulation through PARylation |
| PARP6 | ARTD17 | Other PARPs | Tumor suppression through cell cycle controlCentrosome integrity and mitotic spindle formation Neuronal dendrite morphogenesis and neural development (Shapes nerve cell branches)24 |
| PARP7 | ARTD14, TIPARP | CCCH-type PARPs | Innate immunity and stress response regulation (Controls immune and stress responses) Transcription factor regulation and tumor suppression Microtubule control and cellular organization (Manages cell structural networks) |
| PARP8 | ARTD16 | Other PARPs | Cell proliferation suppression and tumor control Membranous organelle assembly and maintenance Mono-ADP-ribosylation activity |
| PARP9 | ARTD9, BAL1 | Macro-PARPs | Interferon signaling and antiviral immunity (Activates antiviral immune responses) E3 ubiquitin ligase complex formation and protein degradation (Tags proteins for destruction)25 Macrophage activation and STAT1 regulation (Activates immune cells) |
| PARP10 | ARTD10 | Other PARPs | Cellular proliferation and replication stress alleviation G2/M cell cycle transition regulation DNA damage tolerance and genome stability |
| PARP11 | ARTD11 | Other PARPs | Immunosuppression and regulatory T cell activation IFNAR1 regulation and interferon signaling disruption (Disrupts antiviral signaling) Nuclear envelope stabilization and cellular localization |
| PARP12 | ARTD12, ZC3HDC1 | CCCH-type PARPs | Interferon-stimulated antiviral response and translation control (Blocks viral protein synthesis) Mitochondrial function maintenance and AKT signaling regulation Stress granule formation and cellular stress response (Forms protective RNA clusters) |
| PARP13 | ARTD13, ZC3HAV1, ZAP | CCCH-type PARPs | Antiviral RNA targeting and degradation Cellular mRNA stability regulation and apoptosis promotion MicroRNA pathway modulation (Regulates small RNA functions) |
| PARP14 | ARTD8, BAL2 | Macro-PARPs | Macrophage polarization and immune regulation (Directs immune cell specialization) STAT6-dependent transcriptional co-activation (Enhances specific gene expression)26 Cancer progression and therapeutic resistance |
| PARP15 | ARTD7, BAL3 | Macro-PARPs | Mono-ADP-ribosylation and transcriptional regulation Telomere maintenance and stress response Endoplasmic reticulum-associated protein modification |
| PARP16 | ARTD15 | Other PARPs | Endoplasmic reticulum stress response regulation Transmembrane signal transduction (Transfers signals across membranes) Selective ER stress sensor activation |
The family is functionally divided into two groups depending on their catalytic activities: traditional PARPs (PARP1, PARP2, PARP5a, PARP5b) that synthesize poly(ADP-ribose) polymers, and the majority of family members that function as mono-ADP-ribosyl transferases (mARTs)20. PARP1, the founding member, exemplifies the typical structural organization with three functional domains: N-terminal DNA-binding domain containing zinc finger motifs for DNA break recognition, central automodification domain serving as both regulatory region and substrate for auto-ADP-ribosylation, and C-terminal catalytic domain containing the active site for NAD+ binding and ADP-ribose transfer27. PARP family enzymes use NAD+ as a substrate to transfer ADP-ribose moieties to acceptor proteins on aspartate, glutamate, asparagine, arginine, or lysine residues, serving as a signaling mechanism in DNA repair, transcriptional regulation, chromatin remodeling, cell cycle control, and stress responses20.
Within the PARP enzyme family, only a few members are directly implicated in DNA damage repair pathways relevant to BRCA-deficient pancreatic cancer. Among these, PARP-1 plays the biggest role, with PARP-2 and PARP-3 contributing more cooperative roles compared to PARP-128.
PARP inhibitors exert their anticancer effects not only through inhibition of catalytic PARP activity but also through PARP trapping, a mechanism in which PARP enzymes remain bound to sites of DNA damage, obstructing DNA replication and repair29. Trapped PARP–DNA complexes generate replication-associated double-strand breaks that require homologous recombination for repair. In BRCA1- or BRCA2-deficient cancer cells, the inability to perform effective homologous recombination leads to accumulation of lethal DNA damage, forming the mechanistic basis of synthetic lethality exploited by PARP inhibitors29.
