
Abstract
Human immunodeficiency virus (HIV) continues to present a major global health challenge, affecting around 40.8 million people as of 2024. Current technologies like combination antiretroviral therapy (cART) require lifelong medicines, while failing to eliminate latent viral reservoirs. Recent advances in genome editing have positioned CRISPR-based technologies as promising candidates for a functional cure. This review synthesizes findings from peer-reviewed studies on CRISPR-based HIV therapies. Both DNA and RNA-targeting approaches have made strides in combating HIV, including proviral excision through CRISPR-Cas9, CCR5 disruption, and degradation of viral transcripts through Cas13. Evidence from preclinical studies demonstrates the potential that CRISPR systems can effectively confer resistance to infection. Nevertheless, this represents a proof-of-concept rather than clinical evidence of cure, and significant challenges still remain. Barriers include delivery efficiency, potential off-target effects, and the need for conditioning regimens prior to cell transplantation. Socioeconomic constraints further limit potential, particularly in resource-poor regions where HIV is most prevalent. Additionally, high costs pose obstacles to equitable access. Future directions emphasize the integration of CRISPR with other strategies, as well as the development of non-chemotherapy conditioning. Overall, CRISPR technologies hold transformative potential for HIV cure research, but reaching its full potential will require overcoming technical and ethical hurdles with further research.
Introduction
Human immunodeficiency virus (HIV) remains one of the most significant challenges to global public health. As of 2024, around 40.8 million people are living with HIV, including approximately 1.4 million children1. Although the discovery of HIV in the early 1980s was a major milestone in infectious disease history, since then scientists have not developed an FDA approved vaccine or cure. Combination antiretroviral therapy (cART), the current standard therapy for HIV infection, attempts to control the spread of HIV within the host and viral shed. It employs a multi-drug regimen that targets distinct stages of the viral life cycle to suppress viral replication in CD4+ T cells, where HIV replicates within the host2. These drugs lower the viral titer in the blood to undetectable levels, preventing the progression to AIDS, limiting the spread of the virus, and allowing the immune system to recover. However, due to the virus’s ability to hide in latent reservoirs within cells, cART cannot fully eradicate HIV from the body. As a result, the treatment must be continued for life, which may have significant side effects on the patient.
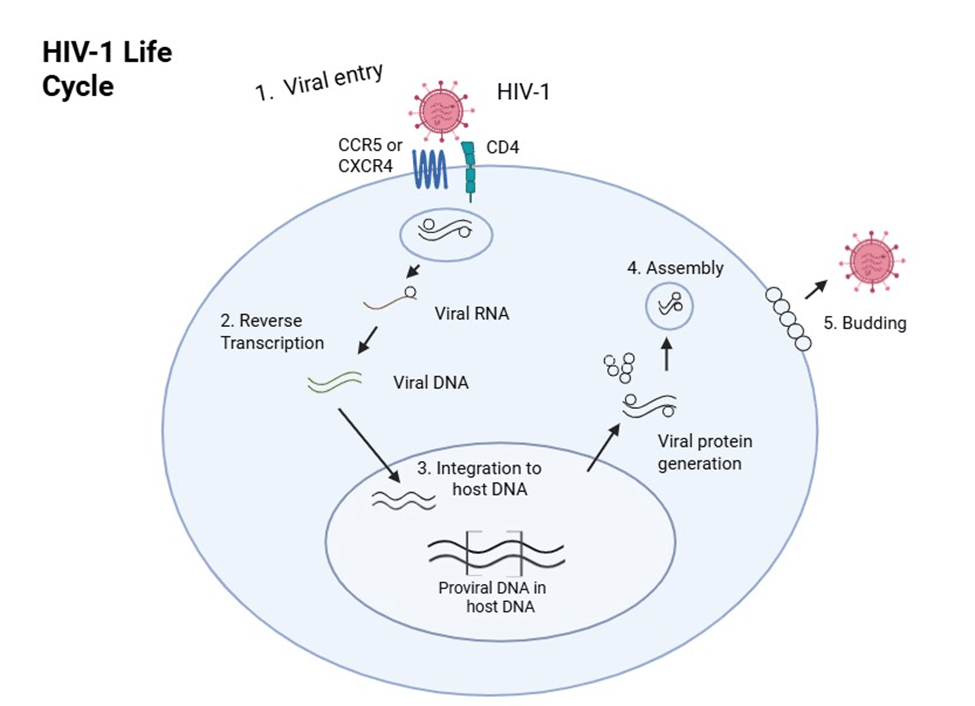
HIV-1 Genome Organization and Latency
HIV-1 is a retrovirus that stores its genetic material as RNA. Once the virus enters a host cell, its RNA is reverse-transcribed into DNA and integrated into the host’s genome. This integrated form is called a provirus. The provirus is flanked by long terminal repeats (LTRs), which are regulatory sequences that control transcription of HIV genes and act as essential promoters for viral replication. The HIV genome consists of several key genes. The gag gene encodes the structural proteins that form a barrier to protect the genetic material, while the pol gene encodes the enzymes needed for the replication cycle, including reverse transcriptase, integrase, and protease. Because these genes are fundamental for building new viral particles and completing the replication cycle, they are central points of vulnerability in the HIV life cycle. Understanding the roles of LTR, gag, and pol helps explain why interfering with these regions can significantly reduce viral replication3.
A central challenge in HIV treatment is viral latency. After integration, the provirus can enter a latent state in resting CD4+ T cells, where it produces little or no virus. These long-lived CD4+ T cells form what are known as latent reservoirs. Latent HIV can also persist in sanctuary sites such as the brain, lymph nodes, and gut-associated lymphoid tissue, where drug penetration is limited and less effective4. These reservoirs allow HIV to persist in the body even when antiretroviral therapy has lowered viral levels in the blood to undetectable levels. These poorly understood latent reservoirs have proven to be a major obstacle for developing HIV therapies. HIV integrates its genetic material into the host gene, creating the long-lived latent reservoirs in resting CD4+ T cells. They are invisible to both the immune system and antiretroviral therapy, allowing the virus to stay in a dormant state for years. Even when replication is suppressed by cART, the passive virus remains intact and can reactivate if treatment is stopped, leading to a rapid rebound5. Furthermore, HIV’s high mutation rate enables it to develop resistance to therapies. These factors make complete eradication of HIV a significant scientific challenge that can only be achieved by targeting reservoir CD4+ T cells.
CRISPR-Cas Approaches to HIV Therapy
The limitations of current antiretroviral therapies, combined with the challenges posed by latent reservoirs, viral diversity, and inefficient delivery of therapeutic molecules, underscore the need for innovative strategies that can directly target HIV’s genome. In recent years, gene-editing technologies, specifically Clustered Regularly Interspaced Short Palindromic Repeat-Cas (CRISPR-Cas), has emerged as a promising approach to directly target and inactivate the HIV genome. Unlike traditional therapies, CRISPR offers the potential to eliminate integrated viral DNA, addressing the infection at its core rather than simply managing it. Originally discovered as part of a bacterial immune defense system, CRISPR functions as a molecular tool that uses guide RNA to direct an enzyme to a specific sequence of DNA or RNA. The nuclease, Cas, binds to a short CRISPR RNA (cRNA) that targets matching viral DNA or RNA sequences, depending on the type of CRISPR-Cas system. Most CRISPR-Cas systems use a short nucleotide sequence that are able to match up with a certain part of the viral genome. Multiple Cas enzymes have been adapted for therapeutic use, each offering distinct mechanisms and advantages depending on the type of target. The three correlating directly to HIV repression and, in time, eradication are the Cas9, Cas12, and Cas13 enzymes. Most CRISPR-Cas systems require a short sequence of nucleotides adjacent to the target site in order to recognize and bind to it. For DNA-targeting systems like Cas9 and Cas12, this sequence is known as a protospacer adjacent motif (PAM), while RNA-targeting systems like Cas13 rely on a similar sequence called the protospacer flanking site (PFS)6. These nucleotide chains prevent the system from mistakenly targeting host sequences, ensuring specificity during genome editing.
