
Abstract
Cancer cells evade immune destruction by overexpressing CD47, a “don’t eat me” signal that blocks macrophage activity. This project proposes the use of small molecule drugs to inhibit CD47, thereby restoring immune surveillance and offering a potential new path for cancer immunotherapy. By targeting CD47 with a computational approach, the study aims to identify viable small molecule drug candidates for further development. Binding site analysis confirmed that CD47 contains several druggable sites with favorable scores. Virtual screening identified 22 candidate compounds with strong alignment to the pharmacophore map. Molecular docking showed that multiple compounds had favorable binding affinities, with some having a SwissParam score below -9.0 kcal/mol. ADME profiling revealed several top candidates satisfied Lipinski’s rule with zero violations, suggesting good drug-likeness. Finally, a toxicity evaluation identified compounds with acceptable profiles, including two drugs that well within the tolerable toxicity classes and LD50 thresholds. Overall, the project demonstrates that a systematic computational workflow can be used to prioritize small molecule candidates predicted to inhibit CD47 for further experimental evaluation. Moving forward, this workflow may be extended to other immune checkpoints as a general strategy for early-stage therapeutic discovery.
Introduction
In 2022, there were an estimated 20 million new cancer cases, and by 2050, this number is expected to increase to 33 million1. Cancer is a disease causing cells to divide uncontrollably, forming lumps of abnormal tissue called tumors2. Cancerous tumors can undergo metastasis by spreading and traveling throughout the body to form new tumors, making them particularly dangerous3.
Chemotherapy is the use of cytotoxic drugs to kill cancerous cells, though it can also kill healthy cells therefore harming the immune system4. On the other hand, immunotherapy works by restoring the body’s immune defenses to recognize and destroy cancer cells, often with fewer side effects5. While immunotherapy is newer and more targeted, not all patients respond to it, primarily due to immune checkpoints that stop white blood cells from effectively killing cancer cells5. Blocking these checkpoints can improve immune activity and improve cancer therapy outcomes6. These immune checkpoints are regulatory pathways that control white blood cell activity by delivering inhibitory or stimulatory signals7. In healthy individuals, these checkpoints maintain balance in the immune system and prevent the body from attacking healthy cells7. For individuals with cancer, tumor cells produce numerous “off” signals which stop white blood cells from attacking them7. Drugs called immune checkpoint inhibitors block these “off” signals, allowing white blood cells to destroy cancerous cells8.
Cluster of Differentiation 47 (CD47) is a protein found on the surface of cells which act as “don’t eat me” signals9. It does this by binding to signal regulatory protein alpha (SIRPα) on macrophages, immune cells responsible for destroying cancer cells10. When CD47 binds to SIRPα, it sends inhibitory signals telling the macrophages not to engulf the cell10. Though this protects healthy cells from being attacked, cancer cells are able to overexpress CD47 to avoid being destroyed10. The overexpression lets tumors grow while being ignored by the immune system, making targeting CD47 a promising approach for cancer immunotherapy10.
Because cancer cells overexpress CD47, blocking CD47 has surfaced as a type of cancer immunotherapy11. By blocking the CD47, the inhibitory signal is disrupted, restoring the immune system’s ability to destroy cancerous cells12. This approach has gained attention because of its potential to improve outcomes in tumors that are resistant to other forms of cancer therapy11.
Small molecules are molecules with a weight less than 500 daltons13. In cancer therapy, small molecules can be used to inhibit proteins in cancer cells14. Antibodies bind to cancer cell antigens, tagging them so the immune system can destroy them15. They trigger immune response by interacting with immune cell receptors, helping the body kill cancerous cells and activate immune defenses16. Therefore, the central aim of this study is to identify druggable rockets on CD47 and apply a hierarchical virtual screening workflow to discover small molecules with the potential to disrupt the CD47-SIRPα immune checkpoint interface. It is hypothesized that structure-guided virtual screening can identify small-molecule candidates that bind CD47 and interface-relevant regions and warrant experimental testing for effects on CD47-SIRPα signaling.
Beyond antibody-based strategies, several non-antibody approaches have demonstrated that disrupting the CD47-SIRPα axis can produce meaningful antitumor effects17. Engineered high-affinity SIRPα variants have been shown to completely antagonize CD47 and enhance macrophage-mediated phagocytosis across multiple tumor models, establishing proof-of-concept for non-antibody CD47 blockade18. In parallel, peptide-based inhibitors targeting the CD47-SIRPα interface have demonstrated both in vitro and in vivo antitumor activity, further validating this interaction as a therapeutically actionable target beyond monoclonal antibodies19.
Regarding antibody-based approaches, in “Targeting CD47 as a Novel Immunotherapy for Breast Cancer,” Chen, Wang, Chen, Hou, and Jiang explore how CD47 helps breast cancer cells evade destruction by the immune system20. They explain that CD47 is highly expressed in aggressive forms of cancer and is associated with poor patient outcomes20. The authors highlight that anti CD47 therapies offer promising results, alone and in combination with targeted drugs or chemotherapy, but note safety concerns and the need to improve effectiveness20. In “Advances in the study of CD47-based bispecific antibody in cancer immunotherapy,” Zhang, Li, Fan, Tian, Zhou, Ji, and Song review the use of bispecific antibodies targeting CD47 for cancer immunotherapy21. The review explores how bispecific antibodies are currently being tested in blood cancers and cancerous tumors and typically function better than single target drugs21. While this is a promising approach, authors discuss challenges like how CD47 is also found on healthy cells, limiting the safety of this treatment21. In “Immunotherapy of endometrial cancer via CD47 blockade-mediated macrophage phagocytosis,” Yucebas et al. explore how endometrial cancer cells are able to escape immune detection by overexpressing CD4722. The study found that blocking CD47 with a monoclonal antibody can increase macrophage activity and decrease tumor size significantly22. The authors also note that delivering the antibody into the uterus is more effective than systemic delivery22. The authors discuss challenges as well stating that CD47 is found on healthy cells as well, however note that there are few side effects22. All three reviews lack long term safety data for CD47 targeting therapies.
Methods
Analysis of Binding Sites in CD47
ProteinPus
It was necessary to determine whether CD47 has binding sites that can accommodate small molecules. Binding sites were prioritized using literature-informed heuristic size criteria for small molecule druggability, with pocket volumes on the order of several hundred to ~1000Å3 and surface areas of ~1000Å2, consistent with standard practice rather than strict cutoffs23. In addition, binding sites were prioritized based on druggability score (0-1), with higher values indicating greater suitability for small-molecule binding24. To evaluate this, the ProteinPlus platform was used25. The protein structure of CD47 was accessed by entering the PDB code 2JJT into the search bar and pressing “Go”26. Next, the DoGSiteScorer tool was selected for binding site detection. After clicking “DoGSiteScorer,” the “Calculate” button was pressed to access binding sites that can accommodate molecules within the volume restriction. ProteinPlus outputted a table with all detected binding sites, including their drug scores and volumes. Visualizations of each site were accessed by clicking the grey eye icons, which display the location of each binding site on CD47.
FTSite
To assess whether binding within these sites would be energetically favorable, the FTSite website was used27. On the website, 2JJT, was entered under “PDB ID,” and an email address was entered under “E-mail.” After clicking “Find my Binding Site,” results were sent via email, typically 2 hours. The email contained an image highlighting all CD47 binding sites predicted to allow energetically favorable interactions between the drug molecule and CD47.
PrankWeb
To identify binding sites that met both criteria, the PrankWeb server was used28. The PDB code 2JJT was entered and the “Submit” button was pressed. A table appeared showing all predicted binding sites that satisfy both conditions: meet the size criteria and support an energetically favorable reaction. A 3D diagram was also generated to see the location of each site on the CD47 structure. Because at least one binding site met both criteria, CD47 contains binding sites suitable for small molecule interaction.
Virtual Screening for Identifying Small Molecules
Virtual screening is a method used to identify small molecule binders to a target protein by predicting how well they fit into the protein’s binding site. This method is significantly more cost effective and time efficient than physical screening. Physical screening involves synthesizing and testing large quantities of compounds in a lab which is extremely expensive. However, virtual screening requires a high-quality protein structure with defined binding pockets. For CD47, the structure and PDB ID 2JJT are suitable. Prior analysis confirms this structure contains pockets capable of accommodating small drug molecules with drug scores greater than 0.7, indicating potential for a positive interaction.
To perform virtual screening, the Pharmit website is used29. After opening the website, the user enters 2JJT as the PDB code and presses “submit”. Under the visualization settings, the ligand and receptor should be set to none, the background color changes to black, and the receptor surface opacity reduced to 0% to improve clarity. Three binding pockets are displayed. Each must be analyzed individually by toggling off interactions for the other pockets. An ideal small molecule binder around 4 interactions with the target protein. These interactions include hydrogen donors, hydrogen acceptors, and hydrophobic reactions. The type, strength, and geometry of these interactions are important to note as they influence binding affinity. The pharmacophore features selected reflect the chemical environment of the chosen CD47 binding pocket. Residues lining this pocket include polar amino acids such as Asparagine 46 and Glutamic Acid 47, which are capable of participating in hydrogen bond donor and acceptor interactions, as well as hydrophobic and aromatic residues including Alanine 45, Isoleucine 49, Phenylalanine 57, and Tyrosine 89. Accordingly, hydrogen bond donor and acceptor features were included to capture polar interactions, while hydrophobic and aromatic features were incorporated to reflect van der Waals and π-mediated contacts expected within this pocket.
Once a binding site is selected, the user starts a compound search by clicking the “Search MolPort” button. MolPort is a chemical supplier database that contains various compounds for drug development. Root mean square deviation (RMSD) is used to quantify the geometric agreements between candidate molecules and the pharmacophore model, with low RMSD values indicating near-ideal geometric alignment between a compound’s functional groups and the hypothesized interaction geometry. A stringent RMSD threshold (<0.1 Å) was applied to prioritize compounds that closely match the spatial arrangement of the pharmacophore features within the pocket. Given the compact and well-defined nature of the selected CD47 pocket, this tight cutoff was chosen to reduce false positives and retain only candidates with high geometric complementarity for subsequent docking. Interactions were examined by rotating the 3D model which can be seen by pressing on any listed small molecule under “Pharmacophore Results”. The 3D model was used to visualize how each compound aligned, providing insight into the spatial fit and the RMSD score that was given.
Molecular Docking Tests
To identify whether the identified small molecule drugs interact favorably with CD47, molecular docking must be performed. Molecular docking is a method used to predict how small molecules bind to a target protein30. The binding affinity is quantified using the estimated binding free energy (∆G, kcal/mol) reported by SwissDock. A lower, more negative ∆G value indicates more energetically favorable and spontaneous binding interactions between the ligand and CD47.
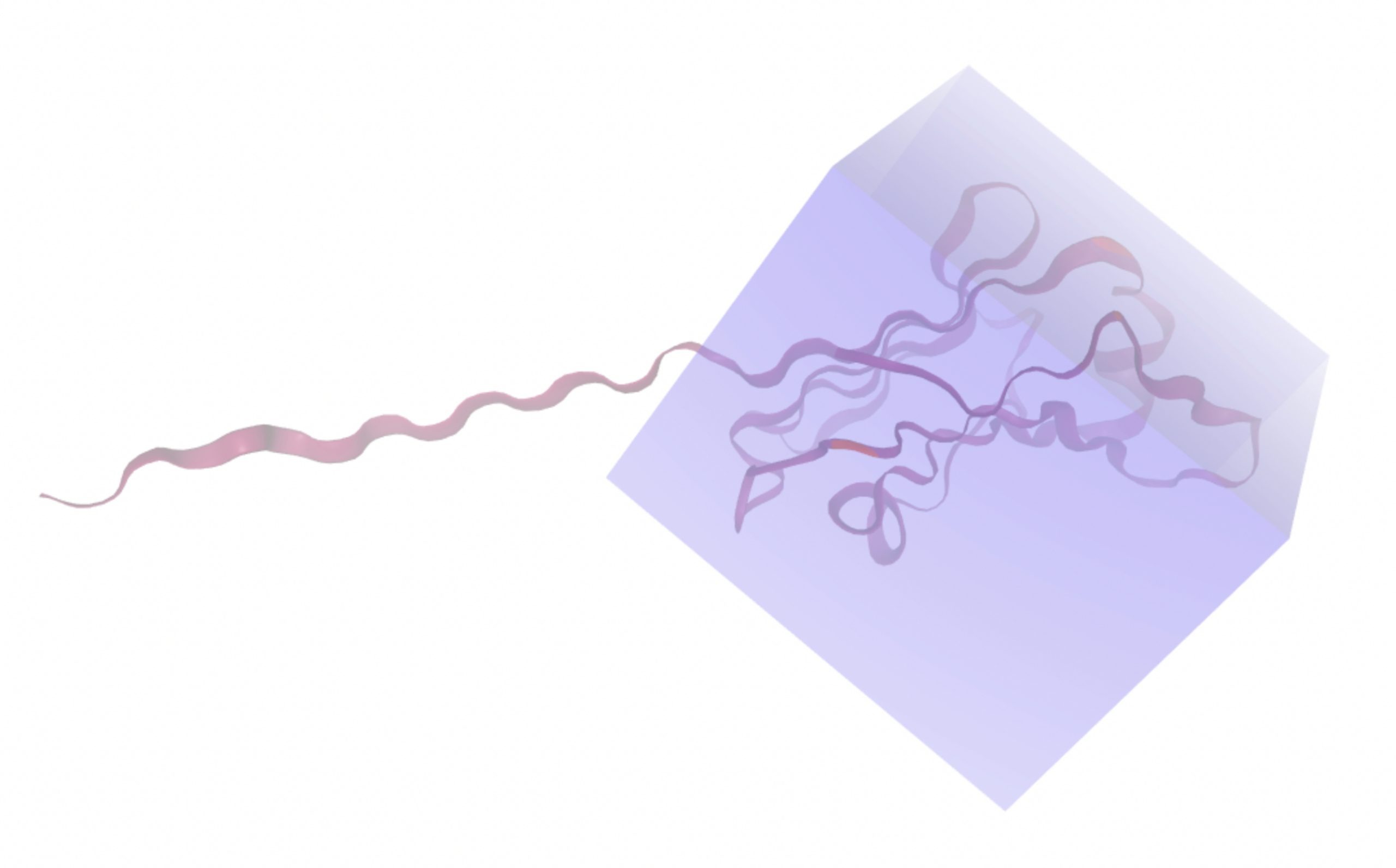
To ensure consistency between pharmacophore-based virtual screening and molecular docking, the SwissDock search space was explicitly aligned to the same binding pocket identified during pocket mapping and Pharmit screening. Docking was performed on CD47 chain A, which corresponds to the chain used for pharmacophore generation. The docking grid center (5, -39, 21 Å) and grid dimensions (33 x 35 x 28 Å) were selected to fully encompass the pharmacophore-defined pocket and its surrounding residuals while excluding the disordered C-terminal tail. The search grid also encompasses the same CD47 pocket used for pharmacophore generation, ensuring structural continuity between virtual screening and docking stages.
Each compound identified in Procedure 2.2 must be docked individually. The process starts by accessing the Molport directory and entering the Molport ID into the search bar. On the resulting page, the SMILES code should be copied. The SwissDock platform will then be used to perform the docking31. Under the “1 – Submit a ligand” section, the SMILES code is pasted into the box labeled “Provide a SMILES”. The “Prepare ligand” button is pressed, and once a green check mark appears, the process can continue. In the “2 – Submit a target” section, the PDB ID 2VSC is entered into the box labeled “Provide a PDB ID (e.g. 5hie)”32. This structure corresponds to the CD47 protein and is compatible with SwissDock, while 2JJT is not. Only Chain A is selected for analysis, as all chains are identical so only one is necessary for docking. Under “Choose heteroatom(s) to keep”, none is selected. The “Prepare target” button is clicked and the process can continue once a green checkmark appears. The search space must then be defined. Under “Search box center”, the coordinates 5, -39, 21 Å are entered. Under “Search box size”, the dimensions 33, 35, 28 Å are entered. These parameters encompass the majority of chain A while excluding its tail. The tail is excluded because it lacks a well defined structure and is unlikely to contain viable binding sites as found in Procedure 3.1. After, the “Check parameters” button is pressed. After confirmation with a green checkmark, an email address and docking name are entered to start the process.
SwissDock processes the docking and returns results via the email provided typically within 10-20 minutes. The link in the email leads to a summary table listing potential binding interactions, each associated with an estimated binding free energy. A lower (more negative) binding free energy represents more energetically favorable interactions so the lowest energy listed is noted. Clicking any of these scores displays a 3D visualization of the interaction. Interaction figures were used for qualitative assessment of pose plausibility rather than quantitative measurement of individual hydrogen-bond distances.
Because docking was performed using CD47 from PBD 2VSC, the experimentally defined CD47-SIRPα interface was mapped onto this structure for comparison. CD47 from the CD47-SIRPα complex (PBD 2JJT) was structurally aligned to CD47 in 2VSC using PyMOL, and residues contacting SIRPα in the complex were transferred into the 2VSC coordinate frame33. This mapping was used to verify that the SwissDock search space encompassed the SIRPα-contacting epitope on CD47.
ADME Profile Evaluation
To further access the top drug candidates identified through molecular docking, it is necessary to evaluate their ADME profile, which stands for absorption, distribution, metabolism, and excretion. A favorable ADME profile is critical for determining whether a drug can be delivered, reach its target, and be safely eliminated from the body while avoiding harmful effects.
Once the drug is taken orally, it must be absorbed through the GI tract to enter the bloodstream. From there, it is distributed to CD47. After binding, the compound must be broken down into non-toxic, excretable forms. If a drug is unable to be excreted, it can accumulate in organs and cause toxic effects. Therefore, it is necessary to predict whether candidate compounds have favorable ADME profiles.
To do this, the top compounds with the lowest SwissParam docking scores will be further analyzed using Lipinski’s rule. This rule provides empirical guidelines to evaluate its profile favorability based on molecular properties34:
- The compound should have no more than 5 hydrogen bond donors as excessive donors can lead to the small molecule drug binding with other proteins.
- The compound should have no more than 10 hydrogen bond acceptors because excessive acceptors can also lead to non-specific binding with other proteins.
- The molecular weight of the compound must be less than 500 daltons so it can be easily absorbed and excreted.
- The calculated partition coefficient (logP) must be between 0 and 5. A LopP between 0 and 5 indicates suitable balance between the aqueous and lipid phase. Values below 0 favor the aqueous phase and values above 5 favor the lipid phase.
SwissADME was used to evaluate drug-likeness and ADME-related properties, including molecular weight, lipophilicity, and hydrogen bonding capacity, which serve as proxies for oral permeability, solubility, and systemic exposure rather than direct measures of toxicity35. Toxicity assessment was therefore performed separately using ProTox, which estimates biological toxicity endpoints rather than pharmacokinetic suitability. For each candidate molecule, the corresponding SMILES code is entered into the input field on the SwissADME website. The user then selects “Run” to start the prediction algorithm. The output includes many molecular descriptors and compound properties. For logP, the “iLOGP” value should be used to evaluate compliance with Lipinski’s rule, as it is considered an accurate predictive model. This step of screening allows for the identification of compounds that are likely to succeed in physiological conditions, increasing their potential as viable small drug candidates for CD47.
Toxicity Prediction
To assess the potential toxicity of each small molecule drug candidate, a toxicity prediction was conducted using the ProTox server36. The aim of the analysis is not to determine whether a compound is toxic in absolute terms, as all substances exhibit some level of toxicity, but to assess the degree of toxicity and whether it falls in a range that is considered tolerable for the human body.
Compounds were evaluated using a uniform set of toxicity acceptance criteria applied identically across all screened candidates. The criteria includes the following:
- Predicted LD50 ≥ 400 mg/kg
- Predicted toxicity class ≥ 3
- Predicted organ-specific toxicity not exceeding the average toxicity of approved drugs across the majority of systems, as addressed using the ProTox radar charts.
