
Abstract
Cystic Fibrosis (CF) is a hereditary disorder caused by mutations in the CFTR gene, resulting in viscous secretions, chronic lung infections, and progressive respiratory failure. Although the CFTR gene has been well-studied, the microbial mechanisms driving lung disease progression remain incompletely understood. Microbial interactions including quorum sensing, biofilm formation, and metabolite exchange, play key roles in colonization and persistence in the CF lung environment. Using 103 sputum samples from 88 adult CF patients and 16S rRNA gene sequencing data from the Qiita database, we analyzed bacterial diversity and composition across age groups. Alpha diversity metrics (Shannon and Simpson indices) revealed a peak in microbial diversity during adolescent age (9–14 years), followed by a gradual decline into adulthood. LEfSe (Linear discriminant analysis Effect Size) analysis identified Pseudomonas as the most significantly age associated genus, increasing sharply after age 19 and dominating in patients over 40. Core microbiome analysis indicated a decrease in shared taxa with age, with Streptococcus and Fusobacterium prevalent in younger groups and diminishing in older individuals. These findings highlight dynamic, age-dependent shifts in microbial diversity and dominance within the CF lung microbiome. Our cross-sectional results are consistent with previous reports suggesting that diversity peaks in adolescence before pathogenic genera, such as Pseudomonas, become dominant and diversity declines thereafter. This pattern raises the hypothesis that adolescence could be an important window for interventions, which should be tested in longitudinal studies.
Keywords: Cystic fibrosis, Microbiome, Microbial diversity, Pseudomonas, Microbial interactions, Pathogenic dominance
Introduction
Cystic Fibrosis (CF) is a genetic disorder defined by variants in the cystic fibrosis transmembrane conductance regulator (CFTR) gene1. According to the American Cystic Fibrosis Foundation patient registry, there are currently more than 30,000 CF patients in the United States and more than 70,000 CF patients throughout the world2. Described by pancreatic insufficiency and chronic endobronchial airway infection, but not limited to these specific organs, the disorder results in progressive bronchiectasis and eventual respiratory failure3. The defect leads to diminished chloride secretion and, in turn, to increased sodium absorption through epithelial sodium channels and removal of water from secretions, which are therefore abnormally viscous4. The CFTR gene has been known for around 30 years, yet an incomplete understanding remains evident in the clinical realm of identifying key genotype-phenotype relationships5,6,7.
Bacteria-bacteria or bacteria-host interactions serve as an essential mechanism of colonizing and establishing in several different habitats. Molecular and genetic information is transferred during microbial contacts, and a variety of processes, including quorum sensing, siderophores, secondary metabolites, biofilm development, and cellular transduction signaling, can be involved, offering insight into numerous health issues8.
The respiratory tract in individuals with CF is colonized by a diverse microbiome from a very young age9. Despite significant advances in treatment, many individuals still die from the chronic lung infections associated with CF. Numerous studies have focused on understanding the factors and mechanisms that lead to increased susceptibility to infection: identifying the relationship between CFTR genotype and respiratory phenotype or exploring the effects of Pseudomonas colonization, the most dangerous pathogen for CF patients. However, our understanding of lung disease progression remains incomplete and does not fully explain the lung phenotypes of CF patients.
Researchers have begun exploring the diverse lung microbiomes apparent in CF patients. In the past, researchers have primarily focused on specific types of pathogens like Pseudomonas aeruginosa (P. aeruginosa species from the Pseudomonas genus), but there has been a shift towards exploring the entire microbiome to understand complex relationships or characteristics –keystone species, mutualism, or commensalism– through next-generation-sequencing (NGS). This research reveals novel age-dependent shifts and microbial relationships, such as quorum sensing, biofilm formation, and metabolite exchange, associated with CF lung disease progression. Proposing the necessity to target these microbial interactions during adolescence, when diversity peaks, may preserve a healthier microbiome and delay pathogenic dominance like Pseudomonas.
Materials and Methods
The dataset utilized was the Neutrophilic Proteolysis in the Cystic Fibrosis Lung Favors a Pathogenic Microbiome from Qiita, composed of 103 unique CF sputum samples using 16S rRNA gene amplicon sequencing. Samples were collected from 88 CF patients during routine clinic visits at UC San Diego, following IRB-approved protocols. Sputum was induced using 7% hypertonic saline, homogenized, aliquoted, and immediately frozen at 80 °C for downstream analyses10. MicrobiomeAnalyst program was used for complete statistical analysis, setting parameters of the low count filter to 10% prevalence, rarefying the data by 10,000, and all other parameters were set to default. MicrobiomeAnalyst comprises four modules, and we used the Marker Data Profiling (MDP) module that is designed for analysis of 16S rRNA marker gene survey data11. The alpha diversity was measured through the Shannon and Simpson indices and utilizing the Welch T-test to identify richness differences between microbial samples. Additionally the LEfSe identifies microbial taxa that are statistically and biologically significant between groups. While the alpha diversity indices quantifies the overall diversity within a single sample, the core microbiome reveals the shared and consistent members of a microbial community.
Our cohort contains a total of 335 samples which were used for the diversity indices, core microbiome, and LEfSe, with some individuals providing multiple samples, which were organized into stratified age groups based on CF disease stages based on a previous study: under 1 (5%), 1 to 2 (7%), 2 to 4 (9%), 4 to 9 (21%), 9 to 14 (7%), 14 to 19 (15%), 20 to 24 (3%), 24 to 29 (13%), 29 to 39 (12%), and over 40 (8%)12. Out of the 335 samples 215 were characterized by ethnicity with Hispanic (20%) and non-Hispanic (80%). Additionally the cohort displayed a sex ratio of male (59%) and female (41%). Additionally, our cohort demonstrated a sex distribution of female (40%) and male (60%). Some patients contributed more than one sputum sample; we treated each sample as independent, which may overestimate the effective sample size and significance of some results.
