
Abstract
Rapid identification of respiratory pathogens is crucial for selecting appropriate treatment, particularly in primary care clinics and settings with limited laboratory resources. In this work, we developed a portable nucleic acid testing device that integrates recombinase polymerase amplification (RPA), CRISPR Cas12a detection, and smartphone-based signal analysis into a single, self-contained system. To simplify sample preparation, an extraction-free nucleic acid releasing reagent was used, enabling sample lysis in approximately one minute. Under optimized experimental conditions, a 6-minute RT-RPA amplification step followed by a 6-minute CRISPR reaction yielded a total assay time of approximately 14 minutes, which is substantially faster than standard qPCR workflows. The device is designed to run five reactions in parallel, allowing simultaneous detection of influenza A virus, influenza B virus, Mycoplasma pneumoniae, an internal reference gene, and a no-template control. Fluorescence signals are captured using a smartphone and analyzed by a custom application that automatically determines positive or negative results, reducing subjectivity in interpretation. Powered by a portable power bank and requiring no fixed laboratory infrastructure, the system achieved a limit of detection of 1 copy/μL, with 100% specificity, 100% positive predictive value, and 100% negative predictive value. Overall, this work presents a rapid, low-cost point-of-care testing approach with potential utility for decentralized respiratory pathogen screening.
Keywords: Isothermal amplification, CRISPR/Cas12a, point-of-care testing, respiratory pathogens
Introduction
Respiratory infectious diseases remain a major threat to global public health security, causing more than 2 million deaths each year from lower respiratory tract infections1,2. They can be caused by a variety of pathogens, including bacteria (e.g., Mycoplasma pneumoniae) and viruses (e.g., influenza A virus (IAV) and influenza B virus (IBV)). Clinical symptoms of these diseases, such as fever, cough, sore throat, runny nose, etc, however, are nonspecific. Thus, detecting the causative pathogens is crucial for determining appropriate treatment strategies3. For example, IAV and IBV infections are usually treated with antiviral medicines such as oseltamivir4, whereas Mycoplasma pneumoniae infections are treated with macrolide antibiotics such as azithromycin5. Inappropriate medication use due to misdiagnosis could lead to antibiotic resistance, eventually delaying the recovery6,7.
Currently, the diagnosis of respiratory infections primarily relies on hospital-based laboratories, which offer high detection accuracy but also have a high cross-infection risk and are highly time-consuming. While antigen-based detection, another diagnosis approach, enables rapid self-diagnosis, its low sensitivity and specificity result in a high false-negative rate8,9. The benchmark for infectious disease detection is real-time fluorescent quantitative PCR (qPCR), a nucleic acid testing (NAT) method, due to its high sensitivity and specificity10,11. For example, multiplex PCR technologies, such as FilmArray (BioFire Diagnostics), can detect up to 21 pathogens simultaneously within 1 hour using a closed system12. The Qiagen QIAstat-Dx system enables sample-to-result analysis of up to 40 pathogen targets in one hour via cassette-based multiplex real-time PCR12,13. However, PCR-based platforms are unsuitable for resource-limited settings and point-of-care testing (POCT) due to their strict temperature requirements, need for trained operators, and high per-test costs.
By contrast, CRISPR-based isothermal amplification has emerged as a powerful tool for POCT due to both speed and high specificity. SHERLOCKv214,15, an exemplified detection system, employs the nucleic acid recognition and cleavage activities of four engineered Cas proteins (LwaCas13a, PsmCas13b, CcaCas13b, and AsCas12a) to enable simultaneous amplification of nucleic acids and multi-target detection in a single reaction. Hu et al.16 employed the cleavage properties of Cas12a and Cas13a for single-tube dual-target detection and achieved RPA reaction followed by CRISPR detection within 45 min, with a sensitivity of 1 copy/μL. In recent years, integrating microfluidic technology with CRISPR also has improved detection capacity. For example, Shen et al. developed an integrated microfluidic RPA-CRISPR/Cas12a chip17; the detection of five pathogen targets in two clinical samples could be completed in parallel within 60 min. Nonetheless, current CRISPR-based detection systems still face challenges such as high chip production costs, the need for large or specialized detection equipment, and complex results that can only be interpreted by trained personnel.
Significant progress has also been made in CRISPR-based portable systems. For instance, Li et al. (2025)18 developed a handheld CRISPR sensing platform for rapid on-site detection of Mycoplasma pneumoniae. However, it only supports single-target detection, with a sensitivity of 100 copies/μL, which is insufficient for low-concentration samples and lacks an internal reference control. Li et al. (2026)19 established SCOPEv2, an integrated, portable, low-cost CRISPR diagnostic platform for single-step dual-target detection of HPV16 and HPV18 for home-use testing. Their CPod2.0 device (52×52×52 mm) captures both FAM and ROX fluorescence signals at a cost of approximately $16.54, but does not achieve extraction-free detection. Tian et al.20 developed the all-in-one POP-CRISPR platform, whose portable device enables sample-to-answer detection within 20 minutes, yet still requires nucleic acid extraction and lacks multiplex detection capability.
To address the need for rapid, sensitive, and low-cost nucleic acid testing in resource-limited settings, we developed a portable integrated detection platform for decentralized respiratory pathogen screening. In contrast to bulky, expensive qPCR systems dependent on sophisticated laboratory infrastructure, our device combines isothermal RT-RPA amplification, CRISPR-Cas12a-mediated specific recognition, and smartphone-based fluorescence readout into a compact, user-friendly system. RT-RPA enables efficient amplification without thermal cycling, reducing hardware complexity, while Cas12a provides high-fidelity target identification to minimize non-specific signals. The smartphone module replaces costly fluorescence detectors, enabling objective, real-time result interpretation. Additionally, the platform integrates extraction-free nucleic acid release for one-step sample processing within 1 minute. The system supports simultaneous detection of IAV, IBV, and M. pneumoniae, along with an internal reference (GAPDH), and a no-template control (NTC). We validated the device’s sensitivity using clinical samples, and the results were comparable to those of standard qPCR.
