
Abstract
Low-dose computed tomography (LDCT) screening has reduced lung cancer mortality but is limited by low specificity and a high rate of indeterminate pulmonary nodules, leading to unnecessary follow-up imaging and invasive procedures. These challenges underscore the need for complementary, noninvasive biomarkers to improve early detection and risk stratification. Circulating tumor DNA (ctDNA), a tumor-derived component of cell-free DNA, has emerged as a promising candidate for early lung cancer detection, although its clinical application is constrained by low tumor DNA abundance in early-stage disease and methodological variability. The objective of this review is to critically assess ctDNA-based approaches for early detection of non–small cell lung cancer (NSCLC), focusing on analytical strategies, clinical validation, and translational considerations. We conducted a systematic review of peer-reviewed studies published between 2015 and 2025 that reported quantitative diagnostic performance of ctDNA assays in early-stage lung cancer or screening-related settings. Studies were grouped by assay modality, including mutation-based sequencing, DNA methylation profiling, fragmentomic analysis, and multi-omic integration, and were synthesized qualitatively. Recent advances in error-corrected next-generation sequencing, methylation-based classifiers, and genome-wide fragmentation analysis have improved ctDNA detection in low-shedding tumors. Reported diagnostic performance ranged from area under the curve values of approximately 0.76 to 0.95, with selected platforms achieving stage I sensitivities of 70–90% in enriched cohorts at high specificity. However, performance varied substantially due to biological factors, preanalytical variability, and confounding from clonal hematopoiesis. In conclusion, ctDNA-based assays hold significant promise as complements to LDCT, particularly for pulmonary nodule evaluation. Prospective validation, assay standardization, and carefully designed clinical integration pathways are required before routine screening implementation.
Keywords: Circulating tumor DNA; Cell-free DNA; Early detection; Non–Small Cell Lung Cancer; Liquid biopsy
Introduction
Lung cancer is the leading cause of cancer mortality worldwide, largely due to late diagnosis. LDCT screening of high-risk individuals can detect early lung cancers and has shown a ~20% mortality reduction1. However, LDCT’s limited specificity results in a high false-positive rate and many indeterminate pulmonary nodules requiring invasive follow-up1. There is a critical need for noninvasive biomarkers to complement LDCT by distinguishing malignant from benign nodules and identifying cancers at an early stage.
Circulating tumor DNA (ctDNA)—fragments of tumor-derived DNA released into the bloodstream by apoptotic and necrotic tumor cells—has shown promise as an early detection biomarker2,3,4. In plasma, ctDNA typically constitutes a tiny fraction of total cell-free DNA, especially in patients with localized tumors. Emerging ultrasensitive techniques can detect these rare tumor-specific DNA fragments amidst abundant normal cfDNA. Recent studies have reported successful detection of early-stage (including stage I) lung cancers via ctDNA analysis3,5,6 leveraging advances in next-generation sequencing (NGS) and machine learning3. At the same time, important challenges remain: not all early lung tumors shed sufficient ctDNA for detection, and various biological and technical factors can confound results.
Objectives: This review provides a comprehensive technical overview and critical evaluation of ctDNA assays for early lung cancer detection. We outline the current ctDNA detection platforms and their performance metrics, assess clinical validation data, and discuss biological and methodological factors influencing ctDNA detection. We also examine how ctDNA-based screening could be integrated with existing practices and highlight future research directions needed to translate these assays into routine clinical use. By explicitly delineating these aims, we categorize our work as a technical review of ctDNA methodologies with a critical assessment of validation studies and translational considerations for early lung cancer detection.
Methods
Literature Search Strategy: We performed a systematic literature search to identify peer-reviewed studies on ctDNA for early lung cancer detection. The search covered PubMed and Web of Science (last search date: August 15, 2025) using keywords such as “ctDNA”, “early lung cancer”, “screening”, “circulating tumor DNA”, “methylation”, and “fragmentomics”. We restricted the search to articles published in 2015–2025 to capture recent technological advances. Additional references were identified by screening bibliographies of relevant papers. This review was conducted in accordance with PRISMA 2020 guidelines7.
Inclusion and Exclusion Criteria: We included primary research articles that reported on ctDNA-based assays for lung cancer early detection or diagnosis (case–control studies, cohort studies, and interventional trials evaluating sensitivity and specificity of ctDNA tests in patients with early-stage lung cancer or in screening populations). Studies focusing on advanced/metastatic disease were included only if they provided specific analyses of early-stage subsets. We excluded review articles, meta-analyses (except for background context), case reports, and studies without performance data (sensitivity/specificity) for lung cancer detection. Non-English articles were excluded.