Results
Genus-Level Taxonomic Composition
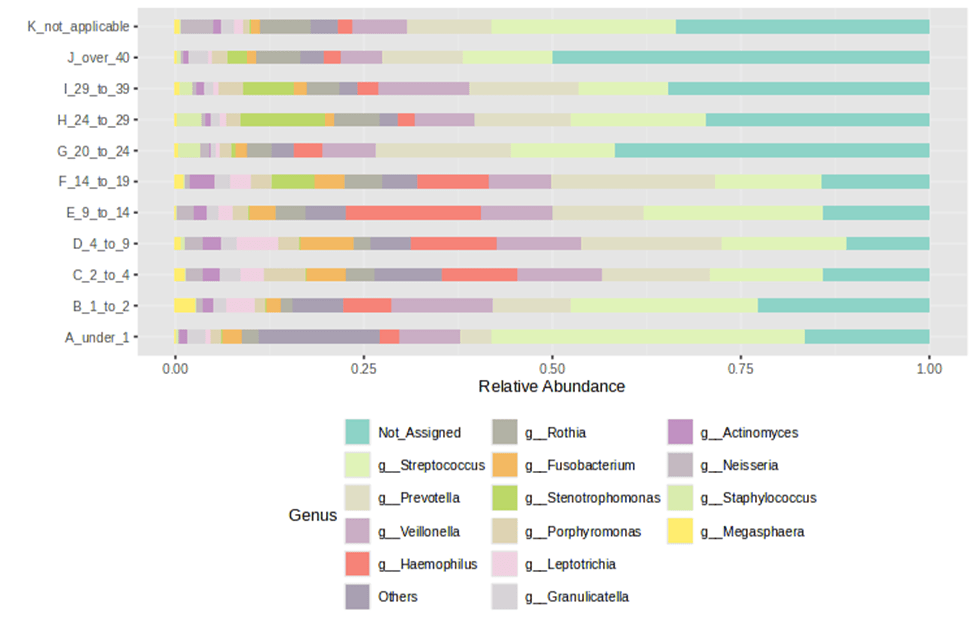
The relative composition of each age group displayed fewer genera as age progressed, clearly emphasizing fluctuations in genus-level diversity over time. Staphylococcus showed high abundance at younger ages and gradually decreased with age. In contrast, Prevotella exhibited an inverse relationship, increasing in abundance as Staphylococcus declined. Haemophilus exhibited a distinct spike during puberty (ages 9 to 14). Stenotrophomonas increased in abundance in older age groups, a trend similarly observed with Rothia. As age increases, the prevalence of dominant genera present at birth declines markedly, indicating a shift in the core microbial community over time. (Figure 1).
Alpha diversity
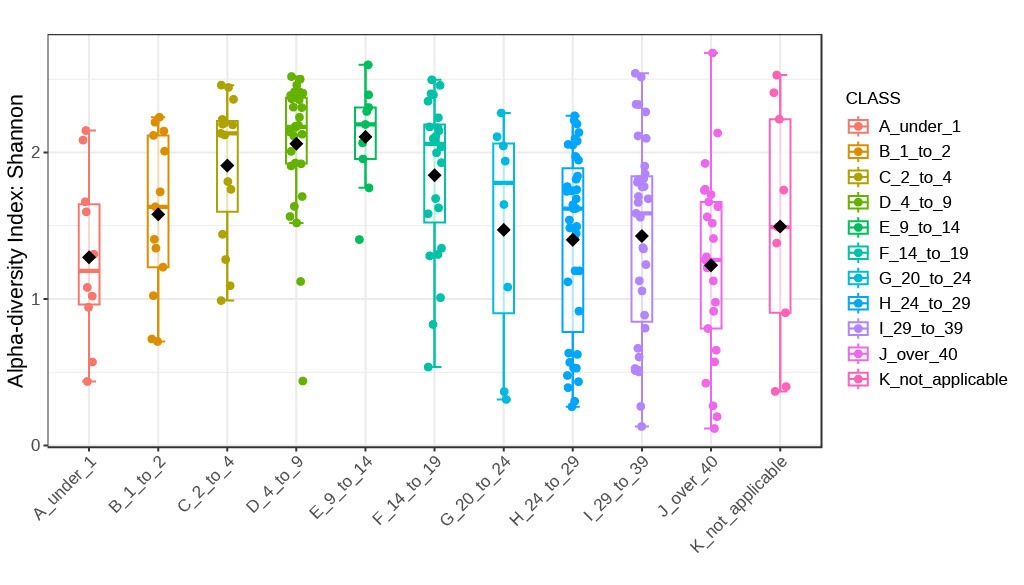
The Shannon index, a measure of diversity commonly used in ecology and other fields, was utilized to evaluate alpha diversity within the CF lung microbiome across different age groups. We assessed alpha diversity using the Shannon index, comparing two age groups with Welch’s t-test and multiple age groups with Welch’s one-way ANOVA, graphing the Shannon index values by age to visualize changes in microbial diversity. Results demonstrated an overall increase in Shannon indices with age. Notably, greater variability was observed within the 20–24 yrs, 25–29 yrs, 30–39 yrs, and over 40 year-old groups. A distinct trend was identified: the Shannon index increased from the 1-year-old group to the 9–14-year-old group, after which it decreased and subsequently stabilized in individuals older than 14 years (Figure 2a).
The Shannon index started low at under 1 and gradually increased through the age groups: 1 to 2, 2 to 4, and 4 to 9-year-old groups. Before eventually reaching the peak at the 9 to 14 -year-old group. Then the Shannon index began gradually decreasing from 14 to 19, 20 to 24, 24 to 29, and 29 to 39-year-old groups, ultimately reaching its lowest at the over 40-year-old group. Interestingly the median for the under 1 and the over 40-year-old groups experienced similar Shannon indices. The Shannon index changes with age, with a Pearson correlation coefficient of -0.33, a 95% confidence interval: -0.44 88209 to -0.20, p0.001 (Figure 2b).