To begin the assessment, the user navigates to the ProTox platform and selects the “TOX PREDICTION” button. The SMILES code of the compound, previously obtained in earlier procedures, is entered into the designated field. After entering the SMILES code, the “smiles” button is selected, followed by the “All” options and then “Start Tox-Prediction”. The platform generates two key outputs. The first is the LD50 value (in mg/kg) that represents the lethal dose at which 50% of test organisms are expected to die. The other is the toxicity class, ranging from 1 to 6, based on classification criteria. For a compound to be considered safe, the LD50 value should be greater than 400mg/kg and the toxicity class should be 3 or higher. These thresholds indicate moderate to low toxicity levels that should be tolerable in drug development.
In addition to these metrics, ProTox provides an overview of predicted toxic systems. While the presence of toxicity in individual systems is noted, it does not necessarily mean the drug is unsuitable for use. Many approved drugs exhibit minor toxicity in one or more systems without consequences.
To further evaluate systemic tolerability, the user should examine the radar chart displayed below the results table. This chart compares the tested compound’s predicted toxicity to the average of known, approved drugs. The blue dots represent the compound’s predicted toxicity across different physical systems, while the orange dot shows the average toxicity of established drugs. If the blue dots fall closer to the center of the chart than the orange, the compound is considered to have an acceptable toxicity profile and is unlikely to be of significant risk to human health.
For reproducibility, core software tools, versions, and key computational parameters used in this study are summarized in Table 1.
| Step | Software/Service | Key Parameters |
| Pocket detection | ProteinPlus (DoGSiteScorer) | Default parameters |
| Binding site prediction | FTSite | Default parameters |
| Pocket ranking | PrankWeb | Default parameters |
| Pharmacophore screening | Pharmit | RMSD cutoff < 0.1 Å |
| Molecular docking | SwissDock | Grid center: (5, -39, 21 Å); Grid size: 33 x 35 x 28 Å |
| ADME filtering | SwissADME | Default settings; Lipinski criteria |
| Toxicity filtering | ProTox | Default model; Predicted LD50 ≥ 400 mg/kg; Predicted toxicity class ≥ 3 |
Results
Analysis of Binding Sites in CD47
ProteinPus
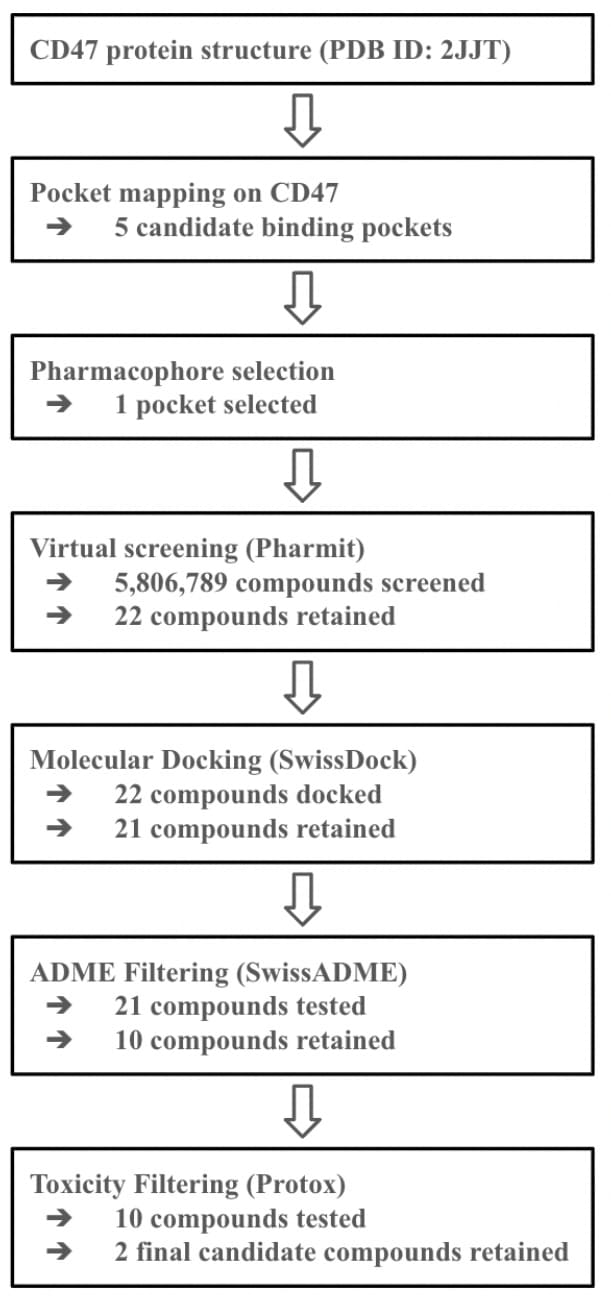
To determine the druggability of CD47, the ProteinPlus platform was used to determine binding sites on CD47 that are capable of accommodating molecules with a weight between 300 and 1000 daltons. DoGSiteScorer detected a total of 14 binding sites. Table 2 summarizes the name, volume, surface area, drug score, and simple score of each binding site. A visual representation of the binding pockets on CD47 is shown in Figure 2.
| Name | Volume Å3 | Surface Area Å2 | Drug Score |
| P_0 | 1382.36 | 1530.85 | 0.8 |
| P_1 | 960.55 | 1147.0 | 0.81 |
| P_10 | 144.32 | 263.3 | 0.26 |
| P_11 | 112.35 | 173.37 | 0.27 |
| P_12 | 105.3 | 221.58 | 0.13 |
| P_13 | 102.08 | 216.36 | 0.28 |
| P_2 | 699.04 | 936.86 | 0.79 |
| P_3 | 495.42 | 825.62 | 0.69 |
| P_4 | 362.24 | 673.6 | 0.6 |
| P_5 | 284.41 | 301.22 | 0.72 |
| P_6 | 197.98 | 386.97 | 0.4 |
| P_7 | 195.36 | 244.28 | 0.44 |
| P_8 | 183.47 | 494.96 | 0.33 |
| P_9 | 149.69 | 234.87 | 0.32 |
FTSite
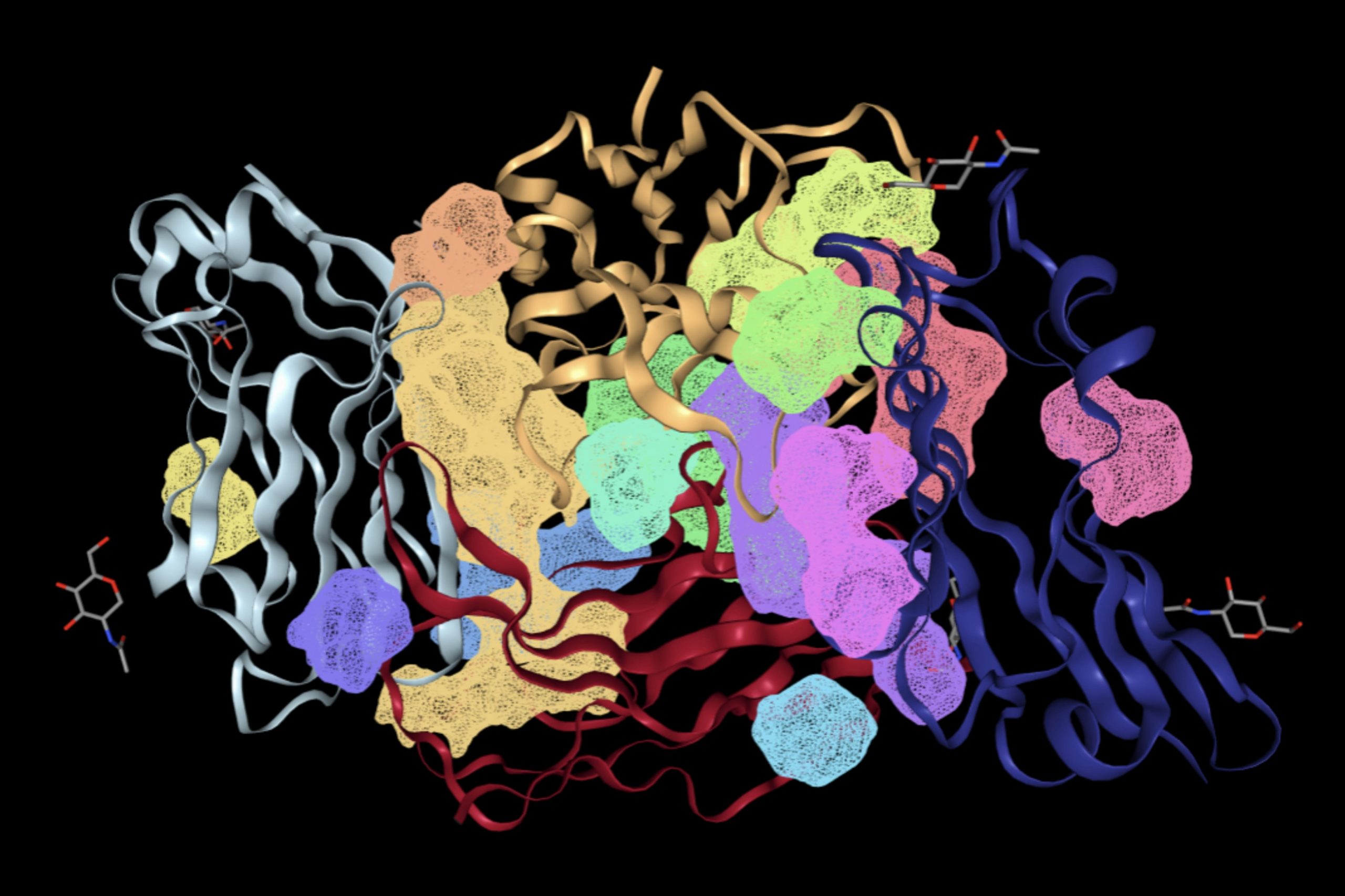
The FTSite website was then used to determine binding sites that would support an energetically favorable reaction. The resulting image of each binding site that meets this condition is shown in Figure 3.
PrankWeb
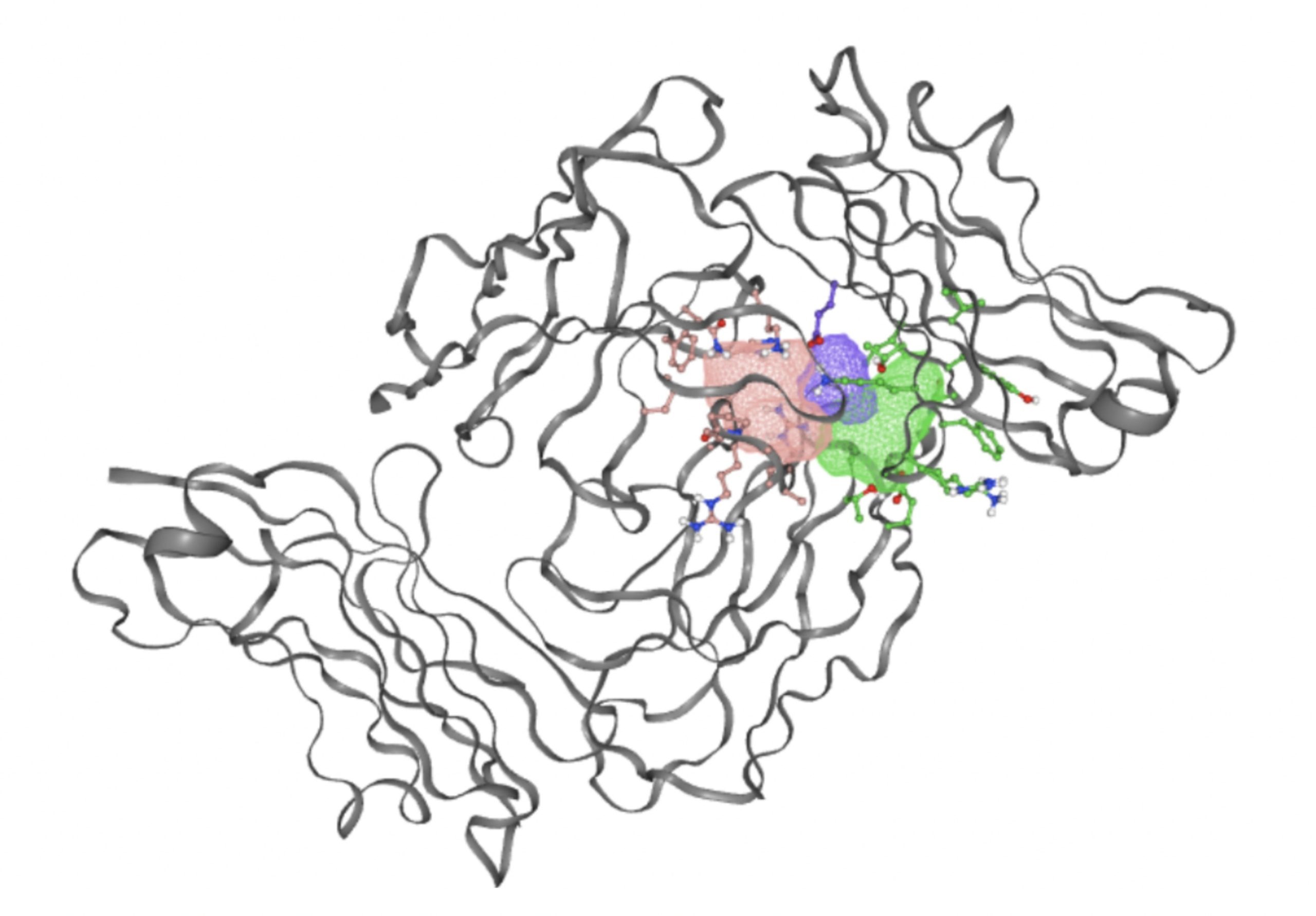
Using the PrankWeb server, 5 binding sites were identified that likely meet both the conditions: an energetically favorable reaction and a suitable volume for small molecules. The server predicted 5 binding sites which satisfy the criteria. These binding sites are listed in Table 3 with their probability scores, number of residues, example amino acids, and conservation values. Binding sites with higher probability scores are more likely to be true druggable sites. Figure 4 shows a 3D visualization of each binding site.
| Score | Ranking | # of Residues | Example Residues |
| 10.67 | 1 | 32 | Asparagine 46, Isoleucine 108, Alanine 45 |
| 9.08 | 2 | 28 | Asparagine 100, Glutamic Acid 97, Proline 35 |
| 3.48 | 3 | 13 | Glutamic Acid 47, Isoleucine 49, Proline 58 |
| 3.36 | 4 | 14 | Isoleucine 49, Tyrosine 89, Phenylalanine 57 |
| 2.18 | 5 | 13 | Phenylalanine 57, Leucine 48, Histidine 56 |
As shown in Table 3, the top ranked binding site, with a score of 10.67, has 32 residues, including Asparagine 46, Isoleucine 108, and Alanine 45. This site has the highest score and the largest number of residues, making it a strong candidate for small molecule interaction. The second binding site, with a score of 9.08, has 28 residues which would contribute to favorable reactions with small molecules. Binding sites ranked third to fifth have significantly lower scores and fewer residues. These sites may still be usable however are less likely to provide a stable binding environment for small molecule drugs due to their size and residues.
Virtual Screening for Identifying Small Molecules
Table 4 shows the results of virtual screening for small molecule binders to CD47 using Pharmit. Each compound reported is retrieved from the MolPort chemical supplier database, These compounds were selected based on their close fit with the pharmacophore model of the binding pockets. They were ranked by their root mean square deviation (RMSD) scores, a metric that quantifies the alignment between a pharmacophore model and compound. A lower RMSD indicates a better spatial fit and a higher chance for binding affinity. Figure 5 shows the basic pharmacophore map prior to the virtual screening process.

All compounds noted in Table 4 had RMSD values between 0.058 and 0.060, indicating close structural alignment between the compound and the pharmacophore model of the binding pockets. Notably, several compounds had an RMSD of 0.058. These compounds likely align with interaction features desired for binding including hydrophobic and donor/acceptor interactions.
The listed 3D figures display the interaction between each molecule and the CD47 binding pocket. Molecules like MolPort-008-326-608 and MolPort-005-280-798, show the compound to be well aligned with the pharmacophore model, explaining the low RMSD values. Compounds with RMSD values of 0.059 or 0.060 still are promising candidates due to their favorable geometry. In all, the compounds identified through Pharmit exhibit promising compatibility to CD47 binding pockets.