Methods
Materials
The basic isothermal rapid amplification kit was purchased from Amp-future Biotechnology. Primers, CRISPR RNA (crRNA), ssDNA fluorescent reporter, and plasmids containing the IAV M gene, IBV M gene, and M. pneumoniae P1 gene were synthesized by Sangon Biotech. The corresponding sequences are listed in Table 1.
| Pathogen | Category | Oligo names | Sequences (5′-3′) |
|---|---|---|---|
| IAV | Primer- crRNA group 1 | Forward primer | GAGGTGTCACTAAGCTATTCAACTGGTGCACTT |
| Reverse primer | ATTCTGTTTCATGCCTGATTAGTGGATTGGTG | ||
| crRNA | GGGGUAAUUUCUACUAAGUGUAGAUGUCUAGUGUGUGCCACUUGUGAAC | ||
| Primer- crRNA group 2 | Forward primer | TGCTAATCAGACTAGGCAGATGGTACATGCAA | |
| Reverse primer | GGCCCTCTTTTCAAACCGTATTTAAAGCGACGA | ||
| crRNA | GGGGUAAUUUCUACUAAGUGUAGAUCAGGCCTACCAGAAGCGAAT | ||
| IBV | Primer- crRNA group 1 | Forward primer | ATGGTCATGTACCTAAACCCTGAAAACTATT |
| Reverse primer | CATTCCATTCATTGTCTTTGCTGTGTTCATA | ||
| crRNA | GGGGUAAUUUCUACUAAGUGUAGAUUCGUCUUACUCCGGUACCGAAGA | ||
| Primer- crRNA group 2 | Forward primer | CTATTCAATGCAAGTAAAACTAGGAACGCTCT | |
| Reverse primer | TGGATTCCTTATTTGTATTTTCAAGTTTACCCCTC | ||
| crRNA | GGGGUAAUUUCUACUAAGUGUAGAUAGCUAUGAACACAGCAAAGA | ||
| M. pneumoni- ae | Primer- crRNA group 1 | Forward primer | AAAGAAATCGGACTCGGAGGACAATGGTCAG |
| Reverse primer | CATAAGGCGCATCGTACAGAATCAGGATCGAG | ||
| crRNA | GGGGUAAUUUCUACUAAGUGUAGAUGCUACACCCGCCCUGACGAG | ||
| Primer- crRNA group 2 | Forward primer | i | |
| Reverse primer | GTACAGAATCAGGATCGAGGCGGATCATTTGG | ||
| crRNA | GGGGUAAUUUCUACUAAGUGUAGAUGUACUGGCCCUUUCCAGUUCG | ||
| ssDNA reporter | FAM-TTATTATT-BHQ1 | ||
RT-RPA-CRISPR/Cas12a Detection
There were three main steps in the experiment. First, the rehydration solution was prepared by mixing 29.5 μL of rehydration buffer, forward primer, and reverse primer, and ddH2O was added to a final volume of 39 μL. The lyophilized RT-RPA powder was dissolved in the rehydration solution, mixed thoroughly, and 9.25 μL of the mixture was transferred to the bottom of the reaction tube. Then, 5 μL of template DNA and 0.75 μL of magnesium acetate solution (280mM) were added. The final volume of the RT-RPA reaction was adjusted to 15 μL. Second, the CRISPR/Cas12a reaction solution was prepared by combining 100 nM LbCas12a (Bio-Lifesci), 200 nM crRNA, 1μM ssDNA fluorescent reporter, 1×Cas12a buffer, and 1 U/μL murine ribonuclease inhibitor (Takara). Third, the “tube-in-tube” consumable developed by our team21 was used. 15 μL of the RPA reaction mixture was added to the bottom of the outer tube, and 10 μL of the CRISPR/Cas12a reaction mixture was added to the inner tube. The reaction tube was incubated at 42 ℃ for 4-12 min and then centrifuged (3000rpm, 30s) to allow the CRISPR/Cas12a reaction solution to flow into the RT-RPA tube. Following the transfer, the mixture was incubated at 42 ℃.
Structure of the Nucleic Acid Detection Device
The developed nucleic acid detection equipment (130×90×101mm) consisted of three core functional units, including a heating unit, a centrifugation unit, and an optical detection unit. The system employs a PD2.0 protocol-compatible decoy (power delivery emulator) and is powered by a portable power bank. It incorporates an STM32F103 microcontroller that connects to a mobile application via Bluetooth 5.0. The heating module comprises a 20W PCB heating plate and a thermistor. The centrifugal module is driven by a high-performance brushless DC motor with an adjustable speed range of 0-3000 rpm. The optical detection unit consists of a solid-state laser source with an excitation wavelength of 450–490 nm and a detection channel covering 515–535 nm. Fluorescence signals are captured using a smartphone camera, and characteristic values are extracted via image processing algorithms. The mobile application enables full-process monitoring, automated result analysis, and data transmission.
Operational Procedures of the Nucleic Acid Detection Device
Firstly, the device was powered on, and Bluetooth on the smartphone was activated to establish a wireless connection via the companion mobile application. Once connected, the device automatically initiated the warm-up process. The desired reaction parameters, including RPA temperature and time, centrifugation speed and time, and CRISPR reaction time, were set via the mobile application. The detection process was then initiated following user confirmation.
qPCR Detection
In sensitivity experiments, the same serial dilutions of plasmid DNA used for the RPA-CRISPR assay were also subjected to qPCR. The primer sequences used for qPCR are listed in Table 2. For real-time qPCR analysis, each reaction mixture consisted of 10 μL of 2× PerfectStart™ Green One-Step qPCR SuperMix, 0.4 μL of TransScript® II One-Step RT/RI Enzyme Mix, 200 nM of each primer, 2 μL of RNA template, and nuclease-free water, for a total volume of 20 μL. Reactions were performed on a Gentier 96E qPCR instrument (from Xi’an Tianlong Technology) under the following cycling conditions: initial incubation at 50°C for 5 min; pre-denaturation at 94°C for 30 s; followed by 40 cycles of denaturation at 94°C for 5 s, annealing at 58°C for 15 s, and extension at 72°C for 10 s. For the detection of clinical samples, the 24 heavy fever respiratory syndrome pathogen nucleic acid detection kit (YP2008, Xi’an Tianlong Technology Co., China) was used.
| Target pathogen | Forward Primer | Reverse Primer |
|---|---|---|
| Influenza A virus | GCCCTAAATGGGAATGGGGAC | TCGTCAACATCCACAGCACTC |
| Influenza B virus | CCTAGACAGGATAGCTGCTGG | ATAACGTTTCTTTGTAATGGTGACA |
| Mycoplasma pneumoniae | TGCCCTTTCCAGTTCGG | CGCCTTTCAGTCCCACA |
Validation in Clinical Samples
A total of 15 clinical samples were obtained from Mengchao Hepatobiliary Hospital. Each liquid sample was mixed with a nucleic acid-releasing reagent and manually shaken for 1 min to ensure complete lysis. After that, 5 μL of the supernatant was withdrawn and used as input for the RPA-CRISPR/Cas12a detection.
For qPCR validation, nucleic acids were extracted using the animal virus DNA/RNA rapid extraction kit (version 5.0, Xi’an Tianlong Biotechnology Co., China). The extracted nucleic acids were then subjected to qPCR.
Results
Design of the Nucleic Acid Detection Device
To address the need for rapid screening and data interconnection in various respiratory infectious disease scenarios, a compact nucleic acid detection device (130×90×101 mm) was developed. The device integrated precise molecular detection and intelligent analysis capacities. It was powered by a portable power bank, which eliminates reliance on fixed power sources. When coupled with a lightweight, 3D-printed bracket, the device could maintain its structural stability while remaining suitable for portable deployment.