Study Selection: The initial database search yielded 512 records. After removal of duplicates, 480 titles/abstracts were screened. Based on relevance and criteria, 87 full-text articles were assessed in detail, of which 53 studies met all inclusion criteria and were included in this review. Study selection and inclusion are summarized in a PRISMA 2020 flow diagram (Figure X).7 Any discrepancies in study inclusion were resolved by consensus among the authors. We focused on studies reporting quantitative performance metrics (sensitivity, specificity, AUC) of ctDNA assays in early-stage lung cancer.
Data Extraction and Synthesis: For each included study, we extracted key data including assay type (mutation panel, methylation, fragmentomic, or multi-omic), study design (prospective vs retrospective; cohort details), and diagnostic performance (sensitivity, specificity, AUC, and limit of detection if available). We also noted reported findings on biological correlates of ctDNA (tumor features) and technical parameters. Findings were synthesized qualitatively and organized by technological approach and clinical context. Given heterogeneity of study designs, a formal meta-analysis was not performed.
Quality Assessment: We prioritized larger studies and those with independent validation cohorts to ensure robustness of conclusions. We noted potential biases (case–control design or enriched high-risk cohorts) when interpreting performance estimates.
Inclusion and Exclusion Criteria
Studies were eligible for inclusion if they met the following criteria:
- Reported primary data on ctDNA- or cfDNA-based assays for early-stage lung cancer detection, pulmonary nodule assessment, or screening-related contexts.
- Provided quantitative performance metrics, including sensitivity, specificity, and/or area under the receiver operating characteristic curve (AUC).
- Included sufficient technical detail on assay methodology, biomarker type, or analytical pipeline.
Studies were excluded if they:
- Focused exclusively on advanced or metastatic disease without early-stage subgroup analysis, lacked diagnostic performance metrics.
- Were non–peer reviewed, non-English, or case reports,
- were review articles or meta-analyses (except for contextual background).
Study Selection
The database search identified 512 records. After removal of duplicate records (n = 32), 480 records were screened by title and abstract. Of these, 454 records were excluded based on relevance and predefined criteria. Twenty-six full-text articles met all inclusion criteria and were included in the qualitative synthesis as representative studies for this systematic technical review.
Any uncertainties regarding eligibility were resolved through consensus among the authors.
Data Extraction and Synthesis
For each included study, data were extracted on:
ctDNA assay type (mutation-based, methylation-based, fragmentomic, or multi-omic), Study design and cohort characteristics, Biomarkers analyzed, Reported diagnostic performance metrics (sensitivity, specificity, AUC).
Findings were synthesized narratively and organized by technological approach and clinical context. Given heterogeneity in study design, patient populations, and assay platforms, a formal meta-analysis was not performed.
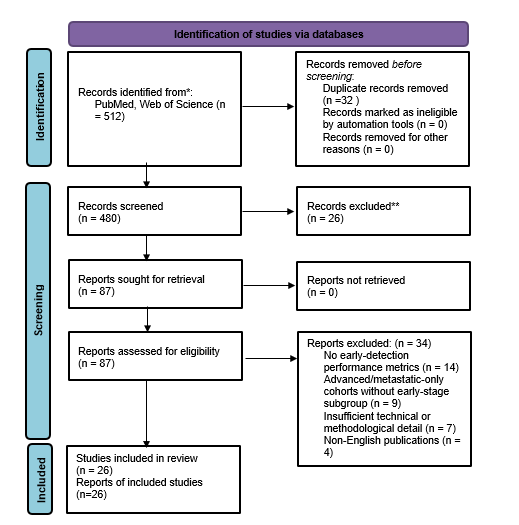
The diagram summarizes the identification, screening, retrieval, eligibility assessment, and inclusion of studies in this systematic technical review conducted in accordance with PRISMA 2020 guidelines8.
Results
Advances in high-sensitivity sequencing and novel biomarkers—including tumor-specific mutations, DNA methylation patterns, fragmentomic features, and copy-number alterations—have improved ctDNA detection in early-stage disease9,10,11,12, where tumor DNA shedding is minimal. Reported AUC values range from approximately 0.76 to 0.95, with some platforms demonstrating stage I sensitivities of 70–90% in enriched cohorts13,14,12. However, performance varies substantially due to biological factors such as tumor size, histology, and proliferation rate, as well as technical variables including preanalytical handling and clonal hematopoiesis-related false positives6,15,15,16.
