
Abstract
Myasthenia Gravis (MG) is a chronic autoimmune disease characterized by fluctuating muscle weakness due to antibody-mediated disruption of neuromuscular transmission. With a prevalence of 150 to 200 cases per million individuals, MG is classified as a rare disease. The most common subtype involves autoantibodies targeting acetylcholine receptors, whereas smaller subsets of patients produce autoantibodies against muscle-specific kinase, low-density lipoprotein receptor-related protein 4, and agrin. While conventional therapies have provided symptom relief for many, they often fall short of achieving sustained remission and are associated with side effects such as increased susceptibility to infections. Advances in the understanding of MG pathophysiology along with the need to address the limitations of conventional treatments have driven the development of targeted therapeutics that modulate specific immune mechanisms. This review focuses on two emerging therapeutic classes, complement and FcRn inhibitors, that have shown promising efficacy in clinical trials. Complement inhibitors mitigate the damage at the neuromuscular junction, while FcRn inhibitors accelerate the degradation of pathogenic autoantibodies. Together, these agents represent a shift in MG management, promising a more effective, safer, and manageable disease control.
Keywords: Autoimmune diseases, Myasthenia Gravis, Neuromuscular junction, autoantibodies, Complement inhibitors, FcRn inhibitors, Membrane Attack Complex, Immunotherapies
Introduction
MG is a rare, chronic autoimmune disorder characterized by neuromuscular dysfunction, such as fluctuating muscle weakness and fatigue. The ocular, bulbar, and limb muscles are most affected1. The pathogenesis of MG has been well characterized over the past several decades. At its core, MG stems from autoantibodies– proteins produced by the immune system that mistakenly target a person’s own tissues or cells– that impair signal transmission at the neuromuscular junction by targeting acetylcholine receptors (AChR), muscle-specific kinase (MuSK), lipoprotein-related protein 4 (LRP4) or agrin on the postsynaptic membrane2.
MG can be improved through therapies, but to date, has not been cured. For decades, treatment has relied on immunosuppressive agents such as corticosteroids, azathioprine, and mycophenolate mofetil along with cholinesterase inhibitors to alleviate symptoms3. While often effective in controlling disease symptoms, the overall benefit of these therapies is limited by several factors such as increased risk of infection and slow onset of action. Moreover, about 10-20% of patients develop refractory disease, meaning that they experience persistent symptoms or relapses despite adequate use of standard treatments4.
After decades in which the treatment options for MG remained largely unchanged, the field has entered a new and transformative era. In recent years, a deeper understanding of MG pathophysiology has enabled the development of targeted therapies that modulate specific pathways without suppressing the entire immune system. As a result, targeted therapies offer new hope for more effective and safer disease management. Two targeted therapeutic strategies have shown promising efficacy in clinal trials: complement inhibitors and FcRn blockers. This efficacy has been demonstrated by reductions in Myasthenia Gravis Activities of Daily Living (MG-ADL) andthe Quantitative Myasthenia Gravis (QMG) scores, along with better patient-reported quality-of-life measures (MG-QoL15). The success of these biologics has reinvigorated research into MG, encouraging the development of additional targeted agents and expanding clinical trials.
This narrative literature review explores the development and clinical impact of new targeted therapies for MG, with a specific focus on complement and FcRn inhibitors. It evaluates their mechanism of action, clinical efficacy, and current limitations, while also considering how these advancements are informing future directions in MG research.
Discussion
MG Pathophysiology
The neuromuscular junction (NMJ) is the primary point of communication between a nerve ending and a muscle fiber. The transmission of the nerve signals at the NMJ initiates muscle contractions needed for bodily functions5. When an action potential reaches the presynaptic terminal of a neuron, it opens voltage-gated calcium channels on its membrane. The rise in intracellular calcium concentration leads to the secretion of neurotransmitter acetylcholine (ACh) into the synaptic cleft. Then, AChs bind to their receptors on the postsynaptic membrane of the muscle fiber, which opens ion channels that let positive ions (mainly sodium) flow into the cell. This small electrical change called local depolarization triggers muscle action potential, which causes the muscle to contract6‘7.
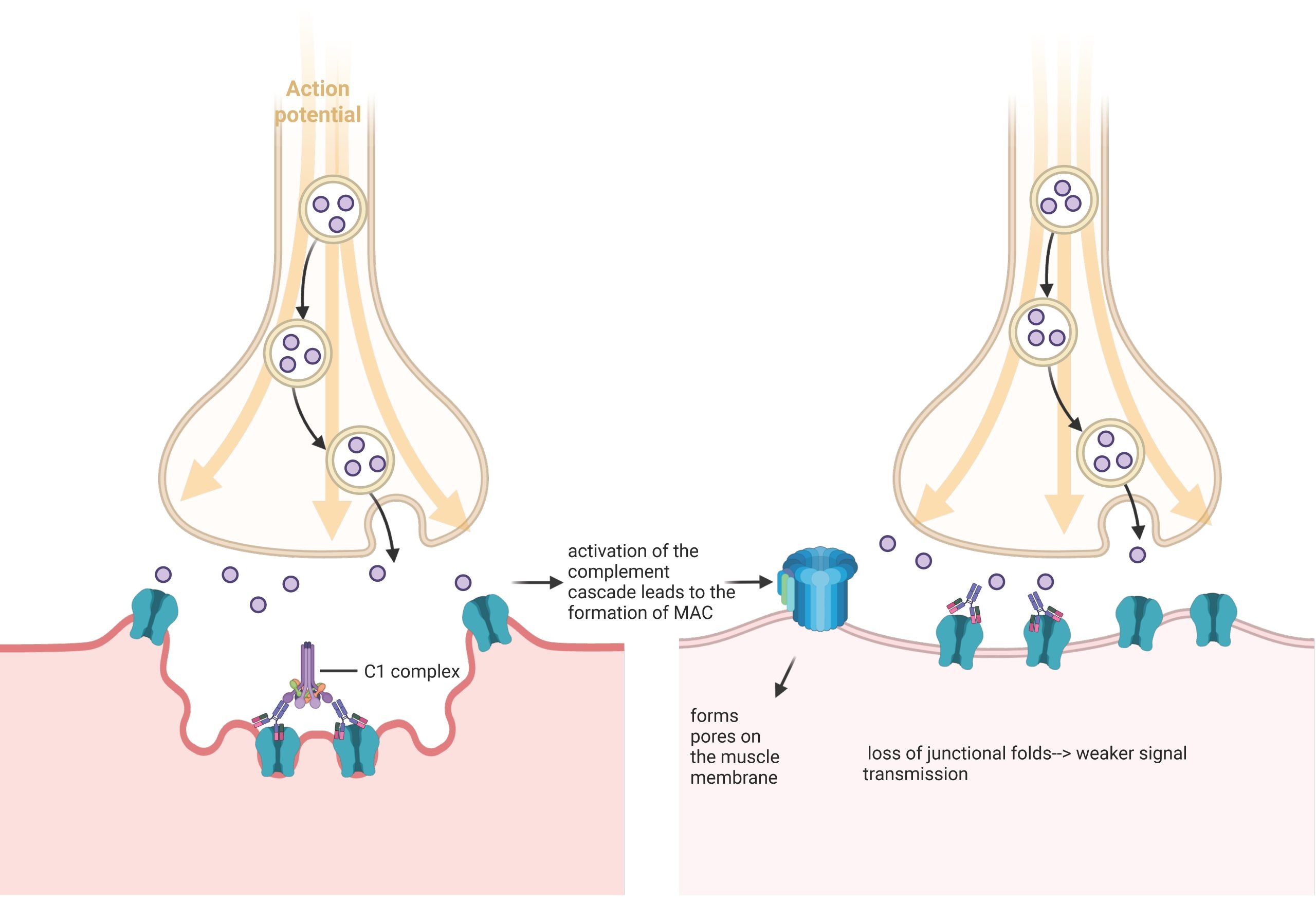
While this process functions properly in a healthy individual, MG causes disruptions. In the most common subtype, AChR+ MG, the immune system misidentifies AChRs as foreign substances and generates IgG1 and IgG3 autoantibodies against them8. These autoantibodies bind to AChRs and activate the complement system. In the final step of the complement activation, the membrane attack complex (MAC) is formed, which punches holes (forms pores) in the muscle membrane at receptor-dense regions. Experimental models show that the muscle cells respond to complement-mediated MAC damage by shedding affected portions of the membrane. This process leads to the loss of junctional folds, which are invaginations that increase the surface area for neurotransmitters to bind to receptors9‘10. As folds flatten and membrane is lost, the density of AChR decreases9‘11. These morphological changes are depicted in Fig 1. The reduced surface area and the loss of AChRs on the muscle membrane cause diminished efficacy of signal transmission, leading to the characteristic muscle weakness and fatigue seen in MG patients12‘11. Thus, inhibiting the complement activation has emerged as a therapeutic approach that has shown clinical benefits by protecting the NMJ from damage.