This study employed a smartphone as the control hub and detection component of the detection device. Once the reaction process was complete, the app issued location commands via the user interface (UI), driving the motor to move the PCR tubes to the detection position using the centrifuge disk, and then invoked the smartphone camera for image analysis. The app was developed in Kotlin and runs on Android.
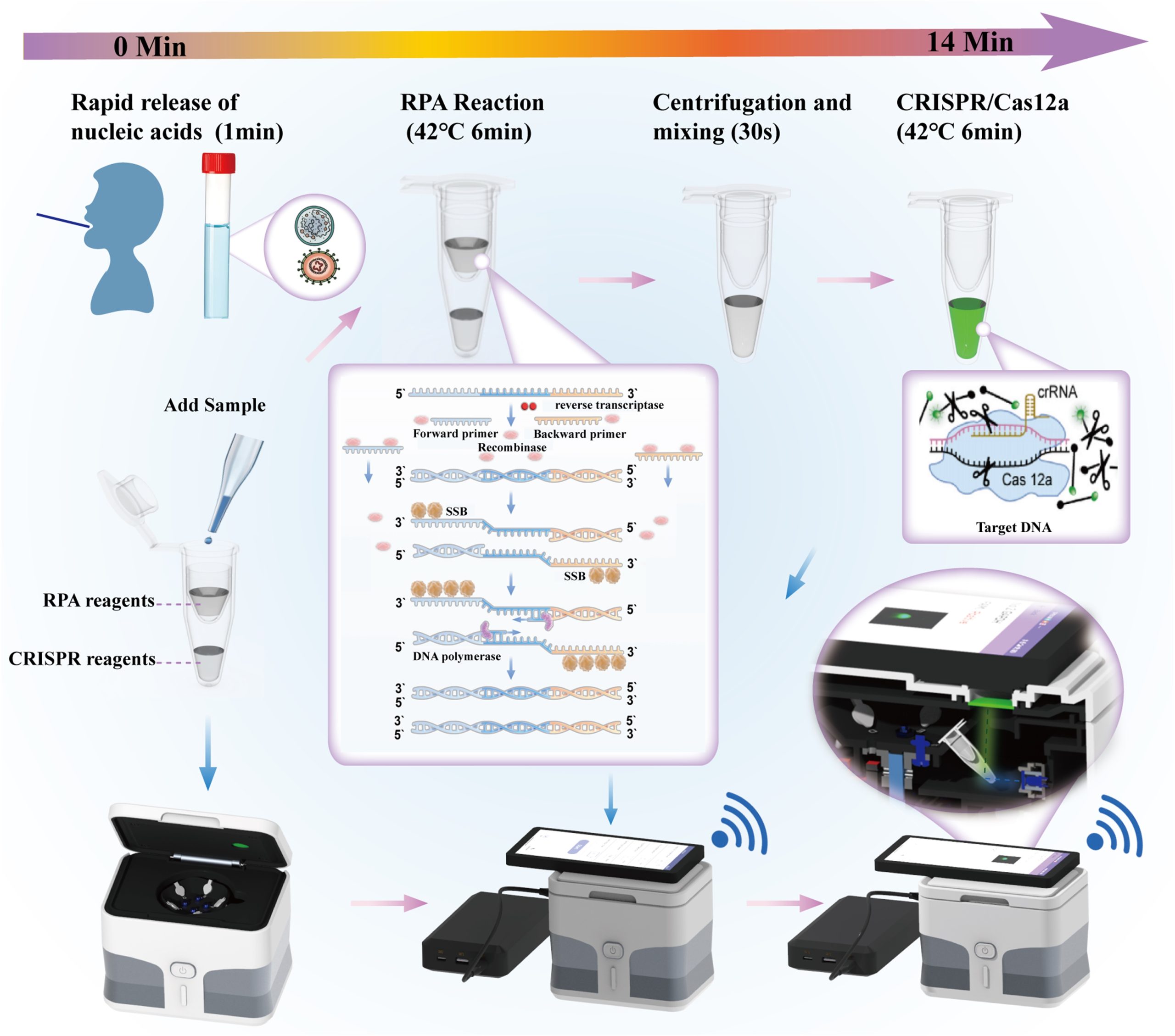
The detection workflow of the device is shown in Fig. 1. The respiratory swab sample was processed with a rapid nucleic acid releasing reagent and then used directly in the RPA reaction. A disposable sleeve was employed to physically separate the RPA reaction system from the CRISPR reaction system. The RPA reaction generated many copies of DNA, which were transferred into the CRISPR reaction solution via centrifugation to proceed with the subsequent reaction. Upon laser irradiation, the reaction mixture emitted a fluorescent signal. The device is equipped with the RPA-CRISPR/Cas12a detection system, enabling simultaneous detection of five nucleic acid targets: IAV M, IBV M, M. pneumoniae P1, the human GAPDH internal reference gene, and NTC. The use of the RPA-CRISPR/Cas12a detection system enhances both specificity and throughput. The accompanying smartphone application further simplifies the operation.