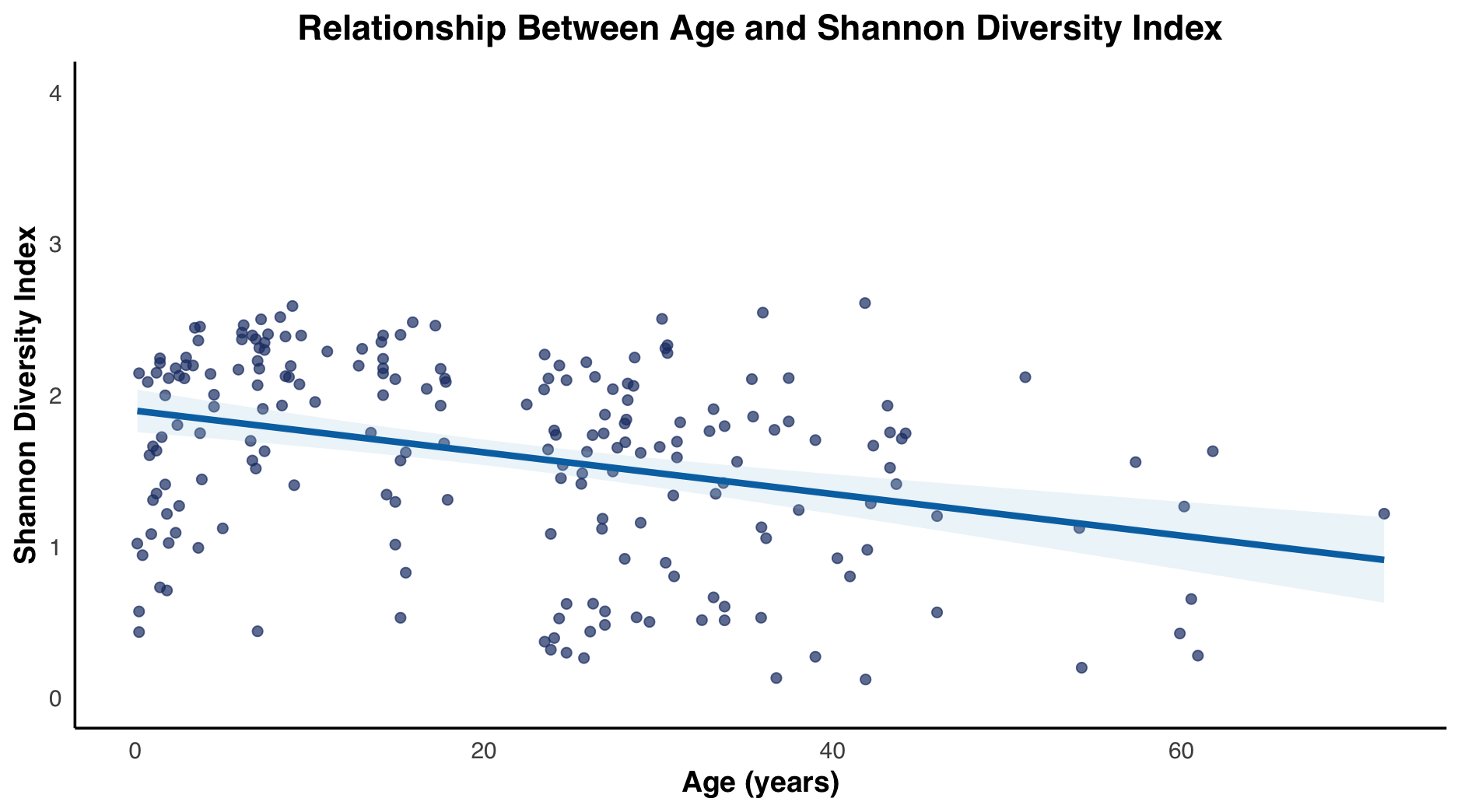
fitted regression line illustrating overall trends.
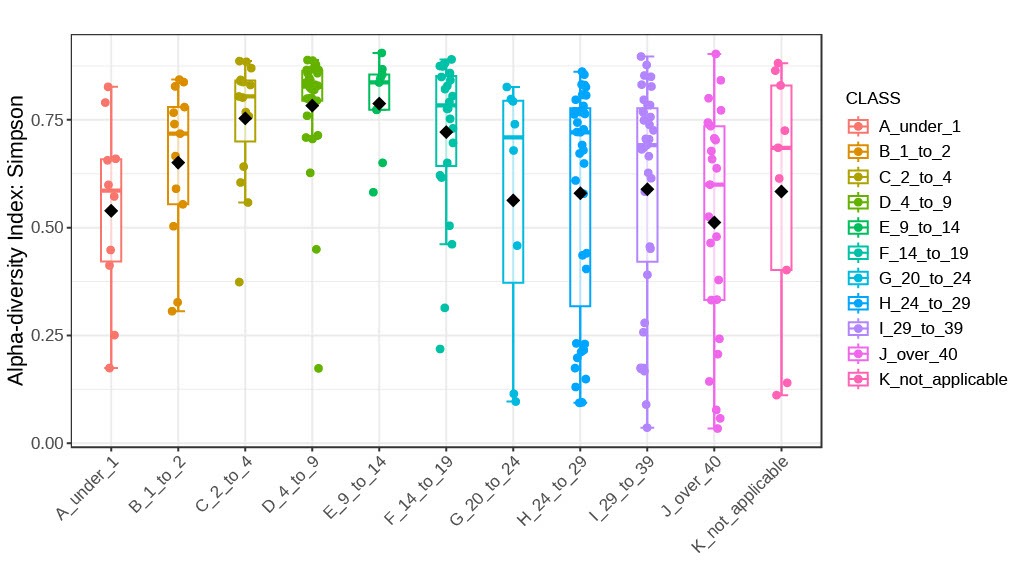
In addition to the Shannon Index, We assessed alpha diversity using the Simpson index, which measures both the richness and evenness of species within a community, comparing two age groups with Welch’s t-test and multiple age groups with Welch’s one-way ANOVA was employed to assess the dominance of bacterial genera across age groups. Results were consistent with those obtained using the Shannon index, highlighting peaks in diversity and dominance during the 4–9, 9–14, and 14–19-year age groups, corresponding to periods of puberty. Additionally, a broader range of Simpson index values became apparent with increasing age. Overall, a trend of initial variability followed by stabilization was observed, with Simpson index values tend to be lower in older age groups compared to younger samples.