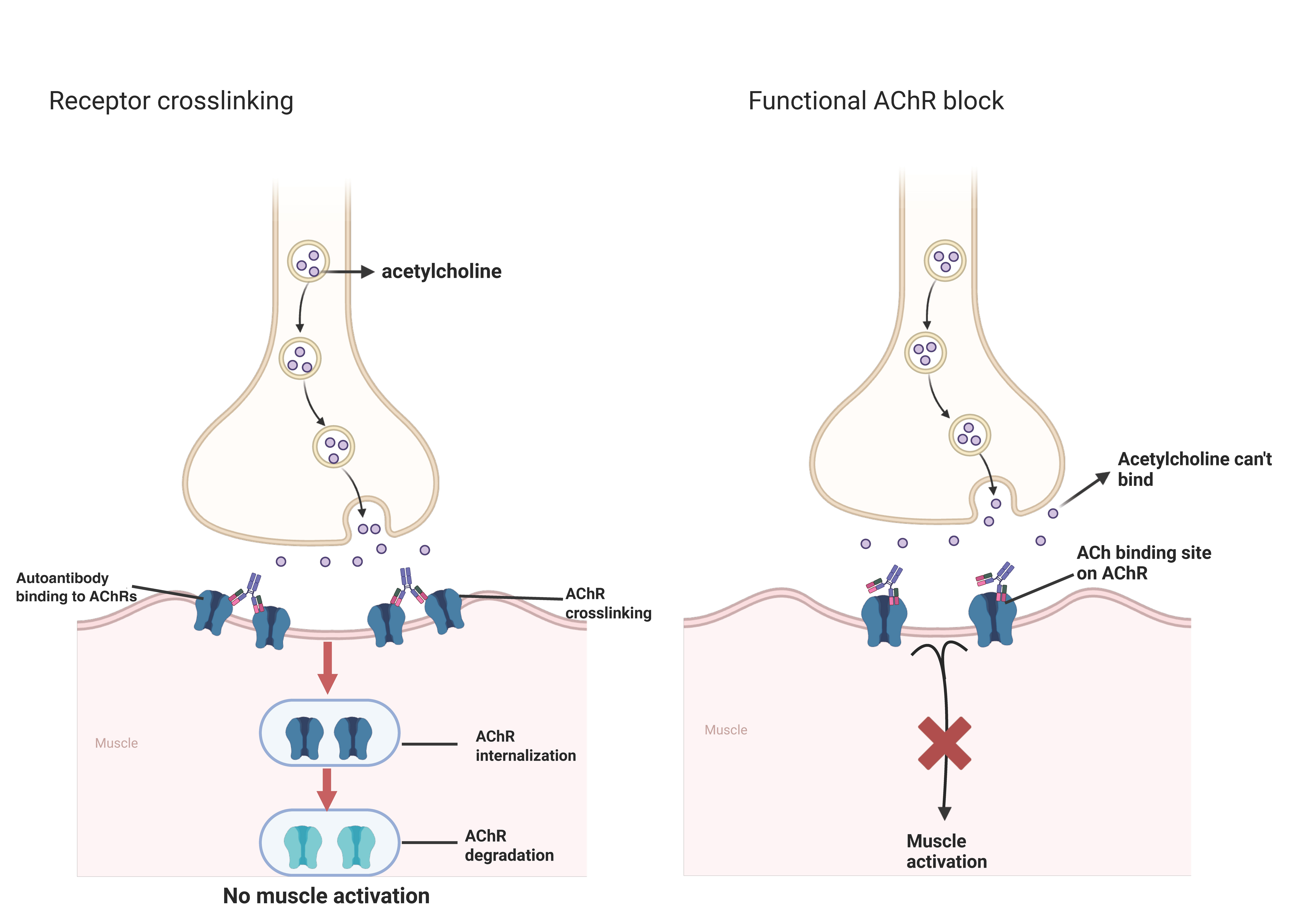
In addition to complement-mediated damage, a mechanism contributing to dysfunction is antibody-induced internalization and degradation of AChRs (illustrated in Figure 2). When multiple antibodies bind closely spaced receptors, they trigger receptor cross-linking, which muscle cells interpret as abnormal. The cell then internalizes these receptor clusters and degrades them13‘14. The gradual decline in receptor availability diminishes the muscle’s ability to respond to ACh. Not all antibodies can cross-link AChR; their ability to do so depends on the exact location of their binding site on the receptor and how the receptors are positioned on the membrane15.
The third pathogenic mechanism by which autoantibodies target AChRs is the direct functional blockade of the receptor (Fig 2). Some anti-AChR antibodies bind to the receptor’s acetylcholine binding sites, blocking acetylcholine from activating the receptor14. As a result, signal transmission is prevented even when the receptor itself is structurally intact. This effect is usually less common than complement-mediated damage and crosslinking of receptors, but it can contribute to muscle weakness13‘8.
Around 80-85% of MG cases involve autoantibodies directly targeting AChRs. A less common (5-8% of MG patients) subtype is MuSK+ MG, which is characterized by worse clinical signs, faster disease progression, and reduced responsiveness to conventional treatments16. In MuSK+ MG, the body produces IgG4 antibodies against MuSK, a protein essential for organizing and stabilizing AChRs at the NMJ. Unlike IgG1 and IgG3 antibodies in AChR+ MG, IgG4 antibodies do not activate complement or cause receptor cross-linking8‘17‘18. Instead, they disrupt the signaling pathway needed to cluster AChRs, leading to functional receptor loss without structural damage19. Other variants of MG (collectively around 1-5%) involve antibodies against proteins such as LRP4 or agrin, which also play critical roles in AChR clustering and maintenance20‘21. However, around 5-15% of cases are classified as seronegative, meaning that no antibodies against the known targets mentioned above are detected in the patient’s blood by using standard tests22. Seronegative MG exposes gaps in our current understanding of the disease. As such, there might be undiscovered antibodies, immune targets or non-antibody-mediated mechanisms at play.
Another important immunological mechanism in MG is the neonatal Fc receptor (FcRn), which regulates the homeostasis and lifespan of IgG antibodies– the most abundant antibody class in the body. IgG molecules are continuously taken up by cells through a process called pinocytosis. Normally, proteins internalized in this way are directed to lysosomes and broken down. However, the body has a protective mechanism to prevent lysosomal degradation of IgG antibodies. Within the cell, IgG antibodies enter acidic endosomal compartments, where FcRn binds to them at low pH. FcRn then recycles the bound IgG back to the cell surface, where the neutral pH of the extracellular environment causes the antibody to dissociate from the receptor and reenter the circulation intact15‘23‘24‘25.This recycling mechanism extends the lifespan of IgG antibodies, maintaining their levels in the bloodstream for up to three weeks. FcRn is so effective that the amount of IgG preserved through recycling is 42% higher than the amount that the body produces on its own 2226. In MG patients, FcRn is unable to distinguish between pathogenic and protective IgG antibodies. Therefore, FcRn continues to recycle and preserve pathogenic IgG antibodies along with normal ones, which makes it an attractive therapeutic target27.
However, recent research has found FcRn to play a much more active role than merely protecting IgG antibodies from lysosomal degradation. When autoantibodies bind their antigen (like AChR), they form immune complexes that can be internalized by antigen-presenting cells (APCs) through surface Fcγ receptors28. Inside endosomes, these complexes dissociate from the surface receptors and bind to FcRn. Whereas individual IgG antibodies are typically recycled and returned to the bloodstream by FcRn, IgG bound to an antigen is sent into vesicles that break down the antigen into smaller peptide fragments. Once degraded, the antigen is presented at the cell surface, specifically to CD4+ T cells23‘29.
T cells are responsible for recognizing and activating B cells that produce autoantibodies when the immune system is triggered. Normally, T cells that target self-antigens are eliminated during development, a process called central tolerance, but in autoimmune diseases like MG, some of the autoreactive T cells escape elimination30. If APCs present self-antigens to these non-eliminated T-cells, it could reactivate them, encouraging the B cells to produce more autoantibodies and thus sustaining the immune attack31. Therefore, blocking FcRn can both decrease circulating IgG levels in the body and lead to suppressed immune responses32.
Complement Inhibitors
Eculizumab
Eculizumab is a monoclonal antibody, a laboratory-produced protein that mimics the structure and targeting ability of natural IgG antibodies. The drug functions by binding to protein C5, a critical component in the terminal phase of the complement cascade. Under normal circumstances, C5 convertase cleaves C5 protein into two fragments: C5a and C5b. C5a triggers inflammation, while C5b facilitates the formation of the MAC33‘34. Eculizumab binds to the C5 protein near where the C5 convertase enzyme would normally bind, preventing the cleavage of the protein and thus blocking the formation of MAC, which would otherwise damage the muscle membrane and AChRs15‘20.
The REGAIN trial was a randomized, double-blind, placebo-controlled phase III study designed to assess the efficacy and safety of Eculizumab. A total of 125 patients who had not benefited from conventional treatments were randomly assigned to receive either Eculizumab or placebo for a 26-week treatment period. Clinical scales used to assess outcomes included the MG-ADL, a scale used to determine whether the patients’ daily symptoms improved or worsened; QMG, which measures muscle strength and fatigue; and MG-QOL15, a patient-reported quality of life measure35‘36. On these scales, negative change from baseline in MG-ADL and QMG and MG-QOL15 indicates clinical improvement, since these patterns reflect reduced functional impairment, stronger muscle performance, and enhanced quality of life from the patient’s perspective. The primary endpoint of REGAIN was defined by the change from the baseline in MG-ADL scores. Although the study did not achieve statistical significance for the primary endpoint (p = 0.0698), secondary analyses demonstrated consistent improvements in the Eculizumab group. These analyses found greater reductions in QMG scores compared to placebo (least square mean difference (LSMD) = -3, 95% CI [-4.6, -1.3]; p= 0.001), and in MG-QoL15 scores (LSMD = -14.3, 95% CI [-26.98, -1.56]; p= 0.0281) and a reduced need for rescue therapies (17 % with eculizumab and 33% with placebo)15‘37‘38‘39. Because the 95% confidence interval stays entirely on one side of zero and the p-value is below 0.05, the observed differences are unlikely to be due to random variation. These results provide statistical support for the clinical relevance of the improvement.