Beyond PARP trapping, PARP inhibition disrupts replication fork protection. In BRCA-deficient cells, stalled replication forks are normally stabilized by BRCA1/2-mediated recruitment of repair proteins30. Loss of this function results in nucleolytic degradation of replication forks, contributing to genomic instability and cell death29. PARP inhibitors further exacerbate this vulnerability by impairing fork restart, amplifying cytotoxic stress in BRCA-mutated pancreatic cancer cells30.
Despite initial sensitivity, resistance to PARP inhibitors can emerge through multiple mechanisms. One well-characterized mechanism is BRCA reversion mutations, which restore the open reading frame of BRCA genes and partially re-establish homologous recombination proficiency31. Additional resistance mechanisms include restoration of homologous recombination through loss of HR suppressors, stabilization of replication forks independent of BRCA function, and reduced PARP trapping31. These adaptations allow cancer cells to tolerate PARP inhibition and survive therapy31.
Clinically, these mechanisms have important implications for patient selection and treatment sequencing. Sensitivity to platinum-based chemotherapy has emerged as a predictive biomarker of PARP inhibitor response, reflecting shared reliance on homologous recombination deficiency32. Conversely, prior exposure to PARP inhibitors may induce cross-resistance to platinum agents, complicating subsequent treatment decisions33. Understanding these resistance pathways is essential for optimizing PARP inhibitor use, identifying patients most likely to benefit, and guiding future combinations or sequential therapeutic strategies in pancreatic cancer.
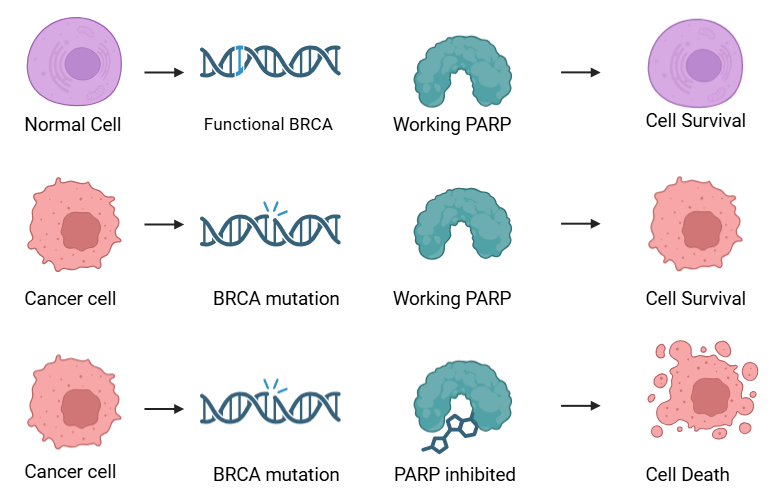
The relationship between PARP and BRCA in pancreatic cancer represents a paradigm of synthetic lethality, where the combination of two non-lethal defects results in cell death (Figure 2)34. In normal cells, both single-strand break repair (via PARP1) and double-strand break repair (via BRCA1/2) function efficiently. However, when BRCA function is lost due to mutations, cells become critically dependent on PARP-mediated repair pathways for survival35.
Methods
Search Terms and Strategy
We conducted a comprehensive search using PubMed’s advanced search interface to identify relevant case studies examining PARP inhibitors in pancreatic cancer treatment. PubMed was selected as the primary database due to its extensive coverage of biomedical literature and robust indexing system using Medical Subject Headings (MeSH) terms. The advanced search function allowed precise query construction using Boolean operators and field-specific searches to maximize the retrieval of relevant studies while discarding irrelevant results. This review was conducted in accordance with PRISMA principles for transparent reporting of literature searches and study selection.
Our search strategy combined controlled vocabulary and free-text terms using the following query: (PARP inhibitors [MeSH] OR olaparib OR niraparib OR talazoparib OR rucaparib) AND pancreatic cancer [MeSH]. The first component captured both the MeSH term “PARP inhibitors” and specific drug names as free-text keywords to ensure comprehensive coverage of all relevant PARP inhibitor studies, including those that might not be indexed under the general MeSH term. The second component utilized the MeSH term “pancreatic cancer” to systematically capture all studies related to pancreatic malignancies. The Boolean operator AND was used to identify studies at the intersection of these two concepts.