The Simpson index fluctuates with age. As age progresses from the under 1 age group, there is a gradual increase in median index values from 1 to 2, 2 to 4, 4 to 9-year-old groups. Eventually reaching its maximum/greatest median at 9 to 14-year-old group. Then gradually decreasing from 14 to 19 and 20 to 24-year-old groups, with a small increase in the 24 to 29 and 29 to 39-year-old groups, before decreasing at the over 40-year-old group. The under 1 and 20 to 24-year-old groups have the similar median Simpson index values. It is also evident that variance of Simpson values increased with age, experiencing the smallest near the peak age groups: 4 to 9 and 14 to 19. Interestingly, the box and whisker plots for every age group experienced a left skew resulting in high median values, and clustering tendency higher Simpson values (Figure 3a). The Simpson index scatterplot, had a correlation coefficient of -0.3002169 and a 95% confidence interval range of -0.4191396 to -0.1711665 (Figure 3b).
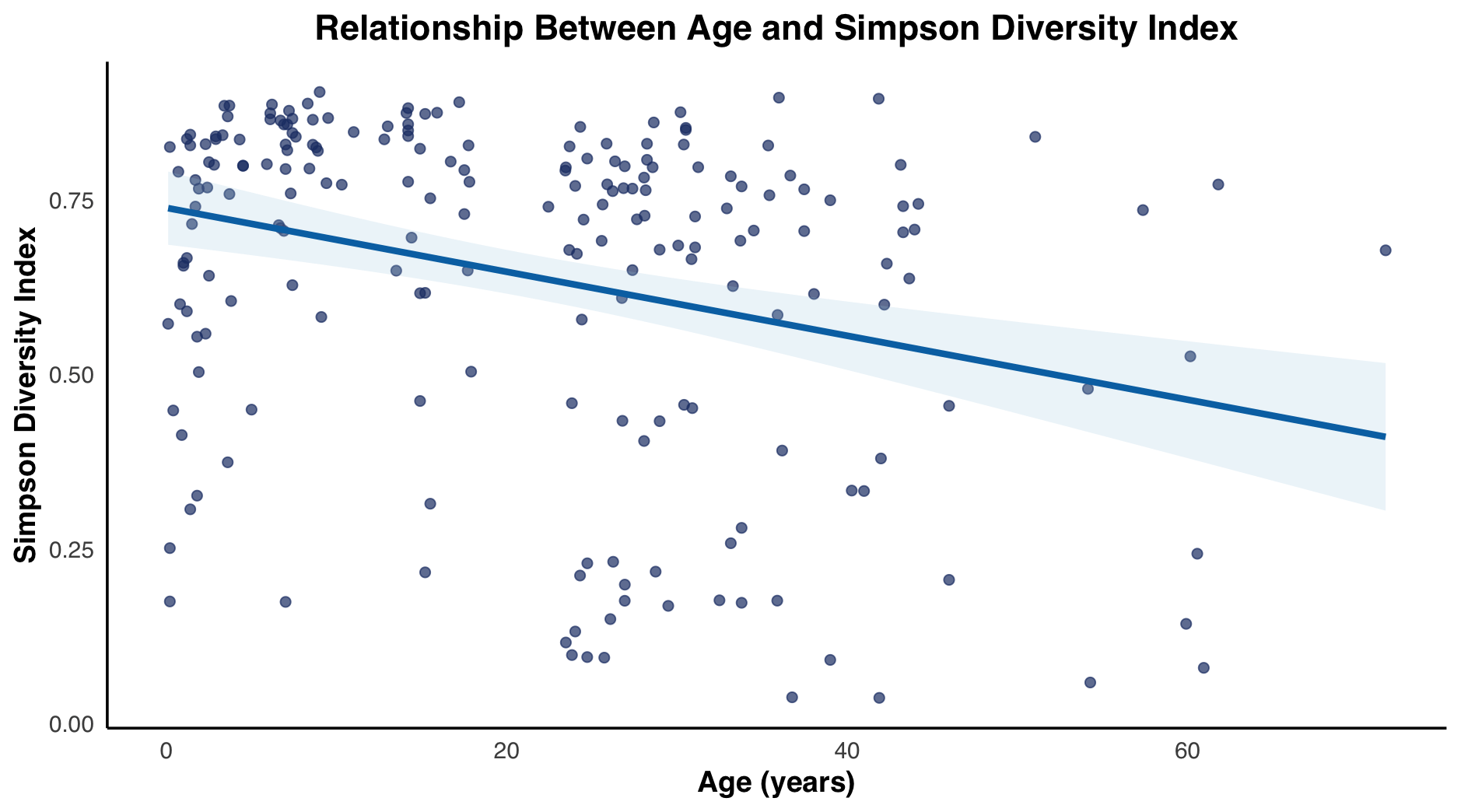
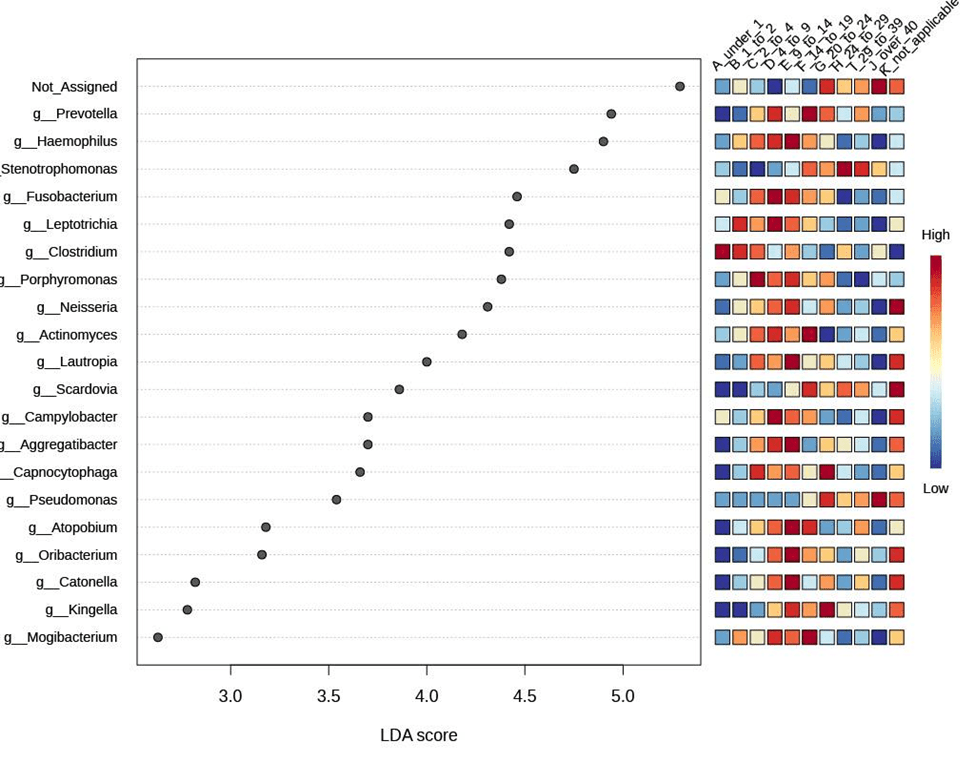
Differential bacteria across age groups
To determine whether there are bacterial strains that are differentially abundant across age groups, we used LEfSE analysis. LEfSe was performed using = 0.05, a constant LDA score threshold of 2.0, and Benjamini–Hochberg FDR–adjusted p ¡ 0.05. Using LEfSE we identified 21 genera with significant age associations (FDR ¡ 0.05). Pseudomonas and Stenotrophomonas were enriched in older age groups (¿40 years), while Streptococcus, Fusobacterium, and Leptotrichia were enriched in younger groups (4–9, 9–14 years). Pseudomonas was the most significant group with the lowest FDR value (3.25 × 10−12), beginning to increase after the 20–24-year group and continuously rising in older age groups. Stenotrophomonas also increased progressively, with a notable spike at 20–24 years (FDR = 0.0595). Interestingly, the prevalence of other genera followed age-specific patterns: Clostridium was highest in infancy (¡1 year) and decreased before rising again in adults over 40 years; Prevotella and several genera including Mogibacterium, Scardovia, and Actinomyces peaked in adolescence (14–19 years); Capnocytophaga and Kingella peaked around 20–24 years; Porphyromonas and Neisseria spiked in 9–14 years. The maximum prevalence of different genera generally spiked around puberty (4–9 to 14–19 years), while Pseudomonas and Stenotrophomonas increasingly dominated older age groups, reflecting the age-associated shift toward opportunistic pathogens in the CF airway microbiome. (Figure 4a).