In the open-label extension phase, Eculizumab continued to show lasting benefits even after 130 weeks of treatment. By that point, 88% of patients had achieved an “improved” status and 57.3% reached “minimal symptom manifestation” according to the Myasthenia Gravis Foundation of America (MGFA) criteria40. Eculizumab is the first complement inhibitor approved by the FDA to treat MG in 2017.
Eculizumab is generally well-tolerated among patients, but like all powerful immunomodulatory drugs, it carries specific risks and side effects. Common side effects include headache, dizziness, and nasopharyngitis. A severe side effect is the increased risk of meningococcal infections. The formation of MAC by the complement system is essential for the body’s defense against the bacteria Neisseria meningitidis. However, because Eculizumab is a complement inhibiting drug, it prevents the body’s defense mechanism against these bacteria38‘39. The use of Eculizumab is associated with 1,000 to 2,000 times increased risk of meningococcal disease39. Therefore, patients are usually vaccinated before taking Eculizumab.
Ravulizumab
Ravulizumab is designed to function similarly to Eculizumab: it hinders the cleavage of complement C5 and thus the immune damage at the NMJ41‘39. Despite this functional similarity, Ravulizumab incorporates structural changes that extend its half-life in the bloodstream compared with Eculizumab42. Specifically, it contains four amino acid substitutions: two in the Fab (fragment antigen-binding) region and two in the Fc region (constant region) of the antibody43. The mutations in the Fab region facilitate the dissociation of the C5-antibody complex in the acidic environment of the endosome. Without this dissociation, the antibody, which in this case is Ravulizumab, would be carried along with C5 into lysosomal degradation and would not bind to FcRn receptors to go through the recycling process. Meanwhile, the changes in the Fc region allow Ravulizumab to bind more tightly to FcRn inside acidic endosomes after detaching from the C5-antibody complex, and it is then released more efficiently once it reaches the neutral pH of the bloodstream44‘45. The modifications in both Fc and Fab regions result in a half-life four times longer than that of Eculizumab43.
A Phase III trial of Ravulizumab (called CHAMPION-MG) was conducted using a study design similar to REGAIN. A total of 175 patients received either the drug or the placebo over a 26-week period. The primary endpoint was change in MG-ADL score from baseline to week 26, which was met with significantly greater improvements in the Ravulizumab group than in the placebo group (LSMD= −1.7, 95% CI [-2.5, -0.9]; p < 0.001)24‘46‘47. The open-label extension phase lasted up to four years and included 161 patients who had completed the initial randomized phase. Improvements observed during the initial treatment phase were not only maintained but, in some cases, further enhanced. Patients who continued Ravulizumab improved by 4 points on the MG-ADL scale (95% CI [-4.8, -3.1]; p=0.0001), while those who switched from placebo to Ravulizumab improved by -1.7 points (95% CI, -2.7 to -0.8, p=0.0007). Importantly, no new safety concerns emerge48. Importantly, no new safety concerns emerged 40. The drug was mostly well-tolerated. Most frequent adverse events included headache, diarrhea, and nausea49.
Up to 20–25% of patients with MG experience at least one myasthenic crisis (MC) during their lifetime. These crises can be triggered by infections, pregnancy, wrong dosage, or certain medications. MCs are characterized by rapid worsening of MG and can even be life-threatening in certain cases. While the long-term effects of targeted therapeutics are assessed, they are rarely tested in these crisis moments. A case series conducted in the University of Augsburg in Germany addressed this gap by observing three patients, all of whom had AChR-positive generalized MG and a history of frequent or treatment-refractory MC. The researchers retrospectively analyzed the treatment response after switching to either Eculizumab or Ravulizumab. Clinical improvement in this case was defined as a ≥2-point change in MG-ADL from baseline. MG-ADL levels of patients were not explicitly mentioned in the paper, but patients at MC normally have MG-ADL levels ranging from 8 to 12. Given that MG-ADL levels declined to 1 for patient 1 and 3 and to 4 for patient 2, all three patients saw rapid and sustained clinical improvements50. While previous studies focused on chronic treatment in relatively stable patients, this recent study underscored that complement inhibitors may also be lifesaving in acute crisis situations, specifically impending Myasthenia crisis (iMC), and not just for long-term maintenance.
Zilucoplan
Zilucoplan is an emerging therapeutic option that expands the scope of complement-targeting treatment in AChR-positive MG. Unlike Eculizumab and Ravulizumab, it is a small, peptide-based complement inhibitor that can be self-administered as a once-daily subcutaneous injection rather than through IV infusions. Zilucoplan has a dual inhibitory effect on the complement cascade: it not only binds to complement protein C5 to prevent its cleavage, but also interferes with C5b binding C6 if a small amount of C5 gets cleaved, which provides an additional blockage of terminal complement activity8‘46‘51. Zilucoplan’s efficacy also stems from its small size that allows it to permeate tissues more rapidly than large monoclonal antibodies and speed up therapeutic onset51. Additionally, Zilucoplan has a different binding site than Eculizumab, which makes it effective in patients who have certain genetic mutations.3 For instance, Zilucoplan has been shown to effectively block complement activation in other C5 variants such as C5 R885C and R885H, which are genetic polymorphisms that render Eculizumab ineffective52.
Phase III RAISE trial recruited 174 patients from 75 hospitals in Europe, Japan, and North America, who showed moderate-to-severe disease despite receiving standard therapies. Participants were randomly assigned to receive either Zilucoplan (0.3 mg/kg) or a placebo over a 12-week treatment period. The primary endpoint was MG-ADL change from the baseline to week 12. At the end of the trial, Zilucoplan demonstrated statistically significant improvements compared with placebo across multiple clinical endpoints: MG-ADL (LSMD = −2.09, 95% CI [-3.24, -0.95]; p=0.0004), QMG (LSMD = -2.94, 95% CI [-4.39, -1.49]; p <0.001), and MG-QoL15r scores (LSMD = −3.28, 95% CI [-5.89, -0.67]; nominal p < 0.05)46‘53. The ability to self-inject at home, along with its compatibility with other immunotherapies like plasma exchange and FcRn inhibitors, makes Zilucoplan an accessible option for patients seeking both clinical benefit and flexibility in disease management46. This drug got approved by the FDA in 2023.
Safety Management of Complement Inhibitors
Safety management is critical to the use of complement inhibitors. All drugs mentioned above carry the risk of meningococcal disease. To mitigate this risk, patients are advised to receive vaccination against Neisseria meningitidis (MenACWY and MenB), ideally completed at least two weeks prior to treatment initiation, with prophylactic antibiotics recommended if therapy must begin earlier. Immunization against Streptococcus pneumoniae and Haemophilus influenzae type b is also recommended to reduce the risk of infections with other encapsulated organisms.Patients and caregivers must be educated to recognize and respond promptly to symptoms indicative of meningitis or sepsis. Beyond infection risk, adverse events are generally mild. Long-term extension studies (REGAIN OLE, CHAMPION-MG OLE, and RAISE extension) have not identified new safety signals, which supports the overall tolerability of these agents when appropriate precautions are taken.54
FcRn Inhibitors
Efgartigimod
While complement inhibitors result in better clinical outcomes than conventional treatments, more than thirty percent of patients receive little or no benefit, which calls for the development of other targeted therapies15. FcRn blockers are one of them. Efgartigimod is the first FcRn antagonist that is approved for the MG treatment in several countries worldwide, including the USA, EU, and Japan. It is an Fc fragment engineered to bind to the FcRn more strongly and stably than normal IgG antibodies. This binding blocks FcRn’s ability to recycle IgGs, reducing total IgGs (including harmful ones), and forcing their cellular degradation55‘56. The selectivity of Efgartigimod to only target IgG, instead of all antibody classes, is a major advantage, because unlike non-specific immunosuppressants, it leaves the rest of the immune system largely intact.
The 26-week, randomized, double-blind, placebo-controlled phase III ADAPT trial investigated the efficacy of Efgartigimod in MG treatment. 167 participants received four weekly intravenous infusions of 10 mg/kg of the medication per cycle, which decreased serum IgG levels and AChR antibody levels from baseline57‘58. One week after the last infusion in the first cycle, there were mean maximum decreases of 61.3% and 57.6% in total IgG and AChR antibody levels, respectively, from baseline. Similar reductions in IgG and AChR antibody levels were seen with each treatment cycle. After the final dose, the drug was cleared from the body over time. Meanwhile, the body naturally produced new IgG antibodies, restoring its level to baseline levels 9 weeks after the last infusion of the cycle59. The primary endpoint was the proportion of MG-ADL responders among AChR+ MG patients during the first treatment cycle. A “responder” was defined as someone who had an improvement of at least 2 points in MG-ADL, sustained for 4 consecutive weeks. At the end of cycle 1, 68% of patients receiving Efgartigimod and 30% receiving placebo met the MG-ADL response criterion. This corresponded to an odds ratio of 4.95 (95% CI [2.21,11.53]; p<0.0001), indicating that patients on Efgartigimod were nearly five times more likely to achieve a response than those on placebo15.