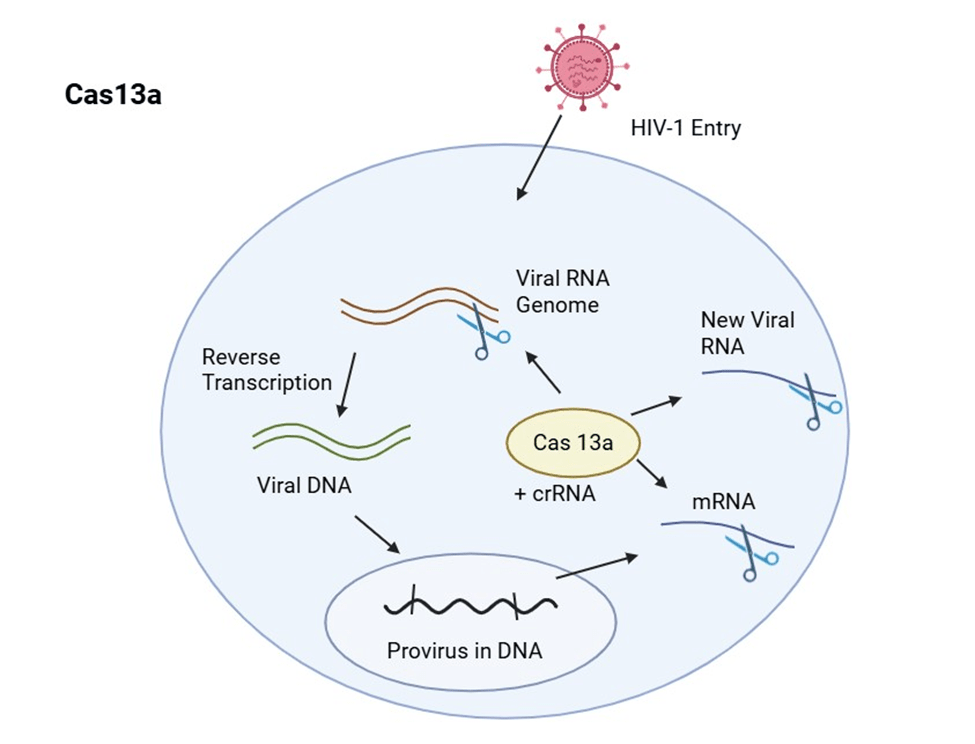
Cas9 and Cas12 nucleases both cleave strands of double-stranded DNA. Cas9 produces blunt ends while Cas12 produces staggered ends, both activating cellular DNA repair mechanisms. In the absence of a repair template, or donor template, the cell uses a quick but error-prone mechanism called non-homologous end joining (NHEJ) to reconnect the broken DNA strands. This method often introduces small insertions or deletions (INDELS) at the cut site, which can disrupt the transcription of the gene, leading to a loss of function. This disruption can be harnessed to inactivate integrated HIV proviral DNA or essential viral genes. Alternatively, by supplying a DNA donor template with specific edits flanked by homologous sequences to match the cut ends of the DNA, the cell can use the more precise homology-directed repair (HDR) pathway. HDR enables researchers to insert desired genetic sequences at the break site with increased precision. In contrast, Cas13 targets single-stranded RNA instead of DNA, making it fundamentally different from Cas9 and Cas12. Guided by cRNS and a PFS, Cas13 can recognize complementary sequences in viral RNA, making it particularly suitable for targeting the HIV genome in its RNA form prior to integration into host DNA. However, an approach implementing only Cas13 will not be able to access the already integrated HIV DNA in latent reservoirs. This review examines the differences and potentials of CRISPR-Cas approaches to inhibit HIV replication and preferably eliminate the viral reservoir.
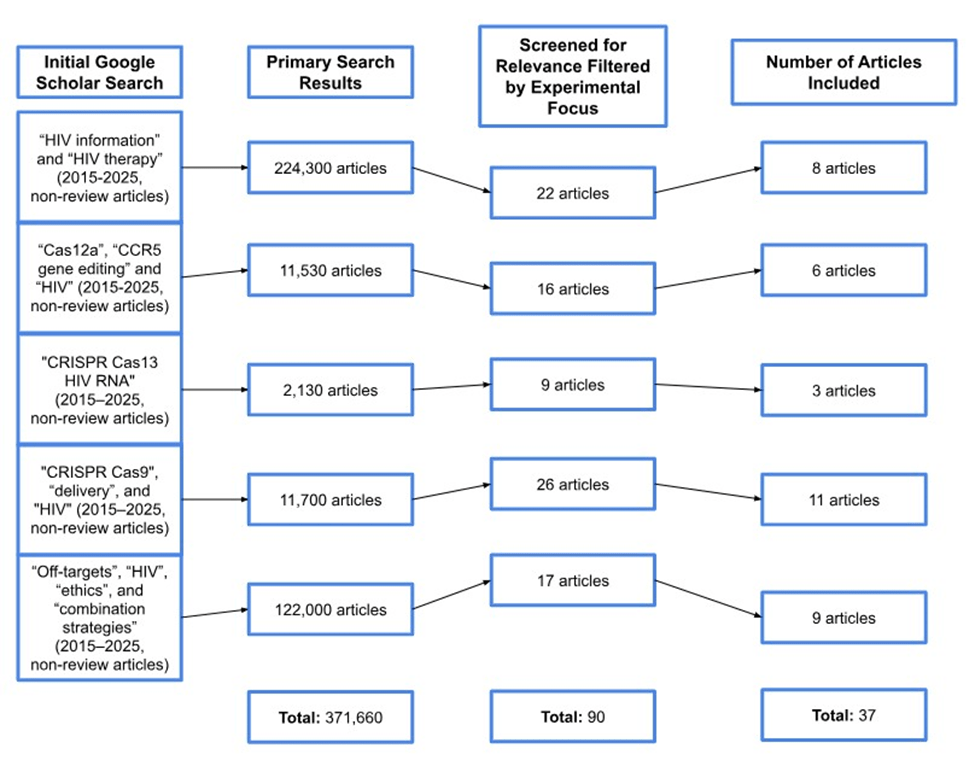
Methods
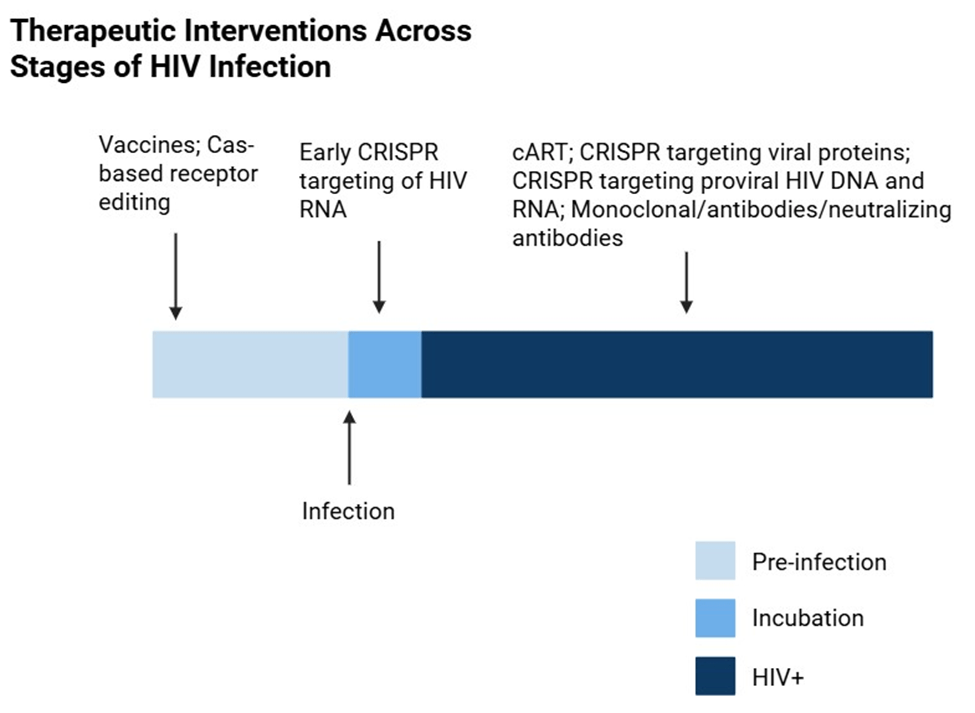
Peer-reviewed articles were used to conduct this literature review. These articles were found using PubMed, MEDLINE, and Google Scholar. Key search terms included “HIV-1”, “CRISPR-Cas9”, “CRISPR-Cas12a”, “CRISPR-Cas13”, and “gene editing”. Inclusion criteria required that papers be published within the last ten years (2015-2025) to ensure relevance to recent advancements in CRISPR-based antiviral research. Studies outside this range were excluded unless they provided essential knowledge on HIV biology or ethical considerations. Both in vitro and in vivo studies were examined, with in vitro studies categorized by human or animal cell models. Preference was given to studies with clearly documented experimental methods, results, and discussion of therapeutic potential. After evaluation of various methods presented in the literature, the decision was made to focus on the technologies of three CRISPR-Cas enzymes. For each therapy, multiple studies were selected to gain a comprehensive understanding of the mechanisms used, as well as to identify and discuss specific limitations and benefits of each system (see Figure 2). Due to the large volume of search results (>370,000 records), a fully exhaustive screening was not feasible. Instead, a targeted, concept-driven approach was used to identify representative experimental studies aligned with the major thematic sections of this review, including host receptor editing, proviral excision, RNA-targeting strategies, and in vivo delivery models.
Viral Escape and the Challenge of HIV Diversity