To further investigate bacterial composition changes with age, a LefSe (Linear Discriminant Analysis Effect Size) analysis was performed. Pseudomonas demonstrated the most significant association with age, exhibiting the lowest p-value across all groups (p = 3.6876 × 10−29), followed by Stenotrophomonas spp. (p = 6.8849 × 10−10), among others.
Core microbiome
To further explore the taxa variation among the different age groups, we performed a core microbiome analysis, revealing shared and consistent members of a microbial community, 20 different bacteria were determined as core microbiome. Core microbiome analysis was performed at the genus level using MicrobiomeAnalyst, defining core taxa as those present in at least 50% of samples per age group with a minimum relative abundance of 0.05%. Including Streptococcus, Stenotrophomonas spp., and Campylobacter. Reinforcing these results, the Simpson and Shannon indexes highlighted how diversity of the microbiome increased during the 4–9, 9–14, and 14–19-year age groups. Core microbiome analysis identified Leptotrichia which had a prevalence in the 29 to 39 age group which is right before the spike of Pseudomonas in LEfSE analysis. An observed trend was that the core microbiome decreased with age, meaning we saw fewer core microbiomes contributing to the lung environment with age. Streptococcus and Granulicatella decreased with age. Fusobacterium is a core microbiome for every age group except over 40. Stenotrophomonas spp. is identified by the core microbiome at 24 to 29 age group only. Actinomyces experiences an increase in core microbiome prevalence at the 29 to 39 age group but then decreases to no prevalence in the over 40 age group (Figure 4b).
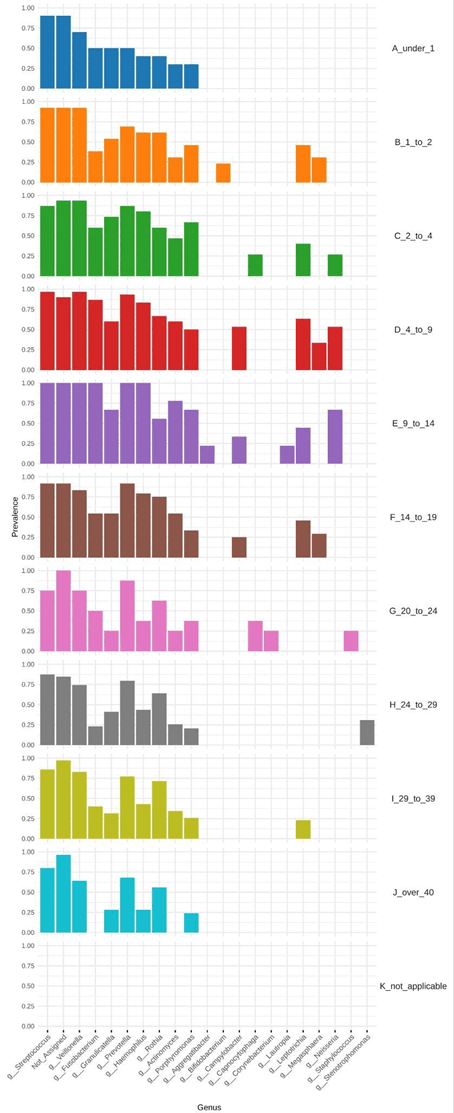
Figure 4. Core microbiome composition across age groups and LEfSe-identified genera showing significant age-associated enrichment, highlighting shifts from diverse commensal-dominated communities to pathogen-enriched profiles with increasing age.