Conclusions
While ctDNA-based assays show substantial promise as complements to LDCT, most supporting evidence derives from retrospective or enriched cohorts. Large prospective trials, standardized protocols, and careful integration into clinical pathways are required before routine screening adoption.
ctDNA Detection Approaches and Platforms
Early lung cancers release only trace amounts of ctDNA into circulation, so detection requires highly sensitive methods that distinguish tumor-specific signals from a high background of normal DNA. Several assay strategies have been developed. These include targeted mutation sequencing, methylation profiling, fragment size analysis, and combinations thereof. Each approach has distinct advantages and limitations in terms of sensitivity and specificity.
Mutation-Based ctDNA Assays
Conventional approaches focus on detecting cancer-specific mutations in plasma DNA. Techniques such as digital PCR and advanced NGS panels with error-suppression methods have been used to query hotspot mutations or personalized tumor mutations. For example, CAPP-Seq and related ultra-deep sequencing approaches can detect mutant allele fractions below 0.1%17. However, in early-stage NSCLC, mutation-based assays often show limited sensitivity because only a subset of tumors carry detectable recurrent mutations and the ctDNA fraction is extremely low. Early-stage cohorts frequently show low detection rates without multi-omic augmentation3,18. Mutation panels are also vulnerable to false positives from clonal hematopoiesis if leukocyte-derived variants are not filtered18,16,19.
Recent advances have improved mutation detection sensitivity. Error-corrected NGS, unique molecular identifiers (UMIs), and deeper sequencing can improve detection, but mutation-only tests remain most effective when integrated with other biomarkers due to limited shedding and genomic heterogeneity of early lung cancers.
DNA Methylation-Based Assays
DNA methylation changes are ubiquitous in cancer and often occur early in tumorigenesis, making them attractive biomarkers. Methylation-based ctDNA assays profile tumor-specific CpG methylation patterns in cfDNA. For example, the LUNG-TRAC assay achieved an AUC of 0.810 with approximately 74% sensitivity and 74% specificity for distinguishing malignant versus benign pulmonary nodules5.
More recently, a multi-center study integrated targeted methylation sequencing with machine learning (ESim-seq) and reported strong performance for early-stage lung cancer detection, including stage I sensitivity around 76.5% at high specificity18. Other genome-wide methylation approaches have reported even higher accuracy. For instance, a large case-control study by Liu et al. used a large methylation marker panel20 and machine learning to detect multiple cancer types including lung; for lung cancer, sensitivity was ~59-86% (stage I-III) at >99% specificity.
These results support the value of methylation signatures in low-tumor-burden disease.
Fragmentomics and Copy Number Analysis
Beyond sequence changes, tumors impart physical and structural differences to cfDNA fragments. Fragmentomics examines cfDNA fragment size distributions, end-motif frequencies, and nucleosome footprint patterns, which can distinguish ctDNA even when mutations are not detected4,6,21. End-motif signatures and genome-wide fragmentation features have been associated with cancer-specific patterns18.
A genome-wide fragmentation approach demonstrated the feasibility of detecting some early cancers years before conventional diagnosis, illustrating the sensitivity gains possible with fragmentation-based signals21.
Copy number alterations can be detected by low-pass whole-genome sequencing, but typically require higher tumor fractions; they are therefore often incorporated as part of integrated models rather than standalone early-screening markers.
Multi-omic Integration
Multi-omic assays integrate mutations, methylation, fragmentomics, and/or copy-number features to improve overall accuracy. Integrated models often achieve higher AUC than single-modality assays, but must be interpreted cautiously because overfitting and limited external validation can inflate performance estimates18,22,23. The SPOT-MAS platform is one example of a multimodal approach validated for multi-cancer detection22.