HIV’s rapid evolution is one of the main reasons it has been challenging to find a persistent and long-lasting cure. The virus replicates at extremely high rates inside the body; in vivo studies estimate an HIV-1 genome-wide spontaneous mutation rate on the order of ~4.1 × 10⁻³ mutations per base per infected cell, one of the highest measured for any biological system. Its reverse transcriptase enzyme also makes frequent copying mistakes7. These errors generate a large pool of genetic variants and mutations. Because many slightly different versions of the virus exist at any given time, some variants can naturally survive pressures that eliminate others. This high mutation rate also enables viral escape, a process where the virus develops changes that allow it to evade immune responses or medical treatments. Even small mutations in critical regions of the viral genome can have major consequences. For example, if the virus mutates the 20-nucleotide target sequence, the short stretch of viral DNA a therapy is designed to recognize, or changes the nearby PAM sequence, a short DNA signal required for binding, the treatment may no longer be able to recognize the proviral DNA. When this occurs, the virus becomes resistant to the targeting strategy, allowing the mutated variant to continue replicating while the original, sensitive version is suppressed. Because HIV exists as a constantly shifting population, strategies that focus on only one site in the viral genome are especially vulnerable. A single mutation at that location is often enough to prevent the therapy from identifying its target, allowing escape variants to replicate. This fragility is an important reason why modern approaches increasingly emphasize addressing several viral regions at once to make it harder for HIV to evade treatment.
Delivery Systems and Their Limitations
Even when CRISPR has a specific target sequence and the correct PAM site, one of the biggest obstacles is delivering that CRISPR/Cas into the target cells. One major challenge is the genome size of the payload. Some gene-editing components are large, and AAV vectors, which are used to deliver these gene-based therapies, can only carry about 4.7 kilobases of genetic material, which makes it difficult to package complete editing systems in a single vector8. Delivery into key immune cells, including resting CD4⁺ T cells that serve as latent HIV reservoirs, is inefficient due to low transduction efficiency and limited gene expression in these cells. Moreover, HIV persists in sanctuary sites such as the central nervous system (CNS), lymph nodes, and gut-associated lymphoid tissue, which are difficult to access because of physical barriers and reduced penetration of therapeutic agents. There are alternative delivery strategies, including lipid nanoparticles (LNPs), electroporation, and ribonucleoprotein (RNP) complexes, but each has limitations. LNPs may not target the right tissues, electroporation is mainly restricted to cells handled outside the body, and RNPs do not stay active long enough to reach all reservoirs9,10,11. These challenges are evident when looking at delivery strategies used in different contexts. When used on cells that were edited outside the body and then reinfused, delivery methods are limited to cell types that can safely be transplanted and removed12. When delivery is used directly in the body, reaching different hard-to-access tissues while maintaining high selectivity for the target cell type can be difficult. Overall, the difficulty of delivering editing tools to all relevant sites remains one of the biggest bottlenecks to achieving complete and durable treatment.