Beta Diversity
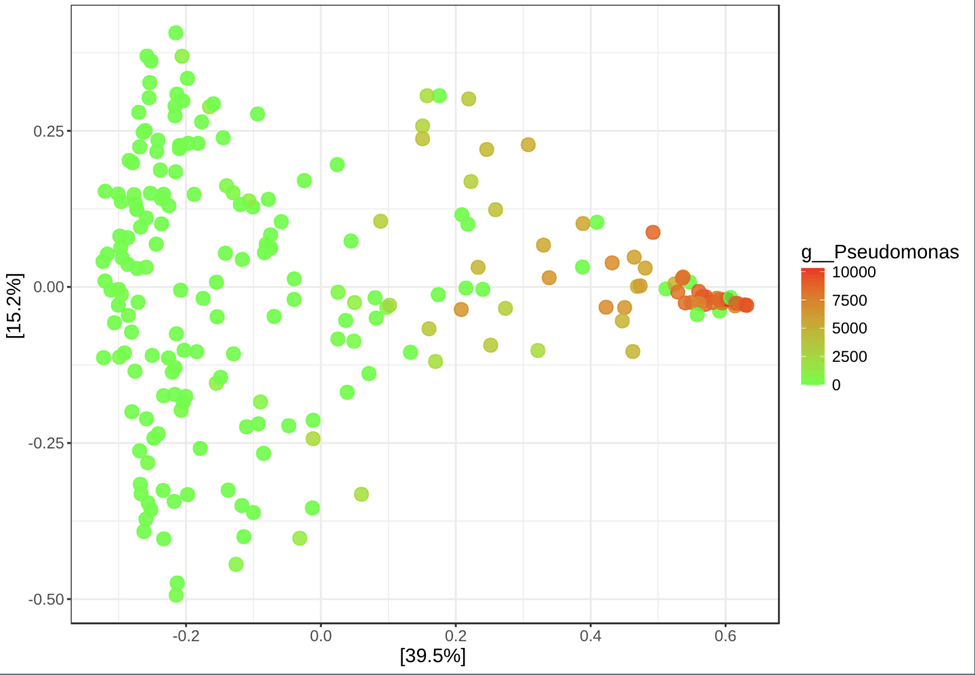
The Bray–Curtis dissimilarity index was used as a metric for quantifying differences in microbial community composition between cystic fibrosis samples based on relative abundances of shared taxa. Pairwise PERMANOVA based on Bray–Curtis dissimilarity revealed significant differences in beta diversity across multiple age-group comparisons after false discovery rate (FDR) correction. Notably, the 4–9 age group differed significantly from all other age categories, with comparatively higher effect sizes (R2 up to  0.20), indicating substantial shifts in microbial community composition during early childhood. Significant ˜ compositional differences were also observed between the youngest cohort (¡1 year) and older age groups, including adults over 40 years, suggesting progressive age-associated restructuring of the microbiome. Collectively, these results demonstrate that microbial community composition varies significantly across age groups, supporting age as an important driver of beta diversity. As shown in Figure 5, separation along PCoA Axis 2 reflects variation in Bray–Curtis community dissimilarity and may be influenced by differences in dominant taxa, such as Pseudomonas; however, taxon-specific analyses are required to confirm this association (Figure 5).
0.20), indicating substantial shifts in microbial community composition during early childhood. Significant ˜ compositional differences were also observed between the youngest cohort (¡1 year) and older age groups, including adults over 40 years, suggesting progressive age-associated restructuring of the microbiome. Collectively, these results demonstrate that microbial community composition varies significantly across age groups, supporting age as an important driver of beta diversity. As shown in Figure 5, separation along PCoA Axis 2 reflects variation in Bray–Curtis community dissimilarity and may be influenced by differences in dominant taxa, such as Pseudomonas; however, taxon-specific analyses are required to confirm this association (Figure 5).
Discussion
We analyzed the bacterial community structure of sputum samples from CF patients across a broad range of ages using both alpha diversity metrics and known CF-associated microbial patterns. Our study confirms previously established microbial patterns while offering additional insights into age-related changes and their potential biological reasonings13,14.
First, consistent with prior studies, we observed that the CF airway microbiome is highly prevalent, with few common taxa shared across patients: Streptococcus, Prevotella, Fusobacterium, Veillonella, Porphyromonas, Haemophilus, and Neisseria5. Diversity metrics, such as the Shannon and Simpson indices, demonstrated dynamic patterns of microbial variation with age. Specifically, the Shannon index increased from infancy to early adolescence, peaking in the 9–14-year age group, before progressively declining with age. This suggests that microbial richness and evenness increase during early childhood and decline as disease progresses. Our Pearson correlation coefficient of -0.33 (p ¡ 0.001) further supports this trend, indicating a possible statistically significant inverse relationship between age and alpha diversity. Simpson index values also revealed similar trends, with peaks in richness and evenness during key developmental windows, notably around puberty (ages 4–14 ), followed by a shift toward dominance by fewer genera in adulthood.
Second, we found a notable increase in the prevalence and relative abundance of Pseudomonas and other pathogenic taxa— Stenotrophomonas spp., Staphylococcus, and Campylobacter— with advancing age. These findings are in line with prior reports of microbial community shifts toward patho-adapted taxa in older CF patients. Several factors may contribute to this transition. The CFTR mutation results in thick mucus and acidic airway surface liquid, both of which hinder bacterial clearance and reduce community diversity. In later disease stages, the lower respiratory tract becomes increasingly micro-anaerobic due to mucus plugging and impaired gas exchange, creating an environment more favorable to Pseudomonas and Streptococcus spp.15.
Pseudomonas may play a particularly central role in reshaping the airway environment through its secretion systems, such as toxin production and Resistance-Nodulation-Cell Division (RND) efflux pumps, which promote survival under hostile conditions and suppress competing microbes. These systems also contribute to its multidrug resistance16. Specifically, P. aeruginosa can further enhance its fitness through siderophore production—such as pyoverdine and pyochelin—which allow it to scavenge iron, even from siderophores like mycobactin J produced by Mycobacterium smegmatis, potentially explaining the observed synergy between Mycobacterium presence and Pseudomonas colonization in older individuals17.
Staphylococcus aureus (S. aureus, species from Staphylococcus genus) has consistently colonized the respiratory tract and the skin of human and animals, yet P. aeruginosa mainly habituated soil and water, but recently there have been developments, especially in patients with CF, of microbial interactions between the two in the lung and respiratory tract18,19. The interactions are mostly antagonistic, however there are instances when they are cooperative, despite these differences their co-existence leads to chronic lung infections and resistance to antimicrobial therapy20. In early years of CF patients there is a possible inverse relationship between S. aureus and P. aeruginosa prevalence as they compete for resources. However, our results highlight there is a viable shift in their polymicrobial interactions with age as they begin to coexist leading to chronic long-term infections, emphasizing age’s feasible effect on infections due to polymicrobial behaviors.