Experimental System Construction and Optimization
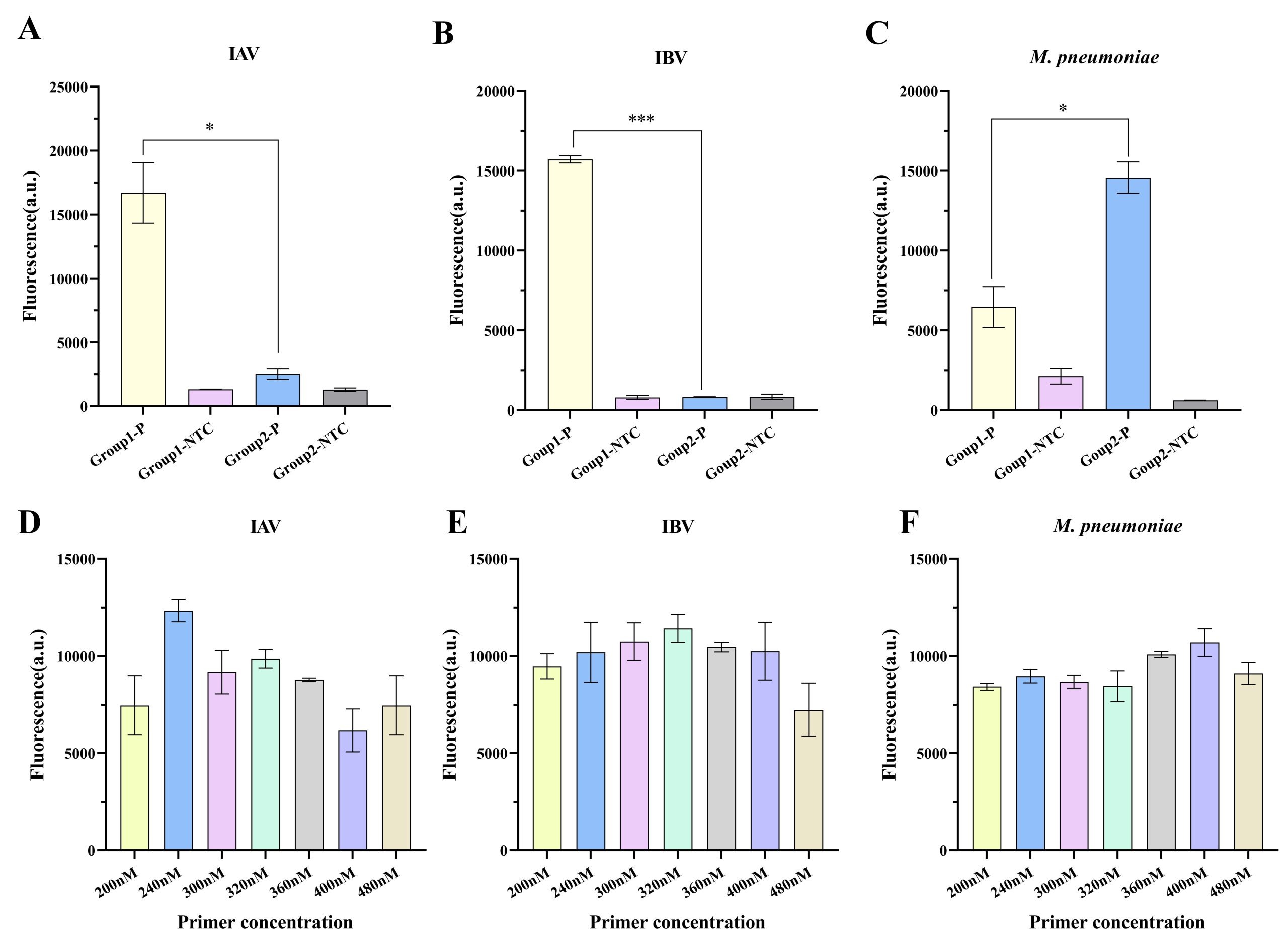
Two sets of specific forward and reverse primers, along with the corresponding crRNA combinations, were designed for each target pathogen: IAV, IBV, and M. pneumoniae. A plasmid at 5 copies/μL was used as the template for comparative evaluation of RPA-CRISPR/Cas12a detection performance.
As shown in Fig. 2A-C, IAV group 1, IBV group 1, and M. pneumoniae group 2 exhibited the strongest fluorescence intensities among all combinations and were therefore selected for subsequent experiments. Given the significant effect of primer concentration on RPA reaction efficiency, optimization experiments were conducted using the selected primer sets (Fig. 2D-F). The results showed that the optimal concentration for IAV group 1 was 240 nM, whereas that for IBV group 1 and M. pneumoniae group 2 was 320 nM and 400 nM, respectively, each of which yielded the highest fluorescence intensities.
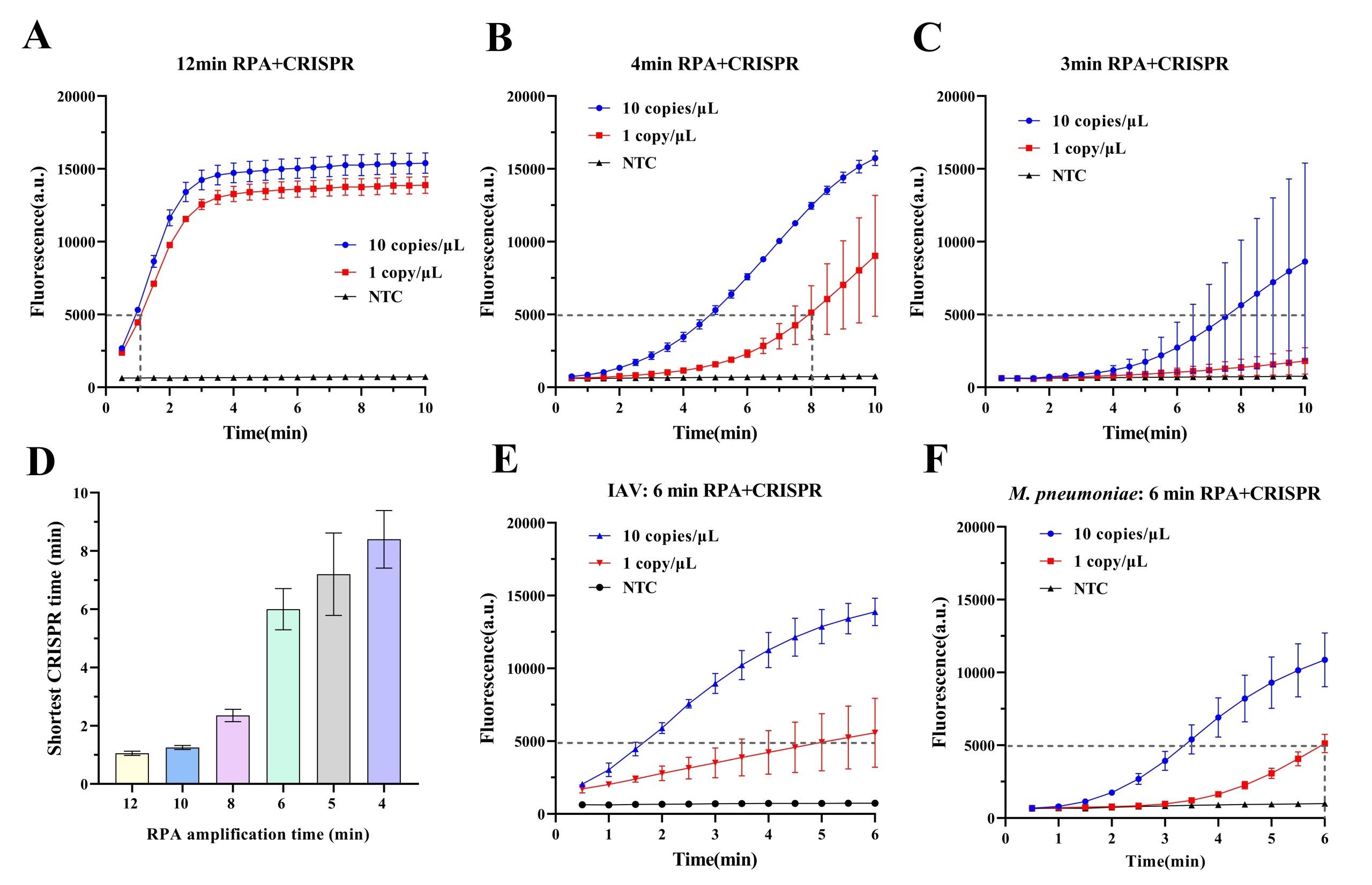
To further optimize the detection efficiency, the effect of RPA pre-amplification duration (1-12 min) on CRISPR detection of IBV was evaluated. Templates at 1 and 10 copies/μL were used, and CRISPR fluorescence intensities were recorded following pre-amplification durations ranging from 1 to 12 min. A fluorescence intensity below 5000 served as the reaction termination endpoint, as negative signals remained stably low at approximately 90016,22. The results showed that detection sensitivity for the 1 copy/μL template was highly time-dependent (Fig. 3A–D): pre-amplification ≥10 min enabled CRISPR detection within ~1 min; reducing pre-amplification to 8, 6, and 5 min prolonged CRISPR reaction time to 2.5, 6, and 7 min, respectively; 4 min pre-amplification required 8 min of CRISPR incubation; 3 min pre-amplification failed to detect the 1 copy/μL template. Comprehensive analysis identified 4 min as the minimum pre-amplification duration for reliable 1 copy/μL template detection. Balancing sensitivity and speed, the optimal protocol was defined as 6 min RPA pre-amplification plus 6 min CRISPR detection. Application of this protocol to IAV and M. pneumoniae detection successfully identified both 1 and 10 copies/μL templates (Fig. 3E–F), confirming the validity of the selected parameters.