Study Selection Criteria
To focus on the most clinically relevant and current evidence, we applied several filters to our search. (1) Study type was limited to clinical trials to capture interventional studies with direct clinical implications; (2) Publication date was restricted to the last 10 years (2015-2025) to ensure contemporary relevance and current treatment approaches; and (3) Language was limited to English to facilitate comprehensive review and analysis. The inclusion criteria are BRCA-mutated pancreatic cancer patients, clinical outcomes, and adverse effects associated with PARP inhibitor therapy. These filters helped refine our results to focus on recent, high-quality clinical evidence regarding PARP inhibitor efficacy and safety in pancreatic cancer treatment. The initial search identified 530 records. After removal of duplicates and title/abstract screening, 15 studies remained for full-text review, of which 5 met the inclusion criteria and were included in the final analysis.
Data Extraction and Outcomes
For each included study, data were extracted on study design, sample size, BRCA mutation status, treatment regimen, progression-free survival, overall survival, secondary endpoints including time to first subsequent therapy (TFST) and time to second subsequent therapy (TSST) and reported adverse events. These data were synthesized qualitatively due to heterogeneity across study designs and outcomes. From this search, we identified four PARP inhibitors that have been investigated in BRCA-mutated pancreatic cancer: Olaparib, Niraparib, Veliparib, and Rucaparib.
Recent Case Studies of PARP Inhibitors in Pancreatic Cancer
PARP inhibitors work through the mechanism of synthetic lethality, selectively targeting cancer cells with deficiencies in DNA repair mechanisms. They are particularly effective in tumors lacking a functional homologous recombination (HR) pathway, a vital system for repairing DNA double-strand breaks. PARP enzymes facilitate the repair of single-strand DNA breaks via base excision repair, becoming critical for the survival of HR-deficient cells. Inhibition of PARP leads to the conversion of unrepaired single-strand breaks into lethal double-strand breaks during DNA replication, resulting in genomic instability and cell death.
Pancreatic cancers often harbor mutations in BRCA1 and BRCA2, key genes in the HR pathway. Loss of BRCA function renders tumor cells heavily reliant on PARP-mediated repair, making these tumors especially susceptible to PARP inhibition. Therefore, PARP inhibitors offer a targeted therapeutic strategy for BRCA-mutated or HR-deficient pancreatic cancers.
Olaparib
In the POLO Phase III study (NCT02184195), Olaparib monotherapy was investigated as a maintenance regimen in patients with metastatic pancreatic cancer who had germline BRCA1/2 mutations and did not show disease progression during at least 16 weeks of platinum-based chemotherapy36,37. Patients were treated with or without Olaparib (300 mg, twice daily). The findings indicate that olaparib monotherapy significantly improved median progression-free survival (PFS) to 7.4 months compared with 3.8 months in the placebo group (hazard ratio [HR], 0.53; 95% CI, 0.35 to 0.82; P = .004)36. The median Time to First Subsequent Therapy (TFST), a secondary end point, was 9.0 months in the Olaparib group and 5.4 months in the placebo group37. The median time to second subsequent therapy was reported to be 14.9 months in the olaparib group and 9.6 months in the placebo group. No statistically significant improvement in overall survival (OS) was observed. Olaparib monotherapy resulted in grade 3 or higher adverse effects, such as anemia, abdominal pain, and fatigue in 40% of the group and 23% in the placebo group36. Because of the adverse events, 5% of the Olaparib group and 2% of the placebo group discontinued the regimen36. While olaparib maintenance therapy is associated with increased toxicity and lacks overall survival benefit, it provides measurable clinical benefit in terms of disease control for BRCA-mutated pancreatic cancer patients.
Niraparib
In this randomized Phase Ib/II study (NCT03404960), niraparib combined with either nivolumab or ipilimumab was investigated as maintenance therapy in 91 patients with advanced pancreatic cancer harboring BRCA or PALB2 variants, who had not progressed after ≥16 weeks of platinum-based chemotherapy38. Patients received niraparib (200 mg PO, daily), plus either nivolumab (240 mg intravenous, every 2 weeks) or ipilimumab (3 mg/kg intravenous, every 3 weeks). In patients with BRCA or PALB2 variants, the median PFS was 3.7 months, and the median OS was 12.2 months in the niraparib plus nivolumab arm. In the niraparib plus ipilimumab arm, the median PFS was 10.4 months, and the median OS was 38.0 months. These outcomes should not be interpreted as direct between-arm comparisons, given the non-comparative design and limited sample sizes of the phase Ib/II study. The overall response rate of patients was 7% in the niraparib plus nivolumab arm and 15% in the niraparib and ipilimumab arm.