| Name | RMSD | Figure |
| MolPort-008-326-608 | 0.058 | 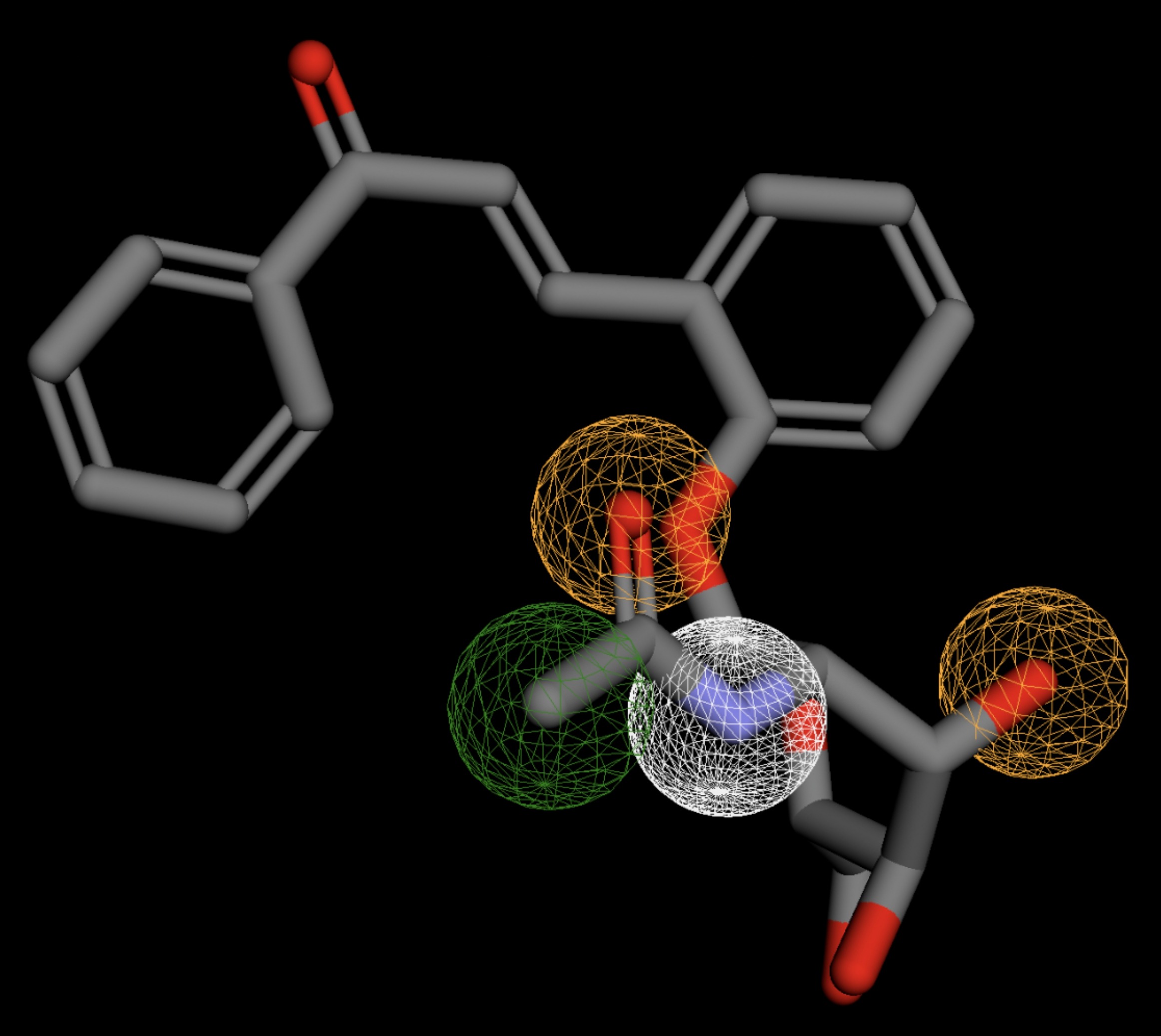 |
| MolPort-005-280-798 | 0.058 | 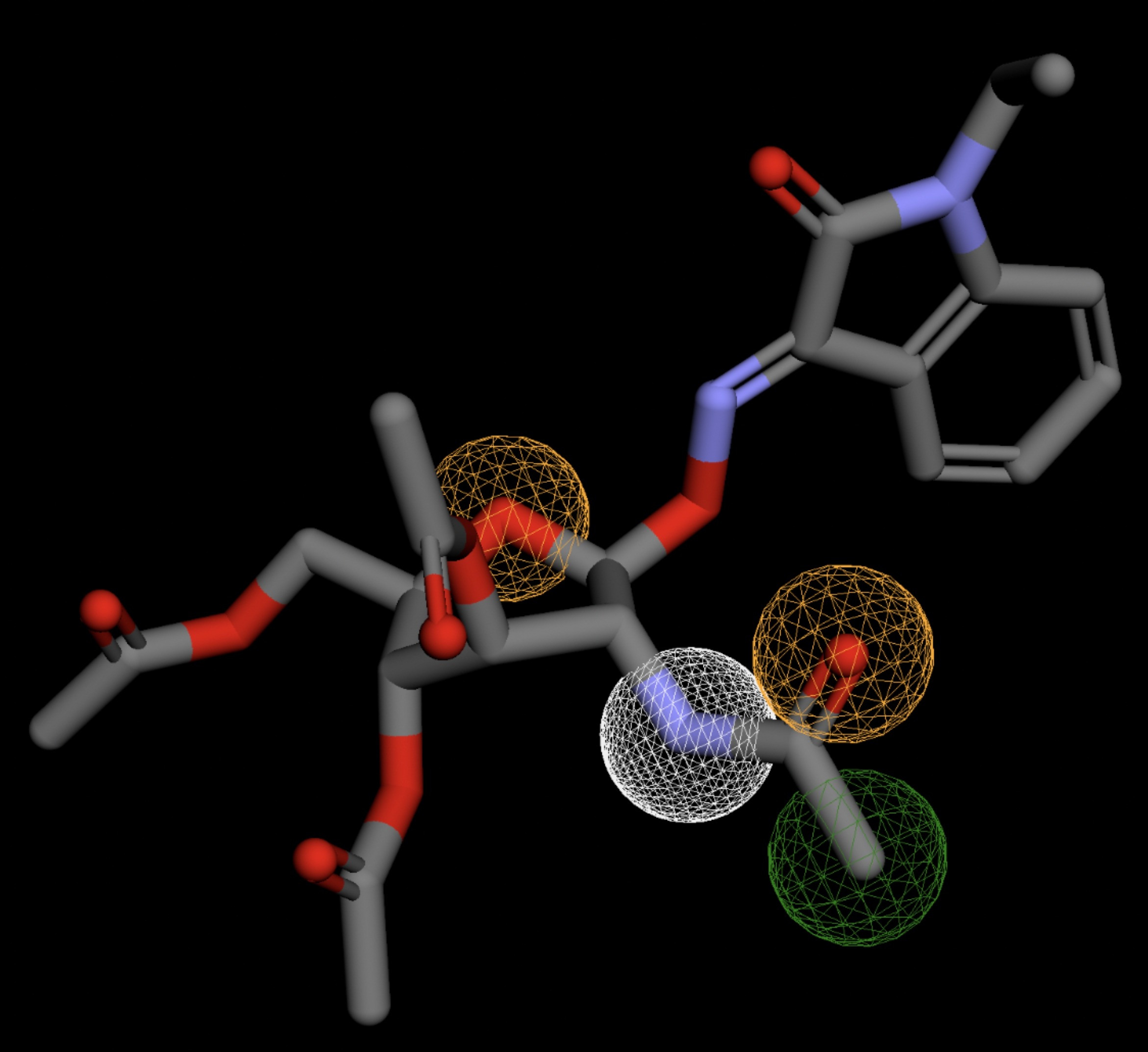 |
| MolPort-002-512-231 | 0.058 | 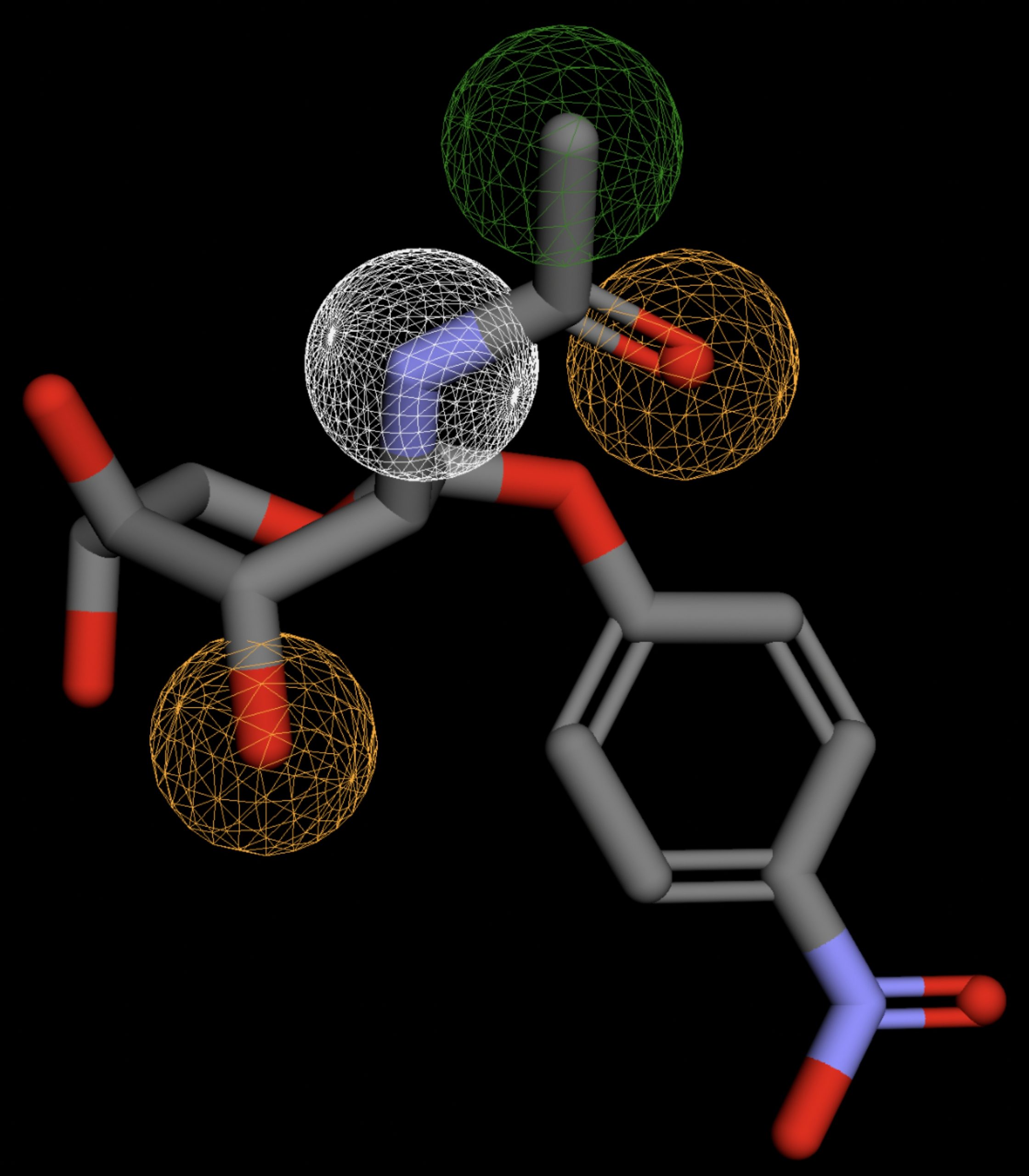 |
| MolPort-006-391-514 | 0.058 | 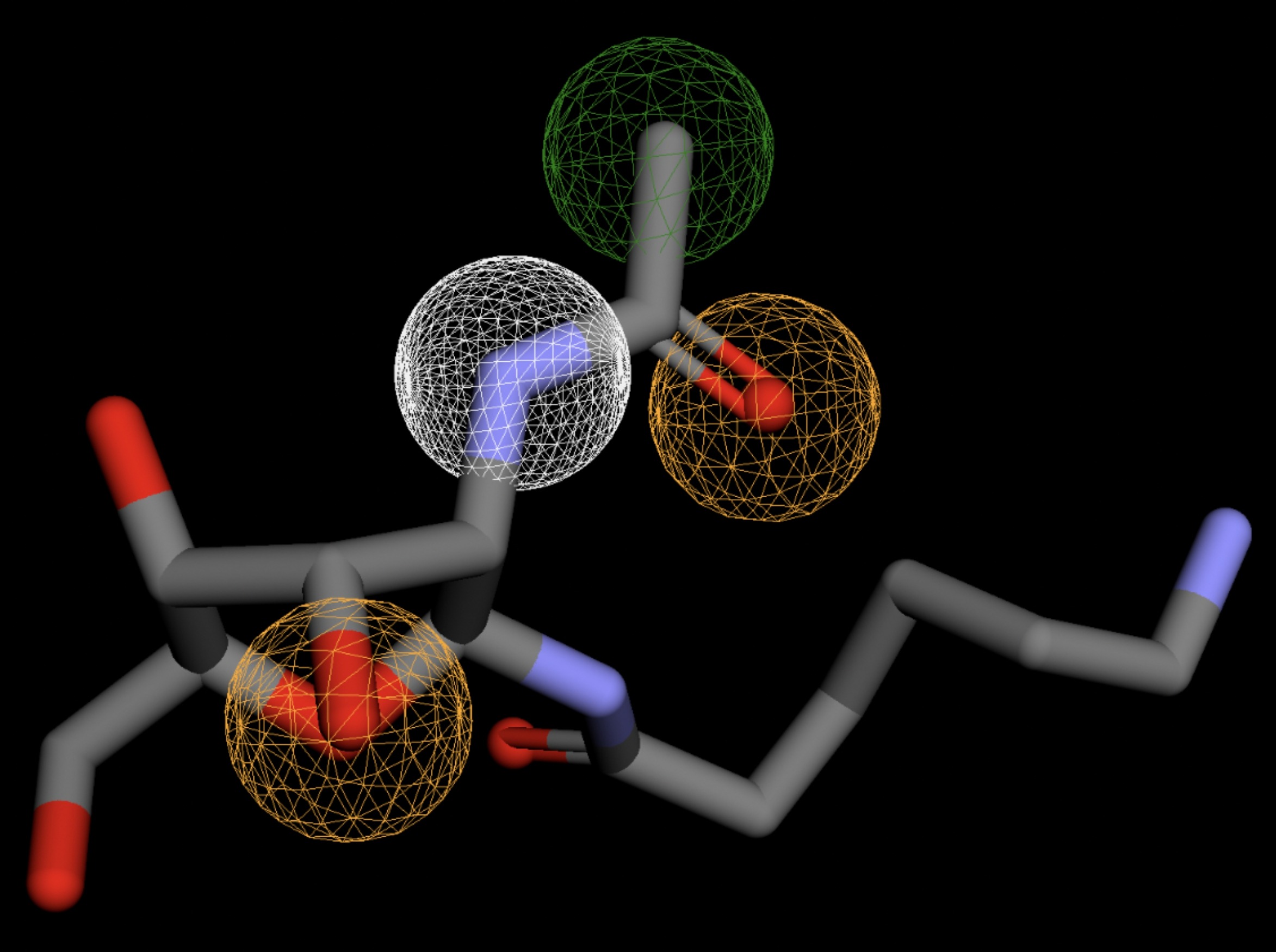 |
| MolPort-051-689-380 | 0.058 | 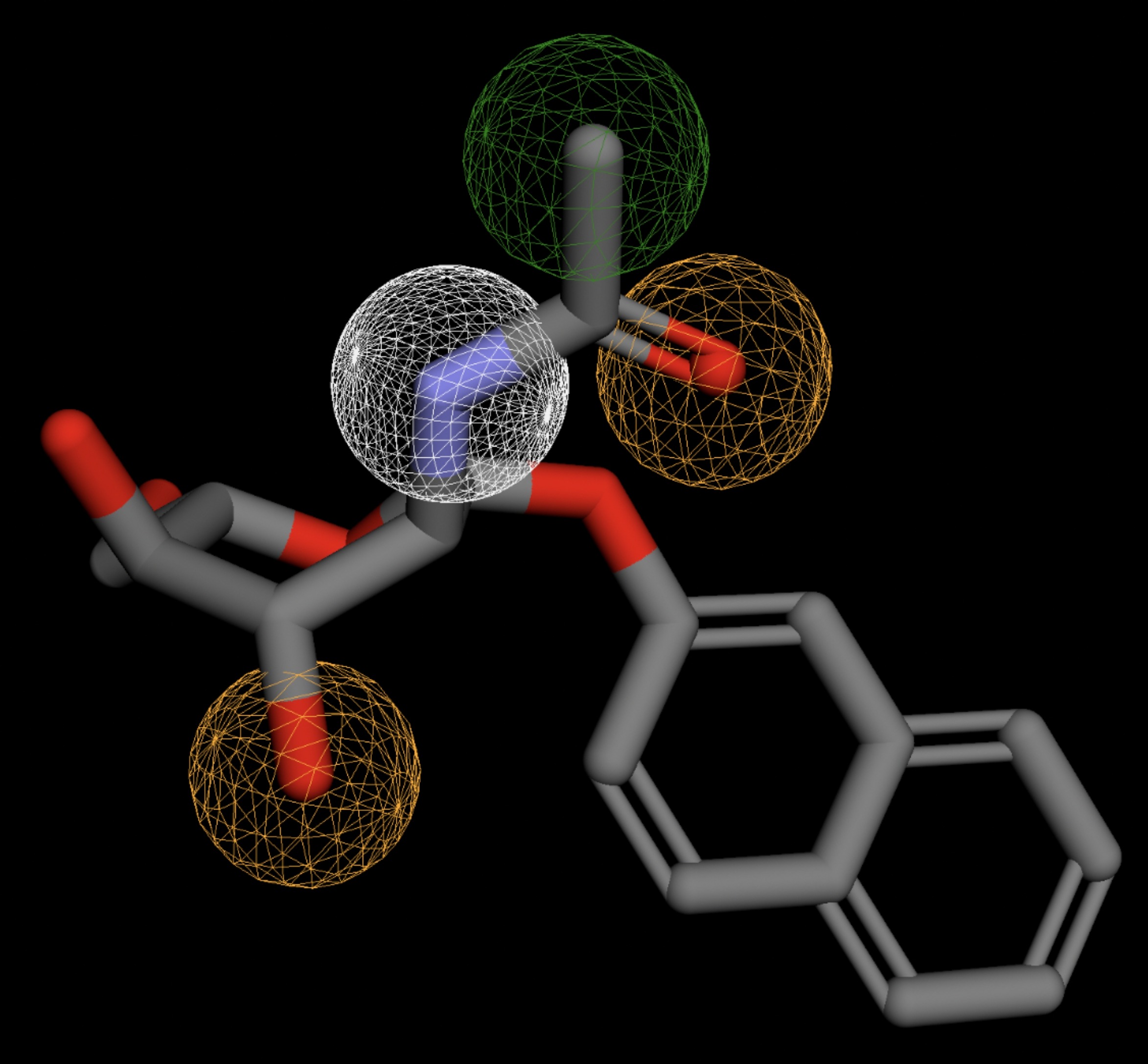 |
| MolPort-023-223-232 | 0.059 | 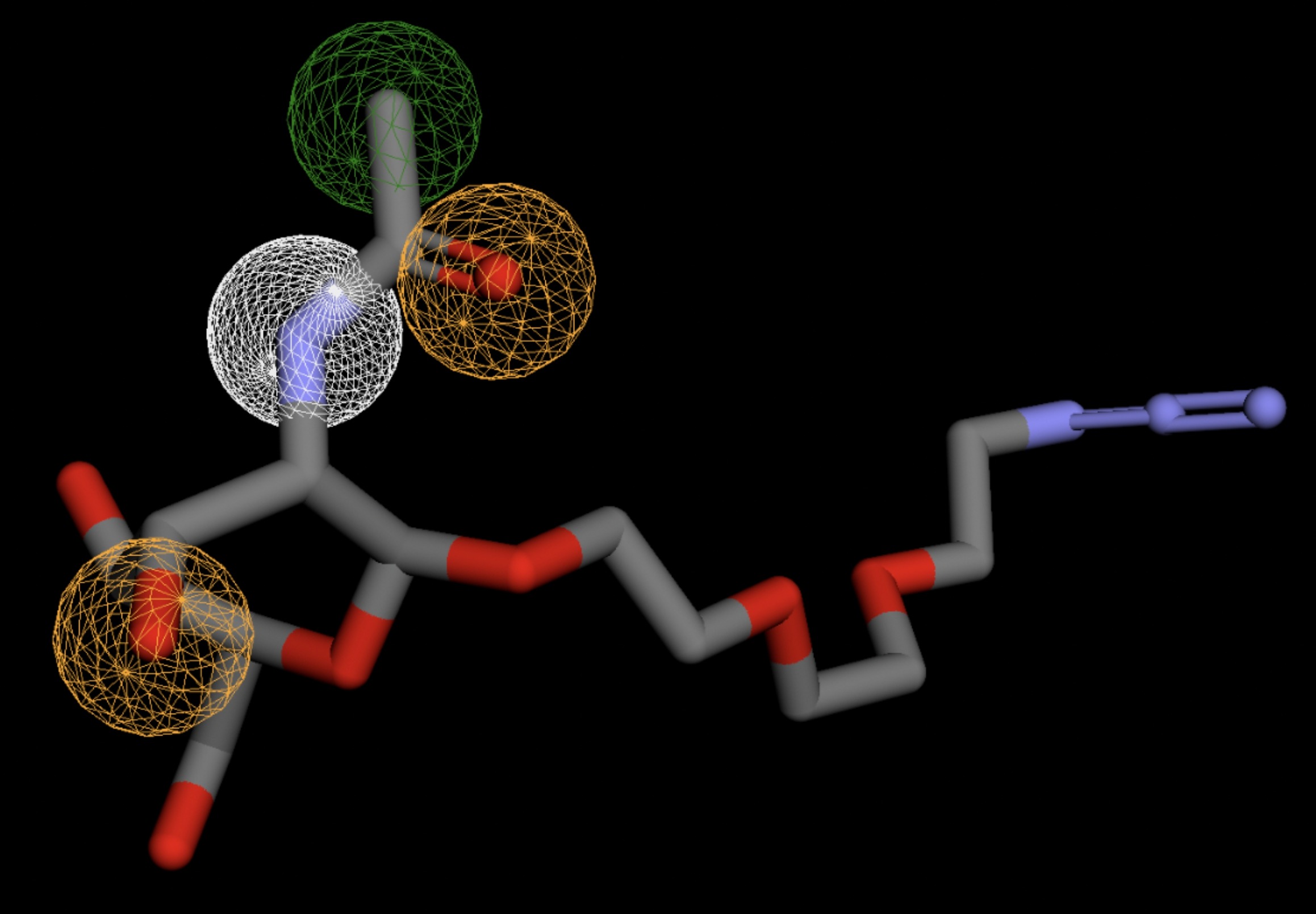 |
| MolPort-000-416-985 | 0.059 | 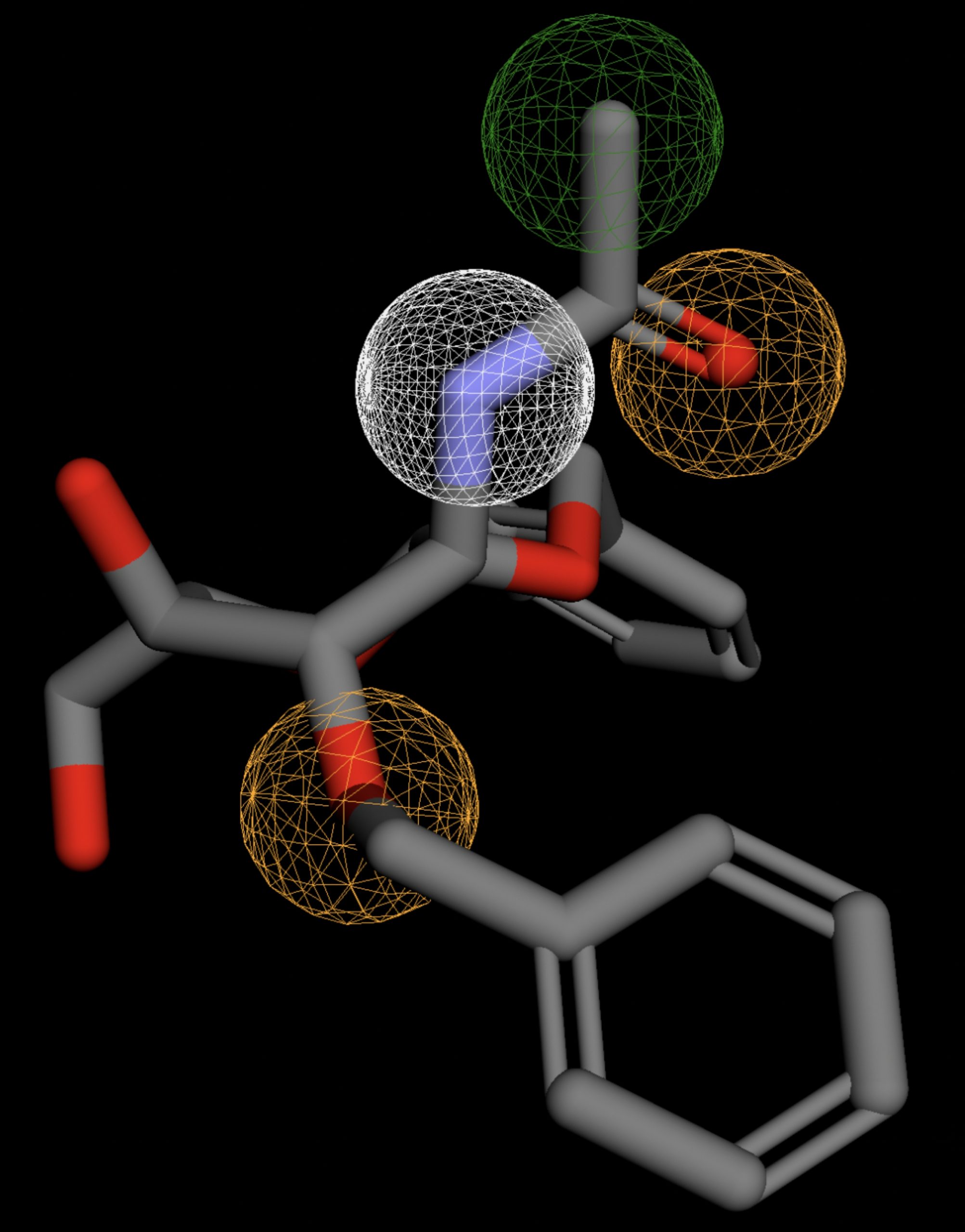 |