Because FcRn inhibitors lower all IgG subclasses, including IgG4 (the predominant pathogenic antibody in MuSK+ MG), Efgartigimod may offer therapeutic flexibility and broaden the treatment options beyond the AChR+ population. Although its use is approved only for the treatment of AChR+ MG for now, there is a growing interest in its potential application for MuSK+ MG. Preclinical studies, in fact, have already explored Efgartigimod’s efficacy in treatment of this subtype. In a MuSK-MG model, 16 mice were randomized to receive either efgartigimod (n=8) or a control Fc fragment (n=8), alongside an additional negative control group. Efgartigimod-treated mice showed clear benefits, with most maintaining significantly greater grip strength and higher compound muscle action potentials, indicating improved neuromuscular transmission. At the end of the study, Efgartigimod led to an approximately eight-fold reduction in MuSK IgG4 levels60‘61.
Phase III trials built on preclinical findings by demonstrating the drug’s effects on humans. To date, most phase III trials aimed at assessing the efficacy of Efgartigimod for this subgroup have focused on patients who were not experiencing exacerbation– consistently worsening symptoms– which provided limited data regarding the drug’s full therapeutic potential62.A study from China addressed this limitation by recruiting four MuSK-positive patients, all of whom experienced exacerbation and two of whom were in MC. Three patients received 10 mg/kg Efgartigimod administered as four infusions per cycle, while the remaining patient received 20 mg/kg Efgartigimod on day 1 and day 5. Medication dosages were gradually reduced or kept constant throughout the treatment process. On average, patients started with an MG-ADL score of 12 and improved to about 6 after one cycle of Efgartigimod, a drop of roughly six points. This level of improvement is considered clinically meaningful, and importantly, every single patient, not just the average, showed this benefit. These findings suggest that Efgartigimod may be beneficial beyond AChR+ MG, even in acute crisis situations, though more research to assess its efficacy and safety is needed.
Rozanolixizumab
Similar to Efgartigimod, Rozanolixizumab is a monoclonal antibody that binds to FcRn, preventing it from binding IgG antibodies. Therefore, antibodies cannot be rescued from cellular degradation and levels of pathogenic IgG antibodies drop significantly63. The phase III trial recruited 200 patients with similar baseline characteristics such as age, disease severity, and treatment history. 90% of the patients had antibodies against AChRs, while 10% had antibodies against MuSK.Patients were administered either 7mg/kg Rozanolixizumab, 10 mg/kg Rozanolixizumab or a placebo once a week for 6 weeks. The primary endpoint was change from baseline to day 43 in MG-ADL score. At the end of the treatment process, both Rozanolixizumab groups showed statistically and clinically significant improvements: Among the overall study population, MG-ADL scores improved significantly versus placebo with both 7 mg/kg (LSMD = –2.59, 95% CI [–4.09, –1.25]; p<0.0001) and 10 mg/kg dosing (LSMD = –2.62, 95% CI [–3.99, –1.16]; p<0.0001). Effects were seen as early as day 8, and the benefit was similar in AChR+ and MuSK+ subgroups64. For the MuSK+ subgroup (n=21) specifically, the MG-ADL score for 7mg/kg group improved by 7.28 points and 10mg/kg group by 4.16, whereas for patients on placebo, it worsened by 2.28. After clinical trials, Rozanolixizumab became the first agent approved by the FDA for the treatment of both AChR+ and MuSK+ MG. Now, its efficacy in different diseases such as Fibromyalgia, LGI1 autoimmune encephalitis, and MOG-antibody disease is being studied65. Side effects were mild and included headache, diarrhea, and sometimes infections.
Clinical and Economic Challenges of MG biologics
Although several new MG treatment options have emerged over the past few years, there is no consensus as to which biologic therapy offers the best balance of efficacy and safety. The absence of direct, head-to-head comparisons between biologics makes it challenging for scientists to determine the most appropriate treatment for specific clinical scenarios. Therefore, a metal analysis was conducted by Sàcca et al, which evaluated and ranked these treatments based on available data. Key findings from this study included: 1) both FcRn inhibitors and complement inhibitors were effective in improving short-term quality of life in gMG patients, with no significant difference between them in MG-ADL scores (p=0.16). 2) FcRn inhibitors may be more potent, particularly in improving muscle strength, given that they had a greater impact on QMG score (LS mean change = -4.78) than complement inhibitors (LS mean change= -2.60). In the network-meta analysis (NMA) rankings, Efgartigimod had the highest probability of being the most effective treatment, followed by Rozanolixizumab36. While these findings are informative, they are not considered definitive due to several important limitations. Most of the included clinical trials were short in duration, which limits their ability to inform on the long-term efficacy and safety of treatments for MG. The trials also varied significantly in design, patient populations, and outcome measures, introducing heterogeneity that undermines the comparability required for reliable NMA66.To address these limitations, a subsequent real-world study analyzed data from 153 MG patients treated with either complement or FcRn inhibitors between 2018 and 2024. Within six months, MG-ADL scores decreased by about 4–5 points, QMG scores by 5–6 points, and MG-QoL15 scores by 8–9 points in both groups. Therefore, the study found no significant difference in clinical outcomes between two treatment groups, refuting the earlier suggestions of FcRn inhibitors’ superiority.
While offering numerous clinical benefits, targeted biologics are often not widely accessible due to their high cost. A cost-effective analysis comparing Eculizumab and Efgartigimod plus conventional treatment versus conventional treatment alone was conducted to determine whether the health benefits provided by these biologics were adequate to justify their high cost. The researchers used findings from earlier clinical trials and entered them into a computer model called Markov Model, which simulated patient outcomes, healthcare costs, and quality-adjusted life years (QALYs)– a measure used to evaluate the value of medical treatments by combining how long a person lives and how well they live– over a lifetime. While both drugs showed improved clinical outcomes and QALYs, they incurred dramatically higher costs. The incremental cost effectiveness ratios (ICERs) were $3.34 million for Eculizumab and $1.99 million for Efgartigimod, which far exceeded accepted cost-effectiveness thresholds (typically $100,000–$200,000 per QALY). When the model incorporated indirect benefits such as reduced caregiver burden and improved work productivity and long-term benefits like reduced hospitalizations, the ICERs decreased only marginally (to $3.31 million and $1.96 million, respectively), underscoring that the therapies remain economically unviable without drastic price reductions (over 88%)67.Therefore, future studies should be aimed at developing more affordable biologics without compromising clinical effectiveness.
Future of Complement and FcRn Inhibitors
The future of MG treatment appears promising with many new biologics currently under development or in trial stage. New generation complement inhibitors include Gefurulimab, which, unlike earlier complement inhibitors, is a small, bispecific nanobody that binds both complement protein C5 and albumin68. Albumin is a naturally abundant blood protein that is recycled by the body through FcRns69. By attaching itself to albumin, Gefurulimab takes advantage of this recycling pathway and extends its lifespan. Gefurulimab’s dual-binding has two major benefits: 1) by staying in circulation longer, it can maintain consistent C5 inhibition. 2) because of the extended lifespan, it doesn’t need to be dosed as often. Gefurulimab can be administered once weekly by subcutaneous injection, making it a practical and convenient alternative for patients. A randomized controlled phase 3 trial for this drug has just been completed22.
The combination of Pozelimab with Cemdisirian represents a novel dual-approach to complement inhibition. Pozelimab is a monoclonal antibody that blocks the complement protein C5 at the site of immune activation, while Cemdisiran is a small interfering RNA (siRNA) that works at the genetic level to reduce the liver’s production of C5 protein24‘39‘70. Together, they target both the source and the action of C5, aiming for more complete and sustained complement inhibition71. Both drugs are given as subcutaneous injections and are well-tolerated at different doses. Pozelimab is already approved for CHAPLE disease, and the Pozelimab–Cemdisiran combination has just been tested in a Phase 3 randomized controlled trial for MG72. Comprehensive data for phase III MG trial has not been released yet.
Iptacopan is an oral, small-molecule inhibitor that targets complement factor B, a component of the alternative complement pathway. This pathway is one of the three distinct activation routes in the complement system, alongside classical and lectin pathways, and plays a central role in amplifying complement activity once it has been triggered21‘73. In AChR-positive MG, the classical pathway is the initiator of immune-mediated damage at the NMJ due to autoantibody binding74. Iptacopan does not block the classical pathway directly, so initial antibody binding and complement activation might still occur. However, by inhibiting factor B, it disrupts the alternative pathway amplification loop, limiting downstream complement activity. As a result, the formation of pro-inflammatory proteins such as C3a and C5a, as well as the membrane attack complex (MAC), is significantly reduced75‘76. This upstream mechanism contrasts with C5 inhibitors, which block the final step of the complement cascade (C5 cleavage and MAC formation) but still allow upstream C3 activation products such as C3a and C3b to drive inflammation. Therefore, factor B inhibition appears more promising than C5 inhibition. Iptacopan is currently being evaluated in a Phase 3 clinical trial for AChR+ MG.