Veliparib
In this single-arm Phase II study (NCT01585805), veliparib monotherapy was investigated in patients with previously treated BRCA-mutated pancreatic ductal adenocarcinoma who had progressive disease on 1-2 prior chemotherapy regimens39. 16 patients with confirmed germline BRCA1/2 mutations and stage III/IV PDAC were enrolled and treated with veliparib (300 mg twice daily for 3 patients, then 400mg twice daily for the remaining patients based on emerging evidence from other trials suggesting improved tolerability and efficacy at the higher dose) continuously until disease progression. The findings indicate that veliparib demonstrated limited efficacy with no confirmed partial responses observed, though 4 patients (25%) achieved stable disease lasting ≥8 weeks. The median PFS was 1.7 months, and the median OS was 3.1 months. Veliparib monotherapy resulted in grade 3 or higher adverse effects in 6 patients (38%). The study was terminated early due to insufficient antitumor activity. The observed response rate did not meet the predefined threshold required for continuation.
Rucaparib
In this single-arm Phase II study (NCT03140670), rucaparib monotherapy was evaluated as maintenance therapy in patients with advanced pancreatic ductal adenocarcinoma harboring germline or somatic pathogenic variants in BRCA1, BRCA2, or PALB2 and had no disease progression during at least 16 weeks of platinum-based chemotherapy40. Patients received rucaparib (600 mg orally, twice daily) until disease progression or development of intolerable toxicity. The study met its primary endpoint, with a 6-month PFS (PFS6) of 59.5%. The median PFS was 13.1 months, and the median OS was 23.5 months. Interestingly, patients with BRCA2 mutations experienced significantly longer median PFS of 18.0 months, compared to 3.7 months in those with BRCA1 mutations. As expected for phase II maintenance studies, subgroup sizes were small, and differences between BRCA1 and BRCA2 mutation carriers should be interpreted cautiously. Treatment was generally well tolerated, with most adverse events being grade 1–2. Grade 3 treatment-related adverse events occurred in a minority of patients, and treatment discontinuation due to toxicity was reported in only one patient (2.2%).
| Clinical Trial | Treatment Regimen | Patients | Therapeutic Benefits |
| POLO (NCT02184195) | Olaparib monotherapy | Metastatic pancreatic adenocarcinoma gBRCAm≥16 weeks platinum-based chemotherapy (no progression) | Olaparib maintenance therapy extended median PFS by 3.6 months longer than placebo (7.4 vs 3.8 months).TFST was 9.0 months in the olaparib arm versus the placebo arm’s 5.4 months.TSST was 14.9 months in the olaparib arm versus the placebo arm’s 9.6 months. |
| Phase Ib/II Study of Niraparib + Nivolumab / Ipilimumab (NCT03404960) | Niraparib + Nivolumab or Ipilimumab | Locally advanced/metastatic PCBRCA or PALB2 variants≥16 weeks platinum-based chemotherapy (no resistance) | The niraparib and ipilimumab combination led to superior outcomes with a median PFS of 10.4 months, a median OS of 38.0 months, and ORR of 15%.In comparison, the niraparib and nivolumab group showed a median PFS of 3.7 months, a median OS of 12.2 months, and ORR of 7%. |
| Phase II Study of Veliparib (NCT01585805) | Veliparib monotherapy | Stage III/IV PDAC gBRCA1/2 mutationNo prior PARP inhibitor reception | 25% of the patients achieved stable disease ≥8 weeks.Veliparib monotherapy led to a median PFS of 1.7 months and a median OS of 3.1 months. |