Simplified Sample Pretreatment Using Nucleic Acid Extraction-Free Method
Nucleic acid extraction-free detection experiments were carried out using IAV virus RNA quality control (QC) material (containing inactivated virus particles at approximately 6.8 × 103 copies/μL).
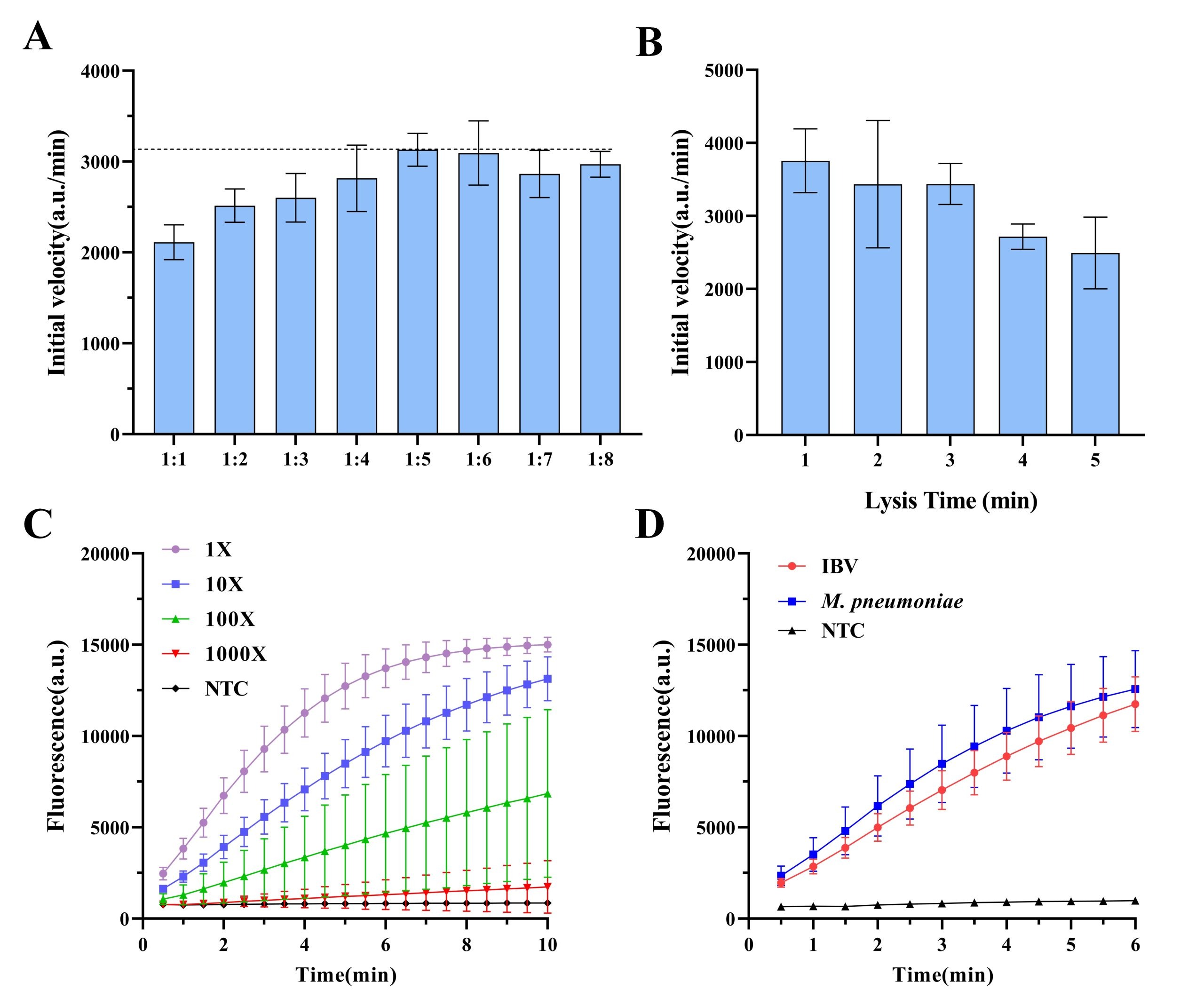
To evaluate the efficiency of different ratios of nucleic acid release, the nucleic acid releasing reagent was mixed with 10-fold dilutions of quality controls at various volumes, and the lysis effects were compared (Fig. 4A). The results showed that a 1:5 volume ratio of nucleic acid releasing reagent to sample yielded the highest fluorescence signal. According to the literature, amplicon concentration within a certain range is positively correlated with the initial CRISPR reaction rate23, justifying the 1:5 ratio for subsequent experiments.
To further assess the effect of lysis duration, the effect of release times (1-5 min) on nucleic acid release was examined (Fig. 4B). The result showed that maximal nucleic acid availability was achieved after only 1 min of lysis.
The performance of the nucleic acid releasing reagent was verified using plasma at varying concentrations (10X, 100X, and 1000X; Fig. 4C). The results showed that the plasma at a 100X dilution was still detectable. By contrast, a 1000X diluted sample did not yield detectable signals, likely due to insufficient viral particles. Finally, the nucleic acid releasing reagent was applied to the IBV and M. pneumoniae QC materials (Fig. 4D). The CRISPR results showed that the reagent was compatible with both IBV and M. pneumoniae, confirming its applicability to a wide range of pathogens.