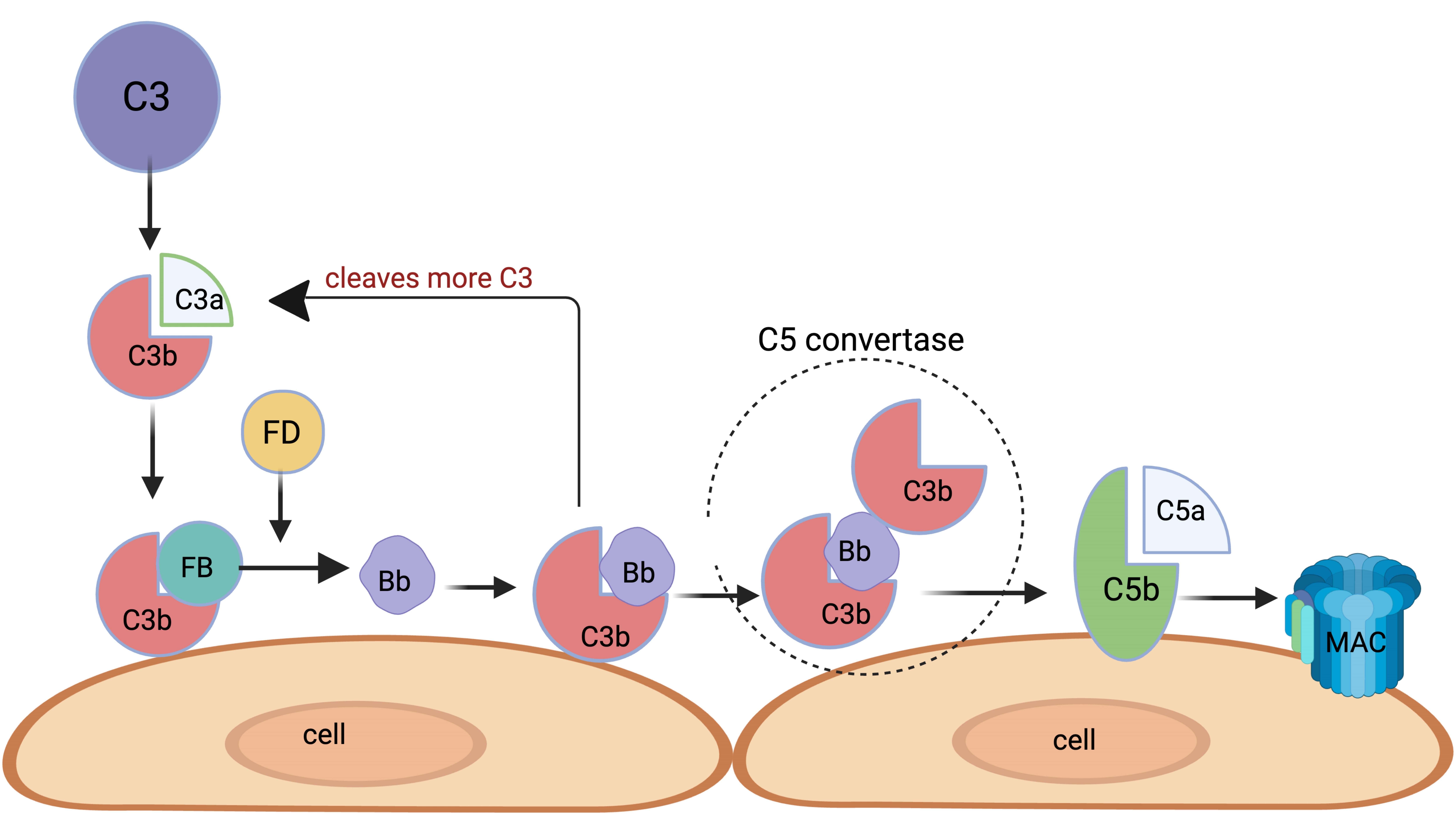
Vemircopan is functionally like Iptacopan in that it blocks the alternative complement cascade. However, instead of inhibiting factor B, Vemircopan inhibits factor D, which is an essential enzyme for activating factor B72‘77. Figure 3 provides a schematic of the alternative complement pathway. Specifically, factor D cleaves factor B when it’s bound to C3b, forming the enzyme complex C3bBb, also known as the C3 convertase of the alternative pathway. This complex amplifies complement activation by cleaving more C3 into C3a and C3b. The resulting C3b helps form C5 convertase, which generates the inflammatory protein C5a and leads to membrane attack complex (MAC) formation78‘33‘79‘80. By blocking factor D, Vemircopan shuts down this amplification loop, reducing inflammation and tissue damage.
The inhibition of factor D and factor B ultimately result in similar clinical outcomes, as they both modulate the same pathway. However, factor D is considered a more efficient pharmacologic target. Factor D circulates through the body at very low levels (~1–2 µg/mL) and functions as the rate-limiting enzyme of the complement pathway, meaning its inhibition effectively shuts down the cascade. Because of this, even modest drug exposure can achieve robust blockade of complement activity. In contrast, factor B is far more abundant and requires higher systemic drug levels for full inhibition.
A study aimed at quantifying alternative complement pathway’s contribution to the classical pathway found that blocking Factor D reduced over 80% of C5a and MAC formation even when activation was through the classical pathway. The classical pathway alone produced limited terminal complement activity unless amplified by the alternative complement pathway81.Despite its promising mechanism, Vemircopan’s clinical development for MG was terminated in early 2025 after Phase II trials failed to demonstrate sufficient clinical benefit72.
Two new generation FcRn inhibitors are Batoclimab and Nipocalimab. Batoclimab is a monoclonal antibody designed as a subcutaneous injection22. In the phase III trial (called FLEX), 132 patients were randomly assigned to take once-weekly injections of 340 mg, 680 mg or a placebo for 12 weeks81. The primary endpoint was mean change from baseline in MG-ADL to week 12. 58.2% of patients who received Batoclimab and 31.3% of placebo patients reached the primary endpoint. Patients receiving the higher dose achieved a 5.6-point improvement in MG-ADL, while those on lower dose showed a 4.7-point improvement. Minimal symptom expression, defined as a near absence of disease symptoms, was reached in up to 42% of patients. Batoclimab also led to a substantial decrease in IgG levels (around 70%), including pathogenic antibodies (Immunovant, Inc., Immunovant announces positive results for Batoclimab myasthenia gravis (MG) and chronic inflammatory demyelinating polyneuropathy (CIDP) studies. Press release, March 19, 2025. https://www.immunovant.com/investors/news-events/press-releases/detail/71/immunovant-announces-positive-results-for-batoclimab). Despite these positive outcomes, the developers chose not to pursue a regulatory approval and instead shifted focus to developing another FcRn inhibitor named IMVT-1402. Table 1 below shows the pipeline status of Batoclimab and IMVT-1402, together with the two next-generation complement inhibitors discussed above.
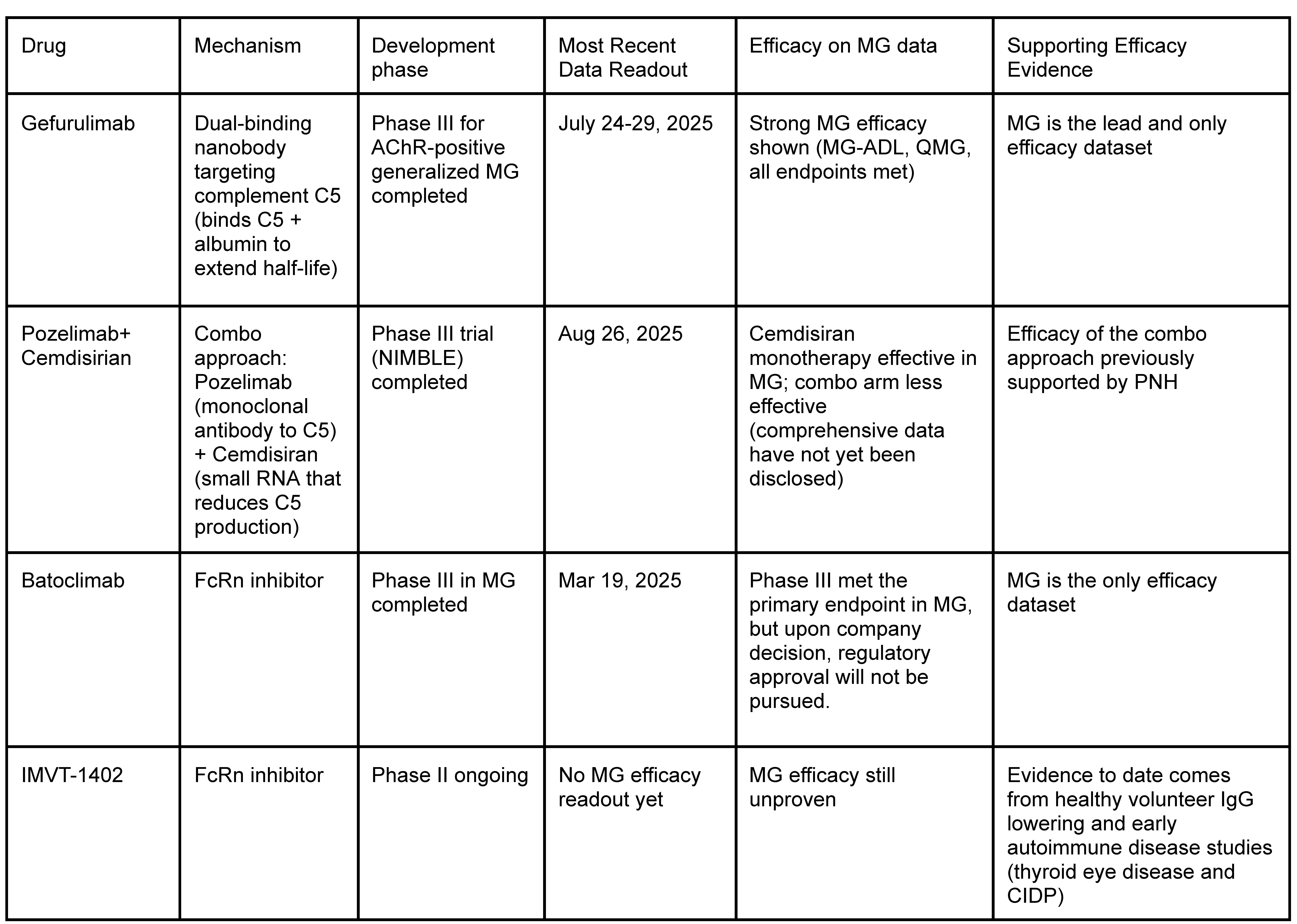
Nipocalimab is an IgG1 monoclonal antibody that targets FcRn and binds with high affinity across a broad pH range24. In the phase 2 VIVACITY-MG trial designed to assess the efficacy of Nipocalimab, 68 MG patients were randomized into five different treatment arms, testing various doses and schedules. While no major issues were identified, and side effects like diarrhea, headache, and nasal congestion were mild, none of the individual dosing groups showed a statistically significant improvement in MG-ADL scores compared to placebo on their own. However, when researchers pooled all the Nipoculimab groups together, the combined group showed a significant improvement in MG-ADL scores compared with placebo (LSMD = -2.2, 95% CI [-3.9, -0.5]; linear trend p = 0.031)72‘82. This finding suggested some clinical effect, but not strong enough to clearly identify an optimal dose, since the p value was only marginally significant (close to 0.05). Later, the phase 3 trial was performed, where patients were either assigned to take 60 mg/kg of the drug every 2 weeks or a placebo. Nipoclimab met its primary endpoint, improvement in MG-ADL (LSMD = −1.45, 95% CI [-2.38, -0.52; p = 0.0024). Both AChR-positive and MuSK-positive MG patients experienced this clinical improvement83. Nipocalimab showed the most prolonged disease control of any FcRn inhibitor in phase III trials84. Table 2 below offers a summary of phase III clinical trials and their results.