| Phase II Study of Maintenance Rucaparib(NCT03140670) | Rucaparib monotherapy | Advanced PDACGermline/somatic BRCA1/2, PALB2≥16 weeks platinum-based chemotherapy (no progression)No evidence of platinum resistance | Rucaparib maintenance therapy led to a 6-month progression-free survival rate of 59.5%.Patients with BRCA2 mutations experienced significantly longer median PFS of 18.0 months, compared to 3.7 months in those with BRCA1 mutations. |
Conclusion and Discussion
PARP inhibitors have demonstrated clinically meaningful activity in pancreatic ductal adenocarcinoma (PDAC) among patients with germline BRCA1/2 mutations, particularly in the maintenance setting following platinum-based chemotherapy. Across multiple clinical trials, including randomized studies, PARP inhibition has consistently improved progression-free survival, supporting the concept that homologous recombination deficiency represents a therapeutically exploitable vulnerability in a subset of pancreatic cancers. These findings validate the biological rationale for targeting DNA damage repair pathways in PDAC, a disease historically resistant to targeted therapies. However, the overall clinical impact of PARP inhibitors in pancreatic cancer remains constrained. While progression-free survival benefits are reproducible, improvements in overall survival have been inconsistent, highlighting the cytostatic rather than curative nature of PARP inhibition in this context. This discrepancy may reflect the rapid emergence of resistance mechanisms, limited durability of response, and the aggressive biology of pancreatic cancer. Importantly, the benefit of PARP inhibitors is restricted to a small molecularly defined population, underscoring the challenge of translating precision oncology approaches to a disease with substantial inter- and intratumoral heterogeneity. Several limitations in the existing clinical evidence further complicate interpretation. Many trials are limited by small sample sizes due to the low prevalence of BRCA1/2 mutations in pancreatic cancer, reducing statistical power and generalizability. Additionally, heterogeneity in trial design, including differences in prior platinum exposure, duration of chemotherapy response, and maintenance strategies, makes cross-study comparisons difficult. The lack of robust predictive biomarkers beyond BRCA1/2 status also limits optimal patient selection, as not all BRCA-mutant tumors derive equivalent benefit from PARP inhibition. From a clinical perspective, the observed overlap between platinum sensitivity and PARP inhibitor responsiveness reinforces the role of functional homologous recombination deficiency as a key determinant of therapeutic efficacy. At the same time, emerging data suggest that prior exposure to PARP inhibitors may induce cross-resistance to platinum-based chemotherapy, raising important questions regarding treatment sequencing. These considerations highlight the need for more refined strategies to integrate PARP inhibitors into the broader therapeutic landscape of pancreatic cancer.
Looking forward, future research should prioritize several key areas. First, improved biomarkers are needed to better identify patients most likely to benefit from PARP inhibition, including functional assays of homologous recombination deficiency and markers of replication fork instability. Second, combination strategies, such as PARP inhibitors with immune checkpoint inhibitors, inhibitors of ATR kinase (ataxia telangiectasia and Rad3-related), or agents targeting replication stress, may enhance efficacy and delay resistance. Finally, expanding investigation beyond germline BRCA mutations to include somatic mutations and other DNA repair defects may broaden the clinical utility of PARP inhibitors in pancreatic cancer. Together, these efforts will be essential for maximizing the therapeutic potential of PARP inhibition and overcoming current limitations in this challenging disease.
