
Abstract
Diabetes mellitus (DM) is a debilitating disease that affects hundreds of millions of patients worldwide. Diabetic neuropathy (DN) and its most common form, diabetic peripheral neuropathy (DPN), are complications that involve nerve dysfunction resulting from DM progression. Metformin, a popular medication used in the treatment of type 2 diabetes mellitus (T2DM), appears to have protective effects on DPN-associated nerve dysfunction, as evidenced by metformin treatment in animal models of DPN. However, the pathways and mechanisms of action through which metformin appears to exert such effects are unclear. This hypothesis-generating review aims to analyze metformin’s mechanisms to ameliorate nerve dysfunction and identify potential pathways utilized by metformin in these mechanisms of action. Metformin appears to ameliorate sciatic nerve dysfunction, induces spinal dorsal horn changes to reduce pain hypersensitivity, and improves peripheral nerve function in preclinical models of DPN. Activation of the AMPK signaling pathway may be a key mechanism behind metformin’s action against DPN, as AMPK activation improves neuronal inflammation, oxidative stress, apoptosis, and pain hypersensitivity. Metformin’s activation of the hedgehog (Hh) signaling pathway and inhibition of the Toll-like Receptor 4 (TLR4) signaling pathway seem to have beneficial effects on apoptosis and inflammation in various tissue types, and these mechanisms appear to improve nerve function and reduce neuronal inflammation, respectively, in animal models of DPN. However, it is unclear if these mechanisms are utilized by metformin in its effect on DPN and require further research to examine this prospect. An understanding of the mechanisms of metformin’s action against DPN can facilitate the examination of the neuroprotection of other similar medications and the development of new drugs that utilize metformin’s neuroprotective mechanisms.
Keywords: Diabetic Neuropathy, Diabetic Peripheral Neuropathy, Metformin, Adenosine Monophosphate-activated Protein Kinase, Nerve Dysfunction
Introduction
Diabetes Mellitus (DM) is a widespread, debilitating group of metabolic diseases that affects approximately 590 million patients worldwide, and this number is projected to reach 853 million by 20501‘2. Diabetic neuropathy (DN) is one of the most common complications of DM and involves nerve damage caused by chronically high blood sugar levels that result from DM. The pathogenesis of DN is multifaceted, though it is roughly understood that hyperglycemia, dyslipidemia, and insulin resistance activate metabolic pathways that lead to neuronal injury3. Over time, at least 50% of diabetes patients develop the most common form of diabetic neuropathy, diabetic peripheral neuropathy (DPN)4. DPN is characterized primarily by distal symmetric polyneuropathy, which involves sensory loss, pain, and motor dysfunction in the extremities, predominantly the hands, lower leg, and feet5. Potential clinical presentations of DPN include painful diabetic neuropathy, asymptomatic diabetic neuropathy, and painless diabetic neuropathy characterized by a loss of sensory nerve function6. While extensive research has shown that medications such as the anticonvulsant pregabalin relieve pain-related symptoms in painful DPN, effective and efficacious treatments to slow or reverse DPN’s progression remain elusive7‘8‘9‘5.
Metformin is one of the most commonly used diabetes medications that increase cellular insulin sensitivity, proving very effective in the treatment of T2DM. Metformin is derived from biguanide, a drug class of herbal origin10. Metformin’s mechanisms of signaling through the Adenosine Monophosphate Protein Kinase (AMPK) pathway and the translocation of glucose-transporter 4 (GLUT4) are key in the reduction of insulin resistance3. In addition to its traditional role as an insulin-sensitizing medication for T2DM, a wealth of studies have found that metformin unexpectedly has protective effects against various cancers, cardiovascular diseases, renal diseases, neurodegenerative disorders, liver diseases, and obesity11. Metformin has also been shown to reduce the severity of diabetes-associated conditions such as diabetic nephropathy and diabetic retinopathy utilizing a variety of mechanisms5‘12‘13‘14‘15. Considering the ameliorative effects of metformin on other diabetes-associated conditions and the significant morbidity caused by DPN, it is necessary that the effects of metformin on DPN severity are evaluated.
Metformin’s therapeutic potential for the treatment of DN has been previously assessed, highlighting the necessity for further research to confirm this prospect as preclinical and clinical studies have yielded conflicting results16. Generally, in rat models of diabetes, metformin appears to have neuroprotective effects on DPN-associated nerve dysfunction. However, the underlying mechanisms through which metformin exerts this effect are unclear. As such, rather than examining the efficacy of metformin as a treatment for DPN, this review aims to discuss the potential mechanisms of action by which metformin exerts protective effects on nerve dysfunction as well as examine metformin’s amelioration of DPN pathophysiology in preclinical and clinical models.
Methods
The purpose of this hypothesis-generating review is to analyze metformin’s effect nerve dysfunction in animal models and identify potential pathways utilized by metformin in these mechanisms. To do so, this paper synthesizes evidence from biomedical literature that discuss the pathogenesis of DPN, metformin’s benefits for DPN pathophysiology, and the potential mechanisms behind metformin’s neuroprotective effects. To search for relevant literature, I used Google Scholar along with PubMed, a database website for biomedical literature. Around 70 studies were selected for use in this review for their relevance to DPN, metformin’s effect on DPN severity, and the mechanisms of metformin that were involved in this effect. Most of these studies presented results from animal models of DPN. Included studies consisted of randomized controlled trials and observational studies and recent publication dates. The majority of the studies used in this review were published primarily in the past 20 years, although a small number of older studies were included that discussed DPN pathogenesis and pathophysiology, foundational concepts that are necessary to understand how metformin appears to exert protective effects on DPN severity. Other review articles were utilized in this review to provide context for certain foundational concepts such as metformin’s activation of Adenosine Monophosphate-activated Protein Kinase (AMPK) and the role of altered voltage-gated ion channel function in DPN pathophysiology. A very small number of studies that did not discuss DPN were utilized to introduce the potential mechanisms of metformin’s action that were discussed in this review. Excluded studies were studies that did not discuss metformin or DPN, studies published more than 25 years ago, and studies from non-peer reviewed sources. In total, nearly 90 articles were screened that included research designs, preclinical or clinical findings, and the effects on DPN severity and pathophysiology as a result of treatment.
To maximize the relevance of the studies discussed in this review, I searched for literature using keywords such as “metformin diabetic peripheral neuropathy”, “AMPK diabetic peripheral neuropathy”, “oxidative stress diabetic peripheral neuropathy”, and “metformin hedgehog pathway”. The key findings of each research article discussed in this paper were extracted through a detailed read-through of the paper’s “results” section. The most important and relevant findings were summarized, and the implications of those findings were discussed in detail. The “discussion” section of each research article was also used to understand the rationale behind the study and the author’s explanation of their results. The key findings of the primary research papers summarized in this review were organized based on common themes such as “metformin’s improvement of sciatic nerve damage in DPN” and “the effect of AMPK activation on TRPA1 expression”. These thematic groups were organized into subtopics such as “Metformin’s Amelioration of Morphological and Physiological Nerve Damage Resulting From DPN” and “Activation of the AMPK Pathway as a Mechanism of Metformin’s Effect on DPN” based on the overarching topic discussed by those groups. Overall, this review article was thematically organized to elucidate the importance and significance of each key finding.
The Pathogenesis and Pathophysiology of DPN
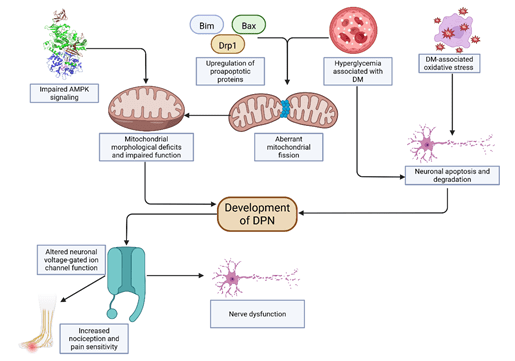
DN is a debilitating disease with very few available treatments and poorly understood pathogenetic mechanisms. It is roughly understood that hyperglycemia, dyslipidemia, and insulin resistance activate metabolic pathways that lead to neuronal injury in the pathogenesis of DN5. DPN is the most common form of DN and has adverse effects on the peripheral nervous system. Several studies have implicated increased mitochondrial biogenesis, oxidative stress, and neuronal apoptosis in DN and DPN pathogenesis. Acute alterations of sodium, potassium, and calcium channels, which play an important role in the generation of action potentials, may have a prominent role in DPN pathophysiology. DPN’s pathogenesis and pathophysiology, and an understanding of the mechanisms of DPN’s action are crucial for the examination of metformin’s effect on DPN severity [Fig. 1].