Biofilms, communities of microbes associated with surfaces of interfaces. In CF patients due to the thick and dehydrated mucus in the lungs; the prevalence of biofilms increases due to the polymicrobial interactions and characteristics of P. aeruginosa21. Through depletion aggregation P. aeruginosa utilizes mucin, a non-absorbing polymer, creating mutual attraction forces between neighboring cells, resulting in an unbalanced osmotic pressure that pushes them together, forming aggregates leading to stronger resistance to antibiotics22,23. These aggregations create micro-environments inside the lungs of aerobic and anaerobic conditions that reduce the diversity of the lung microbiome significantly24. The ESKAPE group is the acronym for Enterococcus faecium, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter spp., which are highly virulent and antibiotic-resistant pathogens25. We observed that Pseudomonas abundance increases with age; previous studies have shown that Pseudomonas can form biofilms with ESKAPE pathogens, which may help explain its clinical impact26. In our data, Pseudomonas and Staphylococcus were consistently detected and increased in prevalence with age.
Interestingly our findings are consistent with studies showing Prevotella evident across all ages of CF patients could possibly inhibit Pseudomonas biofilm formation as it decreases depletion aggregation, illustrating a possible mutualistic relationship with Staphylococcus27. In this regard, recent studies have reported the ability of different bacteria to reduce the biofilm matrix of Pseudomonas, resulting in improved efficacy of currently available antibiotics. For example, some lactobacilli produce matrix-degrading enzymes28, which could favor the diffusion of antibiotics through the biofilm layers and their interaction with biofilm-embedded cells29. Similarly, enzymatic disruption of the extracellular matrix of Pseudomonas biofilms by the extracellular levanase of Bacillus subtilis and the alginate lyase of marine bacteria has been shown to boost the antibiofilm activity of several antibiotics against Pseudomonas30,31.
Hence, it is possible that microaerophilic Campylobacter is able to survive ambient atmospheric oxygen tension by metabolic commensalism with Pseudomonas. This bacterium-bacteria interaction might set the basis for survival of Campylobacter in the lung and thus be the prerequisite step in the pathway toward chronic infection and inflammation due to the production of cytokines32.
These microbial shifts have important clinical implications. Previous studies have reported positive associations between lung function and microbiome diversity; our diversity patterns by age are consistent with these findings, but we did not directly analyze lung function in this dataset, therefore the causal relationship between diversity and lung function remains unclear.
Therapeutically, while antibiotics remain a primary treatment option, their impact on long-term diversity is complex and not always beneficial, as they can reduce microbiome diversity. Emerging strategies such as genetic approaches targeting RND efflux pumps in Pseudomonas represent a novel frontier in managing resistant infections and mitigating the dominance of this pathogen in CF airways or utilizing the antagonistic microbial relationships with Pseudomonas to inhibit their prevalence.
Limitations
Our study has limitations. Sampling was cross-sectional and did not include paired samples during exacerbations or over time, limiting insights into internal patient dynamics. Furthermore, our 16S rRNA gene sequencing approach, while broad in taxonomic scope, lacks species-level resolution and may under-detect certain taxa such as S. aureus without targeted processing. 16S often cannot distinguish P. aeruginosa from other Pseudomonas species, the 10% prevalence filter may remove rare but functionally important taxa, and rarefying to 10,000 reads may discard information and introduce noise. Low-abundance organisms, although excluded from this analysis, may play significant roles in community dynamics and pathogenesis.
Another limitation is this is strictly a correlation study which proposes possible casual relationships between microbes and cystic fibrosis conditions. Additionally, age-associated microbiome patterns in CF can be strongly influenced by potential confounders to this study: CFTR modulator therapy (exp: Kalydeco, Orkambi, Trikafta), antibiotic exposure, hospitalization history, and lung function. Further emphasizing the constraints on a definite causal interpretation of the age trends without the presence of treatment and clinical covariates. In our statistical analysis we performed many pairwise tests without formal correction, which could result in a possible increase in false positives.
Conclusion
This paper highlights the importance in understanding the microbe-to-microbe interactions that stimulate Pseudomonas virulence and aid in creating a niche for P. aeruginosa to persist in the respiratory tract. Additionally cross-sectionally analyzing alterations of the pulmonary microenvironment highlight the possible trend to favor a microbial community dominated by P. aeruginosa seen through the decrease of diversity, providing a warrant for further investigations on microbial relationships to effectively treat CF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
The Author Contributions section is mandatory for all articles, including articles by sole authors. If an appropriate statement is not provided on submission, a standard one will be inserted during the production process. The Author Contributions statement must describe the contributions of individual authors referred to by their initials and, in doing so, all authors agree to be accountable for the content of the work. Please see here for full authorship criteria.
Acknowledgements
I would like to acknowledge Mytien Nguyen’s help in conducting and guidance through this research.
Data Availabilty Statement
The dataset analyzed for this study can be found in the Qiita platform.