Acknowledgement
I would like to thank Dr. Suhui Yang from American University of Health Sciences, for acting my mentor, guiding me through this entire process and editing my final paper. I would also like to thank everyone at Cactidu for assisting me and providing me with this opportunity.
Declaration of conflict of interests
The author declares that there are no conflicts of interest regarding the publication of this article.
References
- S. Wang, Y. Zheng, F. Yang, L. Zhu, X.-Q. Zhu, Z.-F. Wang, X.-L. Wu, C.-H. Zhou, J.-Y. Yan, B.-Y. Hu, B. Kong, D.-L. Fu, C. Bruns, Y. Zhao, L.-X. Qin, Q.-Z. Dong. The molecular biology of pancreatic adenocarcinoma: translational challenges and clinical perspectives. Signal Transduction and Targeted Therapy. Vol. 6, pg. 249, 2021 https://doi.org/10.1038/s41392-021-00659-4 [↩]
- L. Rahib, B. D. Smith, R. Aizenberg, A. B. Rosenzweig, J. M. Fleshman, L. M. Matrisian. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Research. Vol. 74, pg. 2913–2921, 2014 https://doi.org/10.1158/0008-5472.CAN-14-0155 [↩]
- American Cancer Society. Cancer Facts & Figures 2025. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2025/2025-cancer-facts-and-figures-acs.pdf (2025). [↩] [↩] [↩]
- S. Crippa, G. Belfiori, M. Bissolati, S. Partelli, M. Pagnanelli, D. Tamburrino, G. Gasparini, C. Rubini, G. Zamboni, M. Falconi. Recurrence after surgical resection of pancreatic cancer: the importance of postoperative complications beyond tumor biology. HPB. Vol. 23, pg. 1666–1673, 2021 https://doi.org/10.1016/j.hpb.2021.04.004 [↩]
- L. Xing, L. Lv, J. Ren, H. Yu, X. Zhao, X. Kong, H. Xiang, X. Tao, D. Dong. Advances in targeted therapy for pancreatic cancer. Biomedicine & Pharmacotherapy. Vol. 168, pg. 115717, 2023 https://doi.org/10.1016/j.biopha.2023.115717 [↩]
- A. Tubbs, A. Nussenzweig. Endogenous dna damage as a source of genomic instability in cancer. Cell. Vol. 168, pg. 644–656, 2017 https://doi.org/10.1016/j.cell.2017.01.002 [↩]
- A. Ciccia, S. J. Elledge. The dna damage response: making it safe to play with knives. Molecular Cell. Vol. 40, pg. 179–204, 2010 https://doi.org/10.1016/j.molcel.2010.09.019 [↩] [↩] [↩] [↩] [↩]
- S. P. Jackson, J. Bartek. The dna-damage response in human biology and disease. Nature. Vol. 461, pg. 1071–1078, 2009 https://doi.org/10.1038/nature08467 [↩] [↩]
- C. J. Lord, A. N. J. Tutt, A. Ashworth. Synthetic lethality and cancer therapy: lessons learned from the development of parp inhibitors. Annual Review of Medicine. Vol. 66, pg. 455–470, 2015 https://doi.org/10.1146/annurev-med-050913-022545 [↩] [↩] [↩] [↩] [↩]
- H. Li, Z.-Y. Liu, N. Wu, Y.-C. Chen, Q. Cheng, J. Wang. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Molecular Cancer. Vol. 19, pg. 107, 2020 https://doi.org/10.1186/s12943-020-01227-0 [↩] [↩]
- F. Zheng, Y. Zhang, S. Chen, X. Weng, Y. Rao, H. Fang. Mechanism and current progress of poly adp-ribose polymerase (parp) inhibitors in the treatment of ovarian cancer. Biomedicine & Pharmacotherapy. Vol. 123, pg. 109661, 2020 https://doi.org/10.1016/j.biopha.2019.109661 [↩]
- T. Dobzhansky. GENETICS of natural populations. xiii. recombination and variability in populations of drosophila pseudoobscura. Genetics. Vol. 31, pg. 269–290, 1946 https://doi.org/10.1093/genetics/31.3.269 [↩]
- S. M. B. Nijman. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Letters. Vol. 585, pg. 1–6, 2011 https://doi.org/10.1016/j.febslet.2010.11.024 [↩]
- J. Matykiewicz, W. Adamus-Białek, M. Wawszczak-Kasza, B. Molasy, M. Kołomańska, R. Oblap, Ł. Madej, D. Kozieł, S. Głuszek. The known genetic variants of brca1, brca2 and nod2 in pancreatitis and pancreatic cancer risk assessment. Scientific Reports. Vol. 15, pg. 1791, 2025 https://doi.org/10.1038/s41598-025-86249-8 [↩]