| Assay | Biomarkers | Stage I Sensitivity | Specificity | AUC | Notes |
| LUNG-TRAC | Targeted DNA methylation | ≈74% | ≈74% | 0.810 | Pulmonary nodule assessment; cohort-specific performance |
| ESim-seq | Fragmentation + methylation | ≈76.5% | ≈96% | 0.948 | Large training and validation; multi-center study |
| SPOT-MAS | Methylation + fragmentomics + CNA | ≈60% (early-stage) | ≈96% | ≈0.94 | Multi-cancer context; stage-specific sensitivity varies |
| CAPP-Seq | Mutation panel (error-corrected) | Variable; often lower in stage I | High | — | Mutation-only sensitivity limited by low shedding |
| Fragmentomics (genome-wide) | Fragment size/end motifs | Often higher than mutation-only in case–control | Moderate–high | ≈0.90 | Requires careful external validation |
Notes: AUC = area under the ROC curve. NSCLC = non–small cell lung cancer. Cross-study comparisons should be made cautiously due to cohort and protocol differences
Biological Factors Influencing ctDNA Release
ctDNA shedding varies substantially across patients and tumor types. Tumor size and stage correlate strongly with detectability; small (T1) tumors often shed ctDNA near or below assay limits3,23. Histologic subtype, vascular invasion, proliferation rate, and microenvironmental factors can influence ctDNA release, contributing to reduced sensitivity in genuine screening settings.
Discussion
Preanalytical and Technical Variables in ctDNA Testing
Preanalytical variability in blood tube type, processing delays, centrifugation protocols, storage conditions, and DNA extraction methods can dilute ctDNA with leukocyte genomic DNA or alter fragment profiles, reducing assay reproducibility24,15. Controlled comparisons have shown measurable effects of processing choices on cfDNA yield and quality15.
Technical Advances Enabling Early-Stage Detection
Biology that matters for assay design
Early-stage NSCLC is a worst-case scenario for blood-based detection: total cfDNA is dominated by hematopoietic sources, while tumor-derived fragments can be below parts-per-million. ctDNA abundance is shaped by tumor size, vascularity, proliferation/apoptosis rate, necrosis, and anatomic factors that influence drainage into circulation. Clearance is rapid (minutes–hours), meaning timing, handling, and physiologic state can measurably shift signal-to-noise. These constraints explain why mutation-only panels frequently underperform in true stage I screening cohorts, and why orthogonal signals (methylation, fragmentomics) often deliver higher sensitivity at fixed specificity. Fragment length distributions also reflect biology: ctDNA is often shorter and shows distinct end-motif and nucleosome-footprint patterns tied to chromatin organization and nuclease activity, providing signal even when no mutation is detectable3,4,6,21.
Mutation detection: from “deep sequencing” to “ultra-deep with structure-aware error models”
Mutation-based assays have advanced from hotspot ddPCR/BEAMing toward large-panel, error-corrected sequencing using unique molecular identifiers (UMIs) and duplex/consensus read families. The key technical barrier is not simply depth, but distinguishing true low-allele variants from polymerase/sequencing artifacts and context-specific background. Modern pipelines use molecular barcodes, strand consensus, position-specific noise models, and cross-sample artifact blacklists. However, mutation-only strategies remain limited in screening for three reasons: (i) early tumors may shed too little ctDNA for confident variant calling even at extreme depth; (ii) the number of queried loci is finite, limiting sensitivity when ctDNA molecules are scarce; and (iii) biologic confounding from clonal hematopoiesis (CHIP) can produce apparently “actionable” variants in plasma that originate from blood cells, not tumor25,16,17.
DNA methylation: why it often wins in stage I disease
Methylation changes are widespread, occur early in tumorigenesis, and provide high-dimensional signal across many CpGs, making them well-suited for low-shedding disease. Technically, assays span bisulfite conversion panels, enzymatic conversion workflows, and immunoprecipitation-based approaches (e.g., cfMeDIP-seq) that can be more efficient at low input. Compared with mutation detection, methylation readouts can interrogate a much larger effective feature space per molecule, enabling machine-learning classifiers to detect subtle shifts in tumor-derived fragments. Large multi-cancer methylation studies demonstrate that targeted methylation can maintain very high specificity while enabling cancer signal origin prediction, which is operationally important for follow-up workflows26,20,27.
Fragmentomics: genome-wide signal without knowing mutations
Fragmentomics methods analyze fragment size profiles, end-motifs, coverage oscillations, and nucleosome footprints. Their appeal for screening is that they can leverage genome-wide patterns, effectively increasing “signal bandwidth” beyond a limited mutation panel. In lung cancer, fragmentation-based classifiers (including DELFI-style approaches) have shown strong discrimination in validation settings, and fragment features can be integrated with other modalities (methylation, CNA) to improve robustness. A practical advantage is that shallow sequencing can sometimes suffice for certain fragment features, which may be cost-relevant, though performance is highly dependent on consistent preanalytics and stable library prep4,6,21.