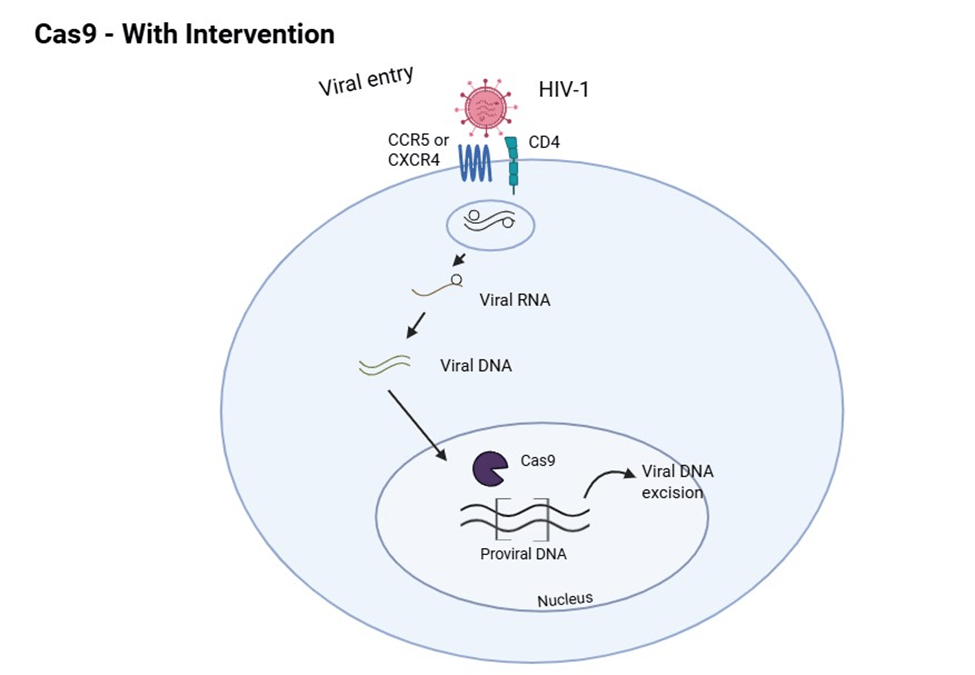
Targeting HIV-1 Proviral DNA
A central approach in CRISPR-Cas based HIV therapy involves directly targeting and disrupting integrated HIV-1 proviral DNA within the host genome. This strategy relies on the CRISPR-Cas9 system to introduce double-stranded DNA breaks at specific locations of the viral genome, guided by short guide RNAs. When the cell repairs these breaks via the NHEJ pathway, it often introduces insertions and deletions that can inactivate essential viral genes by shifting the reading frame or introducing early stop codons. Multiple studies have demonstrated the efficiency of this approach in vitro. For example, in J-Lat10.6 T-cells carrying R7/E-/GFP HIV, CRISPR-Cas9 was used to cut the viral DNA at two places: the LTR (long terminal repeat) and gag gene. These edits significantly reduced viral gene expression and p24 production, a protein that is utilized to mark HIV replication. When dual guide RNAs were used, the system further increased efficiency, editing up to 90% of the HIV DNA in the T-cells13. Similarly, Yin et al. delivered the CRISPR-Cas9 system and 2 guide RNAs into a humanized mouse model and achieved greater than 90% deletion of proviral DNA from multiple tissues, including the spleen, liver, lungs, and brain14. These findings provided early evidence that CRISPRCas9 could reach and edit viral reservoirs in vivo.
The therapeutic potential has also been validated in non-human primates. In a landmark study by Mancuso et al., macaques infected with SIV, a close relative of HIV that causes a similar disease in primates, were first treated with long-acting cART, followed by Cas9 constructs delivered via AAV9, a commonly used virus for safe delivery of genes into a cell15. These constructs targeted and successfully excised SIV DNA from places like lymph nodes and spleen, and showed no detectable viral load in some animals, suggesting the first potential instance of viral clearance in an animal model. More recently, Wang et. al. combined long-acting cART with CRISPR Cas9 targeting both the host CCR5 gene and the HIV provirus16. The CCR5 gene in human CD4+ T-cells is a receptor HIV uses to enter the cell. So, this dual-editing strategy allowed researchers to cut the virus, as well as make the CD4+ T cells harder to infect in the future. In their humanized mouse model, this combination led to viral elimination in 13 out of 23 animals, with no detectable off-target effects or viral rebound post-therapy.
Despite these promising results, several challenges still persist. One major hurdle is the risk of viral escape due to mutations introduced by NHEJ. Since NHEJ allows for the integration of new bases in the repair process, the virus become resistant to CRISPR targeting. For instance, singlesite targeting often leads to escape variants that are immune to the original CRISPR treatment, reducing long-term efficacy. To mitigate this, strategies using multiple gRNAs to simultaneously target multiple regions of the HIV virus have been developed. The targeting of several parts of the HIV genome at the same time reduces the potential for the virus to mutate and escape. Using two guides at once can cause >95% reduction, especially when they target conserved regions like the LTR, gag, or pol genes17. However, adding more guides creates trade-offs: multiplex targeting improves the chance of cutting out the virus, but it also makes the delivery vector larger and may increase the risk of off-target effects. Another limitation is the incomplete and uneven delivery of CRISPR components, particularly to latent reservoirs in sanctuary sites—places in the body that are harder to reach—such as the brain and lymph nodes where dormant HIV can evade the immune system.
While the usage of AAV9 has demonstrated its ability to reach many different tissues, including penetration into the brain, the efficiency of delivery to all infected cells is still substandard. Additionally, proviral excision may result in the formation of circle DNA fragments, which can persist and, under certain circumstances, retain its ability to transcript new proteins18. Safety concerns also remain; although studies to date have not observed widespread offtarget effects in animal models, the potential for Cas9 inducing host immune responses must be addressed before clinical translation. Therefore, while early studies have shown success in vitro and in animal models, future work must overcome the obstacles of delivery and safety to bring this approach closer to human application.
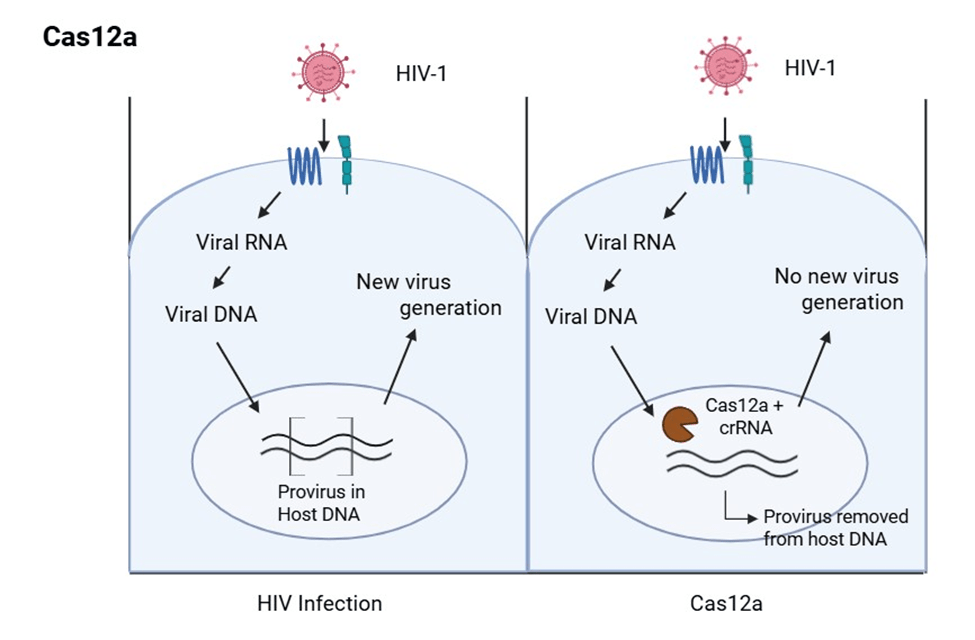
Targeting Host Genes Essential for HIV Entry
An alternative CRISPR-based strategy for combating HIV involves editing host genes required for viral entry, most notably CCR5 and CXCR4, receptors localized on the surface of immune cells that HIV utilizes to gain entry. Unlike proviral deletion strategies, which aim to remove viral integrated DNA, this approach seeks to render host cells resistant to infection by removing genes essential to HIV’s cellular access. It is especially promising because it is unaffected by HIV strain variations and mutations and avoids the challenge of eradicating latent reservoirs. Among the gene-editing tools available, CRISPR-Cas12a has emerged as a particularly powerful enzyme for this purpose. Cas12a differs from Cas9 in several notable ways that make it attractive for host gene targeting. Cas12a recognizes a T-rich PAM sequence, which expands the targetable regions of the genome beyond what Cas9 can access. It also generates staggered double-strand breaks, which may favor certain DNA repair outcomes, and it requires only a single guide RNA for target recognition. These attributes simplify the design and can potentially improve specificity.