Mitochondrial Dysfunction
The alteration of neuronal energy metabolism is key in the development and progression of DN. Nerve mitochondrial dysfunction through increased biogenesis plays a crucial role in the disruption of neuronal energy metabolism and has been observed in animal models of DPN. Mitochondrial biogenesis is the process of mitochondrial DNA (mtDNA) replication followed by mitochondrial division, commonly occurring to replace damaged mitochondria. Mitochondrial division, also known as fission, occurs independently of biogenesis to acutely manage elevated metabolic demand17‘18‘19‘20. Edwards and Vincent et al. found that hyperglycemia leads to increased mitochondrial biogenesis and fission in DRG neurons harvested from DN mice, resulting in morphologically atypical mitochondria with the potential to cause nerve injury. The levels of mtDNA in the DRG neurons were doubled in the 24-week old mice diabetic group compared to their age-matched counterparts in the healthy control group, and increased mtDNA synthesis was noted in diabetic mice17‘21. The expression of biogenesis-associated genes at the level of transcription and translation were generally increased in the diabetic group compared to the control group. Protein levels were also elevated in the DRGs of diabetic mice, indicating increased mitochondrial biogenesis17. 24-week-old mice in the diabetic group had more mitochondria per axon in the unmyelinated and myelinated fibers of the dorsal root than their age-matched counterparts in the healthy control group, indicating an increase in mitochondrial fission21. Mitochondria in the dorsal root neurons of the diabetic mice were observed to be fragmented, asymmetrical, potentially smaller, with lower membrane density, and with altered cristae structure in comparison to those of the healthy control group17‘21. Hyperglycemia likely triggers the observed compensatory mitochondrial biogenesis and fission. With prolonged glucose exposure, fission predominates, producing fragmented mitochondria that injure DRG neurons. However, as a result of the continued exposure to elevated levels of glucose, the mitochondria of the DRG neurons undergo the more rapid process of mitochondrial fission without mtDNA replication as opposed to mitochondrial biogenesis. Prolonged mitochondrial fission results in mitochondria with morphological deficits, leading to mitochondrial dysfunction, a significant loss in energy generation, neuronal injury, and the potential development of DPN17‘21.
Morphologically atypical mitochondria and aberrant mitochondrial fission resulting from DM-related hyperglycemia can underlie DPN pathogenesis and pathophysiology. Mitochondrial dysfunction due to hyperglycemic conditions and neuropathy was found to play a role in altered mitochondrial volume and distribution. The levels of mitochondrial signaling associated with larger volume were increased in DRG neurons treated with elevated glucose levels when compared to control neurons. Similar results were noted in the intraepidermal nerve fibers sampled from the distal thighs and distal legs of DM and DPN patients. These observations are theorized to be associated with mitochondrial fission resulting in morphologically atypical mitochondria that are either swollen or dysfunctional and clumped together6. The findings of Hamid et al. are consistent with the implications of aberrant mitochondrial fission resulting in mitochondrial dysfunction noted in other previously discussed studies17‘21.
An increase in apoptotic stimuli can induce aberrant neuronal mitochondrial fission in DPN pathogenesis. Mitochondrial localization and activation of pro-apoptotic proteins along with the upregulation of the pro-fission protein dynamin-related protein 1 (Drp1) were significant in an in vitro model of hyperglycemic rat DRG neurons. The pro-apoptotic protein Bim was shown to localize with the mitochondria of DRG neurons after high glucose treatment, an effect that grew more pronounced over time. High glucose treatment also increased the number of activated pro-apoptotic Bax proteins, with observed levels being significantly higher than in DRG neurons treated with control media. High glucose treatment induced large clumps of mitochondria in neurites and reduced the number of mitochondria per neuron, indicating mitochondrial degradation and apoptotic mitochondrial fission as a result of high glucose treatment22. This is supported by the increase in Drp1 expression, localization to mitochondrial clumps, and binding to Bax after an extended period of high glucose exposure22‘21. The findings of Leinninger et al. suggest that the localization of pro-apoptotic proteins to neuronal mitochondria leads to mitochondrial levels and distribution indicative of apoptotic fission. This possibility is further supported by the elevated numbers and mitochondrial localization of Drp1. It is noteworthy that the lowered levels of mitochondria in the DRG neurons recorded by Leinninger et al. contradict the findings of increased mitochondrial levels by Vincent and Edwards et al.. However, the instance of mitochondrial fission described in the findings of Vincent et al. likely occurred to accommodate elevated glucose levels while the mitochondrial fission noted by Leinninger et al. was a result of the co-localization of Bax and Drp1, which is an apoptotic event known to trigger fission. The mitochondrial degradation resulting from this fission led to the lower number of mitochondria present in the DRG neurons, resulting in a significant loss in energy generation and the potential development of DN.
The impairment of the Adenosine Monophosphate-activated Protein Kinase (AMPK) signaling pathway may also be a key factor in mitochondrial dysfunction underlying DPN pathogenesis. A key cellular energy receptor, AMPK is present in various organs and tissues and is implicated in the treatment of metabolic diseases such as T2DM. The AMPK signaling pathway and phosphorylated-AMPK (p-AMPK) are implicated in the regulation of redox reaction signaling and ATP levels, resulting in less energy usage and more energy production23‘24‘25. The downregulation of the AMPK and Peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) signaling pathway has been shown to result from diabetes and may be associated with mitochondrial deficits and DPN development. The levels of phosphorylated AMPK (p-AMPK) and PGC-1α were significantly reduced in the DRG neurons of diabetic rodents. The activity of electron transport chain-associated mitochondrial complexes 1 and 4 along with the citric synthase were significantly decreased after an extended period of diabetes affliction, indicating less mitochondrial energy production. Oxygen consumption rate, respiratory control ratio, and spare respiratory capacity were notably decreased in the DRG neurons of diabetic rats, indicating impaired energy production and decreased mitochondrial efficiency in diabetic conditions26. The findings of Chowdhury et al. indicate the lessened capacity of energy production in mitochondria in the diabetic state, which has been shown to result in DRG neuronal injury and the potential development of DN22‘21. The observed reduction in p-AMPK levels may have downstream effects on PGC-1α and cellular mitochondrial dynamics26. The downregulation of AMPK activation may be a result of a lower AMP/ATP ratio occurring in hyperglycemic conditions due to the excess presence of glucose27‘28. Thus, the impairment of AMPK appears to play a significant role in mitochondrial dysfunction and DN pathogenesis.
Oxidative Stress, Hyperglycemia, and Resulting Nerve Deterioration
Further studies have implicated neuronal apoptosis and nerve degradation resulting from hyperglycemia and oxidative stress in the onset and persistence of DN. Schmeichel, Russell, and Vincent et al. have observed increased neuronal apoptosis related to oxidative stress and hyperglycemic injury in animal and cell culture models of DM29‘30‘31. Nerve electrophysiological deterioration was characterized by reduced motor and sensory nerve conduction velocities in the sciatic-tibial and caudal nerves of rats affected by DN29. Apoptosis-associated indicators of mitochondrial dysfunction were observed in diabetic DRG neurons, corroborating the findings of diabetes-associated DRG mitochondrial function discussed previously. High glucose concentrations significantly reduced DRG neurite growth in comparison to control DRG neurons30. The presence of the oxidative stress biomarker 8-Hydroxy-2’-deoxyguanosine increased in diabetic DRG neurons and continued to elevate throughout diabetes progression29. Additionally, reduced cellular enzyme activity and lipid peroxidation were evident in DRG neurons exposed to hyperglycemic conditions, indicating oxidative stress and potential cell death as a result of hyperglycemia31‘32. The percentage of TUNEL-positive DRG neurons and level of caspase-3 activation in DRG neurons were both increased in hyperglycemic and diabetic conditions, indicating neuronal apoptosis29‘30‘31. A significant association was found between 8-Hydroxy-2’-deoxyguanosine, caspase-3, and TUNEL positivity, indicating the correlation between oxidative stress and neuronal injury29.
Changes in Voltage Gated Ion Channel Expression and Function
Voltage-gated ion channels are crucial for the generation of action potentials as well as the regulation of neuronal excitability and resting membrane potential. Increased voltage-gated sodium channel current and expression as well as lowered potassium channel current density elevate neuronal excitability33‘34. In nociceptive neurons such as the A and C fibers of the DRG neurons, increases in neuronal excitability can result in increased pain hypersensitivity and neuropathic pain33. Low voltage-activated calcium channels may also play a role in pain signal amplification35. A number of studies have shown that changes in neuronal voltage-gated sodium, potassium, and calcium channel expression and function may be a key factor in early DPN pathophysiology, particularly in hypersensitivity to mechanical stress. In a study of the DRG neurons of diabetic rats, the mRNA and protein levels of the sodium channel isoforms Nav1.2, Nav1.9, and Nav1.3 were markedly elevated when compared to the control DRGs. The Nav1.9 and Nav1.3 sodium channel isoforms are associated with increased nociception, so the observed elevation in the expression of these channels may indicate increased pain signaling in DRG neurons33‘36. The number of nodes of Ranvier in diabetic sciatic nerves that expressed sodium channels were significantly reduced along with the signal intensity of sodium channels in the nodes of Ranvier when compared to control neurons, an effect that may slow nerve conduction velocity and impair nerve electrophysiology33. The levels of Nav1.8 protein expression were significantly reduced in large DRG neurons of diabetic rats after several weeks of recorded pain hypersensitivity, though the mRNA and protein expression levels of Nav1.9 channels were significantly increased in diabetic DRG neurons36. The total sodium channel current amplitude, ramp current amplitude, and maximum current density were significantly increased in large diabetic DRG neurons, indicating the role of increased Na+ currents in DN-associated pain hypersensitivity35‘37‘38. Similar elevations in current density were noted in the low-voltage gated calcium channel currents of the DRG sensory neurons of diabetic rats35.
In contrast to the observations in channel expression and function in sodium and calcium channels, voltage-gated potassium channels saw decreases in these values. Potassium channel mRNA levels were significantly reduced in diabetic rat DRG neurons in comparison to control neurons. Potassium channel current density was also notably decreased in diabetic DRG neurons34. The observed increase in sodium and calcium current, the decrease in potassium channel current, and increased sodium channel expression may implicate the role of these voltage-gated channels in the development of DPN-associated neuropathic pain.