- T. Golan, R. Casolino, A. V. Biankin, P. Hammel, K. D. Whitaker, M. J. Hall, D. L. Riegert-Johnson. Germline brca testing in pancreatic cancer: improving awareness, timing, turnaround, and uptake. Therapeutic Advances in Medical Oncology. Vol. 15, pg. 17588359231189127, 2023 https://doi.org/10.1177/17588359231189127 [↩]
- W. Zhao, C. Wiese, Y. Kwon, R. Hromas, P. Sung. The brca tumor suppressor network in chromosome damage repair by homologous recombination. Annual Review of Biochemistry. Vol. 88, pg. 221–245, 2019 https://doi.org/10.1146/annurev-biochem-013118-111058 [↩] [↩]
- S. Holter, A. Borgida, A. Dodd, R. Grant, K. Semotiuk, D. Hedley, N. Dhani, S. Narod, M. Akbari, M. Moore, S. Gallinger. Germline brca mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. Journal of Clinical Oncology. Vol. 33, pg. 3124–3129, 2015 https://doi.org/10.1200/JCO.2014.59.7401 [↩]
- M. Rosen, R. A. Goodwin, M. M. Vickers. BRCA mutated pancreatic cancer: a change is coming. World Journal of Gastroenterology. Vol. 27, pg. 1943–1958, 2021 https://doi.org/10.3748/wjg.v27.i17.1943 [↩]
- W.-H. Hou, S.-H. Chen, X. Yu. Poly-adp ribosylation in dna damage response and cancer therapy. Mutation Research/Reviews in Mutation Research. Vol. 780, pg. 82–91, 2019 https://doi.org/10.1016/j.mrrev.2017.09.004 [↩] [↩] [↩]
- R. Gupte, Z. Liu, W. L. Kraus. PARPs and adp-ribosylation: recent advances linking molecular functions to biological outcomes. Genes & Development. Vol. 31, pg. 101–126, 2017 https://doi.org/10.1101/gad.291518.116 [↩] [↩] [↩] [↩]
- F. Wang, Z. Guo, M. J. Carr, W. Shi. PARPs and parp inhibitors: molecular mechanisms and clinical applications. Molecular Biomedicine. Vol. 6, pg. 152, 2025 https://doi.org/10.1186/s43556-025-00385-1 [↩]
- H. Wu, A. Lu, J. Yuan, Y. Yu, C. Lv, J. Lu. Mono-adp-ribosylation, a marylationmultifaced modification of protein, dna and rna: characterizations, functions and mechanisms. Cell Death Discovery. Vol. 10, pg. 226, 2024 https://doi.org/10.1038/s41420-024-01994-5 [↩]
- A. Maniatis, D. Rizopoulou, A.-N. Shaukat, K. Grafanaki, V. Stamatopoulou, C. Stathopoulos. Vault particles in cancer progression, multidrug resistance, and drug delivery: current insights and future applications. International Journal of Molecular Sciences. Vol. 26, pg. 1562, 2025 https://doi.org/10.3390/ijms26041562 [↩]
- J. Y. Huang, K. Wang, A. Vermehren-Schmaedick, J. P. Adelman, M. S. Cohen. PARP6 is a regulator of hippocampal dendritic morphogenesis. Scientific Reports. Vol. 6, pg. 18512, 2016 https://doi.org/10.1038/srep18512 [↩]
- C. Chatrin, M. Gabrielsen, L. Buetow, M. A. Nakasone, S. F. Ahmed, D. Sumpton, G. J. Sibbet, B. O. Smith, D. T. Huang. Structural insights into adp-ribosylation of ubiquitin by deltex family e3 ubiquitin ligases. Science Advances. Vol. 6, pg. eabc0418, 2020 https://doi.org/10.1126/sciadv.abc0418 [↩]
- P. Mehrotra, J. P. Riley, R. Patel, F. Li, L. Voss, S. Goenka. PARP-14 functions as a transcriptional switch for stat6-dependent gene activation. Journal of Biological Chemistry. Vol. 286, pg. 1767–1776, 2011 https://doi.org/10.1074/jbc.M110.157768 [↩]
- M.-F. Langelier, J. M. Pascal. PARP-1 mechanism for coupling dna damage detection to poly(adp-ribose) synthesis. Current Opinion in Structural Biology. Vol. 23, pg. 134–143, 2013 https://doi.org/10.1016/j.sbi.2013.01.003 [↩]
- D. Harrision, P. Gravells, R. Thompson, H. E. Bryant. Poly(adp-ribose) glycohydrolase (parg) vs. poly(adp-ribose) polymerase (parp) – function in genome maintenance and relevance of inhibitors for anti-cancer therapy. Frontiers in Molecular Biosciences. Vol. 7, pg. 191, 2020 https://doi.org/10.3389/fmolb.2020.00191 [↩]