| MolPort-047-134-715 | 0.059 | 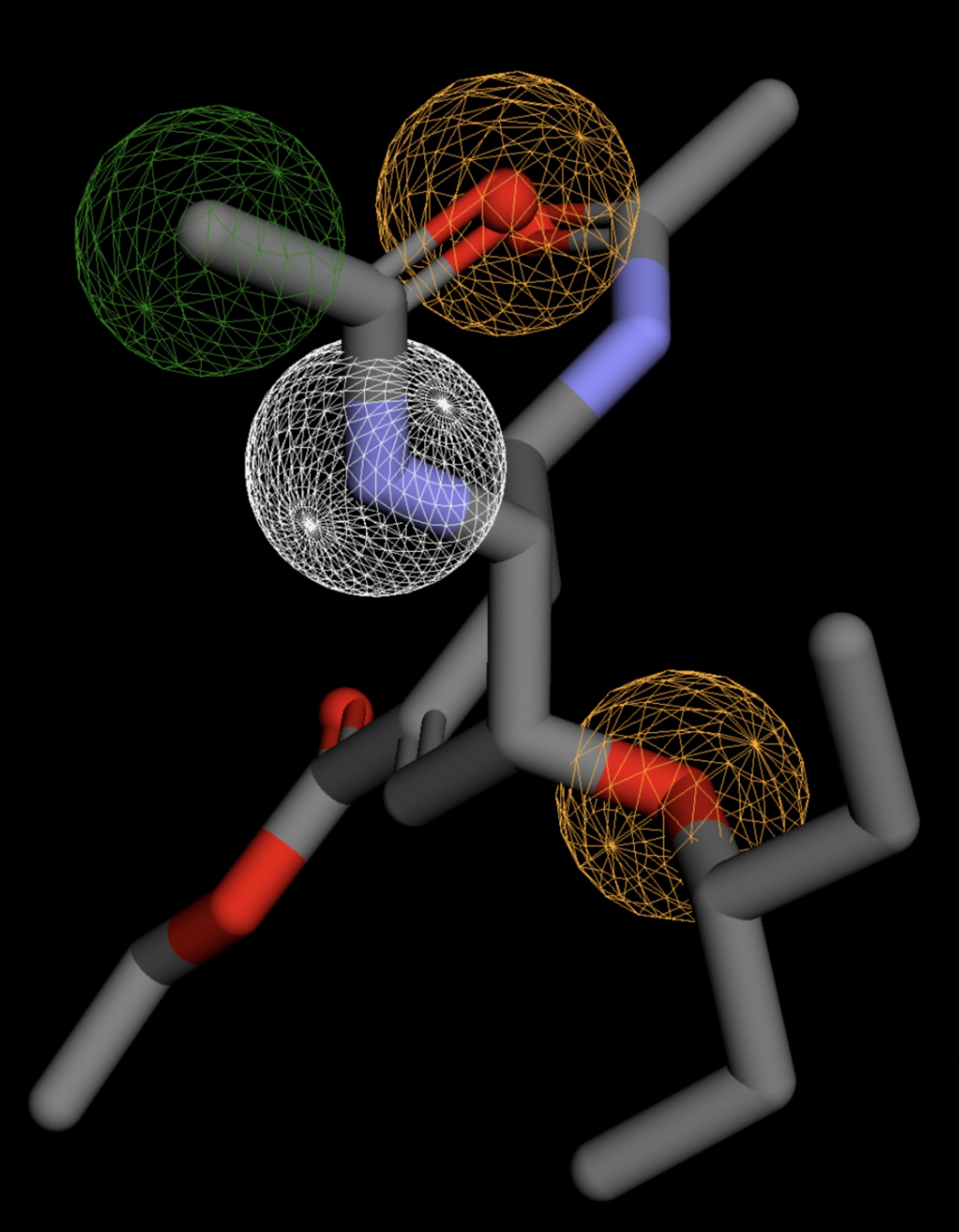 |
| MolPort-051-860-991 | 0.059 | 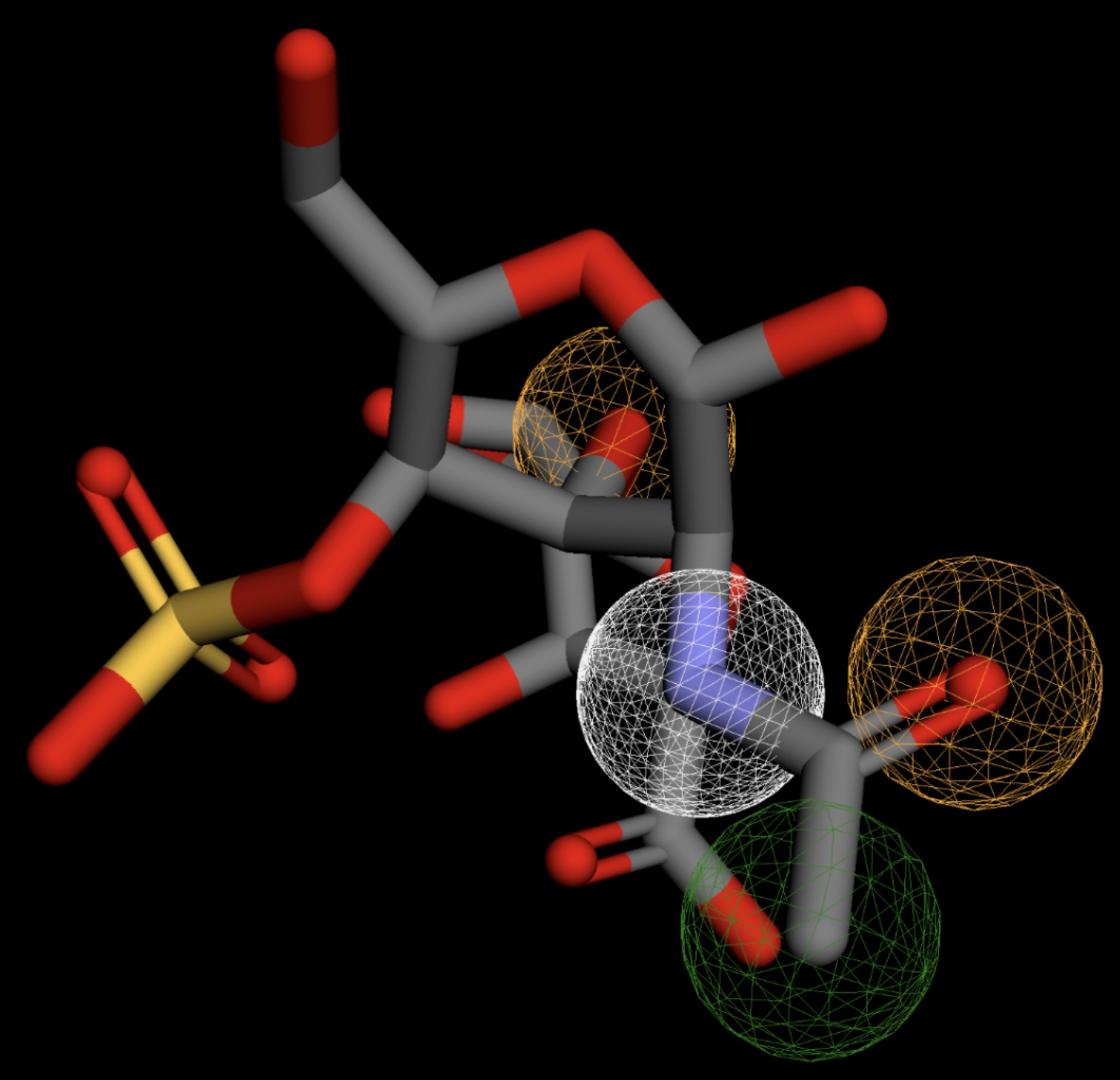 |
| MolPort-006-167-538 | 0.059 | 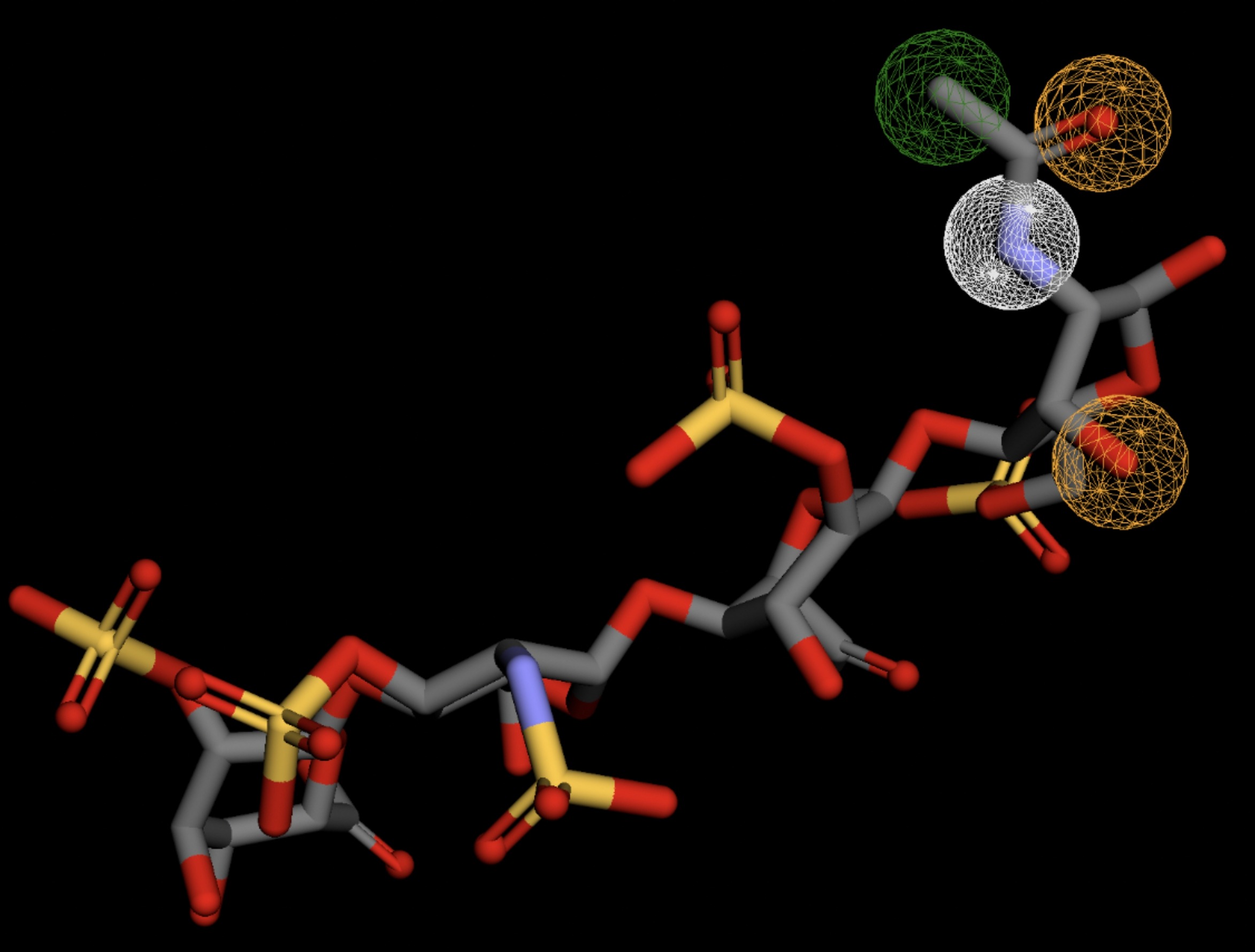 |
| MolPort-004-849-816 | 0.060 | 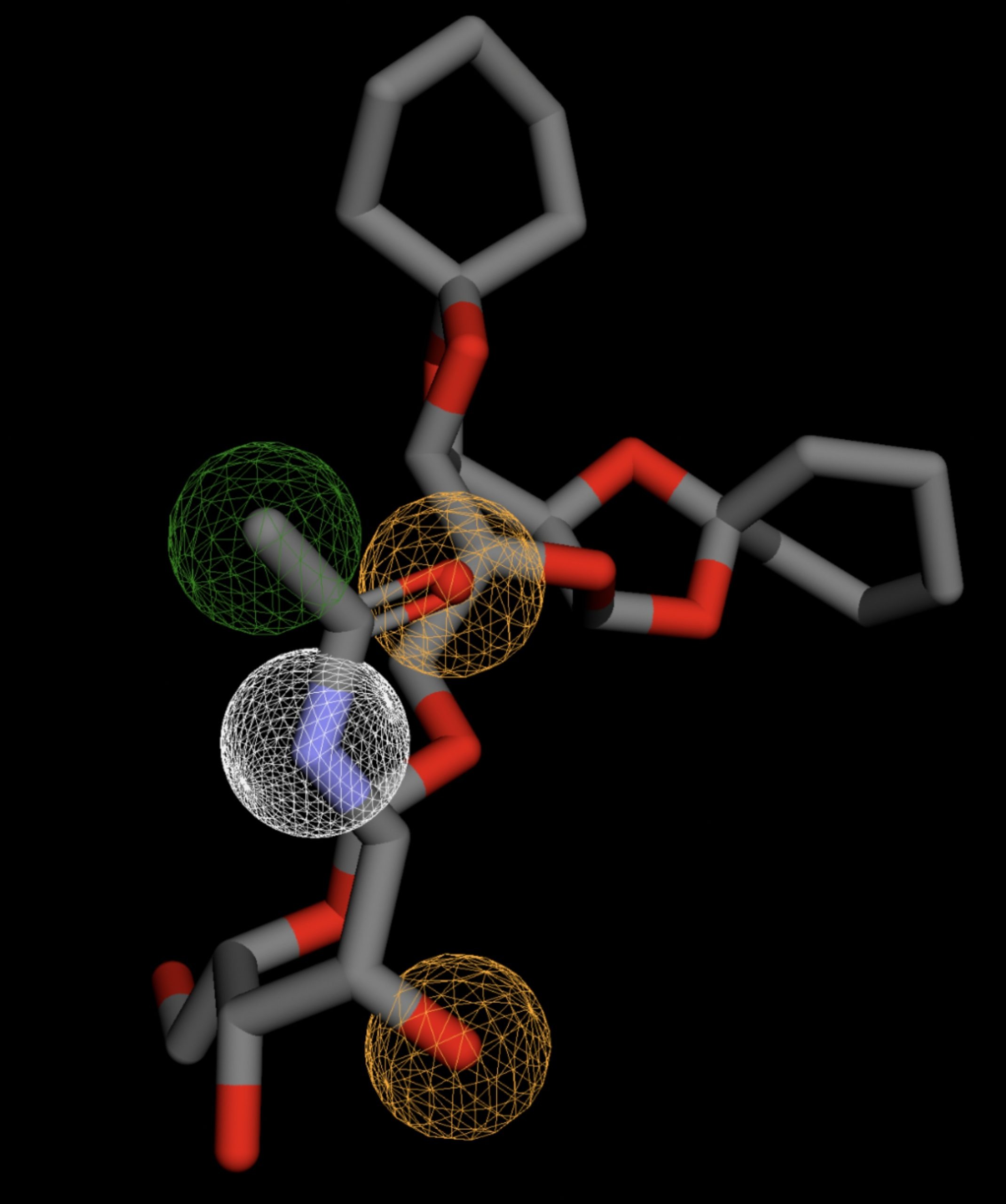 |
| MolPort-008-267-878 | 0.060 | 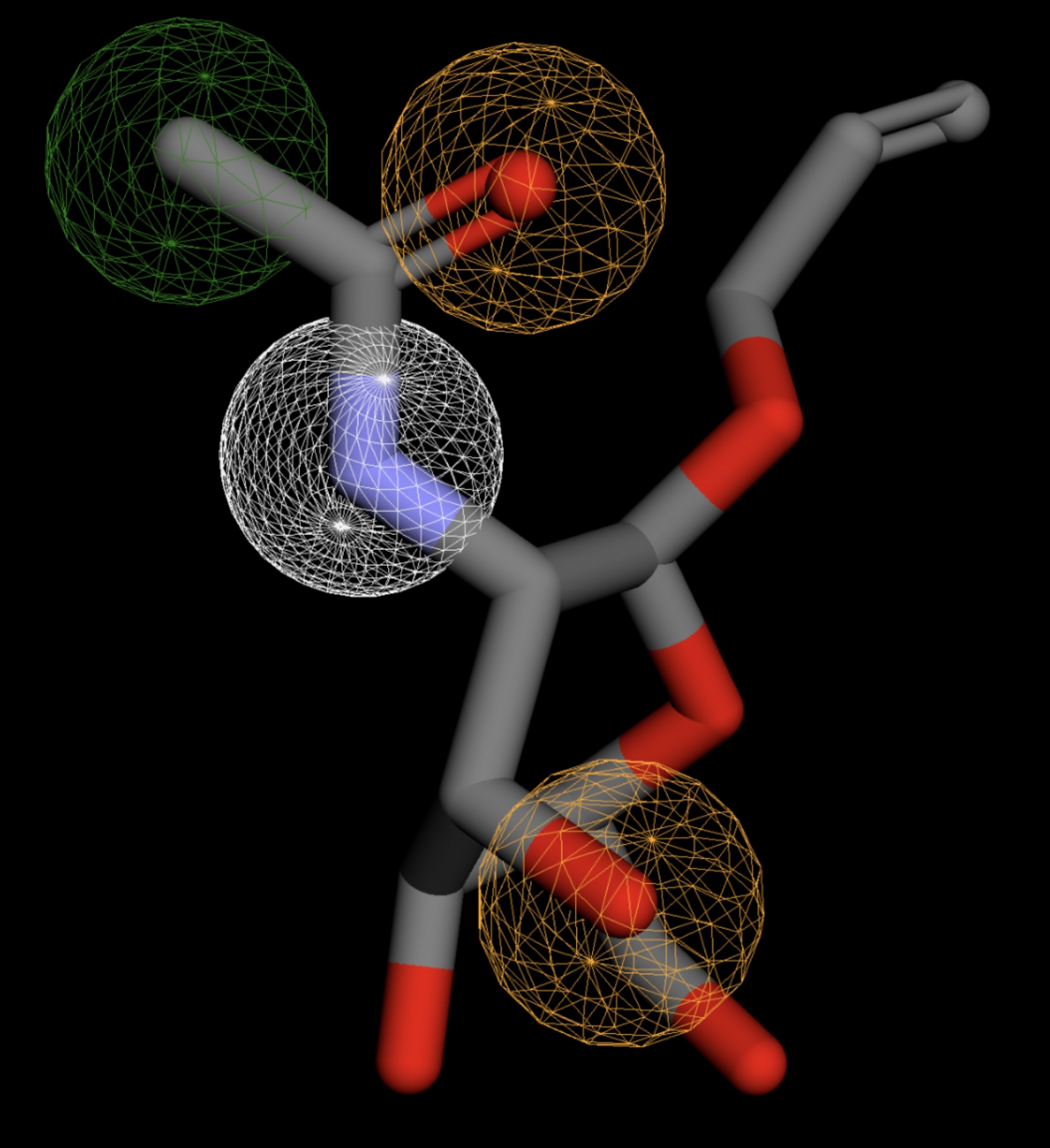 |
| MolPort-002-511-046 | 0.060 | 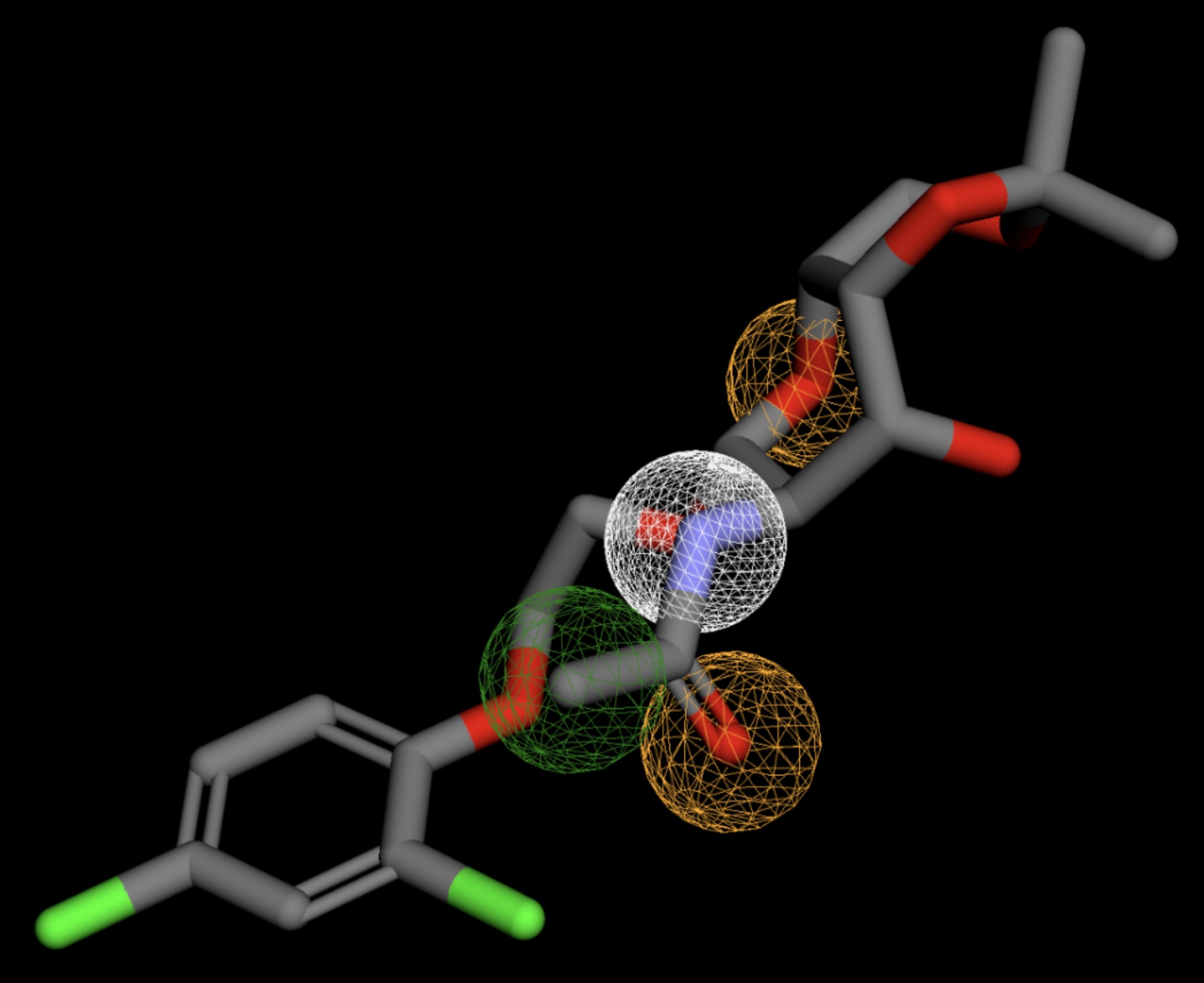 |