Metformin’s Amelioration of Morphological and Physiological Nerve Damage Resulting From DPN
Metformin has been shown to improve various factors of DPN pathogenesis and pathophysiology, thus attenuating DPN severity. Specifically, metformin has been shown to ameliorate sciatic nerve axonal atrophy, inflammation, oxidative stress, and myelin sheath damage. Metformin has also been observed to reduce synapse numbers in the spinal dorsal horn, which is associated with pain hypersensitivity in DPN. Increased axonal excitability, nerve fiber thickness, and the expression of microRNA-146a have been observed after metformin treatment in animals with DPN. In human T2DM patients, metformin treatment reduced DPN severity and improved axonal excitability. In general, metformin may reduce DPN severity by restoring nerve function and attenuating symptoms of DPN.
Improvement of Sciatic Nerve Damage
The sciatic nerve, a major sensorimotor nerve located in the lower back and leg, is a crucial site of DPN-associated metabolic dysregulation40‘41. The sciatic nerves of streptozotocin-induced T1DM mice exhibited lower numbers of myelin fibers and increased axonal atrophy compared to the sciatic nerves of healthy control mice42. Similar findings were observed in male diabetic rats, with significant damage to sciatic nerve myelin sheath structure along with excessive swelling and rupturing of the mitochondria of sciatic nerve Schwann cells. The level of oxidative stress present in the sciatic nerve was also significantly heightened in the diabetic control group when compared to the healthy control group43. In these animal models of DPN, metformin has been found to reduce axonal atrophy, myelin sheath degeneration, neuronal inflammation, and oxidative stress in the sciatic nerves of animals suffering from DPN. Treatment of streptozotocin-induced T1DM mice with both 100 and 200 mg/kg/d of metformin prevented axonal atrophy and potentially reversed myelin fiber degeneration caused by DPN42. Similar effects were noted in doses of 30 mg/kg/d, 200 mg/kg/d, and 500 mg/kg/d, and this effect appears to be dose dependent44‘45. Axonal atrophy and myelin fiber degeneration were prevented through treatment with metformin, and this effect was shown to be dose-dependent42‘44‘45. Diabetic rats treated with 100mg/kg/d of metformin also presented increased sciatic nerve axon diameter and myelin sheath thickness in comparison to untreated diabetic rats, but these values were higher in healthy control rats46. In another study, 400 mg/kg/d of metformin also increased the presence of neurotrophic factors such as NGF and myelin basic protein in Schwann cells and axons, indicating increased axonal maintenance and myelin sheath repair. Decreased expression of the pro-inflammatory cytokine IL-1β and increased expression of the anti-inflammatory cytokine IL-10 were confirmed in this study. Metformin was also shown to increase the expression of microRNA-146a in the sciatic nerve, attenuating inflammation by reducing the expression of inflammatory factors such as TNF-α43. Thus, metformin appears to have significant protective effects on DPN-associated sciatic nerve dysfunction in animal models of DPN, although metformin treatment does not seem to fully ameliorate DPN-induced nerve deficits.
Morphological and Physiological Improvements in the Spinal Cord
Metformin may have the capacity to attenuate DPN-associated alterations in certain regions of the spinal cord. Specifically, metformin has the capacity to lower the number of synapses in the spinal dorsal horn of diabetic rats, an effect associated with lower pain hypersensitivity. A study conducted in streptozotocin-induced diabetic rats noted a higher paw withdrawal threshold (PWT) in rats treated with 200 mg/kg/d of metformin than in untreated diabetic rats, indicating lower pain sensitivity in the metformin-treated rats. However, the PWT in metformin-treated rats was lower than that of healthy control rats, indicating that metformin may not completely attenuate DPN-associated pain hypersensitivity. Both of the observed differences were statistically significant, with a p value of less than 0.05. Additionally, the metformin-treated diabetic rats had a similar number of synapses to the healthy control rats and a significantly lower synaptic number than that of the untreated diabetic rats47. These findings indicate that metformin’s attenuation of synaptic number in the L5 segment of the spinal cord may reduce pain hypersensitivity in animal models of DPN. Sensitization of the sciatic nerve, which is characterized by a decreased PWT, may be a driving mechanism of the correlation between DPN and an increased number of synapses in the L5 segment of the spinal dorsal horn. The PWT is primarily controlled by afferent fibers of the sciatic nerve. The sciatic nerve may transfer pain signals to the L5 segment of the spinal dorsal horn, triggering spinal cord sensitization and hypersensitivity to mechanical stress as a result47‘48. The sciatic nerve’s mechanism of transferring nociceptive signals to the L5 segment of the spinal dorsal horn is supported by the observed increase in the sensitivity of the sciatic nerve to pain in diabetic rats49. Considering the association of the number of synapses in the spinal dorsal horn and DPN severity, metformin’s attenuative effects on the increased synaptic number in the spinal dorsal horn may be an indicator of metformin’s positive effects on DPN-associated nerve damage.
Improved Peripheral Nerve Function in Human Patients
Metformin has been shown to improve aspects of nerve physiology in human studies as well. A cross-sectional study involving 69 T2DM patients conducted by Dhanapalaratnam et al. found that patients taking metformin with an average daily dose of 1,523 mg for 162 months had less severe peripheral neuropathy and higher levels of axonal excitability when compared to patients who did not take metformin. T2DM patients taking metformin had lower modified Toronto Clinical Neuropathy Scale (mTCNS) and total neuropathy scores (TNS) compared to the control group, suggesting metformin treatment lowers DPN symptom severity. Metformin-treated patients also exhibited greater peripheral neuron axonal sensitivity, supporting improved axonal function in the context of DPN50. The statistically significant differences in mTCNS and TNS values between patients who took and did not take metformin coupled with the lowering of key parameters of axonal excitability imply that metformin has the potential to better the impairment of nerve physiology resulting from DPN. The axonal excitability parameters of mean rheobase and stimulation necessary for 50% of maximal response both reflect the functionality of sodium and potassium channels. These channels directly affect axonal depolarization and the potency of action potentials traveling through the axons by extension. Decreased levels of sodium channel expression in sciatic nerve nodes of Ranvier have been theorized to play a role in slowed nerve conduction velocity and impaired nerve physiology33. The lowered levels of these two parameters in patients taking metformin imply greater sodium and potassium channel functionality, leading to greater axonal excitability and improved nerve physiology. However, it is important to note that other key factors of axonal excitability were unchanged between patients who took and did not take metformin50. In conclusion, while metformin shows potential for improving factors of nerve physiology such as axonal excitability, further research is necessary to confirm this prospect.
Activation of the AMPK Pathway as a Mechanism of Metformin’s Effect on DPN
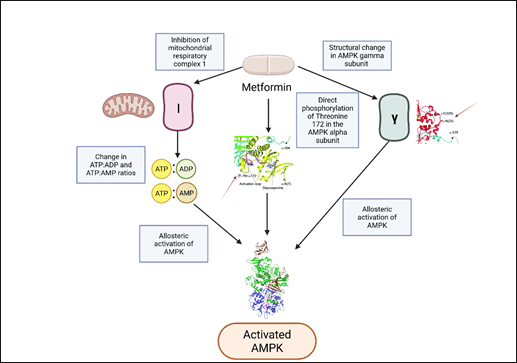
The AMPK pathway is one of the most crucial and well-studied pathways of metformin in the treatment of insulin resistance and T2DM. Metformin has been observed to activate the AMPK signaling pathway directly through AMPK phosphorylation and allosterically through the inhibition of mitochondrial complex 1 and the subsequent change in cellular energy status [Fig. 2]3‘51. In the context of treating insulin resistance, metformin’s activation of the AMPK pathway stimulates the release of glucagon-like peptide 1 (GLP1), enhancing the secretion of insulin and ultimately lowering plasma glucose levels3. Activation of the AMPK pathway has previously been shown to attenuate insulin resistance, reduce abnormal extracellular collagen deposits in white adipose tissue, and improve mitochondrial respiration in obese mice52‘53. Concerning DPN, activation of the AMPK signaling pathway has been shown to attenuate DPN-associated pain hypersensitivity, oxidative stress, apoptosis in DRG neurons, and neuronal inflammation. Metformin’s activation of AMPK improved DPN-associated neuropathic pain and neuronal inflammation, indicating that activation of the AMPK pathway is likely a key mechanism of metformin’s amelioration of DPN.