Above all, Cas12a has demonstrated reduced off-target effects in some contexts compared to Cas9, which is especially valuable when editing human cells. Several recent studies have validated the feasibility of using Cas12a to disrupt the CCR5 protein. In a 2023 study, researchers used an engineered variant of Cas12a to achieve over 90% gene disruption in both human CD4+ T cells and early stem cells (HSPCs)19. This high efficiency was accomplished without detectable off-target effects, making it suitable for therapeutic applications. Another study demonstrated that when CCR5 was edited in HSPCs, followed by a bone marrow transplant of the HSPC cells into humanized mice, the edited stem cells successfully settled in the mice’s tissue and produced immune cells that lacked CCR5. Upon viral exposure, these mice showed significant protection from HIV infection, offering strong in vivo support for this host-targeting strategy20. In addition to stem cells, mature CD4+ T cells have also been successfully edited to prevent HIV entry. A 2021 study showed that using CRISPR editors to disrupt CCR5 in these cells led to over 80% protection against infection from HIV21. These findings underscore the potential of CRISPR-based host gene editing in creating immune cells resistant to HIV.
Regardless of its promise, however, the host gene disruption strategy is not without its own set of challenges. Achieving a complete knockout of CCR5 or CXCR4 in all immune cell populations in organisms remains difficult. Additionally, while individuals without CCR5 are generally healthy, the CXCR4 gene plays crucial roles in the body, and could therefore lead to on-target effects22. Editing CXCR4 could carry significant risks, emphasizing the need for extreme caution and safety testing before being adapted to therapeutic settings. Although Cas12a exhibits lower off-target effects in some contexts, extra care remains essential, particularly while targeting functionally important genomic regions23. Delivery remains another barrier, but advances in non-viral systems are making in vivo applications increasingly viable. For instance, a 2021 study demonstrated that Cas12a enzymes bound to guide RNA could be delivered ex vivo to primary human cells and then transplanted into humanized mice to generate stable gene modifications24. Although the study did not specifically target CCR5 to combat HIV resistance, it still provides evidence that the same delivery method could be applied to CCR5 editing. Disruptions using CRISPR-Cas12a of HIV entry proteins, mainly CCR5 and CXCR4, offer a promising alternative or even complement to traditional proviral targeting strategies.
Emerging RNA-Targeting Strategy with Cas13
While CRISPR-Cas9 and Cas12a have made significant strides in genome editing, the CRISPRCas13 system offers a novel approach by targeting RNA, presenting unique advantages for combating HIV. Cas13, another CRISPR enzyme, is programmed with CRISPR guide RNA (gRNA) to specifically bind and cleave single-stranded RNA targets. Unlike DNA-targeting systems, Cas13’s RNA-editing capabilities allows for temporary moderation of gene expression without permanent genomic changes, potentially reducing the risk of unintended genetic consequences. In the context of HIV, Cas13’s ability to break down the RNA of HIV presents a compelling strategy for inhibiting viral replication. Research has demonstrated that Cas13 can effectively target and cleave HIV RNA, leading to a reduction in viral gene expression and suppression of viral replication in infected cells25. This approach is particularly useful for targeting both newly synthesized viral RNA and RNA from latent reservoirs, which are challenging to eliminate using traditional DNA-targeting methods. Furthermore, Cas13’s programmability and specificity make it a promising tool for developing antiviral therapies that can be tailored to target specific viral strains or variants that may be created from HIV mutations. By designing gRNAs that match certain regions of the HIV genome, Cas13based therapies could cleave RNA from a wide array of HIV variants, potentially overcoming the challenges posed by HIV’s high mutation rate and the emergence of drug-resistant strains.
The CRISPR-Cas13 system represents a promising RNA-targeting strategy for HIV therapy. Its ability to degrade viral RNA, combined with its programmability and temporary effects, offers a new approach to inhibit HIV replication and address challenges associated with latent reservoirs and viral mutations. Continued research and development are essential to investigate the therapeutic potential of Cas13 in the fight against HIV.
The Case for Combination Strategies
While individual CRISPR-Cas enzymes offer powerful tools to target HIV, combining multiple strategies may maximize therapeutic effectiveness and help overcome limitations posed by viral diversity, latent reservoirs, and incomplete editing. For example, Cas9 or Cas12a can excise proviral DNA from infected cells, while Cas13 can simultaneously degrade viral RNA, reducing the production of new viruses26 (see Figure 5). Pairing these targeting strategies could increase the overall reduction of viral load and limit the emergence of escape variants. Additional approaches could be combined with gene editing to further enhance outcomes. CCR5 knockout in HSPCs or T cells can create HIV-resistant immune cells, while latencyreversing agents (“shock-and-kill”) can reactivate dormant virus, making it accessible to editing tools or the immune system27. Broadly neutralizing antibodies (bNAbs) or therapeutic vaccines could complement these strategies by neutralizing circulating virus or boosting immune recognition of infected cells27. Taken together, these combination strategies represent a multilayered approach to HIV therapy. By simultaneously addressing viral DNA, viral RNA, resistant cell populations, and latent reservoirs, such synergistic interventions may overcome the challenges that have limited the effectiveness of single-strategy treatments.
Safety Considerations: Off-Target Effects and Immunogenicity
When evaluating CRISPR-based strategies for HIV, safety remains a top concern. One major concern is off-target editing, which happens when a CRISPR enzyme cuts or modifies a piece of DNA or RNA that is similar to the intended target but not the sequence of interest. These unwanted edits could disrupt normal genes or cellular functions, so researchers use several methods to check where CRISPR cleaved the DNA or RNA. For example, studies assessing HIV-targeting CRISPR tools have used techniques such as whole-genome sequencing, GUIDE-seq, and targeted deep sequencing to scan the genome for possible errors28. In some experiments using Cas9 or Cas12a, off-target events were rare or undetectable with these methods, but other studies have still reported low-level off-target cutting when guide RNAs closely resemble human DNA sequences29 (see Table 1). Cas12a tends to be more selective than Cas9 because it requires a longer guide sequence and shows higher sensitivity to mismatches between the guide and target sequence, which generally results in lower off-target cleavage compared to Cas930. Cas13—which targets RNA instead of DNA—avoids permanent genetic changes but can sometimes cut nearby RNA after activation31. Together, the evidence shows that off-target effects remain a key safety consideration, even if newer systems are improving accuracy.
Another safety issue is immunogenicity, or the risk that the body recognizes CRISPR components as foreign. Some people naturally carry antibodies against certain Cas enzymes, such as SaCas9, because these enzymes come from bacteria humans may have been exposed to before32. Viral delivery systems like AAV vectors or previous exposure to AAV viruses, similar to SaCas9, may also trigger immune responses, which can reduce effectiveness or create inflammation. Managing these risks—through enzyme engineering, temporary delivery systems, or choosing less common Cas proteins—remains essential before CRISPR therapies can be used safely in humans.