Sensitivity and Specificity
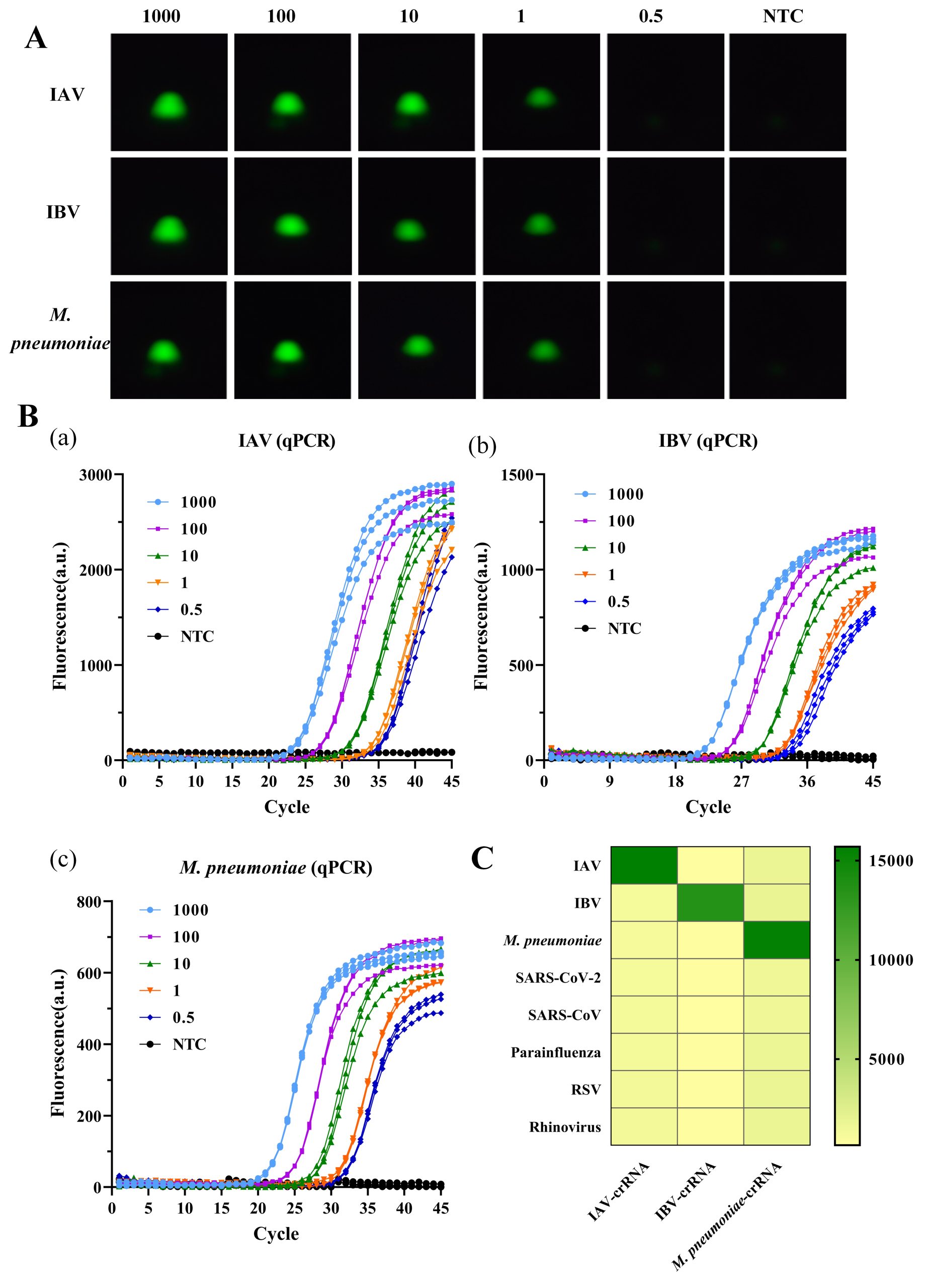
To assess the sensitivity of the detection method, gradient-diluted plasmids (1000 to 0.5 copies/μL) were used. As shown in Fig. 5A-B, in the detection of IAV, IBV, and M. pneumoniae, the device had a limit of detection (LOD) of 1 copy/μL. Specifically, distinct fluorescence signals were consistently observed at plasmid concentrations ≥1 copy/μL, but were not observed at a plasmid concentration of 0.5 copies/μL. Compared to qPCR (LOD = 0.5 copies/μL), the absolute sensitivity of this method was slightly lower. However, the detection limit of 1 copy/μL fully satisfies the minimum requirements for single-copy detection in clinical diagnosis and allows for the accurate identification at the early stages of infection. Notably, the sensitivity of this method exceeded that of several similar previously reported thermostable amplification-CRISPR techniques22,24,25,26,27.
Given the overlapping clinical symptoms of respiratory pathogens, high detection specificity is essential to avoid misdiagnosis. To evaluate specificity, the CRISPR-Cas12a detection system was tested against both target and untargeted respiratory pathogens. As shown in Fig. 5C, the system differentiated the target pathogens (IAV, IBV, and M. pneumoniae) from interfering pathogens (SARS-CoV-2, SARS-CoV, parainfluenza, RSV, and rhinovirus). Significant fluorescence signal enhancement was observed only in the presence of target nucleic acids, with no detectable cross-reactivity for non-target species, confirming a specificity of as high as 100%.
Validation Experiments using Clinical Samples
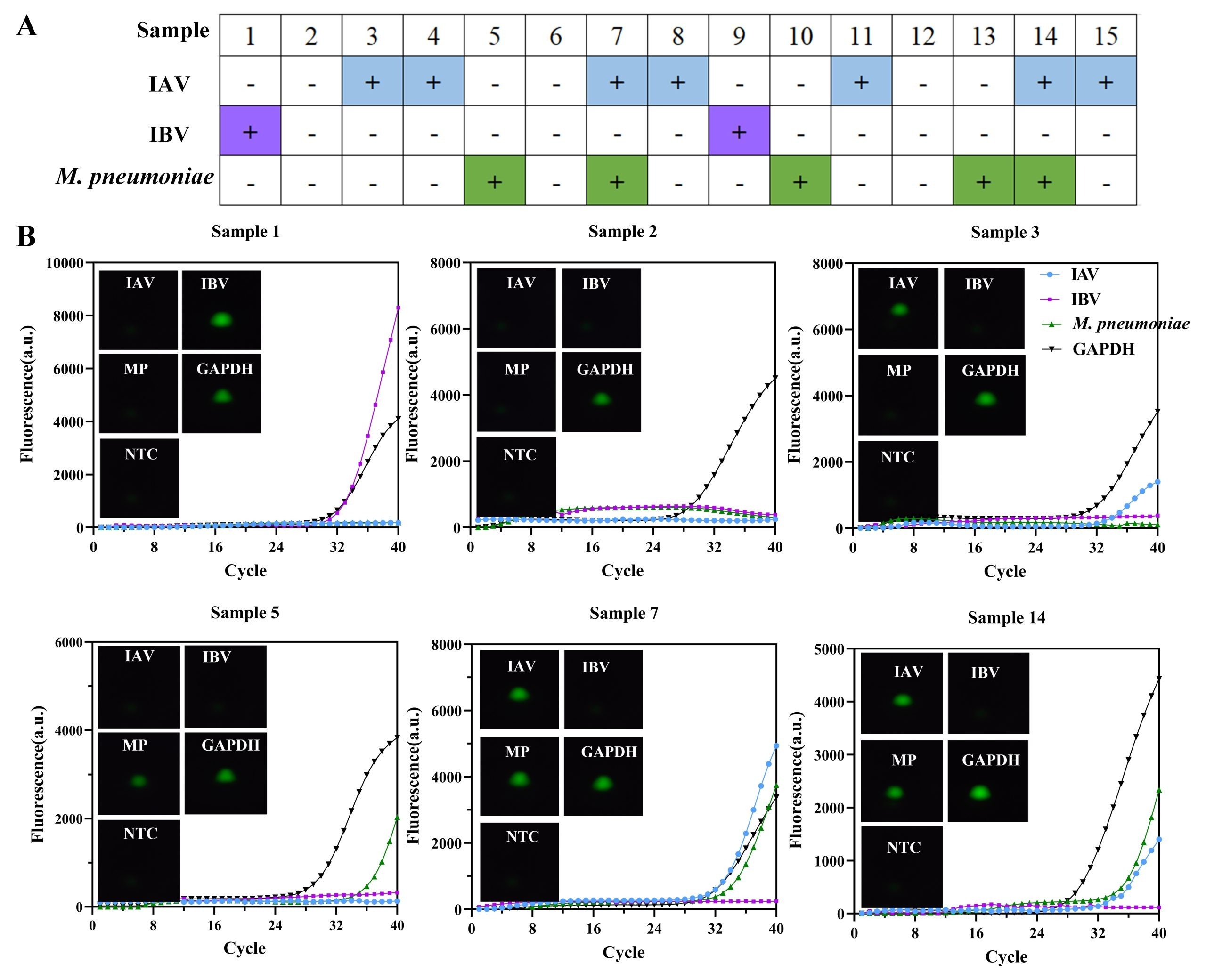
To assess the clinical feasibility of the nucleic acid extraction-free method in combination with the rapid testing device, validation experiments were conducted using fifteen clinical samples obtained from Mengchao Hepatobiliary Hospital. Each sample underwent routine diagnostic testing by standard qPCR in a biosafety laboratory. Fig. 6A showed the results of the diagnosis using the device. Representative images are shown in Fig. 6B. All results were consistent with qPCR results, including samples 3, 4, and 5, which had lower viral loads. These results demonstrate that the device enables detection of viral targets in clinical samples.
Discussion
With the capability to detect pathogen-specific nucleic acid sequences, nucleic acid testing (NAT) plays an essential role in identifying infectious agents and guiding targeted clinical treatment. This is particularly important for respiratory infectious diseases, where overlapping clinical symptoms and diverse etiologies often complicate diagnosis. However, conventional PCR-based diagnostic platforms remain constrained by long turnaround times, reliance on centralized laboratory infrastructure, and the need for trained personnel, limiting their deployment in decentralized and resource-limited settings. To address these challenges, the device developed in this study enables rapid and accurate detection of IAV, IBV, Mycoplasma pneumoniae, and other respiratory tract pathogens within a compact and portable system.
The device integrates isothermal RPA amplification, CRISPR-based detection, and wireless data transmission into a single automated platform. The stand and shell were fabricated using 3D printing, ensuring structural stability while significantly reducing manufacturing costs (total: $52). The per-test reagent cost is approximately $1. The device is lightweight (0.7 kg), compact, and operable through a smartphone application, eliminating the need for professional laboratory equipment. Powered by a portable power bank, the system is fully self-contained and suitable for on-site and field-based testing scenarios (Fig. 1).