Multi-omic fusion and “clinical LOD”: making comparisons less misleading
A recurring issue in the literature is that AUCs across case–control studies are not directly comparable when prevalence, stage mix, and sample handling differ. Multi-omic fusion can improve AUC by combining partially independent signals (e.g., methylation + fragmentation + CNA), but also increases modeling degrees of freedom and risk of overfitting if external validation is limited. Recent MCED work proposed a “clinical limit of detection” concept tying performance to tumor fraction distributions, providing a more realistic way to compare platforms across cohorts and to anticipate performance degradation when moving from enriched studies to population screening23.
Tumor-informed ultrasensitive ctDNA: relevance to “early detection” vs “early intervention”
Tumor-informed approaches (personalized variant sets derived from tumor tissue) can achieve parts-per-million sensitivity and are extremely powerful for MRD and recurrence risk stratification. Translationally, they also inform what is feasible for preoperative detection and risk prediction in early-stage disease, although they are not primary screening tools because they require tumor tissue. Recent early-stage lung cancer analyses using tumor-informed, very-low-LOD strategies illustrate both the promise and the ceiling: even with ppm-level detection and high specificity, a meaningful fraction of stage I patients remain ctDNA-negative preoperatively, reinforcing the need for orthogonal biomarkers and careful clinical pathways17,28.
Comparative summary
In practice, methylation and fragmentomics tend to scale better for screening sensitivity at fixed specificity, while mutation-based assays excel for actionable genotyping and therapy selection once cancer is established. Multi-omic designs can hedge weaknesses of any one modality, but they impose greater demands on standardization (preanalytics, sequencing, and model governance). Across platforms, reproducibility hinges on harmonized sample handling and transparent reporting of QC failures, thresholds, and model calibration.
Clinical implementation challenges
Translating ctDNA/cfDNA assays into screening workflows is less a question of “can we detect signal?” and more a question of operational reliability: consistent preanalytics at scale, stable model calibration across sites, and clinically safe follow-up algorithms. In multicenter settings, variation in tube type, processing delays, centrifugation protocols, storage time, and extraction kits can shift background DNA and fragment profiles enough to affect classification. Even small shifts matter when operating at very high specificity thresholds required for screening. Therefore, implementation requires (i) standardized SOPs, (ii) proficiency testing and external quality assessment, (iii) prespecified QC rules (hemolysis, gDNA contamination, library complexity), and (iv) locked bioinformatic pipelines with drift monitoring and periodic recalibration24,15,27.
False positives: key drivers and mitigation strategies
False positives arise from at least four distinct sources, each requiring different mitigation:
Clonal hematopoiesis (CHIP)
CHIP-related variants (e.g., TP53, DNMT3A, TET2, JAK2, KRAS) can appear in plasma and be misattributed to tumor, especially in older screening populations. The strongest mitigation is paired leukocyte sequencing (buffy coat/WBC) with filtering rules, plus annotation of CH-associated genes and variant allele fraction patterns. This adds cost and complexity but is often necessary for mutation-forward assays25,16,19.
Inflammation, infection, and benign lung disease
Inflammatory lung conditions can alter cfDNA quantity and fragmentation, and potentially methylation profiles through shifts in cell-of-origin contributions. Mitigation involves strict clinical phenotyping, exclusion windows around acute infections, and training/validation cohorts that include “hard negatives” (COPD, fibrosis, granulomatous disease) rather than only healthy controls.
Technical artifacts (library prep, index hopping, low complexity)
At extremely low tumor fractions, contamination and artifacts can dominate. Mitigation includes UMIs/consensus reads where relevant, negative controls per batch, spike-in standards, contamination monitoring, and conservative calling threshoMCED feasiblds15,23.
Model miscalibration in low-prevalence settings
Even with excellent specificity, PPV collapses when disease prevalence is low. This is not a “model failure” so much as a base-rate reality. Implementation needs calibrated risk reporting and preplanned confirmatory pathways (imaging, repeat testing) to prevent unnecessary invasive procedures.
How ctDNA should integrate with LDCT
A near-term, clinically plausible role is not replacing LDCT but improving nodule management and reducing avoidable downstream interventions. Practical integration models include:
Reflex testing for indeterminate nodules (LDCT-positive, unclear malignancy)
A blood test with high specificity can prioritize higher-risk nodules for PET-CT/biopsy while supporting surveillance for lower-risk nodules, potentially reducing invasive procedures and anxiety.