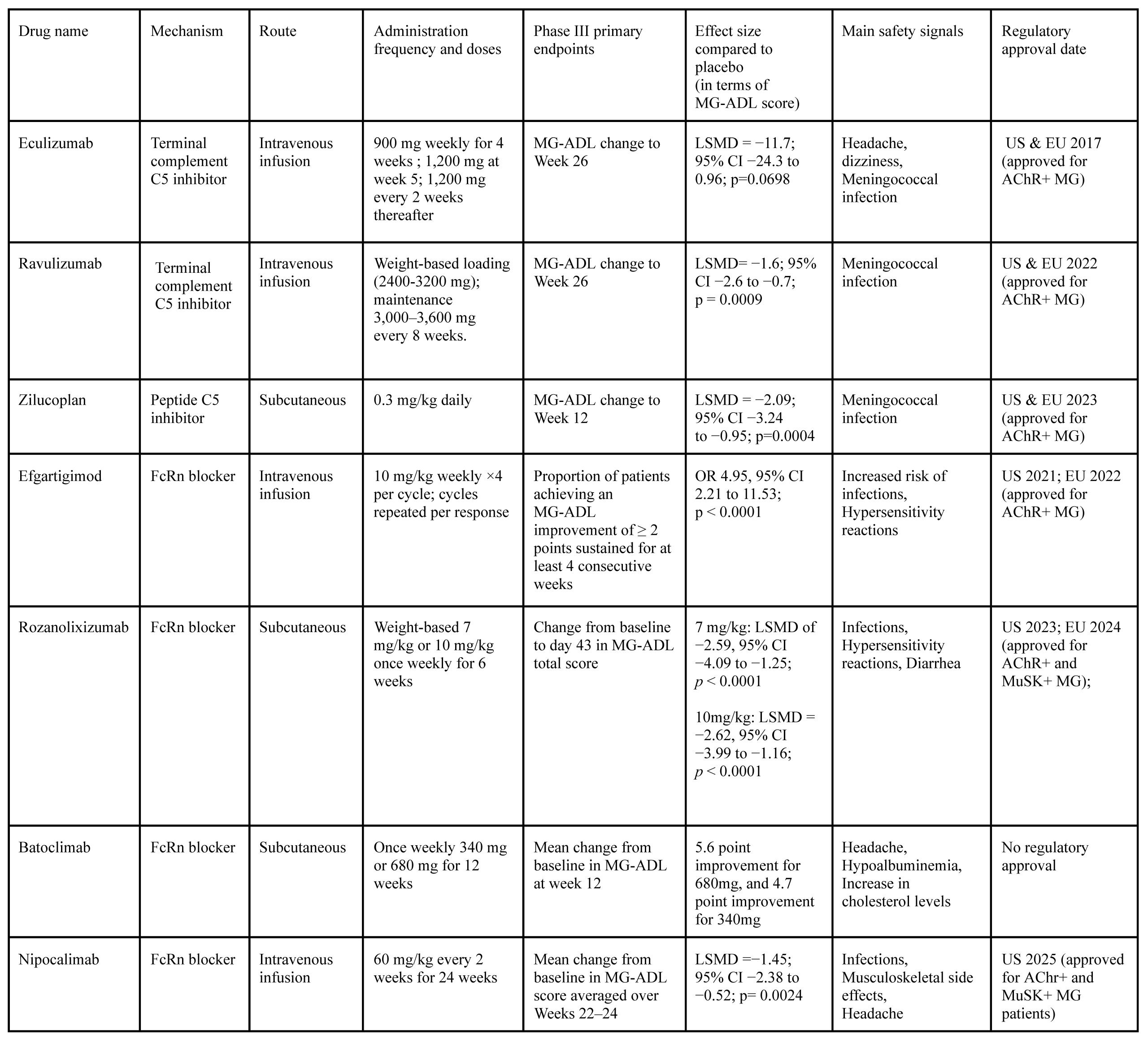
Practical Recommendation to Clinicians
Several factors need to be considered before the decision as to what type of targeted biologics is most suited to a patient is made. These factors include MG subtype, the severity of the patient’s condition, prior treatment history, and comorbidities. Complement inhibitors are currently best suited for refractory AChR+ MG patients, as the phase III RCTs (randomized controlled trials) primarily enrolled this population and the regulatory approvals were granted on that basis. Their use should always be preceded by meningococcal vaccination and infection precautions, given the high risk of serious adverse events. FcRn inhibitors, in contrast, may be considered for patients who require a broader and reversible reduction of pathogenic IgG, including those with frequent relapses or intolerance to traditional therapies. One advantage of these biologics is that they can be used across different antibody subtypes. In clinical practice, complement blockade tends to be reserved for severe, resistant disease, while FcRn blockade offers a more flexible option earlier in the treatment process. Regardless of the choice, close monitoring for infection, durability of effect (open-label extensions), and patient quality of life is essential, and decisions should ideally be made in a multidisciplinary setting with attention to cost, coverage, and long-term safety data.
Conclusion
This review encompasses recent literature on advances in targeted therapeutics for generalized MG, with a specific focus on complement pathway blockers and FcRn inhibitors. The clinical success of these biologics has initiated a wave of innovation in the field. Numerous companies are now directing their efforts to the development of new-generation biologics aimed at enhancing efficacy, and addressing practical limitations such as cost and administration difficulty. Given the accelerating pace of discovery in MG treatment, this review might not capture every emerging therapy. Nonetheless, it offers a comprehensive foundation for understanding the current state and future trajectory of targeted therapeutics in MG treatment.
Complement pathway blockers and FcRn inhibitors represent two mechanistically distinct, yet complementary approaches that provide clinical benefits and symptom relief. FcRn inhibitors reduce the overall burden of pathogenic IgGs by preventing their recycling and constant circulation in the bloodstream. Meanwhile, complement inhibitors prevent downstream tissue damage and amplification loops by blocking activation points in the complement cascade. With the efficacy of these treatment models demonstrated in phase III trials, researchers are now working to make them more accessible, convenient, and affordable for patients.
Treatment strategy in MG is guided by autoantibody subtype and disease severity. One limitation associated with complement inhibitors is its exclusive application to patients with AChR-positive MG. However, as this group comprises 80-85% of all MG patients, these therapies can still benefit the majority. Compared to complement inhibitors, FcRn inhibitors have a broader patient subgroup applicability, because they reduce total IgG antibodies and have shown efficacy in MuSK-positive MG patients as well– for instance, with the FDA approval of Rozanolixumab for this population. Looking ahead, future research and clinical trials should expand to include a more diverse spectrum of MG subtypes, including MuSK-positive, LPR4-positive, and seronegative patients. Additionally, combination strategies, head-to-head comparisons, and biomarker-driven treatment selection will be essential to fully realize the potential of these biologics, better understand the most appropriate therapy for different clinical scenarios, and thus offer the best treatment possible.
Literature Selection
This review draws on peer-reviewed literature published between 2004 and 2025, identified through PubMed, Google Scholar, ScienceDirect, New England Journal of Medicine, and Frontiers. Sources were selected to capture both the original research and the reviews relevant to complement and FcRn inhibition in Myasthenia Gravis, with emphasis on studies providing clear clinical insights. Additionally, articles explaining the drug mechanisms under current investigation for MG were also utilized. Non-peer reviewed sources were excluded.
Acknowledgments
Thank you to Ella Perrault for helping with the critical review process and writing support.