- H. Zhu, M. Wei, J. Xu, J. Hua, C. Liang, Q. Meng, Y. Zhang, J. Liu, B. Zhang, X. Yu, S. Shi. PARP inhibitors in pancreatic cancer: molecular mechanisms and clinical applications. Molecular Cancer. Vol. 19, pg. 49, 2020 https://doi.org/10.1186/s12943-020-01167-9 [↩] [↩] [↩]
- D.-S. Kim, C. V. Camacho, W. L. Kraus. Alternate therapeutic pathways for parp inhibitors and potential mechanisms of resistance. Experimental & Molecular Medicine. Vol. 53, pg. 42–51, 2021 https://doi.org/10.1038/s12276-021-00557-3 [↩] [↩]
- L. M. Jackson, G.-L. Moldovan. Mechanisms of parp1 inhibitor resistance and their implications for cancer treatment. NAR Cancer. Vol. 4, pg. zcac042, 2022 https://doi.org/10.1093/narcan/zcac042 [↩] [↩] [↩]
- T. Golan, J. R. Brody. Targeting homologous recombination addicted tumors: challenges and opportunities. Annals of Pancreatic Cancer. Vol. 3, pg. 6–6, 2020 https://doi.org/10.21037/apc.2020.03.02 [↩]
- A. Mweempwa, M. K. Wilson. Mechanisms of resistance to parp inhibitors – an evolving challenge in oncology. Cancer Drug Resistance. 2019 https://doi.org/10.20517/cdr.2019.50 [↩]
- C. J. Lord, A. Ashworth. PARP inhibitors: synthetic lethality in the clinic. Science. Vol. 355, pg. 1152–1158, 2017 https://doi.org/10.1126/science.aam7344 [↩]
- T. Helleday. The underlying mechanism for the parp and brca synthetic lethality: clearing up the misunderstandings. Molecular Oncology. Vol. 5, pg. 387–393, 2011 https://doi.org/10.1016/j.molonc.2011.07.001 [↩]
- T. Golan, P. Hammel, M. Reni, E. Van Cutsem, T. Macarulla, M. J. Hall, J.-O. Park, D. Hochhauser, D. Arnold, D.-Y. Oh, A. Reinacher-Schick, G. Tortora, H. Algül, E. M. O’Reilly, D. McGuinness, K. Y. Cui, K. Schlienger, G. Y. Locker, H. L. Kindler. Maintenance olaparib for germline brca -mutated metastatic pancreatic cancer. New England Journal of Medicine. Vol. 381, pg. 317–327, 2019 https://doi.org/10.1056/NEJMoa1903387 [↩] [↩] [↩] [↩]
- H. L. Kindler, P. Hammel, M. Reni, E. Van Cutsem, T. Macarulla, M. J. Hall, J. O. Park, D. Hochhauser, D. Arnold, D.-Y. Oh, A. Reinacher-Schick, G. Tortora, H. Algül, E. M. O’Reilly, S. Bordia, D. McGuinness, K. Cui, G. Y. Locker, T. Golan. Overall survival results from the polo trial: a phase iii study of active maintenance olaparib versus placebo for germline brca-mutated metastatic pancreatic cancer. Journal of Clinical Oncology. Vol. 40, pg. 3929–3939, 2022 https://doi.org/10.1200/JCO.21.01604 [↩] [↩]
- K. A. Reiss, R. Mick, U. Teitelbaum, M. O’Hara, C. Schneider, R. Massa, T. Karasic, R. Tondon, C. Onyiah, M. K. Gosselin, A. Donze, S. M. Domchek, R. H. Vonderheide. Niraparib plus nivolumab or niraparib plus ipilimumab in patients with platinum-sensitive advanced pancreatic cancer: a randomised, phase 1b/2 trial. The Lancet Oncology. Vol. 23, pg. 1009–1020, 2022 https://doi.org/10.1016/S1470-2045(22)00369-2 [↩]
- M. A. Lowery, D. P. Kelsen, M. Capanu, S. C. Smith, J. W. Lee, Z. K. Stadler, M. J. Moore, H. L. Kindler, T. Golan, A. Segal, H. Maynard, E. Hollywood, M. Moynahan, E. E. Salo-Mullen, R. K. G. Do, A. P. Chen, K. H. Yu, L. H. Tang, E. M. O’Reilly. Phase ii trial of veliparib in patients with previously treated brca-mutated pancreas ductal adenocarcinoma. European Journal of Cancer. Vol. 89, pg. 19–26, 2018 https://doi.org/10.1016/j.ejca.2017.11.004 [↩]
- K. A. Reiss, R. Mick, M. H. O’Hara, U. Teitelbaum, T. B. Karasic, C. Schneider, S. Cowden, T. Southwell, J. Romeo, N. Izgur, Z. M. Hannan, R. Tondon, K. Nathanson, R. H. Vonderheide, M. M. Wattenberg, G. Beatty, S. M. Domchek. Phase ii study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic variant in brca1 , brca2 , or palb2. Journal of Clinical Oncology. Vol. 39, pg. 2497–2505, 2021 https://doi.org/10.1200/JCO.21.00003 [↩]