AMPK Activation and TRPA1
The AMPK signaling pathway has been found to ameliorate DPN-associated pain hypersensitivity through the negative regulation of transient receptor potential ankyrin 1 (TRPA1)56‘25. TRPA1 is a receptor-potential ion channel first discovered in the dorsal root ganglion and trigeminal ganglion neurons and, as a pain sensor, has been implicated in hypersensitivity to mechanical stress in diabetic rats when stimulated57‘25. In an in vitro study of AMPK and TRPA1 expression levels, RSC96 Schwann cells cultured in hyperglycemic conditions that were treated with 1 mM of the AMPK activator 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) presented significantly reduced TRPA1 expression in comparison to hyperglycemic cells that did not receive treatment. However, this study did not measure TRPA1 expression in cells that were not incubated in hyperglycemic conditions, so it is unclear whether AICAR treatment restores TRPA1 expression to the level found in healthy cells56. Nevertheless, these findings imply that activation of the AMPK signaling pathway has the potential to reduce pain hypersensitivity caused by TRPA1 activation. Another study found that the activation of the AMPK signaling pathway led to the downregulation of TRPA1 and its downstream signaling activity by suppressing allyl isothiocyanate (AITC)-activated inward currents and cells expressing TRPA1 receptors in rat DRG neurons cultured in hyperglycemic conditions. In rat DRG neurons from high-glucose cultures, AMPK activation through the application of 500 μmol/L of metformin and AICAR was discovered to reduce the number of membrane-associated TRPA1 in DRG neurons when compared to neurons that did not receive treatment. Metformin and AICAR both acutely reduced the levels of membrane-associated TRPA1 in DRG neurons, an effect further confirmed through immunofluorescent staining techniques. In diabetic mice, a weekly injection of 250 mg/kg of metformin and 250 mg/kg of AICAR also improved pain hypersensitivity25. The observed effect of AMPK activators on TRPA1 expression provides mechanistic evidence for the potential of AMPK activation to reduce the neuronal presence of TRPA1, thereby attenuating DPN-associated pain hypersensitivity.
Amelioration of Nerve Defects Resulting From DPN
Activation of the AMPK signaling pathway may ameliorate DPN-associated mitochondrial dysfunction, neuroinflammation, neuronal apoptosis, pain hypersensitivity, and impaired nerve function. The treatment of diabetic rats with 15 mg/kg or 30 mg/kg of the AMPK activator A769662 reduced pain hypersensitivity as well as improved tail flick latencies and PWT when compared to diabetic rats that did not receive metformin treatment. This effect was more statistically significant in the diabetic rats treated with 30 mg/kg of A769662. Improved motor and sensory nerve conduction velocities along with reduced expression of the pro-inflammatory factors TNF-α and IL-6 were observed after AMPK activation through A769662 treatment. However, while this effect was statistically significant in the 30 mg/kg-treated rats, it was not in the 15 mg/kg-treated rats. The number of apoptotic cells observed in sciatic nerve microsections was significantly reduced after A769662 treatment, as were unusually elevated levels of mitochondrial membrane depolarization observed in Neuro2a cells exposed to high levels of glucose. These effects were also statistically significant in both 15 mg/kg and 30 mg/kg-treated rats58. The observed attenuative effects of A769662 treatment on pain sensitivity, nerve inflammation, mitochondrial dysfunction, and nerve impairment provide mechanistic evidence for the potential of AMPK activation in ameliorating DPN-associated nerve deficits.
AMPK activation may also have protective effects against DPN-associated neuronal oxidative stress and apoptosis. In DPN rats, treatment with 60 mg/kg of Alpha Lipoic Acid (ALA) was found to increase motor nerve conduction velocity when compared to untreated rats, and this effect was statistically significant. However, the observed increase in sensory nerve conduction velocity was not statistically significant. Immunohistochemistry of the DRG neurons from these ALA-treated DPN rats found statistically significant increases in the p-AMPK to AMPK ratio and the expression of nuclear factor erythroid 2-related factor 2 (NRF2), a transcriptional factor that has well-studied antioxidant effects23’58. This provides correlational evidence for the potential of AMPK activation to protect against neuronal oxidative stress through the upregulation of NRF2. Additionally, the number of TUNEL-positive DRG neurons and the levels of neuronal apoptosis-associated proteins were significantly decreased by AMPK activation, indicating attenuated neuronal apoptosis23.
Metformin’s AMPK Activation and Resulting DPN Attenuation
In several instances, metformin’s activation of the AMPK signaling pathway has improved symptoms of DPN. An example of this is metformin’s attenuation of DPN through the downregulation of excessive autophagy in the sciatic nerve of diabetic rats via the AMPK signaling pathway. Notably, the expression of AMPK between the diabetic metformin and diabetic control groups was generally similar, but the expression of p-AMPK was higher in the diabetic rats treated with 400 mg/kg of metformin than the diabetic control rats. The expression of LC-3 in the sciatic nerve, which reflects the level of autophagy, was significantly higher in the diabetic control rats than the healthy control rats. The level of p-AMPK was markedly low in the sciatic nerve of the diabetic control rats. After treatment with metformin, the diabetic metformin group saw decreased levels of autophagy as well as increased levels of p-AMPK, providing correlational evidence for the positive regulatory effects of metformin’s AMPK activation in the sciatic nerve59.
Metformin’s activation of AMPK may also ameliorate DPN-associated pain hypersensitivity and neuronal inflammation in DRG neurons. Intraperitoneal injection of 200 mg/kg of metformin in diabetic rats increased the PWT from 1 hour to 8 hours in comparison to the diabetic saline injection group, indicating that metformin has a potential analgesic effect on nerve pain in diabetic rats. This effect was blocked when the AMPK antagonist compound C was applied, implying that metformin’s mechanism of nerve pain reduction is caused by metformin’s activation of the AMPK signaling pathway. Further investigation of AMPK activation in the L4-6 DRGs of the diabetic rats showed that, while the total level of AMPK did not change, the level of p-AMPK was significantly increased by treatment with metformin, indicating that metformin positively regulates of p-AMPK in the L4-6 DRGs along with attenuating nerve pain in diabetic rats. This study also noted that NF-κB, a transcription factor for pro-inflammatory cytokines, was markedly present in the DRGs of diabetic rats. Treatment with metformin and injection of 160 mg/kg of AICAR both greatly reduced the expression of NF-κB in these neurons, indicating that metformin’s activation of AMPK has protective effects on neuronal inflammation60.
Metformin was also found to cause the conversion of pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages via the AMPK signaling pathway, improving peripheral nerve regeneration in rats with sciatic nerve injury. Treatment with 50 mg/kg of metformin significantly increased sciatic nerve axonal regeneration, remyelination, and nerve function. The p-AMPK/AMPK ratio was higher in the metformin-treated group than the control group, indicating increased levels of AMPK activation. The distribution and density of M2 macrophages was also higher in the metformin-treated group. The application of compound C predictably reversed these effects, lowering AMPK activation and M2 macrophage distribution61. These findings indicate that metformin uses the AMPK signaling pathway to reduce neuronal inflammation and promote peripheral nerve regeneration through the conversion of M1 macrophages to M2 macrophages.
| Study | Model/Patient | Dosage and Time | Outcome of Treatment |
| Lós42 | Streptozotocin-induced T1DM mice | 100 mg/kg (low-dose group), 200 mg/kg (high-dose group) of metformin daily for 9 weeks | Reduced axonal atrophy, myelin sheath degeneration, neuronal inflammation, and oxidative stress in the sciatic nerve. |
| Liu43 | Streptozotocin-induced DM rats | 400 mg/kg of metformin daily for 8 weeks | Decreased expression of the pro-inflammatory cytokine IL-1β and increased expression of the anti-inflammatory cytokine IL-10. Reduced inflammation. |
| Ma44‘45 | Streptozotocin-induced DM rats | 30 mg/kg (low-dose group), 200 mg/kg (medium-dose group), 500 mg/kg (high-dose group) of metformin daily for 4 weeks | Axonal atrophy and myelin fiber degeneration were prevented. Axonal regeneration was observed. |
| Kim46 | Streptozotocin-induced DM rats | 100 mg/kg of metformin daily for 24 weeks | Increased myelin sheath thickness and axonal diameter were observed. The reduction of the PWT was prevented. |
| Lin47‘48 | Streptozotocin-induced DM rats | 200 mg/kg of metformin daily for 28 days | Attenuation of the DPN-associated increase of the synaptic number in the L5 segment of the spinal dorsal horn. |
| Dhanapalaratnam50 | T2DM patients | 1,523 mg of metformin daily for 162 months | Lower mcTNS scores, which indicate lesser DPN severity. Increased axonal function. |
| Lv56 | RSC96 cells | 1 mM of AICAR | Significantly reduced TRPA1 expression. |
| Wang25 | Rat DRG neurons cultured in hyperglycemic conditions and db/db mice | AICAR and 500 μmol/L of metformin in DRG neurons. 250 mg/kg of metformin and 250 mg/kg of AICAR in db/db mice | AMPK activation along with significantly reduced TRPA1 expression and lowered pain hypersensitivity. |
| Yerra58 | Streptozotocin-induced DM rats | 15 mg/kg (low-dose group) and 30 mg/kg (high-dose group) of A769662 dissolved in saline | AMPK activation. Reduction of pain hypersensitivity, neuronal expression of inflammatory factors, and neuronal apoptosis. Improvement of sensory and motor nerve conduction. |
| Zhang23 | Streptozotocin-induced DM rats | 60 mg/kg of Alpha Lipoic Acid daily for 12 weeks | AMPK activation along with attenuation of neuronal oxidative stress and apoptosis. |
| You59 | Streptozotocin-induced DM rats | 400 mg/kg of metformin daily for 8 weeks | AMPK activation and reduced neuronal autophagy. |
| Cao60 | Streptozotocin-induced DM rats | 200 mg/kg of metformin daily for 6 days in metformin group. 160 mg/kg of AICAR daily for 6 days in AICAR group. | Reduced pain hypersensitivity in both metformin and AICAR groups, suggesting that metformin’s AMPK activation attenuates pain hypersensitivity. |
| Zhou61 | Rats with sciatic nerve crush injury | 50 mg/kg of metformin | Axonal regeneration and remyelination along with increased M2 macrophage polarization. Increased AMPK activation. |
Other Potential Mechanisms of Metformin’s Action Against DPN
Outside of AMPK activation, metformin’s mechanisms of action to attenuate DPN severity remain unclear. One mechanism that may be utilized by metformin to improve nerve dysfunction is activation of the hedgehog (Hh) signaling pathway. The Hh signaling pathway is a critical means of intercellular communication during embryogenesis in mammals62. The pathway plays a critical role in organogenesis during embryonic development along with tissue regeneration and adult stem cell differentiation after birth62‘63. A large number of studies have shown that metformin inhibits the Hh signaling pathway to reduce tumorigenesis and brain tumor formation, although metformin’s activation of the Hh pathway to ameliorate tissue-specific dysfunction is a prospect that requires further investigation. Additionally, metformin’s inhibition of the Toll-like Receptor 4 (TLR4) signaling pathway may also reduce cellular apoptosis and improve cell viability. Activation of the Hh signaling pathway and inhibition of the TLR4 signaling pathway have been shown to exhibit neurotrophic benefits for DPN-associated nerve damage, potentially implicating the activation of these pathways as mechanisms of metformin’s reduction of DPN severity.