Funding: None
Disclosures: None
References
- I. J. AL‑Zwaini, A. AL‑Mayahi. Introductory chapter: Myasthenia gravis – an overview. In Selected Topics in Myasthenia Gravis; IntechOpen: London, UK, 2019. [↩]
- K. Lazaridis, S. J. Tzartos, Myasthenia gravis: Autoantibody specificities and their role in MG management. Front. Neurol. 11, 596981 (2020). [↩]
- A. M. Lascano, P. H. Lalive, Update in immunosuppressive therapy of myasthenia gravis. Autoimmun. Rev. 20(1), 102712 (2021). [↩]
- Y.-A. Heo, Efgartigimod alfa in generalized Myasthenia Gravis: A profile of its use. CNS Drugs 37(4), 337–344 (2023). [↩]
- P. M. Rodríguez Cruz, J. Palace, A. Vincent, Molecular mechanisms of the neuromuscular junction and their role in myasthenia gravis. Front. Mol. Neurosci. 13, 610964 (2020). [↩]
- J. F. Howard Jr., Myasthenia gravis: The role of complement at the neuromuscular junction. Ann. N. Y. Acad. Sci. 1412(1), 113–128 (2018. [↩]
- B. M. Conti-Fine, M. Milani, H. J. Kaminski, Myasthenia gravis: Past, present, and future. J. Clin. Invest. 116(11), 2843–2854 (2006). [↩]
- L. Dresser, R. Wlodarski, K. Rezania, B. Soliven, Myasthenia gravis: Epidemiology, pathophysiology and clinical manifestations. J. Clin. Med. 10(11), 2235 (2021). [↩] [↩] [↩] [↩]
- N. S. R. Sanderson, Complement and myasthenia gravis. Mol. Immunol. 151, 11–18 (2022). [↩] [↩]
- Y. Zou, J. Pan, Mechanisms of neuromuscular junction pathology in aging and disease. Cell Biosci. 12, 93 (2022). [↩]
- A. Martinez Salazar, S. Mokhtari, E. Peguero, M. Jaffer, The role of complement in the pathogenesis and treatment of myasthenia gravis. Cells 14, 739 (2025). [↩] [↩]
- B. M. Conti-Fine, M. Milani, H. J. Kaminski, Myasthenia gravis: Past, present, and future. J. Clin. Invest. 116(11), 2843–2854 (2006). [↩]
- B. M. Conti-Fine, M. Milani, H. J. Kaminski, Myasthenia gravis: Past, present, and future. J. Clin. Invest. 116(11), 2843–2854 (2006). [↩] [↩]
- M. L. Fichtner, R. Jiang, A. Bourke, R. J. Nowak, K. C. O’Connor, Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front. Immunol. 11, 776 (2020). [↩] [↩]
- D. Ma, B. Liu, Y. Wang, R. Yang, R. Zhu, Advancements and prospects of novel biologicals for myasthenia gravis: Toward personalized treatment based on autoantibody specificities. Front. Pharmacol. 15, 1370411 (2024). [↩] [↩] [↩] [↩] [↩] [↩]
- A. G. Vakrakou, E. Karachaliou, et al., Immunotherapies in MuSK-positive myasthenia gravis; an IgG4 antibody-mediated disease. Front. Immunol. 14, 1212757 (2023). [↩]
- D. Ma, B. Liu, Y. Wang, R. Yang, R. Zhu, Advancements and prospects of novel biologicals for myasthenia gravis: Toward personalized treatment based on autoantibody specificities. Front. Pharmacol. 15, 1370411 (2024). [↩]
- R. Herbst, MuSK function during health and disease. Neurosci. Lett. 716, 134676 (2020). [↩]
- M. L. Fichtner, R. Jiang, A. Bourke, R. J. Nowak, K. C. O’Connor, Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front. Immunol. 11, 776 (2020). [↩]
- H. J. Kaminski, P. Sikorski, S. I. Coronel, L. L. Kusner, Myasthenia gravis: The future is here. J. Clin. Invest. 134(12), e179742 (2024). [↩] [↩]
- C. Schneider-Gold, N. E. Gilhus, Advances and challenges in the treatment of myasthenia gravis. Ther. Adv. Neurol. Disord. 14, 17562864211065406 (2021). [↩] [↩]
- L. Gerischer, P. Doksani, S. Hoffmann, A. Meisel, New and emerging biological therapies for myasthenia gravis: A focussed review for clinical decision-making. BioDrugs 39(2), 185–213 (2025). [↩] [↩] [↩]
- W. M. Baldwin III, A. Valujskikh, R. L. Fairchild, The neonatal Fc receptor: Key to homeostatic control of IgG and IgG-related biopharmaceuticals. Am. J. Transplant. 19(7), 1881–1887 (2019). [↩] [↩]
- A. Nair, S. Jacob, Novel immunotherapies for myasthenia gravis. Nat. Rev. Neurol. 19(6), 339–354 (2023). [↩] [↩] [↩] [↩]
- C. Nelke, M. Spatola, C. B. Schroeter, H. Wiendl, J. D. Lünemann, Neonatal Fc receptor–targeted therapies in neurology. Neurotherapeutics 19(3), 729–740 (2022). [↩]
- L. N. Zhu, H. M. Hou, S. Wang, S. Zhang, G. G. Wang, Z. Y. Guo, J. Wu, FcRn inhibitors: A novel option for the treatment of myasthenia gravis. Neural Regen. Res. 18(8), 1637–1644 (2022). [↩]
- T. T. Gjølberg, S. Mester, G. Calamera, J. S. Telstad, I. Sandlie, J. T. Andersen, Targeting the neonatal Fc receptor in autoimmune diseases: Pipeline and progress. BioDrugs 39(3), 373–409 (2025). [↩]
- Y. Zhou, S. Jiang, Roles of FcRn in antigen-presenting cells during autoimmunity and a clinical evaluation of efgartigimod as an FcRn blocker. Pathogens 12(6), 817 (2023). [↩]
- K. Baker, T. Rath, M. Pyzik, R. S. Blumberg, The role of FcRn in antigen presentation. Front. Immunol. 5, 408 (2014). [↩]
- Y. Xing, K. A. Hogquist, T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 4(6), a006957 (2012). [↩]
- M. J. Shlomchik, Activating systemic autoimmunity: B’s, T’s and tolls. Curr. Opin. Immunol. 21(6), 626–633 (2009). [↩]
- L. N. Zhu, H. M. Hou, S. Wang, S. Zhang, G. G. Wang, Z. Y. Guo, J. Wu, FcRn inhibitors: A novel option for the treatment of myasthenia gravis. Neural Regen. Res. 18(8), 1637–1644 (2022). [↩]
- P. F. Zipfel, T. Hallström, K. Riesbeck, Human complement control and complement evasion by pathogenic microbes: Tipping the balance. Mol. Immunol. 56(3), 152–160 (2013). [↩] [↩]
- C. Giorgio, M. Zippoli, P. Cocchiaro, V. Castelli, G. Varrassi, A. Aramini, M. Allegretti, L. Brandolini, M. C. Cesta, Emerging role of C5 complement pathway in peripheral neuropathies: Current treatments and future perspectives. Biomedicines 9, 399 (2021). [↩]
- J. L. S. Thomsen, H. Andersen, Outcome measures in clinical trials of patients with myasthenia gravis. Front. Neurol. 11, 596382 (2020). [↩]
- F. Saccà, C. Pane, P. E. Espinosa, M. P. Sormani, A. Signori, Efficacy of innovative therapies in myasthenia gravis: A systematic review, meta-analysis and network meta-analysis. Eur. J. Neurol. 30(18), 3854–3867 (2023). [↩] [↩]
- J. F. Howard Jr., K. Utsugisawa, M. Benatar, et al., REGAIN Study Group, Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): A phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 16(12), 976–986 (2017). [↩]
- K. Albazli, H. J. Kaminski, J. F. Howard Jr., Complement inhibitor therapy for myasthenia gravis. Front. Immunol. 11, 917 (2020). [↩] [↩]
- P. P. San, S. Jacob, Role of complement in myasthenia gravis. Front. Neurol. 14, 1277596 (2023). [↩] [↩] [↩] [↩] [↩]
- S. N. M. Binks, I. M. Morse, M. Ashraghi, A. Vincent, P. Waters, M. I. Leite, Myasthenia gravis in 2025: Five new things and four hopes for the future. J. Neurol. 272(3), 226 (2025). [↩]
- A. Nair, S. Jacob, Novel immunotherapies for myasthenia gravis. Nat. Rev. Neurol. 19(6), 339–354 (2023). [↩]
- L. Gerischer, P. Doksani, S. Hoffmann, A. Meisel, New and emerging biological therapies for myasthenia gravis: A focussed review for clinical decision-making. BioDrugs 39(2), 185–213 (2025). [↩]
- P. M. Ladwig, M. A. V. Willrich, Ravulizumab: Characterization and quantitation of a new C5 inhibitor using isotype specific affinity purification and high-resolution mass spectrometry. J. Mass Spectrom. Adv. Clin. Lab. 21, 10–18 (2021). [↩] [↩]
- R. M. Stern, N. T. Connell, Ravulizumab: A novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Ther. Adv. Hematol. 10, 2040620719874728 (2019). [↩]
- C. Gurnari, I. Nautiyal, S. Pagliuca, Current opinions on the clinical utility of ravulizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Ther. Clin. Risk Manag. 17, 1343–1351 (2021). [↩]
- P. P. San, S. Jacob, Role of complement in myasthenia gravis. Front. Neurol. 14, 1277596 (2023). [↩] [↩] [↩] [↩]
- T. Vu, A. Meisel, R. Mantegazza, D. Annane, M. Katsuno, R. Aguzzi, A. Enayetallah, K. N. Beasley, N. Rampal, J. F. Howard Jr., for the CHAMPION MG Study Group, Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid. 1(5) (2022). [↩]