| MolPort-003-011-602 | 0.060 | 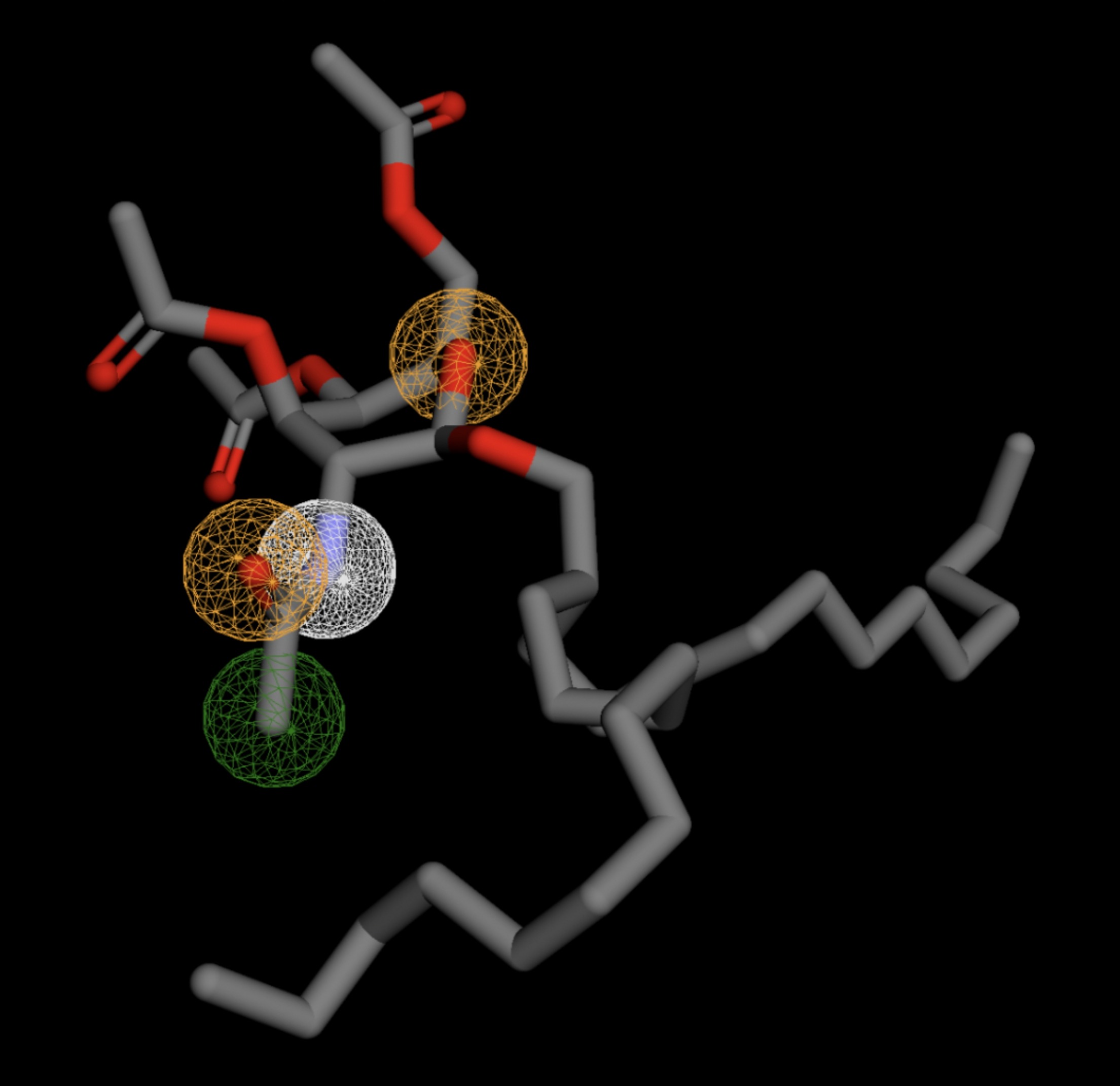 |
| MolPort-038-394-503 | 0.061 | 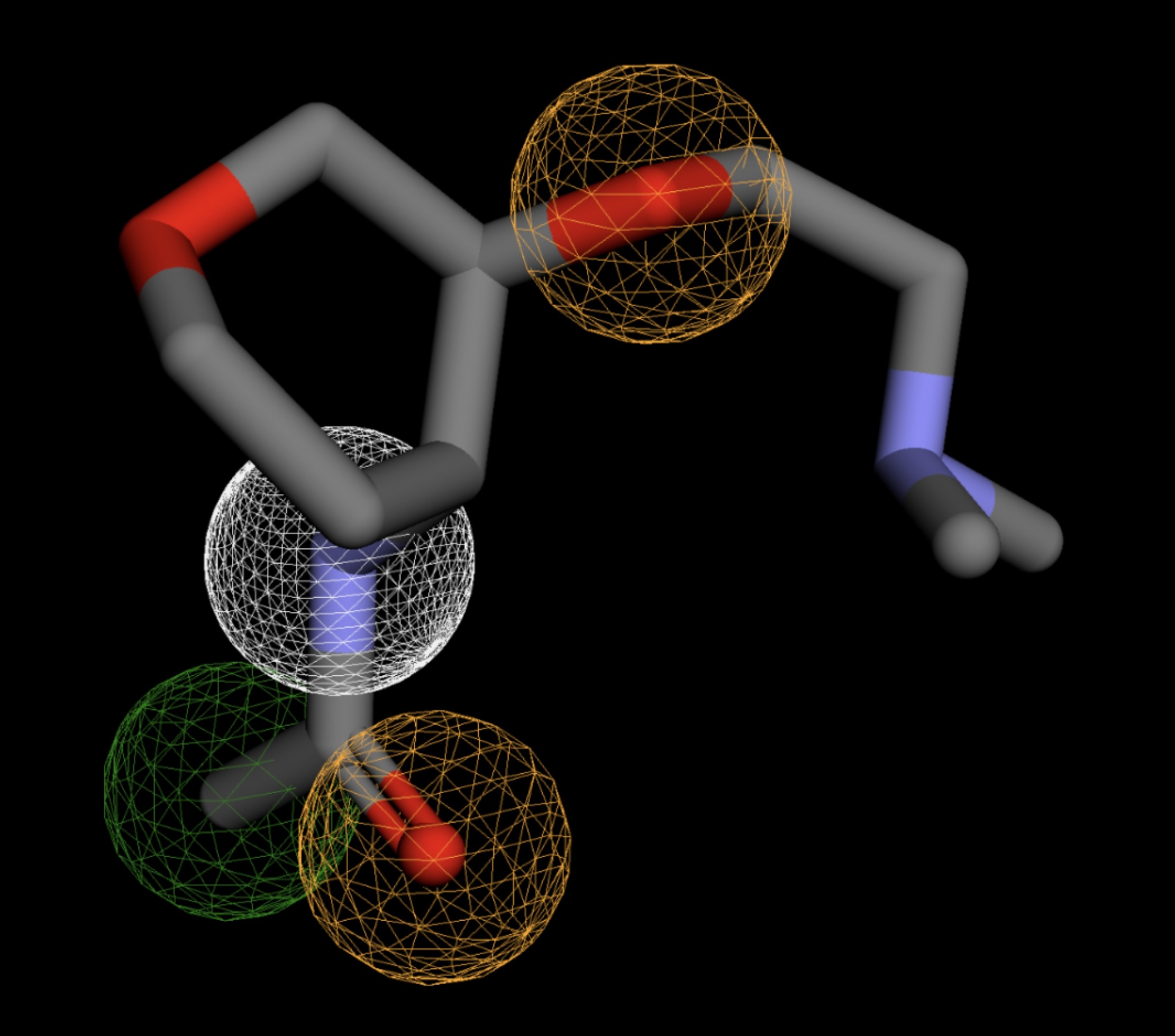 |
| MolPort-052-043-087 | 0.061 | 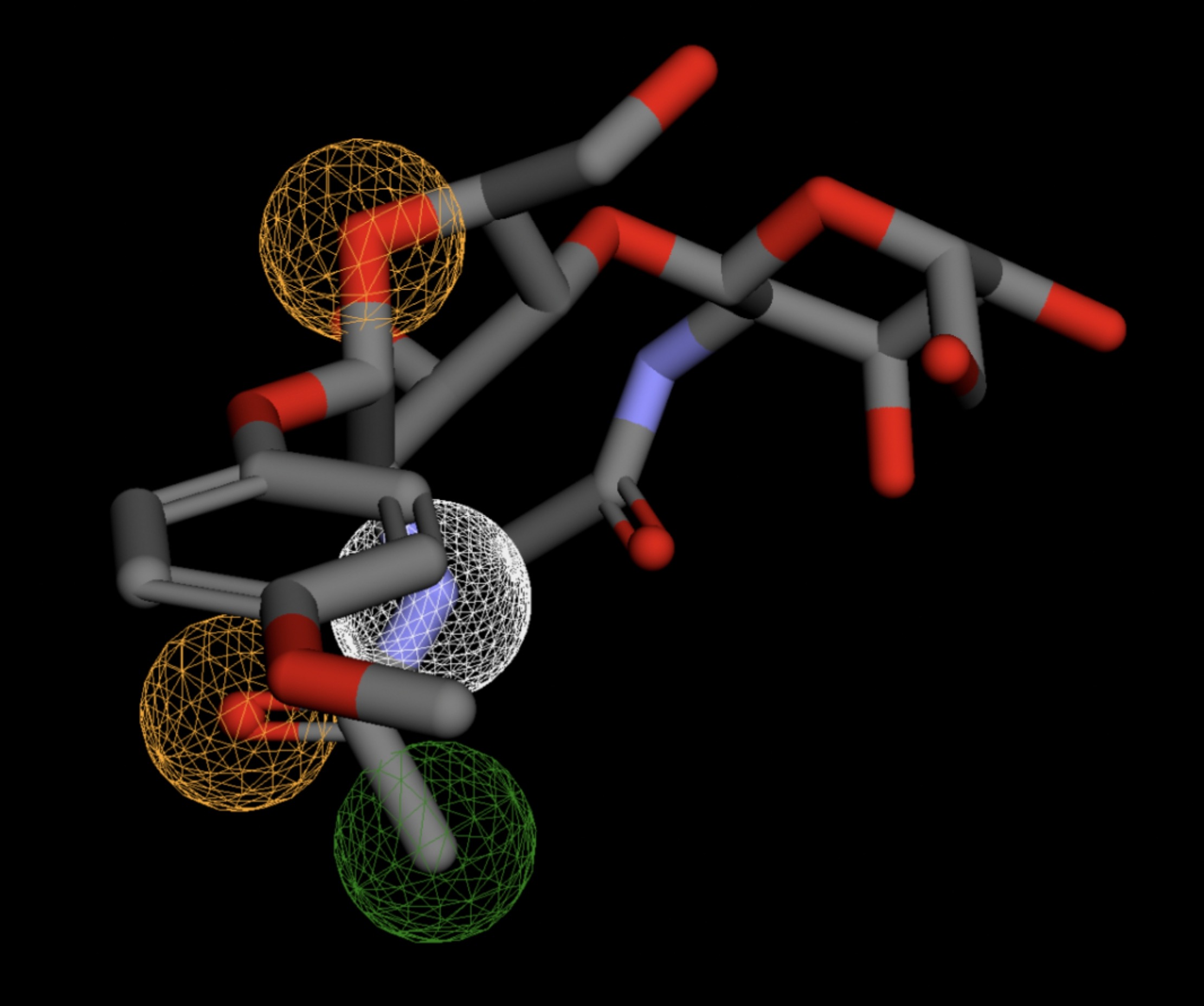 |
| MolPort-005-289-910 | 0.061 | 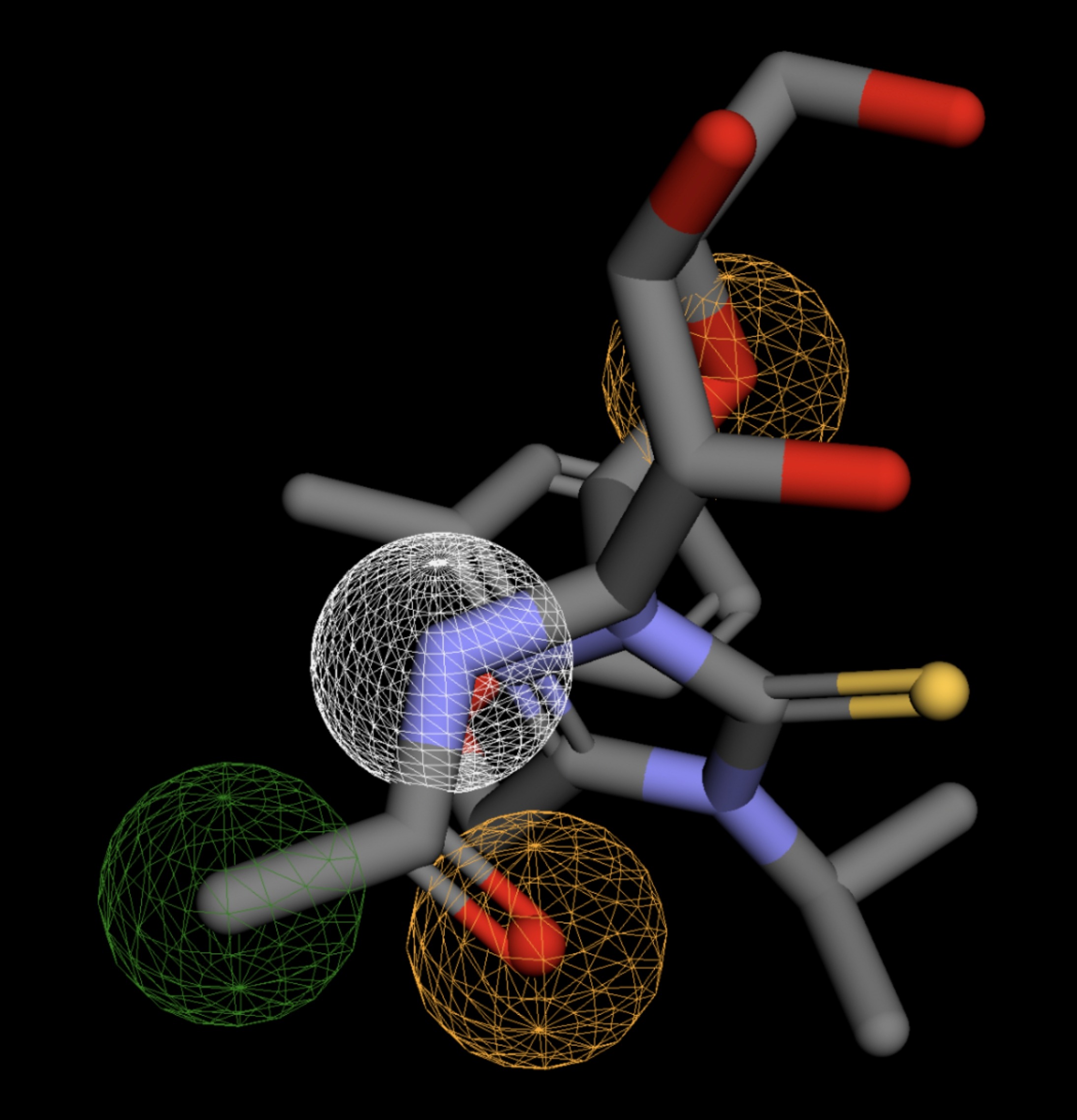 |
| MolPort-051-856-633 | 0.061 | 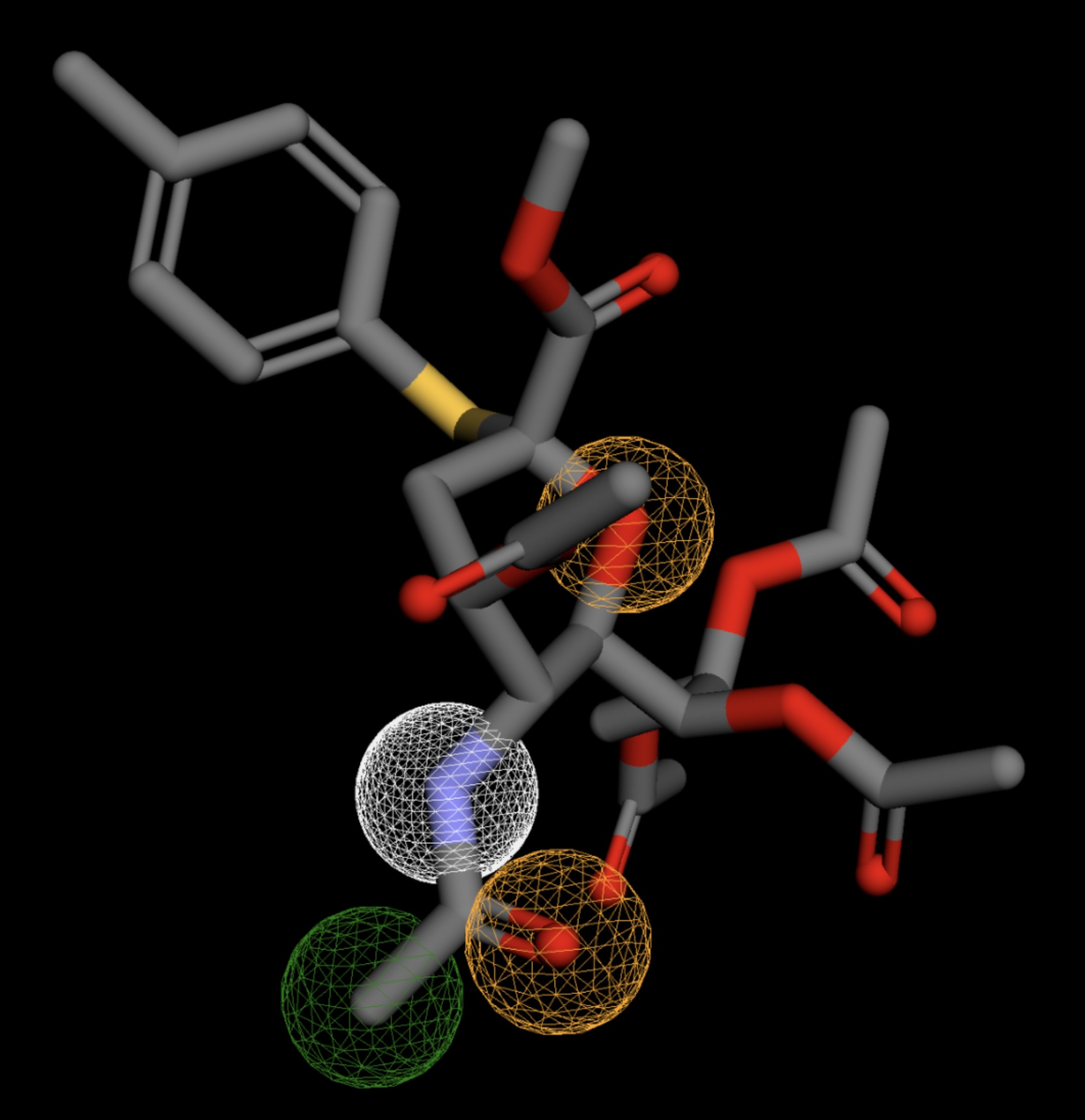 |
| MolPort-047-590-656 | 0.061 | 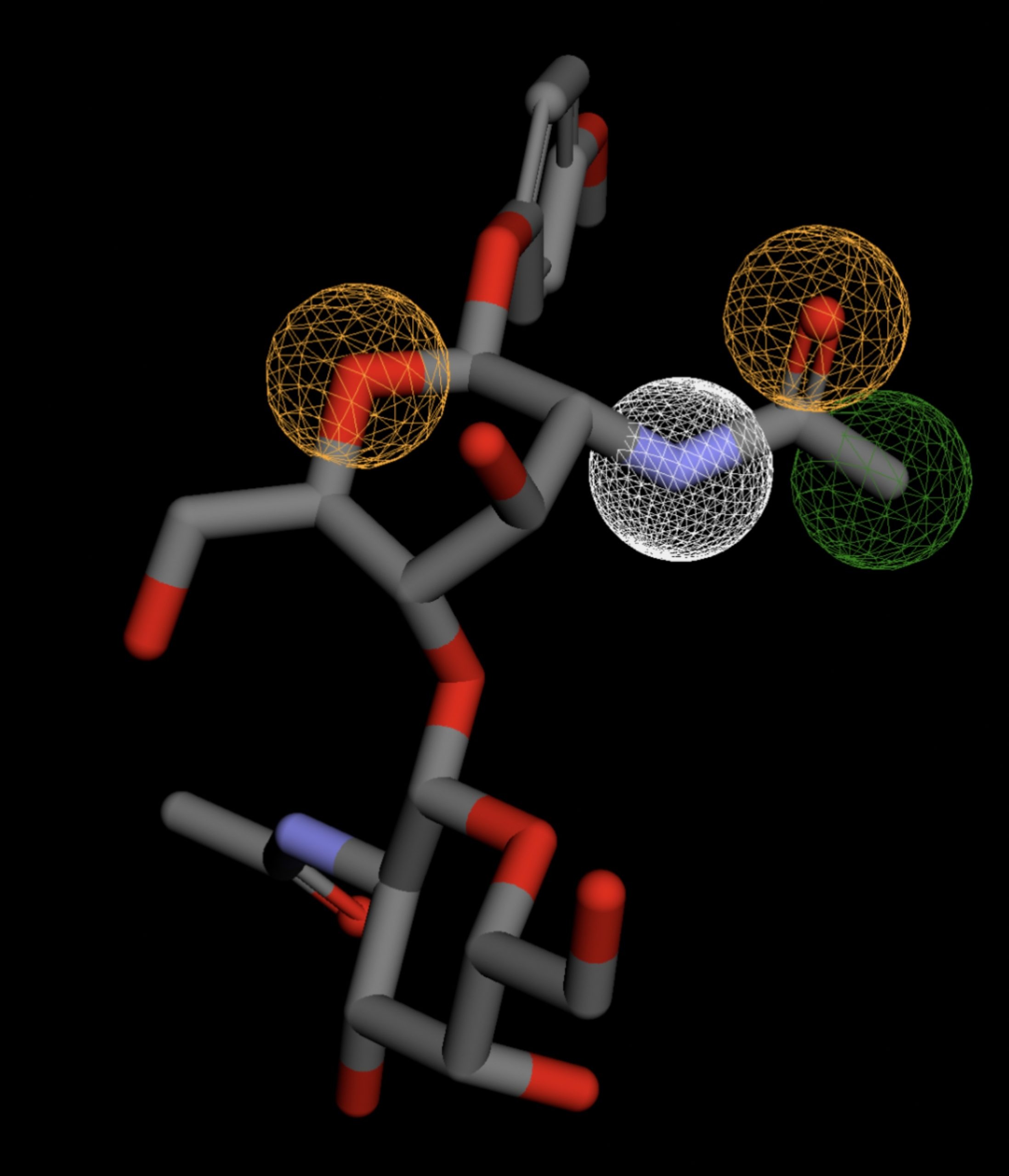 |
| MolPort-046-763-086 | 0.061 | 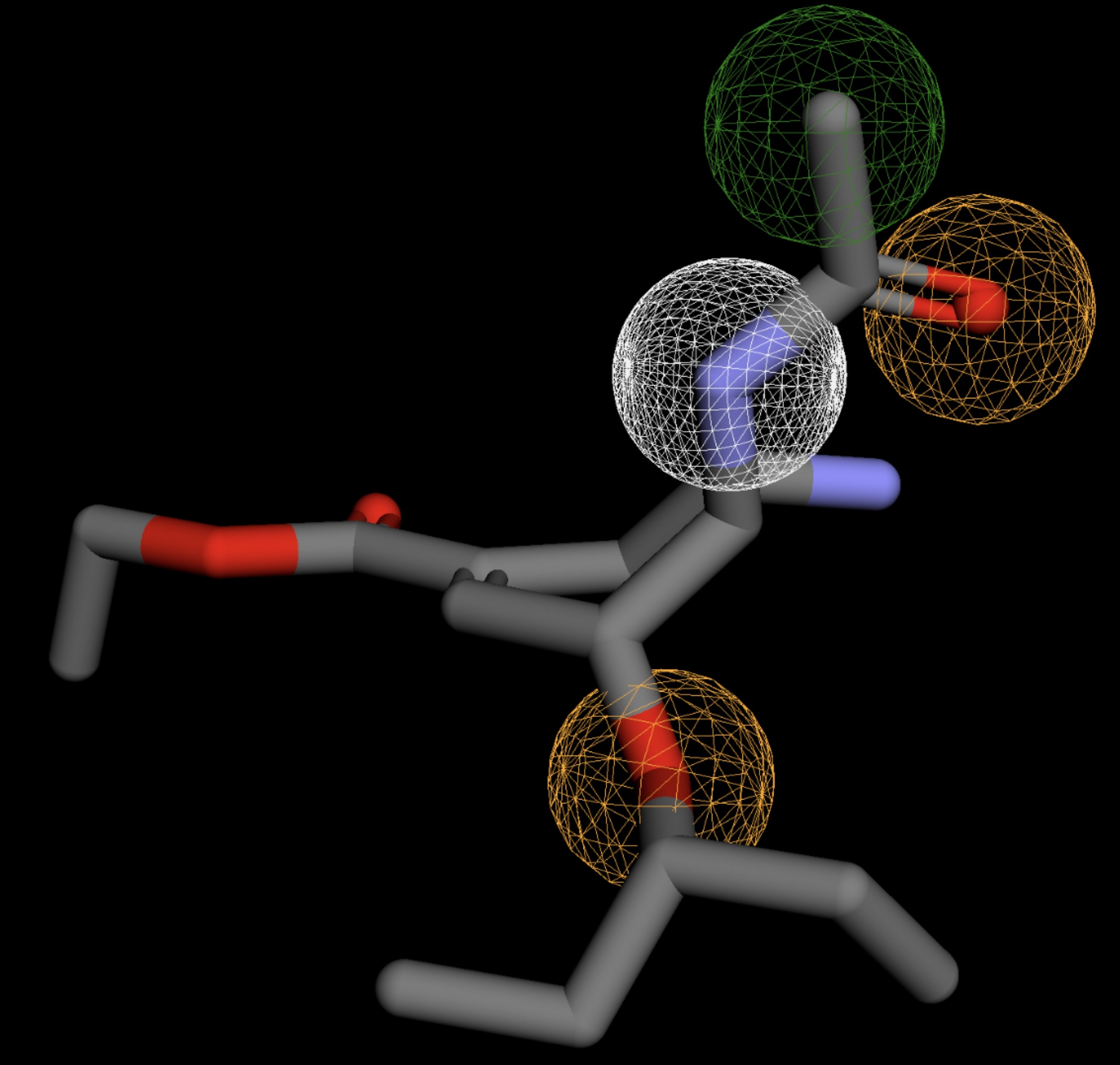 |