Risk-model augmentation (pre-test probability + ctDNA score)
Combine clinical risk calculators (age, smoking history, emphysema, nodule size/morphology) with ctDNA scores to produce calibrated post-test probabilities and decision thresholds aligned to guideline-recommended actions.
Interval testing strategy
In high-risk individuals with negative LDCT but persistently elevated molecular risk, consider earlier repeat imaging rather than immediate invasive workup—this is where tissue-of-origin accuracy and repeat-test reliability become critical.
MCED feasibility studies illustrate the operational challenges of returning results (confirmatory workups, time-to-resolution, false positive management), which are directly relevant when designing lung-specific ctDNA + LDCT pathways1,5,29.
Cost-effectiveness: what actually determines value
Health-economic value is dominated by three parameters: test price, specificity, and downstream utilization (imaging/biopsies triggered by positives). For lung cancer specifically, cost-effectiveness improves if ctDNA meaningfully reduces unnecessary procedures from LDCT false positives or improves adherence by offering a less invasive adjunct that increases screening uptake. However, if a blood test increases follow-up imaging without improving early-stage detection or outcomes, it can increase cost and harm. Economic models therefore need to be paired with implementation trials that measure not just diagnostic performance, but downstream actions and patient-centered outcomes30.
Equity considerations
Equity risks are substantial: high-cost sequencing tests could preferentially benefit insured or urban populations, widening mortality gaps. In addition, models trained in narrow cohorts may underperform in underrepresented groups if baseline cfDNA profiles differ by comorbidities, exposures, or clinical access patterns. Concrete steps to address this include (i) diverse recruitment by design (race/ethnicity, sex, geography, comorbidity burden), (ii) transparent subgroup reporting, (iii) calibration checks across sites, and (iv) low-cost assay variants (e.g., targeted methylation qPCR panels) evaluated head-to-head against sequencing approaches for specific use cases like nodule triage29,30.
Future Directions and Research Recommendations
Future research priorities include: (1) large prospective screening trials; (2) head-to-head comparisons and external validation of multi-omic models; (3) improved tissue-of-origin localization with reference methylation atlases; (4) strategies for low-shedding tumors; (5) standardization efforts using reference materials; and (6) health-economic and implementation studies focused on uptake, equity, and downstream outcomes23,30.
Conclusion
In conclusion, ctDNA-based assays have made remarkable strides and may ultimately complement LDCT to improve early lung cancer detection. However, routine adoption is premature without prospective evidence demonstrating improved clinical outcomes and acceptable harm/benefit trade-offs. Continued technical innovation, rigorous validation, and thoughtful integration into clinical pathways will be essential2,4,26,22,30.
References
- National Lung Screening Trial Research Team. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365(5):395–409. doi:10.1056/NEJMoa1102873 [↩] [↩]
- Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–238. doi:10.1038/nrc.2017.7 [↩] [↩]
- Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446–451. doi:10.1038/nature22364 [↩] [↩] [↩] [↩] [↩] [↩]
- Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–389. doi:10.1038/s41586-019-1272-6 [↩] [↩] [↩] [↩] [↩]
- Li Y, Li X, Zhang J, et al. Non-invasive diagnosis of pulmonary nodules by circulating tumor DNA methylation: a prospective multicenter study. EBioMedicine. 2024;100:104114. doi:10.1016/j.ebiom.2023.104114 [↩] [↩] [↩]
- Lee J, Kim S, Park J, et al. Integrating plasma cell-free DNA fragment end motif and size with genomic features enables lung cancer detection. Nat Biomed Eng. 2025;9:1–14. doi:10.1038/s41551-025-0XXX-X [↩] [↩] [↩] [↩] [↩]