- T. H. Vu, R. Mantegazza, D. Annane, M. Katsuno, et.al, CHAMPION MG Study Group, Long-term efficacy and safety of ravulizumab in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: Final results from the phase 3 CHAMPION MG open-label extension. Eur. J. Neurol. 32, e70158 (2025). [↩]
- A. Meisel, D. Annane, T. Vu, R. Mantegazza, M. Katsuno, R. Aguzzi, G. Frick, L. Gault, J. F. Howard Jr., CHAMPION MG Study Group, Long-term efficacy and safety of ravulizumab in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: Results from the phase 3 CHAMPION MG open-label extension. Eur. J. Neurol. 32(4), 70158 (2025). [↩]
- M. Menacher, M. Ellssel, I. Kwiedor, M. Naumann, A. Bayas, Complement inhibition in seropositive generalized myasthenia gravis as rescue therapy in impending and effective treatment in frequently recurring impending myasthenic crisis—A case series. Ther. Adv. Neurol. Disord. 17, 1–8 (2024). [↩]
- G.-Q. Tang, Y. Tang, K. Dhamnaskar, M. D. Hoarty, R. Vyasamneni, D. D. Vadysirisack, Z. Ma, N. Zhu, J.-G. Wang, C. Bu, B. Cong, E. Palmer, P. W. Duda, C. Sayegh, A. Ricardo, Zilucoplan, a macrocyclic peptide inhibitor of human complement component 5, uses a dual mode of action to prevent terminal complement pathway activation. Front. Immunol. 14, 1213920 (2023). [↩] [↩]
- J.-i. Nishimura, M. Yamamoto, et al., Genetic variants in C5 and poor response to eculizumab. N. Engl. J. Med. 370(7), 632–639 (2014). [↩]
- J. F. Howard Jr., S. Bresch, A. Genge, et. al., RAISE Study Team, Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol. 22(5), 395–406 (2023). [↩]
- CDC, Clinical guidance for managing meningococcal disease risk in patients receiving complement inhibitor therapy. Meningococcal Disease [Internet], (2024). Available from: https://www.cdc.gov/meningococcal/hcp/clinical-guidance/complement-inhibitor.html. [↩]
- Y. A. Heo, Efgartigimod: First approval. Drugs 82(3), 341–348 (2022). [↩]
- K. G. Gwathmey, C. M. Broome, M. Goebeler, H. Murai, Z. Bata-Csörgo, et al., Safety profile of efgartigimod from global clinical trials across multiple immunoglobulin G-mediated autoimmune diseases. Expert Rev. Clin. Immunol. 21(5), 627–638 (2025). [↩]
- Y.A. Heo, Efgartigimod: First approval. Drugs 82(3), 341–348 (2022). [↩]
- V. Bhandari, V. Bril, FcRn receptor antagonists in the management of myasthenia gravis. Front. Neurol. 14, 1229112 (2023). [↩]
- J. F. Howard Jr., V. Bril, T. Vu, C. Karam, et al., ADAPT Investigator Study Group, Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): A multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 20(7), 526–536 (2021). [↩]
- Y. A. Heo, Efgartigimod alfa in generalised myasthenia gravis: A profile of its use. CNS Drugs 37(4), 337–344 (2023). [↩]
- M. G. Huijbers, J. J. Plomp, I. E. van Es, Y. E. Fillié-Grijpma, et al., Efgartigimod improves muscle weakness in a mouse model for muscle-specific kinase myasthenia gravis. Exp. Neurol. 317, 133–143 (2019). [↩]
- F. Shi, J. Chen, L. Feng, R. Lai, H. Zhou, et al., Efgartigimod treatment in patients with anti-MuSK-positive myasthenia gravis in exacerbation. Front. Neurol. 14, 1486659 (2024). [↩]
- L. Gerischer, P. Doksani, S. Hoffmann, A. Meisel, New and emerging biological therapies for myasthenia gravis: A focussed review for clinical decision-making. BioDrugs 39(2), 185–213 (2025). [↩]
- V. Bril, A. Drużdż, J. Grosskreutz, A. A. Habib, R. Mantegazza, et al., Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): A randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. 22(5), 383–394 (2023). [↩]
- S. M. Hoy, Rozanolixizumab: First approval. Drugs 83(14), 1341–1347 (2023). [↩]
- N. Huntemann, L. Gerischer, M. Herdick, C. Nelke, F. Stascheit, et al., C5 complement inhibition versus FcRn modulation in generalised myasthenia gravis. J. Neurol. Neurosurg. Psychiatry 96(4), 310–321 (2025). [↩]
- P.-W. Lien, M. Joshi, J. A. Tice, F. Agboola, et al., Cost-effectiveness of eculizumab and efgartigimod for the treatment of anti–acetylcholine receptor antibody–positive generalized myasthenia gravis. J. Manag. Care Spec. Pharm. 30(6), 517–527 (2024). [↩]
- S. Jindal, D. V. Pedersen, N. Gera, J. Chandler, R. Patel, A. Neill, et al., Characterization of the bispecific VHH antibody Gefurulimab (ALXN1720) targeting complement component 5, and designed for low volume subcutaneous administration. Mol. Immunol. 165, 29–41 (2024). [↩]
- K. M. K. Sand, M. Bern, J. Nilsen, H. T. Noordzij, I. Sandlie, J. T. Andersen, Unraveling the interaction between FcRn and albumin: Opportunities for design of albumin-based therapeutics. Front. Immunol. 5, 682 (2015). [↩]
- P. Badri, X. Jiang, A. Borodovsky, N. Najafian, J. Kim, V. A. Clausen, V. Goel, B. Habtemariam, G. J. Robbie, Pharmacokinetic and pharmacodynamic properties of cemdisiran, an RNAi therapeutic targeting complement component 5, in healthy subjects and patients with paroxysmal nocturnal hemoglobinuria. Clin. Pharmacokinet. 60(3), 365–378 (2021). [↩]
- K. Devalaraja-Narashimha, C. Huang, M. Cao, Y. P. Chen, A. Borodovsky, W. C. Olson, L. G. Morton, M. W. Retter, Pharmacokinetics and pharmacodynamics of pozelimab alone or in combination with cemdisiran in non-human primates. PLoS One 17(6), e0269749 (2022). [↩]
- L. Gerischer, P. Doksani, S. Hoffmann, A. Meisel, New and emerging biological therapies for myasthenia gravis: A focused review for clinical decision-making. BioDrugs 39(2), 185–213 (2025). [↩] [↩] [↩] [↩]
- V. Perkovic, J. Barratt, B. Rovin, N. Kashihara, B. Maes, et al., Alternative complement pathway inhibition with iptacopan in IgA nephropathy. N. Engl. J. Med. 392(6), 531–543 (2025). [↩]
- I. Michailidou, A. Patsiarika, E. Kesidou, M. K. Boziki, D. Parisis, C. Bakirtzis, E. Chroni, N. Grigoriadis, The role of complement in the immunopathogenesis of acetylcholine receptor antibody-positive generalized myasthenia gravis: Bystander or key player? Front. Immunol. 16, 1526317 (2025). [↩]
- V. Perkovic, J. Barratt, B. Rovin, N. Kashihara, B. Maes, H. Zhang, H. Trimarchi, D. Kollins, O. Papachristofi, S. Jacinto-Sanders, et al., Alternative complement pathway inhibition with iptacopan in IgA nephropathy. N. Engl. J. Med. 392(6), 531–543 (2025). [↩]
- A. Schubart, K. Anderson, N. Mainolfi, H. Sellner, T. Ehara, C. Adams, et al., Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. U. S. A. 116(16), 7926–7931 (2019). [↩]
- V. R. Gadhachanda, J. A. Wiles, et al., First-in-class clinically investigated oral factor D inhibitors for the treatment of complement-mediated diseases. Pharm. Res. 42(7), Article 88 (2025). [↩]
- J. F. Howard Jr., Myasthenia gravis: The role of complement at the neuromuscular junction. Ann. N. Y. Acad. Sci. 1412(1), 113–128 (2018). [↩]
- A. Schubart, K. Anderson, N. Mainolfi, H. Sellner, et al., Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. U. S. A. 116(16), 7926–7931 (2019). [↩]
- V. R. Gadhachanda, J. A. Wiles, S. D. Podos, D. Boyer, J. Thanassi, D. Patel, Y. Zhao, L. Wang, M. Huang, First-in-class clinically investigated oral factor D inhibitors for the treatment of complement-mediated diseases. Pharm. Res. 42(7) (2025). [↩]
- M. Benatar, H. Wiendl, R. Nowak, Y. Zheng, W. Macias, Batoclimab as induction and maintenance therapy in patients with myasthenia gravis: Rationale and study design of a phase 3 clinical trial. BMJ Neurol. Open 6(1), e000536 (2024). [↩] [↩]
- C. Antozzi, J. Guptill, V. Bril, et al., Vivacity-MG Phase 2 Study Group, Safety and efficacy of Nipocalimab in patients with generalized myasthenia gravis: Results from the randomized phase 2 Vivacity-MG study. Neurology 102(2), e207937 (2024). [↩]
- C. Antozzi, T. Vu, S. Ramchandren, R. J. Nowak, C. Farmakidis, et al., Vivacity-MG3 Study Group, Safety and efficacy of nipocalimab in adults with generalised myasthenia gravis (Vivacity-MG3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet Neurol. 24(2), 105–116 (2025). [↩]
- D. Menon, V. Bhandari, FcRn inhibitors in the context of myasthenia gravis. Expert Opin. Emerg. Drugs 30(1), 7–10 (2025). [↩]



{kind=link}