References
- Ong, T., & Ramsey, B. W. (2023). Cystic fibrosis: a review. Jama, 329(21), 1859-1871. [↩]
- Chen, Q., Shen, Y., & Zheng, J. (2021). A review of cystic fibrosis: Basic and clinical aspects. Animal models and experimental medicine, 4(3), 220-232. [↩]
- Goetz, D., & Ren, C. L. (2019). Review of cystic fibrosis. Pediatric annals, 48(4), e154-e161. [↩]
- Naehrig, S., Chao, C. M., & Naehrlich, L. (2017). Cystic fibrosis: diagnosis and treatment. Deutsches Ärzteblatt International, 114(33-34), 564. [↩]
- Françoise, A., & Héry-Arnaud, G. (2020). The microbiome in cystic fibrosis pulmonary disease. Genes, 11(5), 536. [↩] [↩]
- Travert, G., Heeley, M., & Heeley, A. (2020). history of newborn screening for cystic fibrosis—The early years. International journal of neonatal screening, 6(1), 8. [↩]
- Sosnay, P. R., Raraigh, K. S., & Gibson, R. L. (2016). Molecular genetics of cystic fibrosis transmembrane conductance regulator: genotype and phenotype. Pediatric Clinics, 63(4), 585-598. [↩]
- Braga, R. M., Dourado, M. N., & Araújo, W. L. (2016). Microbial interactions: ecology in a molecular perspective. Brazilian journal of microbiology, 47, 86-98. [↩]
- Cox, M. J., Allgaier, M., Taylor, B., Baek, M. S., Huang, Y. J., Daly, R. A., et al. (2010). Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PloS one 5, e11044 [↩]
- Quinn, R. A., Adem, S., Mills, R. H., Comstock, W., DeRight Goldasich, L., Humphrey, G., … & Dorrestein, P. C. (2019). Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome, 7, 1-13. [↩]
- Nardelli, C., Scaglione, G. L., Testa, D., Setaro, M., Russo, F., Di Domenico, C., et al. (2023). Nasal microbiome in covid-19: a potential role of corynebacterium in anosmia. Current Microbiology 80, 53 [↩]
- Coburn, B., Wang, P. W., Diaz Caballero, J., Clark, S. T., Brahma, V., Donaldson, S., et al. (2015). Lung microbiota across age and disease stage in cystic fibrosis. Scientific reports 5, 10241 [↩]
- Cuthbertson, L., Walker, A. W., Oliver, A. E., Rogers, G. B., Rivett, D. W., Hampton, T. H., et al. (2020). Lung function and microbiota diversity in cystic fibrosis. Microbiome 8, 1–13 [↩]
- Zhao, J., Schloss, P. D., Kalikin, L. M., Carmody, L. A., Foster, B. K., Petrosino, J. F., et al. (2012). Decade-long bacterial community dynamics in cystic fibrosis airways. Proceedings of the National Academy of Sciences 109, 5809–5814 [↩]
- Huffnagle, G. B., & Dickson, R. P. (2015). The bacterial microbiota in inflammatory lung diseases. Clinical Immunology, 159(2), 177-182. [↩]
- Yang, L., Chen, L., Shen, L., Surette, M., & Duan, K. (2011). Inactivation of MuxABC-OpmB transporter system in Pseudomonas aeruginosa leads to increased ampicillin and carbenicillin resistance and decreased virulence. The Journal of Microbiology, 49, 107-114. [↩]
- Tang, X. X., Ostedgaard, L. S., Hoegger, M. J., Moninger, T. O., Karp, P. H., McMenimen, J. D., … & Welsh, M. J. (2016). Acidic pH increases airway surface liquid viscosity in cystic fibrosis. The Journal of clinical investigation, 126(3), 879-891. [↩]
- Short, F. L., Murdoch, S. L., & Ryan, R. P. (2014). Polybacterial human disease: the ills of social networking. Trends in microbiology, 22(9), 508-516. [↩]
- Hogan, D. A., & Kolter, R. (2002). Pseudomonas-Candida interactions: an ecological role for virulence factors. Science, 296(5576), 2229-2232. [↩]
- Trivedi, U., Parameswaran, S., Armstrong, A., Burgueno-Vega, D., Griswold, J., Dissanaike, S., & Rumbaugh, K. P. (2014). Prevalence of multiple antibiotic resistant infections in diabetic versus nondiabetic wounds. Journal of pathogens, 2014(1), 173053. [↩]
- Schwarz-Linek, J., Winkler, A., Wilson, L. G., Pham, N. T., Schilling, T., & Poon, W. C. (2010). Polymer-induced phase separation in Escherichia coli suspensions. Soft Matter, 6(18), 4540-4549. [↩]
- Secor, P. R., Michaels, L. A., Ratjen, A., Jennings, L. K., & Singh, P. K. (2018). Entropically driven aggregation of bacteria by host polymers promotes antibiotic tolerance in Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences, 115(42), 10780-10785. [↩]
- Arias, S. L., & Brito, I. L. (2021). Biophysical determinants of biofilm formation in the gut. Current opinion in biomedical engineering, 18, 100275. [↩]
- Worlitzsch, D., Tarran, R., Ulrich, M., Schwab, U., Cekici, A., Meyer, K. C., … & Döring, G. (2002). Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. The Journal of clinical investigation, 109(3), 317-325. [↩]
- Aloke, C., & Achilonu, I. (2023). Coping with the ESKAPE pathogens: Evolving strategies, challenges and future prospects. Microbial pathogenesis, 175, 105963. [↩]
- Ciofu, O., & Tolker-Nielsen, T. (2019). Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents—how P. aeruginosa can escape antibiotics. Frontiers in microbiology, 10, 913. [↩]
- Grassi, L., Asfahl, K. L., Van den Bossche, S., Maenhout, I., Sass, A., Vande Weygaerde, Y., … & Crabbé, A. (2024). Antibiofilm activity of Prevotella species from the cystic fibrosis lung microbiota against Pseudomonas aeruginosa. Biofilm, 7, 100206. [↩]
- Chappell, T. C., & Nair, N. U. (2020). Engineered lactobacilli display anti-biofilm and growth suppressing activities against Pseudomonas aeruginosa. npj Biofilms and Microbiomes, 6(1), 48. [↩]
- Batoni, G., Catelli, E., Kaya, E., Pompilio, A., Bianchi, M., Ghelardi, E., … & Maisetta, G. (2023). Antibacterial and antibiofilm effects of Lactobacilli strains against clinical isolates of Pseudomonas aeruginosa under conditions relevant to cystic fibrosis. Antibiotics, 12(7), 1158. [↩]
- Trizna, E., Bogachev, M. I., & Kayumov, A. (2019). Degrading of the Pseudomonas aeruginosa biofilm by extracellular levanase SacC from Bacillus subtilis. Bionanoscience, 9, 48-52. [↩]
- Daboor, S. M., Rohde, J. R., & Cheng, Z. (2021). Disruption of the extracellular polymeric network of Pseudomonas aeruginosa biofilms by alginate lyase enhances pathogen eradication by antibiotics. Journal of Cystic Fibrosis, 20(2), 264-270. [↩]
- Hilbert, F., Scherwitzel, M., Paulsen, P., & Szostak, M. P. (2010). Survival of Campylobacter jejuni under conditions of atmospheric oxygen tension with the support of Pseudomonas spp. Applied and environmental microbiology, 76(17), 5911-5917. [↩]



{kind=link}