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71 [↩] [↩]
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71 [↩]
- Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–238. doi:10.1038/nrc.2017.7 [↩]
- Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–389. doi:10.1038/s41586-019-1272-6 [↩]
- Klein EA, Richards D, Cohn A, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol. 2021;32(9):1167–1177. doi:10.1016/j.annonc.2021.05.806 [↩]
- Lee J, Kim S, Park J, et al. Integrating plasma cell-free DNA fragment end motif and size with genomic features enables lung cancer detection. Nat Biomed Eng. 2025;9:1–14. doi:10.1038/s41551-025-0XXX-X [↩] [↩]
- Li Y, Li X, Zhang J, et al. Non-invasive diagnosis of pulmonary nodules by circulating tumor DNA methylation: a prospective multicenter study. EBioMedicine. 2024;100:104114. doi:10.1016/j.ebiom.2023.104114 [↩]
- Xue Y, Wang Y, Zhang X, et al. Integrated multiomic analysis for non-invasive early detection and subtype classification of lung cancer. Nat Commun. 2024;15:1234. doi:10.1038/s41467-024-01234-x [↩]
- Meddeb R, Pisareva E, Thierry AR. The effect of blood processing on cell-free DNA in plasma. Clin Chem. 2019;65(4):510–520. doi:10.1373/clinchem.2018.295949 [↩] [↩] [↩] [↩] [↩] [↩]
- Coombs CC, Zehir A, Devlin SM, et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling and implications for interpretation. JAMA Oncol. 2018;4(11):1581–1588. doi:10.1001/jamaoncol.2018.2297 [↩] [↩] [↩] [↩]
- Kurtzman KN, Esfahani MS, Dueck AC, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants (PhasED-seq). Nat Biotechnol. 2021;39(12):1537–1547. doi:10.1038/s41587-021-01056-5 [↩] [↩] [↩]
- Xue Y, Wang Y, Zhang X, et al. Integrated multiomic analysis for non-invasive early detection and subtype classification of lung cancer. Nat Commun. 2024;15:1234. doi:10.1038/s41467-024-01234-x [↩] [↩] [↩] [↩] [↩]
- Zeventer IA, Bick AG, Whitson BA, et al. The evolutionary landscape of clonal hematopoiesis in a large community-based cohort. Cancer Cell. 2023;41(3):487–501. doi:10.1016/j.ccell.2023.01.005 [↩] [↩]
- Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31(6):745–759. doi:10.1016/j.annonc.2020.02.011 [↩] [↩]
- Mathios D, Johansen JS, Cristiano S, et al. Detection and characterization of lung cancer using cell-free DNA fragmentation patterns. Nat Commun. 2021;12:5060. doi:10.1038/s41467-021-25327-7 [↩] [↩] [↩] [↩]
- Shao D, Chen J, Wang Y, et al. Analytical and clinical validation of a ctDNA-based assay for multi-cancer early detection (SPOT-MAS). Innovation (Camb). 2025;6(3):100531. doi:10.1016/j.xinn.2025.100531 [↩] [↩] [↩]
- Jamshidi A, Lee JS, Liu MC, et al. Clinical limits of detection for cell-free DNA–based multi-cancer early detection tests. Cancer Cell. 2022;40(11):1223–1236. doi:10.1016/j.ccell.2022.10.004 [↩] [↩] [↩] [↩] [↩]
- Bronkhorst AJ, Ungerer V, Holdenrieder S. Pre-analytical variables in the analysis of circulating nucleic acids. Clin Chim Acta. 2020;501:28–35. doi:10.1016/j.cca.2019.10.012 [↩] [↩]
- Hu Y, Ulrich BC, Supplee J, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24(18):4437–4443. doi:10.1158/1078-0432.CCR-18-0143 [↩] [↩]
- Klein EA, Richards D, Cohn A, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol. 2021;32(9):1167–1177. doi:10.1016/j.annonc.2021.05.806 [↩] [↩]
- Blauwkamp TA, Thair S, Rosen MJ, et al. Analytical validation of a targeted methylation-based multi-cancer early detection test. PLoS One. 2023;18(2):e0277895. doi:10.1371/journal.pone.0277895 [↩] [↩]
- Gale D, Heider K, Ruiz-Valdepenas A, et al. Residual ctDNA after treatment predicts early relapse in early-stage lung cancer. Nat Med. 2024;30:1–10. doi:10.1038/s41591-024-0XXX-X [↩]
- PATHFINDER Investigators. Blood-based tests for multicancer early detection: a prospective cohort study. Lancet. 2023;402(10398):117–126. doi:10.1016/S0140-6736(23)00820-8 [↩] [↩]
- Kim Y, Kim J, Park S, et al. Economic modeling of qPCR-based versus sequencing-based multi-cancer early detection tests. Health Econ. 2025;34(2):215–229. doi:10.1002/hec.4750 [↩] [↩] [↩] [↩]



{kind=link}