Inhibition of the TLR4 Signaling Pathway
Metformin has been documented to inhibit TLR4 signaling pathway, and this mechanism of action may attenuate nerve damage in animal models of DPN . Studies in animal models show that metformin appears to exert protective effects in various tissues, and inhibition of the TLR4 signaling pathway appears to play a role in metformin’s action64‘65‘66‘67‘68‘69‘70. In animal models of DPN, inhibition of the TLR4 signaling pathway appears to reduce neuronal inflammation, and increased expression of TLR4 in monocytes was found to be associated with nerve inflammation in DPN patients71‘72. Animal studies of DPN have also noted that metformin appears to exert anti-inflammatory effects in damaged sciatic nerves43. As such, metformin’s inhibition of the TLR4 signaling pathway could potentially attenuate DPN-associated neuronal inflammation and apoptosis, although further research is necessary to confirm this prospect.
Activation of the Hedgehog Signaling Pathway
Activation of the Hh signaling pathway may also be a mechanism of metformin’s potential betterment of nerve damage in animal models of DPN, although this prospect remains unclear. In vivo and In vitro studies of diabetic animals have shown that daily treatment with 300 mg/kg of metformin appears to alleviate apoptosis and excessive autophagy in vascular endothelia, and the activation of the hedgehog signaling pathway may be a key mechanism of this action73. However, little research currently exists to confirm activation of the Hh pathway as a mechanism of metformin. Nonetheless, activation of the Hh pathway also seems to have neuroprotective effects in animal models of DPN, highlighting its potential as a possible mechanism for metformin’s protection against DPN in animal models. Hh pathway activity has been shown to be significantly lowered in diabetic rats, leading to sciatic nerve damage, lowered nerve conduction velocity, and decreased threshold of nociception. Mediation of the Hh signaling pathway generally improved DPN74. Exogenous SHH treatment had neuroprotective effects on damaged retinal ganglion cells in diabetic rats, and the inhibition of SHH signaling reversed this effect75. Exogenous SHH treatment also restored peripheral nerve conduction velocities and maintained axonal function in diabetic rats, further highlighting the neuroprotective effects of Hh signaling. The mRNA levels of Desert hedgehog (DHH), another important protein in Hh signaling, were reduced in the sciatic nerves of diabetic rats not treated with SHH, indicating that Hh signaling is impaired in nerve dysfunction resulting from DN76. Considering the ameliorative effects of Hh signaling on DPN-associated nerve dysfunction and the impairment of Hh signaling in dysfunctional nerves, activation of the Hh signaling pathway may exert neuroprotective effects on DPN-associated peripheral nerve damage. It has been previously noted that metformin appears to increase peripheral nerve conduction velocity and axonal function in human patients50. Thus, it is possible that activation of the Hh signaling pathway may be a mechanism of metformin’s protective effects against DPN in animal models. However, little research exists that indicates that activation of the Hh signaling pathway is certainly a mechanism of metformin’s action. As such, it is unknown whether metformin utilizes this mechanism to potentially attenuate DPN-associated nerve dysfunction. Thus, further research is necessary to confirm metformin’s activation of the Hh signaling pathway as a potential mechanism of treating nerve damage.
| Model/Patient Group | Outcome of Metformin Treatment | Proposed Mechanisms | Reasoning |
| Diabetic mice and rats | Reduced sciatic nerve axonal atrophy, increased axonal diameter. | Hh activation (requires further investigation) | The Hh pathway plays a role in tissue regeneration and has been shown to improve nerve function, though it is unknown if metformin activates Hh signaling while treating DPN. |
| Diabetic mice and rats | Restored sciatic nerve myelin sheath thickness, reduced Schwann cell mitochondrial dysfunction. | AMPK activation | Impaired AMPK signaling results in mitochondrial dysfunction, so AMPK may play a role in improved mitochondrial function. |
| Diabetic rats | Reduced neuronal inflammation | AMPK activation, TLR4 inhibition (requires further investigation) | Metformin’s AMPK activation and have been shown to reduce neuronal inflammation in DPN. TLR4 inhibition has been found to reduce neuronal inflammation in DPN, but it is unknown if metformin inhibits TLR4 signaling while treating DPN. |
| Diabetic rats | Reduced sciatic nerve oxidative stress | AMPK activation | AMPK activation has been shown to reduce oxidative stress and apoptosis in diabetic DRG neurons. |
| Diabetic rats | Reduced pain hypersensitivity through lower synaptic number in spinal dorsal horn. | AMPK activation (requires further investigation) | Metformin’s AMPK activation has been shown to improve pain hypersensitivity, and metformin activates AMPK in the spinal dorsal horn . It is unknown whether AMPK reduces the synaptic number in the spinal dorsal horn. |
| T2DM patients | Improved nerve conduction and axonal excitability. Reduced DPN severity. | AMPK activation, Hh activation (requires further investigation) | Hh and AMPK activation have been shown to improve nerve conduction velocity, although it is unclear if metformin utilizes Hh activation to improve axonal excitability. |
| Diabetic rats | Reduced sciatic nerve autophagy | AMPK activation, Hh activation (requires further investigation) | Metformin was shown to reduce sciatic nerve autophagy via AMPK activation. Metformin has been found to activate Hh signaling to reduce autophagy in adipose tissue. |
Discussion
DPN is the most common form of DN, affecting sensorimotor and autonomic function in the peripheral nervous system1‘2. Metformin, one of the most popular medications for T2DM, was shown to potentially attenuate DPN severity in a large number of preclinical studies through the improvement of sciatic nerve damage and morphological changes in the spinal dorsal horn. These neuroprotective effects resulted in increased peripheral nerve conduction velocity, lower pain hypersensitivity, and improved nerve function. Metformin’s effect on DPN severity is most likely the result of direct neuroprotection rather than glucose control. This neuroprotection may be carried out by utilizing the following mechanisms of action. Activation of the AMPK signaling pathway is likely a key mechanism of metformin’s neuroprotective effects on DPN pathophysiology, having been shown to improve pain hypersensitivity, neuronal oxidative stress, apoptosis, and inflammation. Other possible mechanisms of metformin’s action against DPN include inhibition of the TLR4 pathway and activation of the Hh signaling pathway, as both of these mechanisms have been utilized by metformin to yield tissue-specific benefits and have been shown to exhibit neuroprotective effects in animal models of DPN. However, the role of these mechanisms in metformin’s action against DPN is currently hypothetical and requires further research for confirmation.
The main objective of this paper was to identify potential pathways that play a role in metformin’s potential amelioration of DPN pathophysiology. In that respect, this paper discussed the well-defined role of the AMPK signaling pathway in the betterment of DPN and hypothesized that inhibition of the TLR4 pathway and activation of the Hh pathway may be additional mechanisms of metformin’s neuroprotective effects. Metformin’s neurotrophic utilization of the Hh and TLR4 pathways is not well-studied, so this review provides a basis for further research to investigate this prospect. Overall, this review was successful in identifying potential pathways behind metformin’s action against DPN.
However, this review and the study of metformin in DPN as a whole suffer from major limitations. One such limitation that affects the clinical study of metformin and neuropathy in DM patients is the inconsistency of results reported by clinical studies and the focus of these studies on metformin-induced vitamin B12 deficiency. While some clinical studies have shown that metformin treatment reduces the severity and prevalence of DPN among DM patients, others state that metformin treatment may increase the prevalence of neuropathy among DM patients16‘50‘79. As summarized thoroughly by Wei et al., the majority of clinical studies about metformin in neuropathy focus on how metformin-induced vitamin B12 deficiency leads to neuropathy in DM patients, although the results of these studies are largely inconsistent. A large number of studies have found an association between metformin, vitamin B12 deficiency, and neuropathy, but others report little association between the risk of neuropathy and metformin-induced vitamin B12 deficiency. Certain studies also noted an increased prevalence of neuropathy among DM patients who took metformin, although there was no association between metformin-induced B12 deficiency and neuropathy. The inconsistency of these results as well as the difficulty of distinguishing between B12 deficiency-induced neuropathy and DPN make the correlation between metformin-induced B12 deficiency and increased prevalence of neuropathy controversial. However, as it stands, the results from clinical studies of metformin and neuropathy seem to indicate that metformin exerts less neuroprotection in DM patients than in animal models of DM. This may be due to the fact that, unlike the animal studies of metformin in DPN, very few clinical studies exist that examine metformin’s potential neuroprotective effects and mechanisms in DM patients, and the majority of clinical studies focus on the implications of metformin-induced B12 deficiency for neuropathy instead16. As such, further clinical trials and studies in DM patients are necessary to investigate metformin’s effect on DPN and the mechanisms behind said effect.