Finally, ex vivo approaches that edit hematopoietic stem and progenitor cells (HSPCs) require patients to undergo conditioning, a process that prepares the bone marrow to integrate the edited HSPC cells. Conditioning is usually done with agents like busulfan or with total body irradiation, which reduce or eliminate the patient’s existing HSPC cells to “make space” for the edited HSPCs to take hold. While necessary for long-term engraftment, conditioning carries significant risks, including neutropenia, increased infection risk, liver and lung toxicity, infertility, and even the possibility of secondary cancers later in life33. These risks make full-intensity conditioning difficult to justify for people who are stable on cART. Emerging alternatives, such as reduced-intensity conditioning, antibody-based approaches that selectively clear stem cells (like anti-CD117 or anti-CD45), or in vivo delivery methods that edit cells directly inside the body, aim to reduce or eliminate the need for chemotherapy, but these strategies are still under investigation.
| Study | Cas | System | Delivery Method | Sample Size | Target | Editing Efficie ncy | Off Targets | Considera tions |
| Soria no (2017 ) | SpCas9 | J-Lat 10.6 T- cell line | Lentiviral vectors | 40,000 J-Lat cells | Integrat ed HIV proviral DNA (LTR and gag regions) | Up to ~90% editing with dual gRNAs | Limited offtarget analysis | In vitro latency model; dual gRNAs increase efficiency and reduce viral escape, but clinical translation limited by delivery and model system |
| Yin et. al. (2016 ) | SaCas9 | Transge nic mice and rats | rAAV9 vectors | Multiple animals per cohort (not explicitly number ed) | Integrat ed HIV1 proviral DNA (LTR and Gag) | Greater than 90% decrea se in gag/LT R; percent age varies by types of cell | Not fully genome- wide offtarget analysis; local PCR/seque ncing suggests minimal non-target cutting | Model uses transgenic HIV sequences , not natural infection |
| Manc uso et. al. (2020 ) | SaCas9 | SIVmac 239infected rhesus macaqu es on ART | AAV9 vectors | n = 45 tissues collecte d | Integrated SIV proviral DNA (5′LTR & gag) | Excisio n in vivo up to ~92% (varies by animal & tissue) | Top predicted off-targets tested; limited genome- wide offtarget profiling | Small cohort and limited statistics; |
| Wang et. al. (2025 ) | SpCas9 | HIV-1 infected humaniz ed mice with longacting cART | Viral vectors | n = 23 mice | Host CCR5 receptor & integrat ed HIV1 proviral DNA | Elimina tion in 58% of mice; evidenc e in multiple tissues | No evidence of off-target toxicities | Dual CRISPR therapy demonstrat ed statistically significant improveme nts from single treatments |
| Ham mad et. al. (2023 ) | Cas12a Ultra | Human CD4+ T cells, HSPCs, and iPSCs | Cas12a Ultra ribonucleop rotein (RNP) delivery via electroporat ion; AAV donor vectors for knock-in | Multiple indepen dent donorderived cell samples | Non-HIV genomic loci | >90% gene disrupti on in both CD4⁺ T cells and stem cells | Limited offtarget analysis | Not an HIV- specific study; demonstrat es highefficiency editing in HIVrelevant immune cells, supporting feasibility of Cas12abased strategies for future HIV application s |
| Pal et. al. (2025 ) | SpCas9 | Human CD34+ HSPCs; humaniz ed mice | Electropora tion | ~5–9 mice per group; 30,000 cells per mouse | Host genes (CCR5, IFNAR, RAG2, TCF7, HLA-A2) | >90– 95% gene knocko ut in vivo | Limited offtarget analysis | Study investigate s multiple host immune genes using a CRISPRCas9 humanized mouse platform; however, the discussion here focuses specifically on CCR5 knockout |
| Knippi ng et. al. (2022 ) | Cytosin e and Adenin e base editors | Primary human CD4+ T cells & CD34+ HSPCs | Electropora tion | Multiple donors | Host co‑rece ptors CCR5, CXCR4 | Up to ~95% (CCR5 ABE), ~89% (CBEs for stop codons ), ~88% (CXCR 4) | Low numbers of off-target mutations that were introduced by base editing | Focuses on editing host coreceptors (CCR5/CX CR4) to prevent HIV entry, not on targeting the virus itself |
| Yin et. al. (2020 ) | LbuCas 13a | HEK293 T & Jurkat cells | Plasmid transfection | n = 3 biologic al replicate s per conditio n | HIV‑1 RNA (viral transcrip ts & incomin g genome ) | ~50% reducti on in p24; ~60% reducti on in HIV-1 mRNA | Not comprehen sively assessed | Targets viral RNA to suppress infection in vitro |
The table includes key studies referenced and integrated into the review, summarizing basic information and statistics, as well as considerations in terms of the review.Ethical and Global Health Considerations
While gene-editing technologies offer hope for more durable HIV therapies, they also raise important ethical and socioeconomic questions. CRISPR-based treatments are extremely expensive—similar to current gene and cell therapies such as CAR-T treatments, which often cost hundreds of thousands to over a million dollars per patient when factoring in manufacturing, hospital care, and supportive treatments34. These high costs make it important to consider who will realistically benefit from CRISPR therapies if they reach the clinic. Most people living with HIV reside in low- and middle-income countries, where access to advanced medical infrastructure, specialized equipment, and long-term follow-up care is limited. This creates a mismatch between where HIV is most prevalent and where high-tech therapies can realistically be delivered.
Scaling up CRISPR-based HIV treatments also competes with other global health priorities. For many regions, investing in broader access to cART, improving prevention strategies, expanding diagnostic testing, and addressing social determinants of health may save more lives than an extremely costly curative approach available to only a small fraction of patients. Ethical concerns also arise around socioeconomic equality: if CRISPR cures become available, it is uncertain whether low-income populations or countries will have timely access, or whether these therapies will widen existing healthcare inequalities. Additionally, complex procedures like stem-cell editing or in vivo gene delivery require long-term monitoring and specialized facilities, raising questions about feasibility in resource-limited settings. Considering these issues early is essential. As CRISPR technologies continue to develop, researchers, clinicians, and policymakers will need to balance scientific progress with practical and ethical responsibility. Ensuring that future HIV cures are not only effective but also accessible and globally relevant remains a critical goal.