To optimize detection performance, the system was used to screen primer–crRNA combinations for IAV, IBV, and M. pneumoniae, allowing identification of the most effective target designs (Fig. 2). The relationship between RPA pre-amplification time and CRISPR detection efficiency was further investigated. When template concentration was low (1 copy/μL), extending the RPA pre-amplification time from 1 to 12 minutes markedly shortened the subsequent CRISPR detection time (Fig. 3). At an RPA pre-amplification time of 6 minutes, CRISPR reactions could be completed within approximately 6 minutes, balancing speed and sensitivity.
To further simplify the workflow, a nucleic acid extraction-free sample preparation method requiring only mixing and shaking was adopted28. Optimization experiments identified a 1:5 volume ratio of releasing reagent to sample as optimal, corresponding to the highest onset rate of the CRISPR reaction (Fig. 4). A lysis time of 1 minute was sufficient to fully release nucleic acids, and dilution gradient analysis showed that detection capacity was maintained up to a 100-fold dilution. Sensitivity testing using plasmid samples demonstrated accurate detection across concentrations ranging from 1 to 1000 copies/μL, with results closely matching those obtained by qPCR (Fig. 5). Specificity experiments confirmed the absence of cross-reactivity with non-target pathogens. In blinded testing of 15 pharyngeal swab samples, results generated by the device were consistent with qPCR, demonstrating the feasibility of combining extraction-free nucleic acid preparation with a rapid molecular testing device (Fig. 6).
Compared with established commercial point-of-care testing platforms (e.g., QIAstat-Dx, Cepheid Xpert® Xpress), the proposed system exhibits distinct advantages in workflow simplification and operational simplicity by obviating the need for complex nucleic acid extraction procedures. Nevertheless, several key limitations of the present study warrant consideration. The single-plex design of the developed device constrains both analytical throughput and multiplex detection capability, restricting its utility in high-throughput screening and clinical differential diagnosis. Meanwhile, the current clinical validation is based on a limited sample cohort, which undermines the statistical confidence and generalizability of the reported diagnostic performance. In addition, the stability of reagents under field-relevant conditions, including fluctuating temperatures and long-term storage, has not been systematically assessed. All these limitations will be addressed in future studies.
Conclusion
By streamlining key steps in nucleic acid testing, this platform addresses several limitations of standard qPCR workflows. A rapid nucleic acid–releasing step of approximately 1 minute, followed by 6 minutes of RPA pre-amplification and 6 minutes of CRISPR detection, reduces the total assay time to 14 minutes. The system supports parallel sample processing and smartphone-based result readout, removing dependence on bulky instrumentation and fixed laboratory infrastructure. Although these results confirm the feasibility of the prototype for rapid detection of respiratory pathogens, this study has several limitations, including a relatively small number of validated clinical samples and single-target detection per reaction. Future work will focus on expanding clinical cohorts, improving automation, and enhancing multiplex detection capability in a single reaction to further translate this platform into practical point-of-care diagnostic applications. Overall, this work demonstrates that the integration of isothermal amplification, CRISPR-based detection, and portable device engineering enables rapid, accurate, and decentralized molecular diagnosis of respiratory pathogens, laying a foundation for future clinical and field-based applications.