| MolPort-004-849-824 | 0.061 | 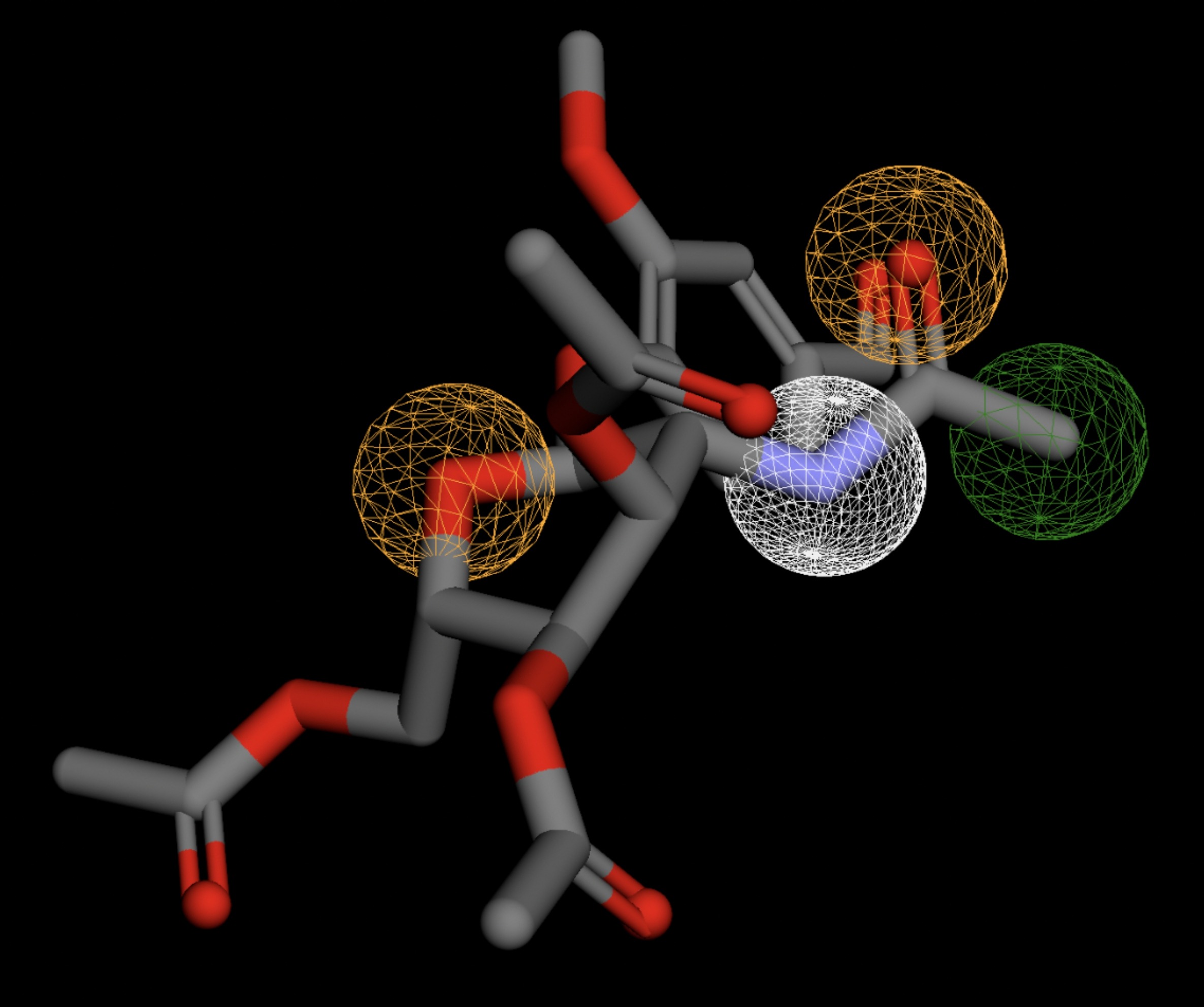 |
| MolPort-003-060-590 | 0.061 | 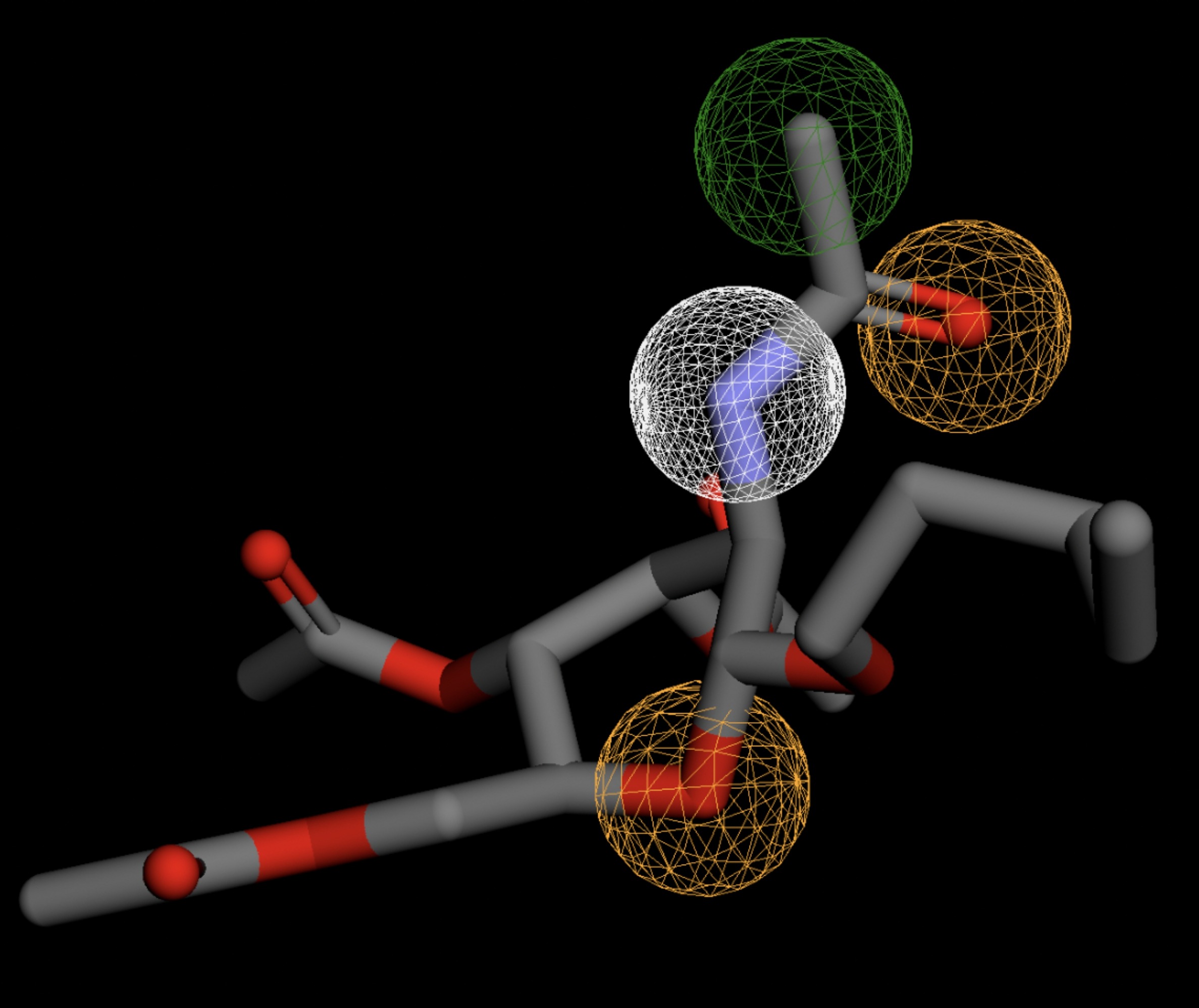 |
Molecular Docking Tests
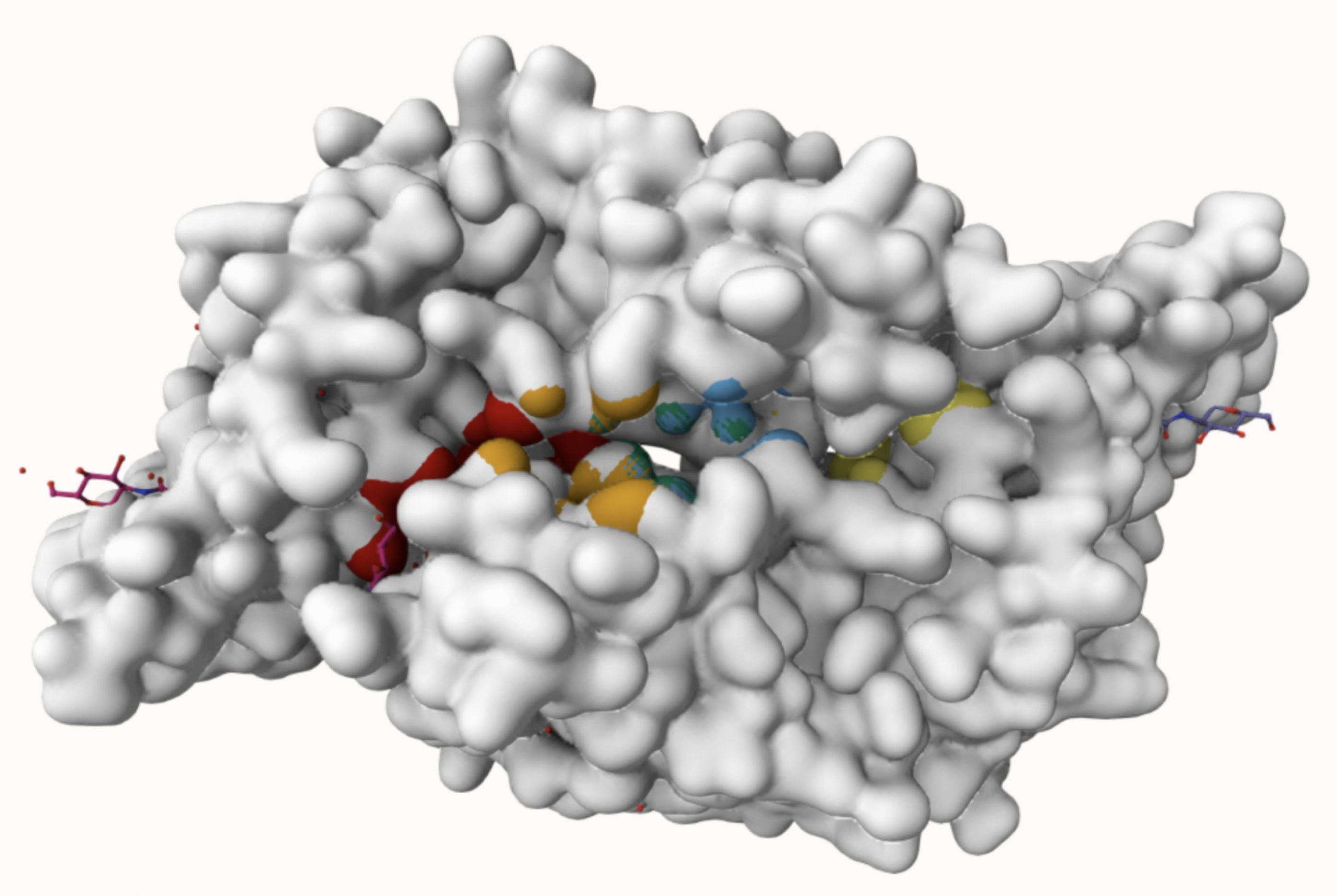
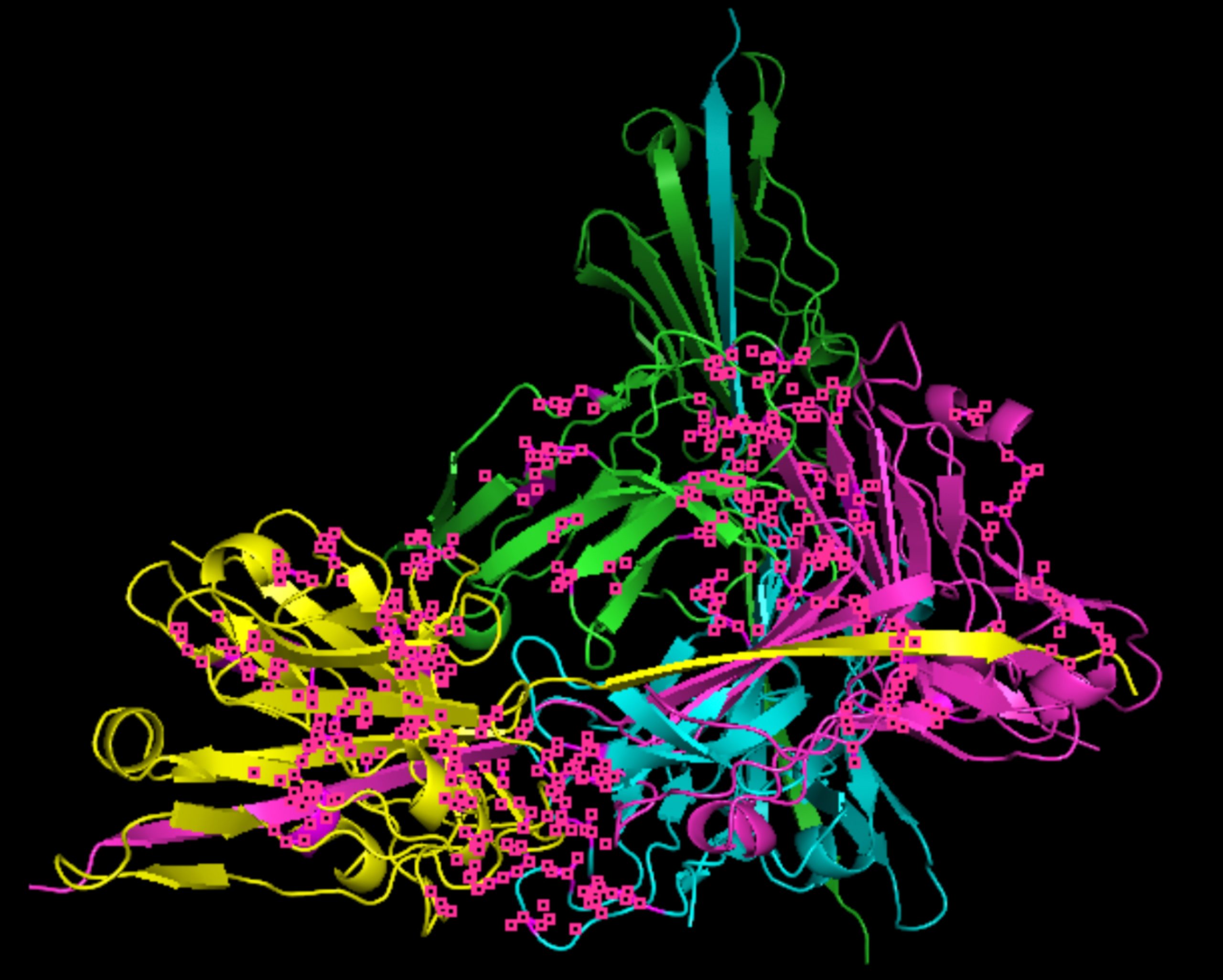
Molecular docking was performed through SwissDock. Each compound was docked individually and docking scores were evaluated based on the SwissParam scores. The search space center was defined as 5, -39, 21 Å and the search space size was defined as 33, 35, 28 Å. A visual of this search