Within this review, a key limitation is the possibility of selection bias in this review due to the limited number of findings reported that did not support metformin’s potential for protection against DPN. However, this is a result of how the study of metformin’s neuroprotective mechanisms against DPN is relatively small and requires further research to develop a wholistic view of metformin’s neuroprotective potential. The majority of the primary research articles that were found using the search keywords mentioned in the “Methods” section were preclinical studies, and very few clinical studies that discussed metformin’s potential neuroprotective mechanisms were found. As mentioned previously, the majority of clinical studies about metformin and neuropathy focus on how metformin-induced B12 deficiency may cause neuropathy, and this unclear prospect is discussed in detail above. The consensus from all of the preclinical studies that the author was able to find is that metformin appears to significantly attenuate the severity of DPN in diabetic animals, but metformin may not completely reverse DPN-induced nerve deficits. However, further preclinical and clinical research is necessary to provide a complete view of metformin’s potential to ameliorate DPN. Another key concern is the validity of the animal models utilized in this review, which indicates the validity of the results presented in this paper. The animal studies of DPN presented in this review predominantly use streptozotocin and db/db models of DM. The streptozotocin model of DM excels in modeling DM-associated hyperglycemia and resulting conditions such as DPN, although its chemically induced approach to replicating the diabetic state does not accurately capture the complex pathogenetic mechanisms that lead to DM onset80. On the other hand, the db/db model utilizes a genetically induced approach in its near-perfect model of T2DM, although its spontaneous induction of hyperglycemia and insulin resistance may not accurately represent DM progression80‘81. As this review analyzes metformin’s effect on DPN, a downstream complication of DM, both the streptozotocin and db/db models of DM are valid models for studying the potential mechanisms behind metformin’s effect on DPN.
In conclusion, DPN is a debilitating complication that causes significant morbidity in DM patients. Efficacious medications that can halt or reverse the progression of DPN remain elusive. The evaluation of metformin as a potential therapeutic drug for DPN and identification of possible mechanisms of metformin’s treatment of nerve dysfunction provide a basis for further research to examine the neuroprotective effects of these mechanisms in the betterment of DPN. This review uniquely identifies inhibition of the TLR4 pathway and activation of the Hh pathway as other potential mechanisms behind metformin’s effect on DPN in animal models, but their potential currently remains hypothetical and requires further research to explore their role. Identification of the pathways behind metformin’s betterment of DPN can facilitate the examination of the neuroprotection of other similar medications and the development of new drugs that utilize the same pathways as metformin. In a world where the prevalence, incidence, and morbidity of DM continues to rise at a worrying rate, the identification of mechanisms of action that protect against DM-associated conditions as well as the subsequent development of efficacious medications for these complications are crucial for the reduction of DM’s heavy burden on society.
Acknowledgments
Thank you to the Lumiere team for providing mentorship through the research and writing process. Special thanks to Yoo Jin Jung and Bharat Sharma for providing insightful and valuable feedback. An additional thank you to BioRender for facilitating the creation of my figures.
References
- J. Zenker, D. Ziegler, & R. Chrast (2013). Novel pathogenic pathways in diabetic neuropathy. Trends in Neurosciences, 36(8), 439–449. [↩] [↩]
- International Diabetes Federation (IDF). (2025). Diabetes Facts and Figures | International Diabetes Federation. IDF Diabetes Atlas 11th Edition. [↩] [↩]
- R. Herman, N. A. Kravos, M. Jensterle, A. Janež, & V. Dolžan (2022). Metformin and Insulin Resistance: A Review of the Underlying Mechanisms behind Changes in GLUT4-Mediated Glucose Transport. In International Journal of Molecular Sciences (Vol. 23, Issue 3). MDPI. [↩] [↩] [↩] [↩]
- E. L. Feldman, B. C. Callaghan, R. Pop-Busui, D. W. Zochodne, D. E. Wright, D. L. Bennett, V. Bril, J. W. Russell, & V. Viswanathan (2019). Diabetic neuropathy. In Nature Reviews Disease Primers (Vol. 5, Issue 1). Nature Publishing Group. [↩]
- Y. Yang, B. Zhao, Y. Wang, H. Lan, X. Liu, Y. Hu, & P. Cao (2025). Diabetic neuropathy: cutting-edge research and future directions. Signal Transduction and Targeted Therapy, 10(1). [↩] [↩] [↩]
- H. S. Hamid, C. M. Mervak, A. E. Münch, N. J. Robell, J. M. Hayes, M. T. Porzio, J. R. Singleton, A. G. Smith, E. L. Feldman, & S. I. Lentz (2014). Hyperglycemia- and neuropathy-induced changes in mitochondria within sensory nerves. Annals of Clinical and Translational Neurology, 1(10), 799–812. [↩] [↩]
- R. Freeman, E. Durso-DeCruz, & B. Emir (2008). Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: Findings from seven randomized, controlled trials across a range of doses. Diabetes Care, 31(7), 1448–1454. [↩]
- J. Rosenstock, M. Tuchman, L. Lamoreaux, & U. Sharma (2004). Pregabalin for the treatment of painful diabetic peripheral neuropathy: a double-blind, placebo-controlled trial. Pain, 110(3), 628–638. [↩]
- J. Ahn, R. Shahriarirad, K. Kwon, L. Bejarano-Pineda, G. Waryasz, , & S. Ashkani-Esfahani, (2025). Comparative analysis of the therapeutic effects of pregabalin, gabapentin, and duloxetine in diabetic peripheral neuropathy: A retrospective study. Journal of Diabetes and Its Complications, 39(4), 109001. [↩]
- J. Flory, & K. Lipska, (2019). Metformin in 2019. JAMA, 321(19), 1926–1927. [↩]
- Z. Lv, & Y. Guo (2020). Metformin and Its Benefits for Various Diseases. In Frontiers in Endocrinology (Vol. 11). Frontiers Media S.A. [↩]
- A. Alhaider, H. M. Korashy, M. M. Sayed-Ahmed, M. Mobark, H. Kfoury, & M.A. Mansour (2011). Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through modulation of oxidative stress genes expression. Chemico-Biological Interactions, 192(3), 233–242. [↩]
- Y. Zhang, F. Chen, & L. Wang (2017). Metformin inhibits development of diabetic retinopathy through microRNA-497a-5p. In Am J Transl Res (Vol. 9, Issue 12). [↩]
- Y. Li, C. Ryu, M. Munie, S. Noorulla, S. Rana, P. Edwards, H. Gao, & X. Qiao (2018). Association of Metformin Treatment with Reduced Severity of Diabetic Retinopathy in Type 2 Diabetic Patients. Journal of Diabetes Research, 2018. [↩]
- Q.-Y. Yi, G. Deng, N. Chen, Z.-S. Bai, J.-S. Yuan, G.-H. Wu, Y.-W. Wang, & S.-J. Wu (2016). Metformin inhibits development of diabetic retinopathy through inducing alternative splicing of VEGF-A. In Am J Transl Res (Vol. 8, Issue 9). [↩]
- Wei, J., Wei, Y., Huang, M., Wang, P., & Jia, S. (2022). Is metformin a possible treatment for diabetic neuropathy? In Journal of Diabetes (Vol. 14, Issue 10, pp. 658–669). John Wiley and Sons Inc. [↩] [↩] [↩]
- J. L. Edwards, A. Quattrini, S. I. Lentz, C. Figueroa-Romero, F. Cerri, C. Backus, Y. Hong, & E. L. Feldman (2010). Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia, 53(1), 160–169. [↩] [↩] [↩] [↩] [↩] [↩]
- W. Yin, A. P. Signore, M. Iwai, G. Cao, Y. Gao, & J. Chen (2008). Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke, 39(11), 3057–3063. [↩]