Acknowledgments
I sincerely thank Dr. Hu for his guidance in topic selection, research design, experimentation, and the writing of this paper. I am also grateful to all members of the Institute of Bio-Detection and Instrumentation Laboratory at Xi’an Jiaotong University for trusting me and providing access to the laboratory. I would like to give special thanks to Xingcheng Wang for his help with the image analysis algorithm and embedded system software; Qin Ma for contributing to the schematic diagram in Figure 1 and patiently answering my questions about structural design; and Yunyun Zhang for supervising me during laboratory validation experiments. I am especially thankful to Cece and Mr. Denison, my advisor and genetics teacher, who trained me through ADIs and encouraged me to pursue research.
References
- A. C. Moy; Kimmoun, A. Performance evaluation of a PCR panel (FilmArray(R) Pneumonia Plus) for detection of respiratory bacterial pathogens in respiratory specimens: A systematic review and meta-analysis. Anaesth Crit Care Pain Med. Vol. 42, pg.101300, 2023. [↩]
- M. Lanaspa; Bassat, Q.; Medeiros, M. M.; Munoz-Almagro, C. Respiratory microbiota and lower respiratory tract disease. Expert Rev Anti Infect Ther. Vol. 15, pg.703-711, 2017. [↩]
- J. H. Kim; Kwon, J. H. Clinical features of Mycoplasma pneumoniae coinfection and need for its testing in influenza pneumonia patients. J Thorac Dis. Vol. 10, pg.6118-6127, 2018. [↩]
- M. Javanian; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.; Ebrahimpour, S. A brief review of influenza virus infection. J Med Virol. Vol. 93, pg.4638-4646, 2021. [↩]
- L. Gao; Sun, Y. Laboratory diagnosis and treatment of Mycoplasma pneumoniae infection in children: a review. Ann Med. Vol. 56, pg.2386636, 2024. [↩]
- J. Chen; Wang, Y. Synergistic impact of macrolide resistance and H3N2 infection on M. pneumoniae outbreak in children. Microbiol Spectr. Vol. 13, pg.e0184424, 2025. [↩]
- S. Baumgart; Gray, D.; Holland, J.; Rockett, R.; Sintchenko, V.; Kok, J. Mycoplasma pneumoniae: evolving diagnostic methods for a known pathogen. Pathology. Vol. 57, pg.415-424, 2025. [↩]
- H. Han; Xu, Z. Retrospective Study of the Clinical Characteristics of Asymptomatic COVID-19 Patients. mSphere. Vol. 5, 2020. [↩]
- G. J. Kost. The Impact of Repeating COVID-19 Rapid Antigen Tests on Prevalence Boundary Performance and Missed Diagnoses. Diagnostics (Basel). Vol. 13, 2023. [↩]
- P. Rai; Kumar, B. K. Detection technologies and recent developments in the diagnosis of COVID-19 infection. Appl Microbiol Biotechnol. Vol. 105, pg.441-455, 2021. [↩]
- S. A. Baylis; Nubling, C. M. Nucleic Acid Testing of Viral Pathogens: Traceability of the International Unit. Clin Chem. Vol. 68, pg.257-258, 2021. [↩]
- M. A. Poritz; Blaschke, A. J. FilmArray, an automated nested multiplex PCR system for multi-pathogen detection: development and application to respiratory tract infection. PLoS One. Vol. 6, pg.e26047, 2011. [↩] [↩]
- S. A. Boers; van Houdt, R. A multicenter evaluation of the QIAstat-Dx meningitis-encephalitis syndromic test kit as compared to the conventional diagnostic microbiology workflow. Eur J Clin Microbiol Infect Dis. Vol. 43, pg.511-516, 2024. [↩]
- J. S. Gootenberg; Abudayyeh, O. O. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. Vol. 356, pg.438-442, 2017. [↩]
- J. S. Gootenberg; Abudayyeh, O. O. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. Vol. 360, pg.439-444, 2018. [↩]
- F. Hu; Zhang, Y. A rapid and ultrasensitive RPA-assisted CRISPR-Cas12a/Cas13a nucleic acid diagnostic platform with a smartphone-based portable device. Biosens Bioelectron. Vol. 280, pg.117428, 2025. [↩] [↩]
- J. Shen; Chen, Z.; Xie, R.; Li, J.; Liu, C.; He, Y.; Ma, X.; Yang, H.; Xie, Z. CRISPR/Cas12a-Assisted isothermal amplification for rapid and specific diagnosis of respiratory virus on an microfluidic platform. Biosens Bioelectron. Vol. 237, pg.115523, 2023. [↩]
- J. J. Li; Huang, H. Q. Palm-sized CRISPR sensing platform for on-site detection. Biosensors & Bioelectronics. Vol. 281, 2025. [↩]
- L. Li; Wang, Y. Miniaturized Single-Step Duplex CRISPR Diagnostic Platform for At-Home Molecular Testing of HPV16 and HPV18. ACS Sens. Vol. 11, pg.1117-1128, 2026. [↩]
- T. Tian; Zhang, T. Identification of thermotolerant non-canonical PAMs for robust one-pot CRISPR-Cas12a detection. Nature Communications. Vol. 17, 2026. [↩]
- F. Hu; Liu, Y. A one-pot CRISPR/Cas13a-based contamination-free biosensor for low-cost and rapid nucleic acid diagnostics. Biosensors and Bioelectronics. Vol. 202, pg.113994, 2022 . [↩]
- J. Fu; Mo, R.; Li, Z.; Xu, S.; Cheng, X.; Lu, B.; Shi, S. An extraction-free one-pot assay for rapid detection of Klebsiella pneumoniae by combining RPA and CRISPR/Cas12a. Biosens Bioelectron. Vol. 267, pg.116740, 2025. [↩] [↩]
- Y. Shan; Zhou, X. High-Fidelity and Rapid Quantification of miRNA Combining crRNA Programmability and CRISPR/Cas13a trans-Cleavage Activity. Anal Chem. Vol. 91, pg.5278-5285, 2019. [↩]
- D. Xu; Zeng, H.; Wu, W.; Liu, H.; Wang, J. Isothermal Amplification and CRISPR/Cas12a-System-Based Assay for Rapid, Sensitive and Visual Detection of Staphylococcus aureus. Foods. Vol. 12, 2023 [↩]
- K. Zhang; Sun, Z. RPA-CRISPR/Cas12a-Based Detection of Haemophilus parasuis. Animals (Basel). Vol. 13, 2023 [↩]
- S. Huang; Liu, Y. A Rapid RPA-CRISPR/Cas12a Detection Method for Adulteration of Goat Milk Powder. Foods. Vol. 12, 2023 [↩]
- K. Zhang; Li, Q. RPA-CRISPR-Cas-Mediated Dual Lateral Flow Assay for the Point-of-Care Testing of HPV16 and HPV18. Bioconjug Chem. Vol. 35, pg.1797-1804, 2024 [↩]
- D. J. Delgado-Diaz; Sakthivel, D. Strategies That Facilitate Extraction-Free SARS-CoV-2 Nucleic Acid Amplification Tests. Viruses. Vol. 14, 2022 [↩]