Discussion
CRISPR-based gene editing strategies, including Cas9, Cas12a, and RNA-targeting Cas13 systems, collectively represent a leading change in HIV therapeutic research. These technologies offer specificity, programmability, and the potential to cure HIV by disrupting proviral DNA or disabling proteins used for HIV cell entry. Their success both in vitro and in vivo underscores the high potential for a functional strategy. At the same time, even what is considered high efficiency in CRISPR Cas editing still rarely reaches 100% efficiency, leaving the possibility for unedited HIV+ T-cells, and CRISPR tools can trigger immune responses against Cas proteins or the viral vectors used to deliver them. These remaining cells, along with the immune reactions, show why more refinement is needed before CRISPR can fully overcome the limitations of cART. Furthermore, significant challenges still stand in the way of clinical translation. Key barriers remain, like off-target effects, inefficient delivery to latent reservoir sites, and the need for chemotherapy-based conditioning prior to gene editing. All of these factors raise the cost, complexity, and risk of the treatments. For example, autologous stem-cell transplants—when the patient’s own stem cells are collected, edited outside the body, then returned to the same patient— often require high-dose chemotherapy or radiation to wipe out the patient’s existing HSPC cells, so the CRISPR-edited cells will have room to engraft35. This adds toxicity and resource demands, which burden accessibility, particularly for regions lacking resources where HIV is most prevalent. Socioeconomic and real-world factors further complicate equitable implementation. As therapies like CRISPR approaches develop, high costs—such as those stemming from chemotherapy conditioning—will likely limit global access unless addressed. Achieving fairness in access remains a critical ethical goal. A synergistic approach may help overcome some of these obstacles. Combination strategies, like pairing proviral excision with Cas13 RNA targeting or combining gene-editing with cART therapies, may provide additive effects. For example, anti-HIV strategies coupling CCR5 knockout in HSPC cells with the secretion of antibodies that neutralize HIV from engineered plasma B cells have been shown to have a combined effect on long-term HIV resistance in animal models36. Similarly, pairing CRISPR strategies with drugs that can shock HIV cells in latent reservoirs out of the dormant phase may target both reservoirs and active infection. These examples prove that combining CRISPR edits with various other strategies can strengthen resistance more than one strategy alone, but latent reservoirs remain difficult to fully eliminate even with multiple therapies, which is why combination strategies will still need continued refinement.
Beyond technical barriers and safety considerations, future CRISPR-based interventions must also be evaluated through an ethical and global health lens. Clinical trials will need to balance potential benefits with risks, including off-target mutations, immune responses, and the long-term consequences of genome editing. Ensuring meaningful informed consent is essential, given the scientific complexity of gene-editing and the vulnerability of many populations affected by HIV. Fairness and global access must also guide the development of these therapies: mechanisms such as tiered pricing models and technology transfer to low-resource regions may help ensure that any successful CRISPR-based strategies do not remain accessible only to wealthy healthcare settings.
Looking forward, research must prioritize improving delivery methods, refining specificity (minimizing off-target risks), and minimizing conditioning toxicity. Researchers are exploring safer alternatives to full-intensity chemotherapy. Approaches like reduced-intensity regimens, antibody-based conditioning (such as antibodies targeting CD117 or CD45), and direct in-vivo delivery of gene-editing tools aim to prepare the body for edited cells with much less toxicity. These developments matter because the risks of full-intensity conditioning are difficult to justify for people who are stable on cART, since the current treatment already keeps them healthy without exposing them to major side effects. The development of non-chemo approaches or delivering CRISPR directly into the body could reduce the need for chemotherapy altogether. Evidence of progress in delivery innovation comes from recent CRISPR trials using AAV9 vectors in humanized mice, signaling movement toward more practical in vivo therapy37. Ultimately, for CRISPR-based strategies to evolve from experimental promise to reality, they must navigate not only technical complexities, but also financial and ethical concerns as well.
Conclusion
CRISPR technologies represent a shift in HIV therapy. They hold the unique possibility of eradicating HIV from reservoirs, rendering HIV+ cells HIV-resistant, or suppressing viral replication at the RNA level. Yet, persistent challenges such as delivery inefficiency, safety concerns, high treatment costs, and the need for conditioning via chemotherapy must be overcome, and translation from preclinical models to safe, accessible therapies will likely require at least another decade of research, clinical trials, and health systems innovationFuture progress hinges on innovative delivery methods, safer and cheaper conditioning protocols, and treatment regimens that combine gene editing with cART and other existing methods. Moreover, to assure equality, global access and ethical concerns will be essential to analyze and address. While no CRISPRbased HIV therapy has yet been proven safe or effective in humans, these emerging technologies may—pending successful clinical trials and regulatory approval—contribute to the development of a functional cure. Their continued refinement offers hope for a future in which some individuals may achieve long-term remission without daily antiretroviral therapy.
References
- World Health Organization. HIV data and statistics. https://www.who.int/teams/global-hiv-hepatitis-and-stis-programmes/hiv/strategicinformation/hiv-data-and-statistics. (2025). [↩]
- M. Weichseldorfer, M. Reitz, O. S. Latinovic. Past HIV-1 medications and the current status of combined antiretroviral therapy options for HIV-1 patients. Pharmaceutics. 13 (2021). [↩]
- German Advisory Committee Blood (Arbeitskreis Blut), Subgroup ‘Assessment of Pathogens Transmissible by Blood’. Human immunodeficiency virus (HIV). Transfusion Medicine and Hemotherapy. 43, 203-222 (2016). [↩]
- J. Chen, T. Zhou, Y. Zhang, S. Luo, H. Chen, D. Chen, C. Li, W. Li. The reservoir of latent HIV. Frontiers in Cellular and Infection Microbiology. 12 (2022). [↩]
- E. K. Maina, A. A. Adan, H. Mureithi, J. Muriuki, R. M. Lwembe. A review of current strategies towards the elimination of latent HIV-1 and subsequent HIV-1 cure. Current HIV Research. 19, 14-26 (2021). [↩]
- M. Hussein, M. A. Molina, B. Berkhout, E. Herrera-Carrillo. A CRISPR-Cas cure for HIV/AIDs. International Journal of Molecular Sciences. 24 (2023). [↩]