- D. C. Chan (2006). Mitochondrial fusion and fission in mammals. Annual Review of Cell and Developmental Biology, 22(Volume 22, 2006), 79–99. [↩]
- A. Garnier, D. Fortin, J. Zoll, B. N’Guessan, B. Mettauer, E. Lampert, V. Veksler, & R. Ventura-Clapier (2005). Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. The FASEB Journal, 19(1), 43–52. [↩]
- A. M. Vincent, J. L. Edwards, L. L. McLean, Y. Hong, F. Cerri, I. Lopez, A. Quattrini, & E. L. Feldman (2010). Mitochondrial biogenesis and fission in axons in cell culture and animal models of diabetic neuropathy. Acta Neuropathologica, 120(4), 477–489. [↩] [↩] [↩] [↩] [↩] [↩] [↩]
- G. M. Leinninger, C. Backus, A. M. Sastry, Y. B. Yi, C. W. Wang, & E. L. Feldman (2006). Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiology of Disease, 23(1), 11–22. [↩] [↩] [↩]
- T. Zhang, D. Zhang, Z. Zhang, J. Tian, J. An, W. Zhang, & Y. Ben (2023). Alpha-lipoic acid activates AMPK to protect against oxidative stress and apoptosis in rats with diabetic peripheral neuropathy. Hormones, 22(1), 95–105. [↩] [↩] [↩] [↩]
- C. Rodríguez, M. Muñoz, C. Contreras, & D. Prieto (2021). AMPK, metabolism, and vascular function. The FEBS Journal, 288(12), 3746–3771. [↩]
- S. Wang, K. Kobayashi, Y. Kogure, H. Yamanaka, S. Yamamoto, H. Yagi, K. Noguchi, & Y. Dai (2018). Negative regulation of TRPA1 by AMPK in primary sensory neurons as a potential mechanism of painful diabetic neuropathy. Diabetes, 67(1), 98–109. [↩] [↩] [↩] [↩] [↩]
- S. K. R. Chowdhury, D. R. Smith, A. Saleh, J. Schapansky, A. Marquez, S. Gomes, E. Akude, D. Morrow, N. A. Calcutt, & P. Fernyhough (2012). Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain, 135(6), 1751–1766. [↩] [↩]
- J. N. Feige, & J. Auwerx (2007). Transcriptional coregulators in the control of energy homeostasis. Trends in Cell Biology, 17(6), 292–301. [↩]
- D. G. Hardie (2008). AMPK: A key regulator of energy balance in the single cell and the whole organism. International Journal of Obesity, 32(4), S7–S12. [↩]
- A. M. Schmeichel, J. D. Schmelzer, & P. A. Low (2003). Oxidative Injury and Apoptosis of Dorsal Root Ganglion Neurons in Chronic Experimental Diabetic Neuropathy. In DIABETES (Vol. 52). [↩] [↩] [↩] [↩] [↩]
- J. W. Russell, K. A. Sullivan, A. J. Windebank, D. N. Herrmann, & E. L. Feldman (1999). Neurons Undergo Apoptosis in Animal and Cell Culture Models of Diabetes. Neurobiology of Disease, 6(5), 347–363. [↩] [↩] [↩]
- A. M. Vincent, L. L. Mclean, C. Backus, & E. L. Feldman (2005). Short‐term hyperglycemia produces oxidative damage and apoptosis in neurons. The FASEB Journal, 19(6), 1–24. [↩] [↩] [↩]
- M. A. Babizhayev, I. A. Strokov, V. V. Nosikov, E. L. Savel’yeva, V. F. Sitnikov, E. Y. Yegor, & V. Z. Lankin (2015). The Role of Oxidative Stress in Diabetic Neuropathy: Generation of Free Radical Species in the Glycation Reaction and Gene Polymorphisms Encoding Antioxidant Enzymes to Genetic Susceptibility to Diabetic Neuropathy in Population of Type I Diabetic Patients. Cell Biochemistry and Biophysics, 71(3), 1425–1443. [↩]
- S. Hong, & J. W. Wiley (2006). Altered expression and function of sodium channels in large DRG neurons and myelinated A-fibers in early diabetic neuropathy in the rat. Biochemical and Biophysical Research Communications, 339(2), 652–660. [↩] [↩] [↩] [↩] [↩]
- X. H. Cao, H. S. Byun, S. R. Chen, Y. Q. Cai, & H. L. Pan (2010). Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: Role of brain-derived neurotrophic factor. Journal of Neurochemistry, 114(5), 1460–1475. [↩] [↩]
- M. M. Jagodic, S. Pathirathna, M. T. Nelson, S. Mancuso, P. M. Joksovic, E. R. Rosenberg, D. A. Bayliss, V. Jevtovic-Todorovic, & S. M. Todorovic (2007). Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. Journal of Neuroscience, 27(12), 3305–3316. [↩] [↩] [↩]
- M. J. Craner, J. P. Klein, M. Renganathan, J. A. Black, & S. G. Waxman (2002). Changes of sodium channel expression in experimental painful diabetic neuropathy. Annals of Neurology, 52(6), 786–792. [↩] [↩]
- C. G. Faber, G. Lauria, I. S. J. Merkies, X. Cheng, C. Han, H. S. Ahn, A. K. Persson, J. G. J. Hoeijmakers, M. M. Gerrits, T. Pierro, R. Lombardi, D. Kapetis, S. D. Dib-Hajj, & S. G. Waxman (2012). Gain-of-function Nav1.8 mutations in painful neuropathy. Proceedings of the National Academy of Sciences of the United States of America, 109(47), 19444–19449. [↩]
- J. Huang, C. Han, M. Estacion, D. Vasylyev, J. G. J. Hoeijmakers, M. M. Gerrits, L. Tyrrell, G. Lauria, C. G. Faber, S. D. Dib-Hajj, I. S. J. Merkies, S. G. Waxman (2014). Gain-of-function mutations in sodium channel NaV1.9 in painful neuropathy. Brain, 137(6), 1627–1642. [↩]
- Lbattist. (2022). Active AMPK – Wikimedia Commons. In AMP-activated protein kinase. Wikimedia Foundation. [↩] [↩]
- B. A. Giuffre, A. C. Black, & R. Jeanmonod (2023). Anatomy, Sciatic Nerve. In StatPearls. StatPearls Publishing. [↩]
- O. J. Freeman, R. D. Unwin, A. W. Dowsey, P. Begley, S. Ali, K. A. Hollywood, N. Rustogi, R. S. Petersen, W. B Dunn, G. J. S. Cooper, & N. J. Gardiner (2016). Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy. Diabetes, 65(1), 228–238. [↩]
- D. B. Lós, W. H. de Oliveira, E. Duarte-Silva, W. W. D. Sougey, E. S. R. de Freitas, A. G. V. de Oliveira, C. F. Braga, M. E. R. de França, S. M. R. Araújo, G. B. Rodrigues, S. W. S. Rocha, C. A. Peixoto, & S R. A. de Moraes (2019). Preventive role of metformin on peripheral neuropathy induced by diabetes. International Immunopharmacology, 74, 105672. [↩] [↩] [↩] [↩]
- F. Liu, F. You, L. Yang, S. Wang, & D. Xie (2024). Metformin improves diabetic neuropathy by reducing inflammation through up-regulating the expression of miR-146a and suppressing oxidative stress. Journal of Diabetes and Its Complications, 38(6), 108737. [↩] [↩] [↩] [↩]
- J. Ma, J. Liu, Y. Chen, H. Yu, & L. Xiang (2022). Metformin Promotes Axonal Regeneration and Functional Recovery in Diabetic Rat Model of Sciatic Nerve Transection Injury. NeuroSci, 3(3), 366–375. [↩] [↩] [↩]
- J. Ma, J. Liu, H. Yu, Y. Chen, Q. Wang, & L. Xiang (2016). Beneficial Effect of Metformin on Nerve Regeneration and Functional Recovery After Sciatic Nerve Crush Injury in Diabetic Rats. Neurochemical Research, 41(5), 1130–1137. [↩] [↩] [↩]
- S. H. Kim, T. S. Park, & H. Y. Jin (2020). Metformin preserves peripheral nerve damage with comparable effects to alpha lipoic acid in streptozotocin/high-fat diet induced diabetic rats. Diabetes and Metabolism Journal, 44(6), 842–853. [↩] [↩]
- J. Y. Lin, Y. N. He, N. Zhu, & B. Peng (2018). Metformin attenuates increase of synaptic number in the rat spinal dorsal horn with painful diabetic neuropathy induced by type 2 diabetes: a stereological study. Neurochemical Research, 43(12), 2232–2239. [↩] [↩] [↩]
- J. Y. Lin, X. L. Huang, J. Chen, Z. W. Yang, J. Lin, S. Huang, & B. Peng (2017). Stereological study on the number of synapses in the rat spinal dorsal horn with painful diabetic neuropathy induced by streptozotocin. NeuroReport, 28(6), 319–324. [↩] [↩]
- D. Fuchs, F. Birklein, P. W. Reeh, & S. K. Sauer (2010). Sensitized peripheral nociception in experimental diabetes of the rat. Pain, 151(2), 496–505. [↩]
- R. Dhanapalaratnam, T. Issar, L. L. Wang, D. Tran, A. M. Poynten, K.-L. Milner, N. C. G. Kwai, & A. V. Krishnan (2024). The effect of metformin on peripheral nerve morphology in type 2 diabetes: a cross-sectional observational study. Diabetes. [↩] [↩] [↩] [↩] [↩]
- G. Zhou, R. Myers, Y. Li, Y. Chen, X. Shen, J. Fenyk-Melody, M. Wu, J. Ventre, T. Doebber, N. Fujii, N. Musi, M. F. Hirshman, L. J. Goodyear, & D. E. Moller (2001). Role of AMP-activated protein kinase in mechanism of metformin action. Journal of Clinical Investigation, 108(8), 1167–1174. [↩]
- T. Luo, A. Nocon, J. Fry, A. Sherban, X. Rui, B. Jiang, X. J. Xu, J. Han, Y. Yan, Q. Yang, Q. Li, & M. Zang (2016). AMPK activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes, 65(8), 2295–2310. [↩]
- Y. Wang, H. An, T. Liu, C. Qin, H. Sesaki, S. Guo, S. Radovick, M. Hussain, A. Maheshwari, F. E. Wondisford, B. O’Rourke, & L. He (2019). Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Reports, 29(6), 1511-1523.e5. [↩]
- R. Herman, N. A. Kravos, M. Jensterle, A. Janež, & V. Dolžan (2022). Metformin and Insulin Resistance: A Review of the Underlying Mechanisms behind Changes in GLUT4-Mediated Glucose Transport. In International Journal of Molecular Sciences (Vol. 23, Issue 3). MDPI. [↩]
- B. Xiao, M. J. Sanders, E. Underwood, R. Heath, F. V. Mayer, D. Carmena, C. Jing, P. A. Walker, J. F. Eccleston, L. F. Haire, P. Saiu, S. A. Howell, R. Aasland, S. R. Martin, D. Carling, & S. J. Gamblin (2011). Structure of mammalian AMPK and its regulation by ADP. Nature, 472(7342), 230–233. [↩]
- J. Lv, L. Cao, R. Zhang, F. Bai, & P. Wei (2018). A curcumin derivative J147 ameliorates diabetic peripheral neuropathy in streptozotocin (STZ)-induced dpn rat models through negative regulation AMPK on TRPA1. Acta Cirurgica Brasileira, 33(6), 533–541. [↩] [↩] [↩]
- H. Wei, M. M. Hämälä, M. Saarnilehto, A. Koivisto, & A. Pertovaara (2009). Attenuation of Mechanical Hypersensitivity by an Antagonist of the TRPA1 Ion Channel in Diabetic Animals. In Anesthesiology (Vol. 111). [↩]
- V. G. Yerra & A. Kumar (2017). Adenosine Monophosphate-Activated Protein Kinase Abates Hyperglycaemia-Induced Neuronal Injury in Experimental Models of Diabetic Neuropathy: Effects on Mitochondrial Biogenesis, Autophagy and Neuroinflammation. Molecular Neurobiology, 54(3), 2301–2312. [↩] [↩]
- F. You, D. Xie, C. Li, L. Yang, & F. Liu (2024). Metformin ameliorates peripheral neuropathy in diabetic rats by downregulating autophagy via the AMPK pathway. Archives of Endocrinology and Metabolism, 68, e240137. [↩] [↩]
- X. J. Cao, R. Wu, H. Y. Qian, X. Chen, H. Y. Zhu, G. Y. Xu, Y. Z. Sun, & P. A. Zhang (2021). Metformin attenuates diabetic neuropathic pain via AMPK/NF-κB signaling pathway in dorsal root ganglion of diabetic rats. Brain Research, 1772, 147663. [↩] [↩]
- Z. Zhou, G. Luo, C. Li, P. Zhang, W. Chen, X. Li, J. Tang, & L. Qing (2023). Metformin induces M2 polarization via AMPK/PGC-1α/PPAR-γ pathway to improve peripheral nerve regeneration. American Journal of Translational Research, 15(5), 3778. [↩] [↩]
- G. B. Carballo, J. R. Honorato, G. P. F. de Lopes, & T. C. L. D. S. E. Spohr (2018). A highlight on Sonic hedgehog pathway. In Cell Communication and Signaling (Vol. 16, Issue 1). BioMed Central Ltd. [↩] [↩]
- J. Jing, Z. Wu, J. Wang, G. Luo, H. Lin, Y. Fan, & C. Zhou (2023). Hedgehog signaling in tissue homeostasis, cancers, and targeted therapies. In Signal Transduction and Targeted Therapy (Vol. 8, Issue 1). Springer Nature. [↩]
- L. Zheng, X. Shen, J. Ye, Y. Xie, & S. Yan (2019). Metformin alleviates hyperglycemia-induced apoptosis and differentiation suppression in osteoblasts through inhibiting the TLR4 signaling pathway. Life Sciences, 216, 29–38. [↩]
- Alzokaky, A. A., Al-Karmalawy, A. A., Saleh, M. A., Abdo, W., Farage, A. E., Belal, A., Abourehab, M. A. S., & Antar, S. A. (2023). Metformin ameliorates doxorubicin-induced cardiotoxicity targeting HMGB1/TLR4/NLRP3 signaling pathway in mice. Life Sciences, 316, 121390. [↩]
- Autumn, C., Vaez, H., Najafi, M., Seyed Toutounchi, N., Barar, J., Barzegari, A., & Garjani, A. (2016). IN PRESS Metformin Alleviates Lipopolysaccharide-induced Acute Lung Injury through Suppressing Toll-like Receptor 4 Signaling. In Iran J Allergy Asthma Immunol (Vol. 15, Issue 6). [↩]
- Lai, X., Liu, B., Wan, Y., Zhou, P., Li, W., Hu, W., & Gong, W. (2025). Metformin alleviates colitis-associated colorectal cancer via inhibition of the TLR4/MyD88/NFκB/MAPK pathway and macrophage M2 polarization. International Immunopharmacology, 144, 113683. [↩]
- Li, X., Wang, L., Yang, X., & Huang, C. (2020). Metformin attenuates ischemia/reperfusion injury of fatty liver in rats through inhibition of the tlr4-nf-κb axis. Balkan Medical Journal, 37(4), 196–202. [↩]
- Peixoto, L. G., Teixeira, R. R., Vilela, D. D., Barbosa, L. N., Caixeta, D. C., Deconte, S. R., de Assis de Araújo, F., Sabino-Silva, R., & Espindola, F. S. (2017). Metformin attenuates the TLR4 inflammatory pathway in skeletal muscle of diabetic rats. Acta Diabetologica 2017 54:10, 54(10), 943–951. [↩]
- Zhang, Y., Liu, W., Zhong, Y., Li, Q., Wu, M., Yang, L., Liu, X., & Zou, L. (2021). Metformin Corrects Glucose Metabolism Reprogramming and NLRP3 Inflammasome-Induced Pyroptosis via Inhibiting the TLR4/NF- κ B/PFKFB3 Signaling in Trophoblasts: Implication for a Potential Therapy of Preeclampsia. Oxidative Medicine and Cellular Longevity, 2021. [↩]
- B. Zhao, Q. Zhang, X. Liang, J. Xie, & Q. Sun (2021). Quercetin reduces inflammation in a rat model of diabetic peripheral neuropathy by regulating the TLR4/MyD88/NF-κB signalling pathway. European Journal of Pharmacology, 912, 174607. [↩]
- T. Zhu, Q. Meng, J. Ji, L. Zhang, & X. Lou (2017). TLR4 and Caveolin-1 in Monocytes Are Associated With Inflammatory Conditions in Diabetic Neuropathy. Clinical and Translational Science, 10(3), 178–184. [↩]
- C. Niu, Z. Chen, K. T. Kim, J. Sun, M. Xue, G. Chen, S. Li, Y. Shen, Z. Zhu, X. Wang, J. Liang, C. Jiang, W. Cong, L. Jin, & X. Li (2019). Metformin alleviates hyperglycemia-induced endothelial impairment by downregulating autophagy via the Hedgehog pathway. Autophagy, 15(5), 843–870. [↩]
- Q. Sun, J. Zeng, Y. Liu, J. Y. Chen, Q. C. Zeng, Y. Q. Chen, L. L. Tu, P. Chen, F. Yang, & M. Zhang (2020). microRNA-9 and -29a regulate the progression of diabetic peripheral neuropathy via ISL1-mediated sonic hedgehog signaling pathway. Aging, 12(12), 11446–11465. [↩]
- X. Zhao, Y. Li, S. Lin, Y. Cai, J. Zhang, X. Yu, H. Yang, L. Yang, X. Chen, Y. Luo, & L. Lu (2015). The effects of sonic hedgehog on retinal müuller cells under high-glucose stress. Investigative Ophthalmology and Visual Science, 56(4), 2773–2782. [↩]
- N. A. Calcutt, K. L. Allendoerfer, A. P. Mizisin, A. Middlemas, J. D. Freshwater, M. Burgers, R. Ranciato, J.-D. Delcroix, F.R. Taylor, R. Shapiro, K. Strauch, H. Dudek, T. M. Engber, A. Galdes, L. L. Rubin, & D. R. Tomlinson (2003). Therapeutic efficacy of sonic hedgehog protein in experimental diabetic neuropathy. Journal of Clinical Investigation, 111(4), 507–514. [↩]
- A. Hasanvand, H. Amini-khoei, M. R. Hadian, A. Abdollahi, S. M. Tavangar, A. R. Dehpour, E. Semiei, & S. E. Mehr (2016). Anti-inflammatory effect of AMPK signaling pathway in rat model of diabetic neuropathy. Inflammopharmacology, 24(5), 207–219. [↩]
- A. Ge, S. Wang, B. Miao, & M. Yan (2018). Effects of metformin on the expression of AMPK and STAT3 in the spinal dorsal horn of rats with neuropathic pain. Molecular Medicine Reports, 17(4), 5229–5237. [↩]
- M. D. Farooq, F. A. Tak, F. Ara, S. Rashid, & I. A. Mir (2022). Vitamin B12 Deficiency and Clinical Neuropathy with Metformin Use in Type 2 Diabetes. Journal of Xenobiotics, 12(2). [↩]
- BenchChem Technical Support Team. (2025). The Streptozotocin Model: A Critical Evaluation for Human Diabetes Research. [↩] [↩]
- Szabadfi, K., Pinter, E., Reglodi, D., & Gabriel, R. (2014). Neuropeptides, trophic factors, and other substances providing morphofunctional and metabolic protection in experimental models of diabetic retinopathy. International Review of Cell and Molecular Biology, 311, 1–121. [↩]



{kind=link}