- J. M. Cuevas, R. Geller, R. Garijo, J. Lopez-Aldeguer, R. Sanjuan. Extremely high mutation rate of HIV-1 in vivo. PLoS Biology. 13 (2015). [↩]
- B. Kantor, B. O’Donovan, J. Rittiner, D. Hodgson, N. Lindner, S. Guerrero, W. Dong, A. Zhang, O. ChibaFalek. The therapeutic implications of all-in-one AAV-delivered epigenome-editing platform in neurodegenerative disorders. Nature Communications. 15, 7259 (2024). [↩]
- K. Chen, H. Han, S. Zhao, B. Xu, B. Yin, A. Lawanprasert, M. Trinidad, B. W. Burgstone, N. Murthy, J. A. Doudna. Lung and liver editing by lipid nanoparticle delivery of a stable CRISPR–Cas9 ribonucleoprotein. Nature Biotechnology. 43, 1445-1457 (2024). [↩]
- S. Demirci, K. Essawi, P. Germino-Watnick, X. Liu, W. Hakami, J. F. Tisdale. Advances in CRISPR delivery methods: perspectives and challenges. The CRISPR Journal. 5, 660-676 (2022). [↩]
- Z. Molaei, Z. Jabbarpour, A. Omidkhoda, N. Ahmadbeigi. Exploring non-viral methods for the delivery of CRISPR-Cas ribonucleoprotein to hematopoietic stem cells. Stem Cell Research and Therapy. 15, 233 (2024). [↩]
- F. Uddin, C. M. Rudin, T. Sen. CRISPR gene therapy: applications, limitations, and implications for the future. Frontiers Oncology. 10 (2020). [↩]
- V. Soriano. Gene therapy with CRISPR/Cas9 coming to age for HIV cure. AIDs Reviews. 19, 167-172 (2017). [↩]
- R. Kaminski, R. Bella, C. Yin, J. Otte, P. Ferrante, H. E. Gendelman, H, Li, R. Booze, J. Gordon, W. Hu, K. Khalili. Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene therapy. 23, 690-695 (2016). [↩]
- P. Mancuso, C. Chen, R. Kaminski, J. Gordon, S. Liao, J. A. Robinson, M. D. Smith, H. Liu, I. K. Sariyer, R. Sariyer, T. A. Peterson, M. Donadoni, J. B. Williams, S. Siddiqui, B. A. Bunnell, B. Ling, A. G. MacLean, T. H. Burdo, K. Khalili. CRISPR based editing of SIV proviral DNA in ART treated non-human primates. Nature Communications. 11 (2020). [↩]
- J. Wang, J. Liu, J. Xun. CCR5 gene editing and HIV immunotherapy: current understandings, challenges, and future directions. Frontiers in Immunology. 16 (2025). [↩]
- G. Wang, N. Zhao, B. Berkhout, A. T. Das. A combinatorial CRISPRCas9 attack on HIV-1 DNA extinguishes all infectious provirus in infected T Cell cultures. Cell reports. 17, 2819-2826 (2016). [↩]
- M. Lai, E. Maori, P. Quaranta, G. Matteoli, F. Maggi, M. Sgarbanti, S. Crucitta, S. Pacini, O. Turriziani, G. Antonelli, J. L. Heeney, G. Freer, M. Pistello. CRISPR/Cas9 ablation of integrated HIV-1 accumulates proviral DNA circles with reformed long terminal repeats. Journal of Virology. 95 (2021). [↩]
- R. Hammad, J. Alzubi, M. Rhiel, K. O. Chmielewski, L. Mosti, J. Rositzka, M. Heugel, J. Lawrenz, V. Pennucci, B. Gläser, J. Fischer, A. Schambach, T. Moritz, N. Lachmann, T. I. Cornu, C. Mussolino, R. Schäfer, T. Cathomen. CRISPR-Cas12a for highly efficient and marker-free targeted integration in human pluripotent stem cells. International Journal of Molecular Sciences. 25 (2024). [↩]
- P. Pal, S. Gao, H. Gao, X. Qin, M. Cella, Q. Wang, L. Shan. Establishment of a reverse genetics system for studying human immune functions in mice. Science Advances. 11 (2025). [↩]
- F. Knipping, G. A. Newby, C. R. Eide, A. N. McElroy, S. C. Nielsen, K. Smith, Y. Fang, T. I. Cornu, C. Costa, A. Gutierrez-Guerrero, S. P. Bingea, C. J. Feser, B. Steinbeck, K. L. Hippen, B. R. Blazar, A. McCaffrey, C. Mussolino, E. Verhoeyen, J. Tolar, D. R. Liu, M. J. Osborn. Disruption of HIV-1 co-receptors CCR5 and CXCR4 in primary human T cells and hematopoietic stem and progenitor cells using base editing. Molecular Therapy. 30, 130-144 (2021). [↩]
- G. Alkhatib. The biology of CCR5 and CXCR4. Current Opinion in HIV and AIDs. 4, 96-103 (2009). [↩]
- S. V. Baranova, P. V. Zhdanova, P. E. Pestryakov, A. A. Chernonosov, V. V. Koval. Key thermodynamic characteristics of Cas9 and Cas12a endonucleases’ cleavage of a DNA substrate containing a nucleotide mismatch in the region complementary to RNA. Biochemical and Biophysical Research Communications. 768 (2025). [↩]
- N. Kruglova, M. Shepelev. Increasing gene editing efficiency via CRISPR/Cas9- or Cas12a-mediated knock-in in primary human T cells. Biomedicines. 12, 119 (2024). [↩]
- L. Yin, F. Zhao, H. Sun, Z. Wang, Y. Huang, W. Zhu, F. Xu, S. Mei, X. Liu, D. Zhang, L. Wei, S. Cen, S. Hu, C. Liang, F. Guo. CRISPR-Cas13a inhibits HIV-1 infection. Molecular Therapy – Nucleic Acids. 21, 147-155 (2020). [↩]
- N. Zhao, G. Wang, A. T. Das, B. Berkhout. Combinatorial CRISPR-Cas9 and RNA interference attack on HIV-1 DNA and RNA can lead to cross-resistance. Antimicrobial Agents and Chemotherapy. 61, (2017). [↩]
- A. J. Atkins, A. G. Allen, W. Dampier, E. K. Haddad, M. R. Nonnemacher, B. Wigdahl. HIV-1 cure strategies: Why CRISPR? Expert Opinion on Biological Therapy. 21, 781-793 (2021). [↩] [↩]
- C. Chung, A. G. Allen, A. J. Atkins, N. T. Sullivan, G. Homan, R. Costello, R. Madrid, M. R. Nonnemacher, W. Dampier, B. Wigdahl. Safe CRISPR-Cas9 inhibition of HIV-1 with high specificity and broad-spectrum activity by targeting LTR NF-κB binding sites. Molecular therapy, Nucleic acids. 21, 965-982 (2020). [↩]
- L. Yang, D. Grishin, G. Wang, J. Aach, C. Zhang, R. Chari, J. Homsy, X. Cai, Y. Zhao, J. Fan, C. Seidman, J. Seidman, W. Pu, G. Church. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nature Communications. 5, 5507 (2014). [↩]
- S. Khan, E. Sallard. Current and prospective applications of CRISPR-Cas12a in pluricellular organisms. Molecular Biotechnology. 65, 196-205 (2023). [↩]
- N. Gunitseva, M. Evteeva, A. Borisova, M. Patrushev, F. Subach. RNADependent RNA targeting by CRISPR-Cas systems: characterizations and applications. International Journal of Molecular Science. 24, 6894 (2023). [↩]
- C. T. Charlesworth, P. S. Deshpande, D. P. Dever, J. Camarena, V. T. Lemgart, M. K. Cromer, C. A. Vakulskas, M. A. Collingwood, L. Zhang, N. M. Bode, M. A. Behlke, B. Dejene, B. Cieniewicz, R. Romano, B. J. Lesch, N. Gomez-Ospina, S. Mantri, M. Pavel-Dinu, K. I. Weinberg, M. H. Porteus. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nature Medicine. 25, 249-254 (2019). [↩]
- B. Gyurkocza, B M. Sandmaier. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood. 124, 344-353 (2014). [↩]
- J. Rueda, I. M. Beriain, L. Montoliu. Affordable pricing of CRISPR treatments is a pressing ethical imperative. The CRISPR Journal. 7, 220-226 (2024). [↩]
- V. Buffa, J. B. A. Vargas, A. Galy, S. Spinozzi, C. J. Rocca. Hematopoietic stem and progenitors cells gene editing: Beyond blood disorders. Frontiers in Genome Editing. 4 (2023). [↩]
- W. N. Feist, S. E. Luna, K. Ben-Efraim, M. V. Filsinger Interrante, A. Amorin, N. M. Johnston, T. U. J. Bruun, A. Utz, H. Y. Ghanim, B. J. Lesch, T. M. McLaughlin, A. M. Dudek, M. H. Porteus. Multilayered HIV-1 resistance in HSPCs through CCR5 knockout and B cell secretion of HIV-inhibiting antibodies. Nature Communications. 16 (2025). [↩]
- O. Humbert, C. Samuelson, H. Kiem. CRISPR/Cas9 for the treatment of haematological diseases: A journey from bacteria to the bedside. British Journal of Haematology. 192, 33-49 (2020). [↩]



{kind=link}