space is pictured in Figure 6. The results from the molecular docking process are shown in Table 5. To relate the docking search space to the experimentally defined CD47-SIRPα interface, SIRPα-contacting residues from the CD47-SIRPα complex (PDB 2JJT) were mapped onto the CD47 structure used for docking (PDB 2VSC), as shown in Figure 7.
| MolPort ID | Lowest Observed Estimated ∆G (Kcal/mol) | Visualization of Ligand Binding Using the Interacting Residue Pose | Visualization of Ligand Binding with Protein Surface |
| MolPort-008-326-608 | -8.1251 | 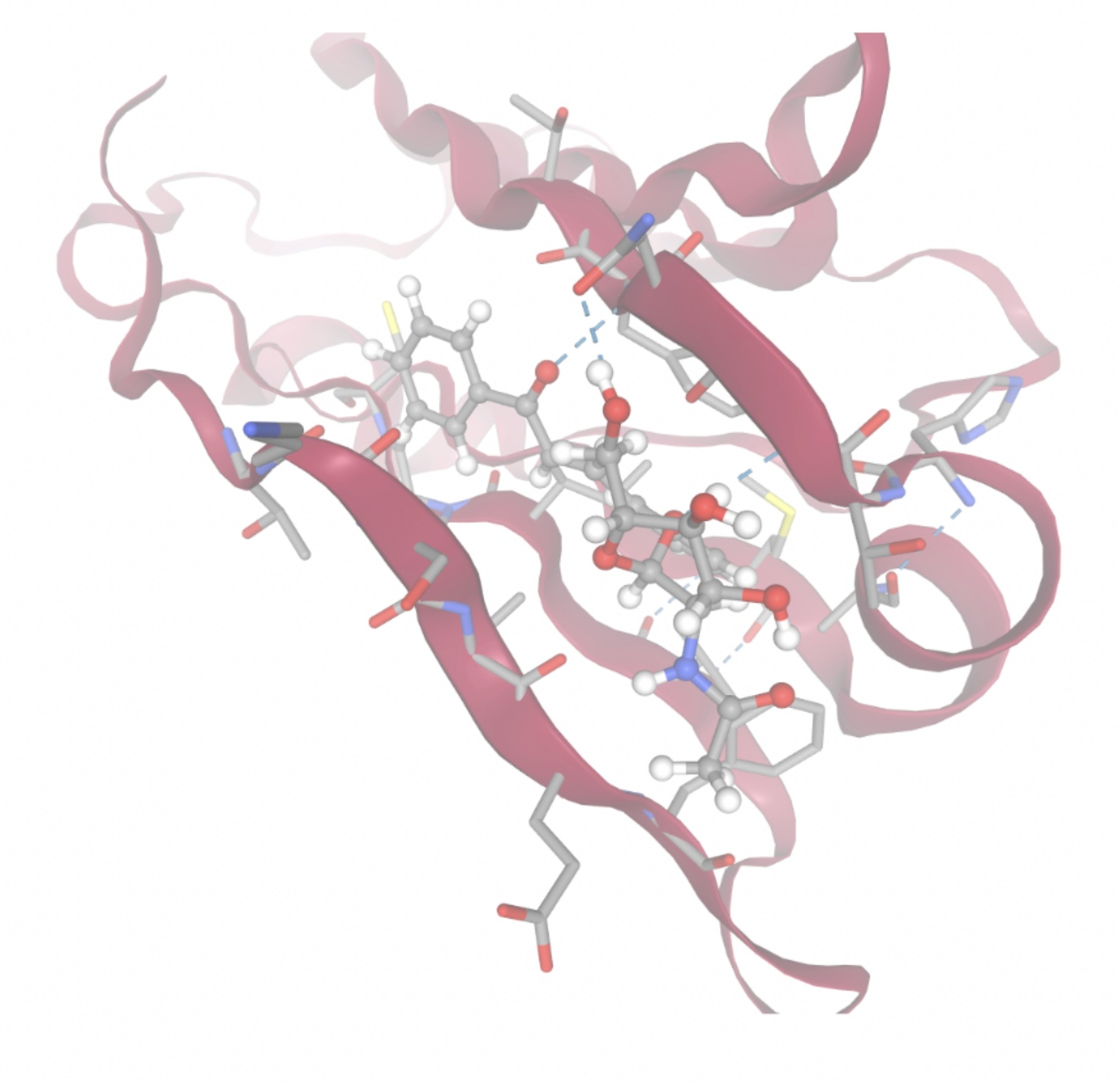 | 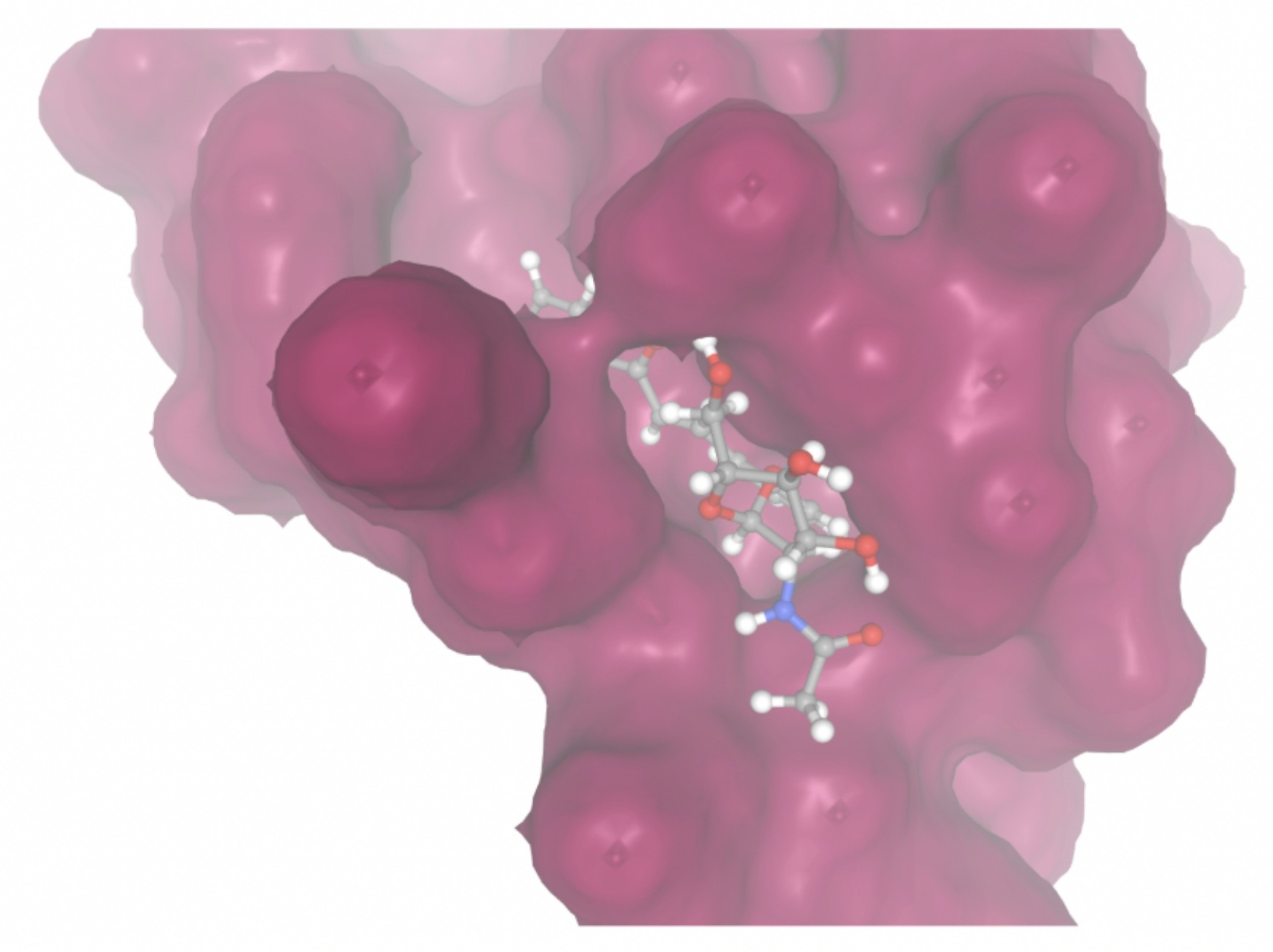 |
| MolPort-005-280-798 | -8.7131 | 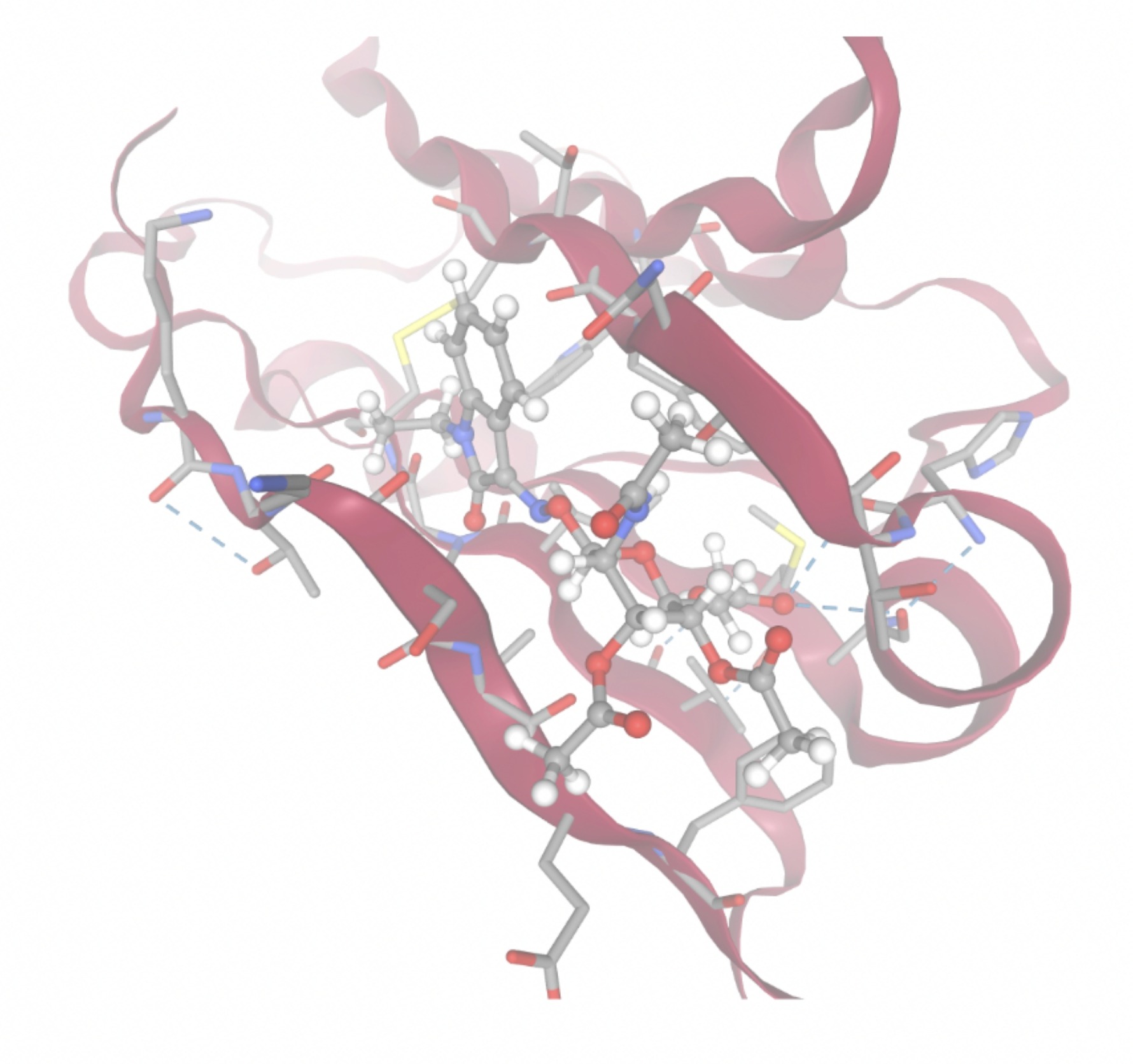 | 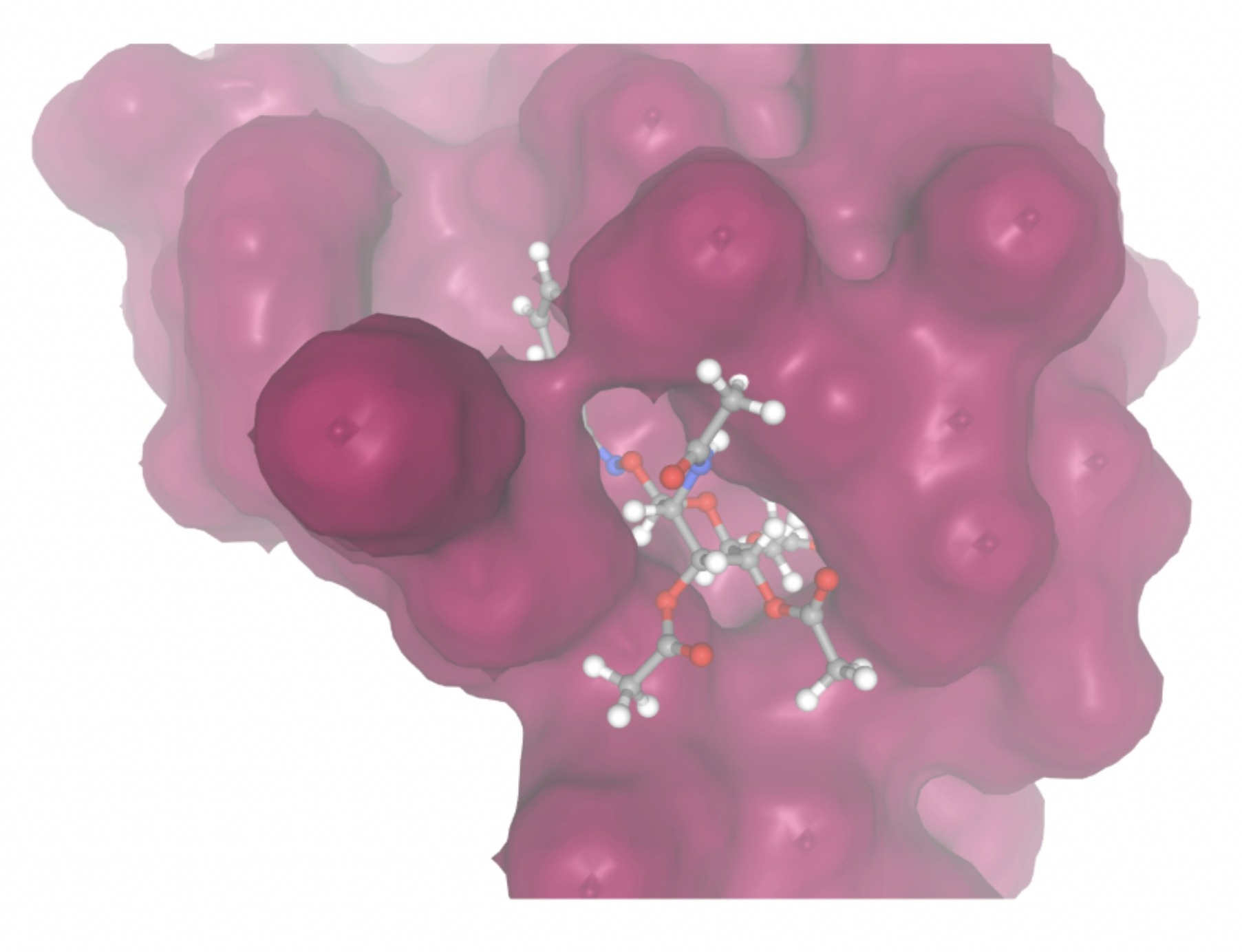 |
| MolPort-002-512-231 | -7.6195 | 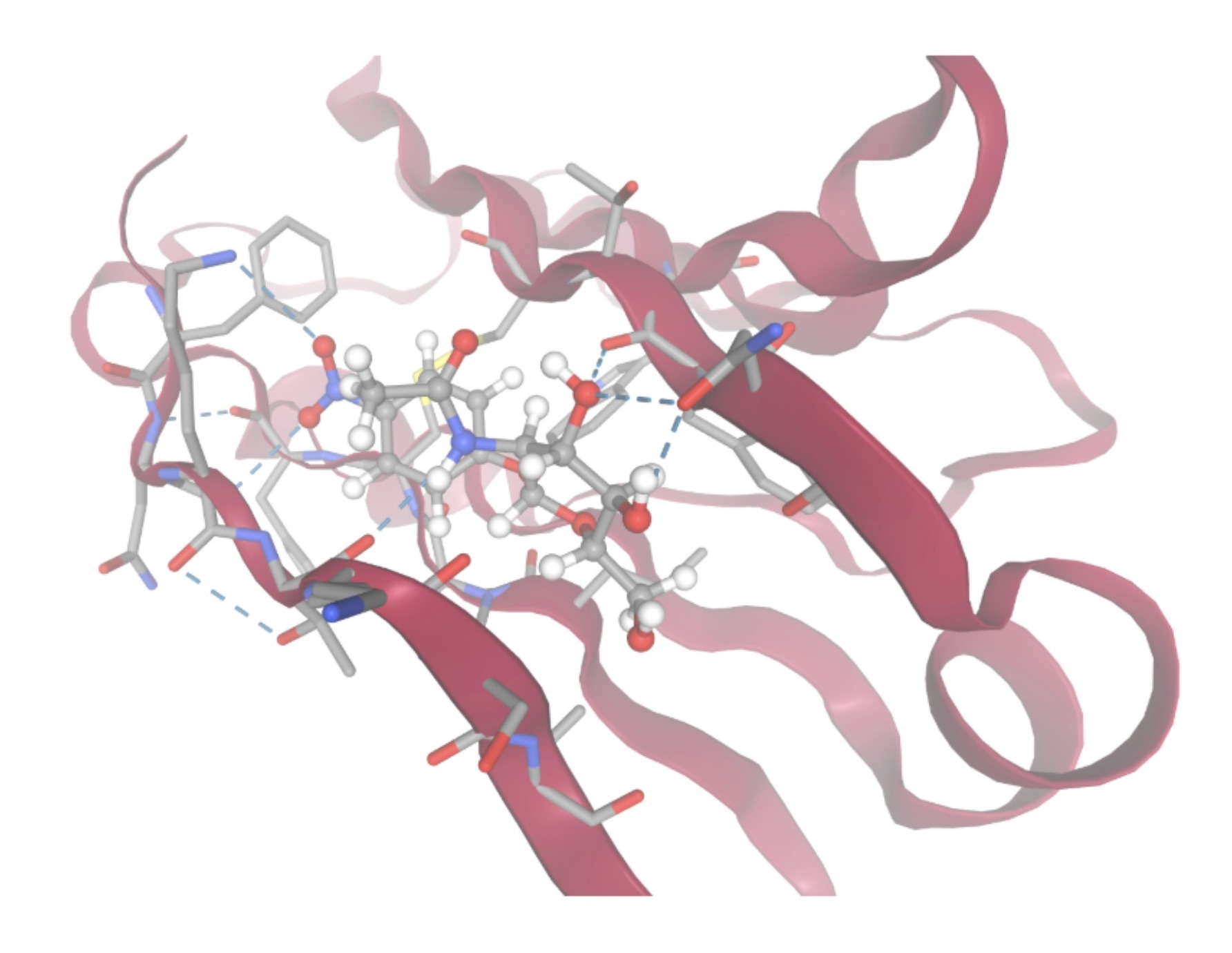 | 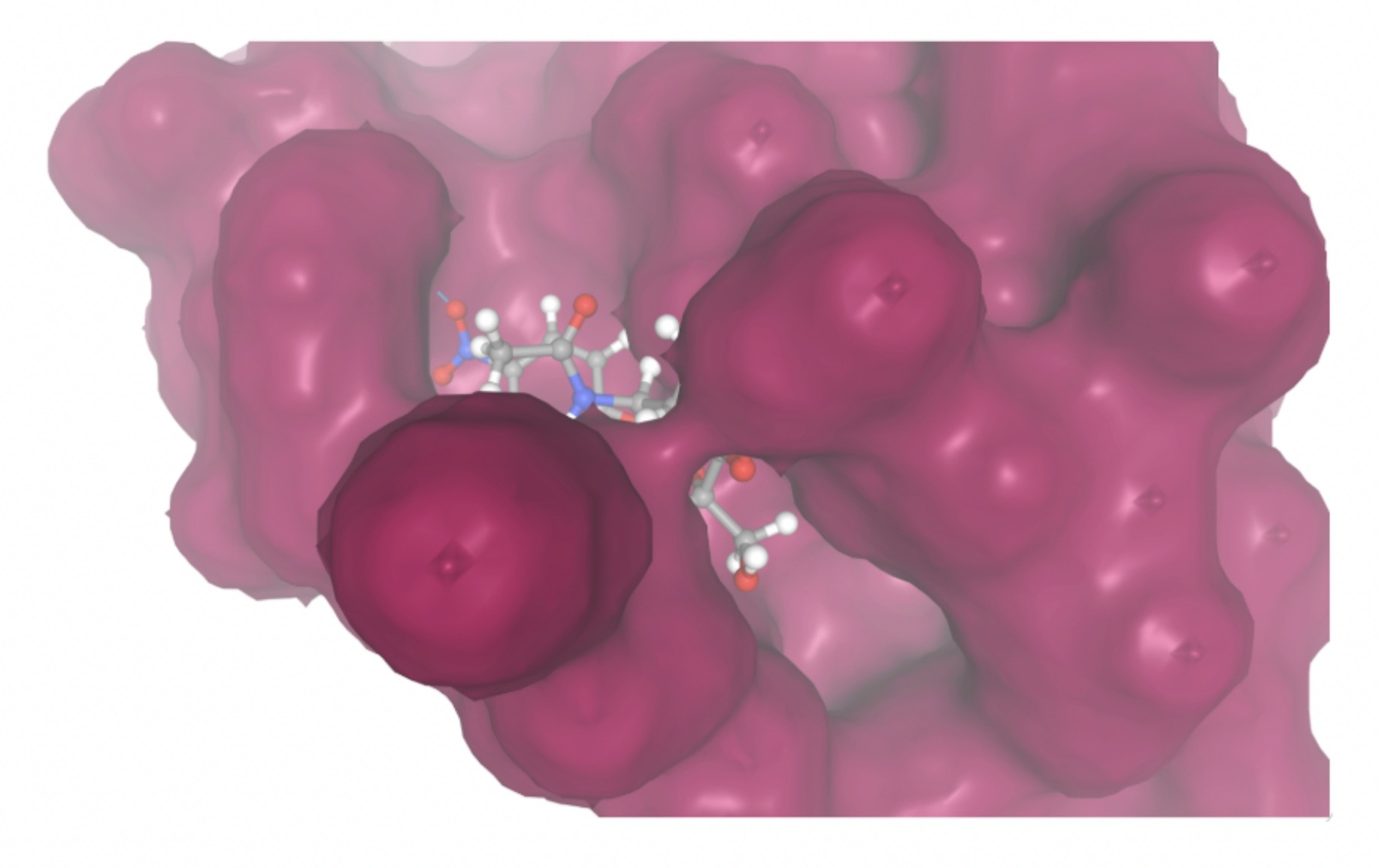 |
| MolPort-006-391-514 | -7.5954 | 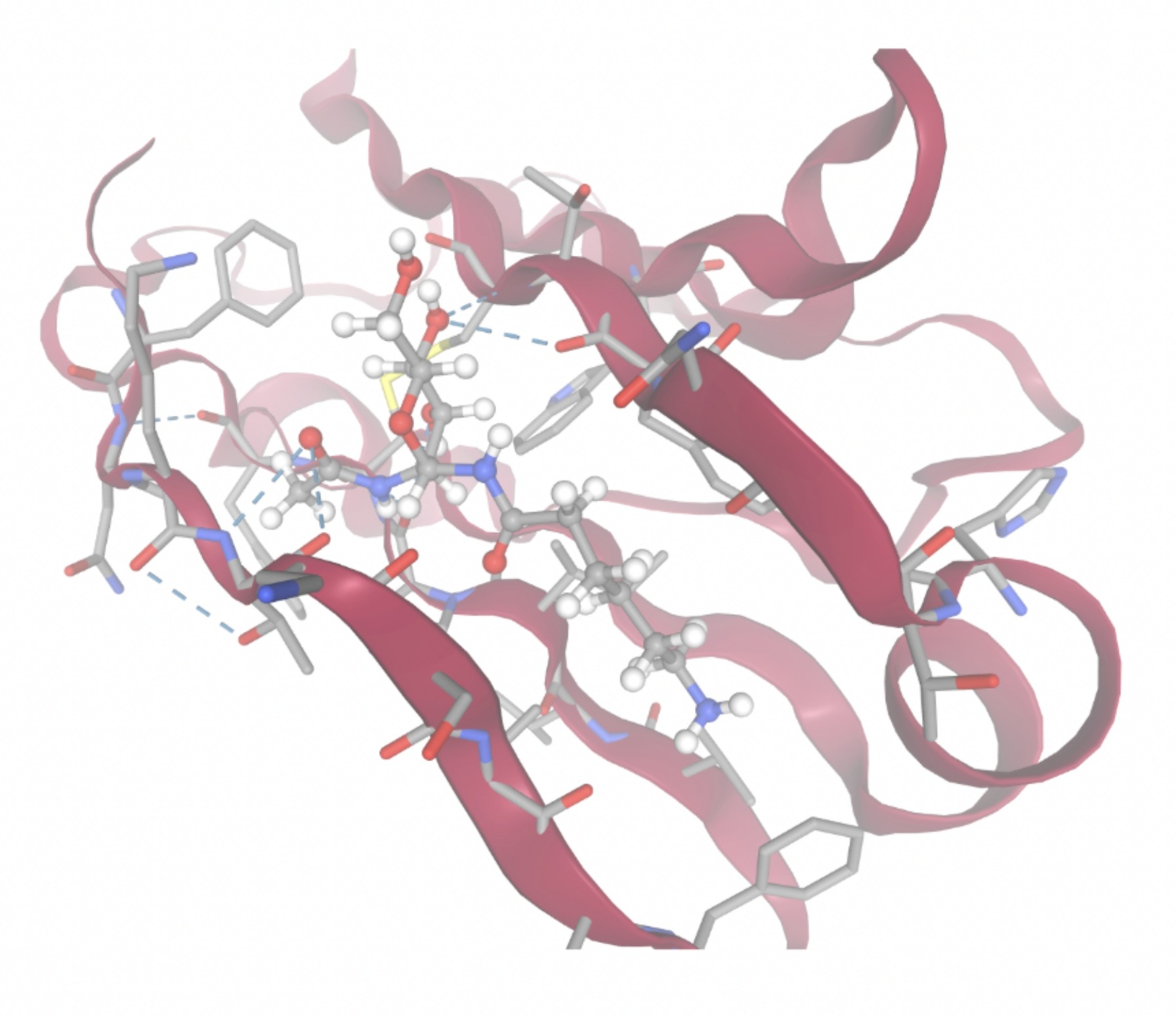 | 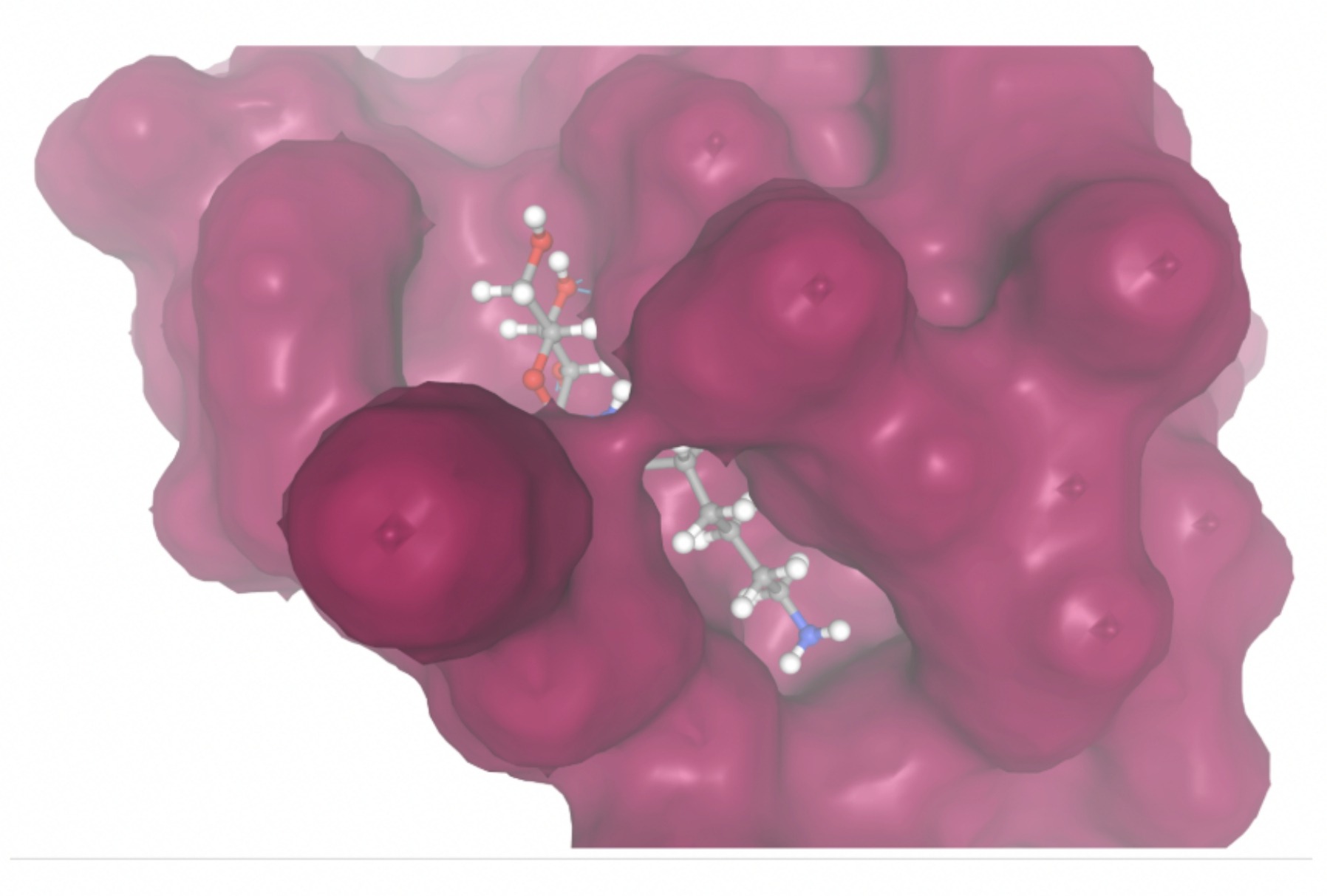 |
| MolPort-051-689-380 | -7.5941 | 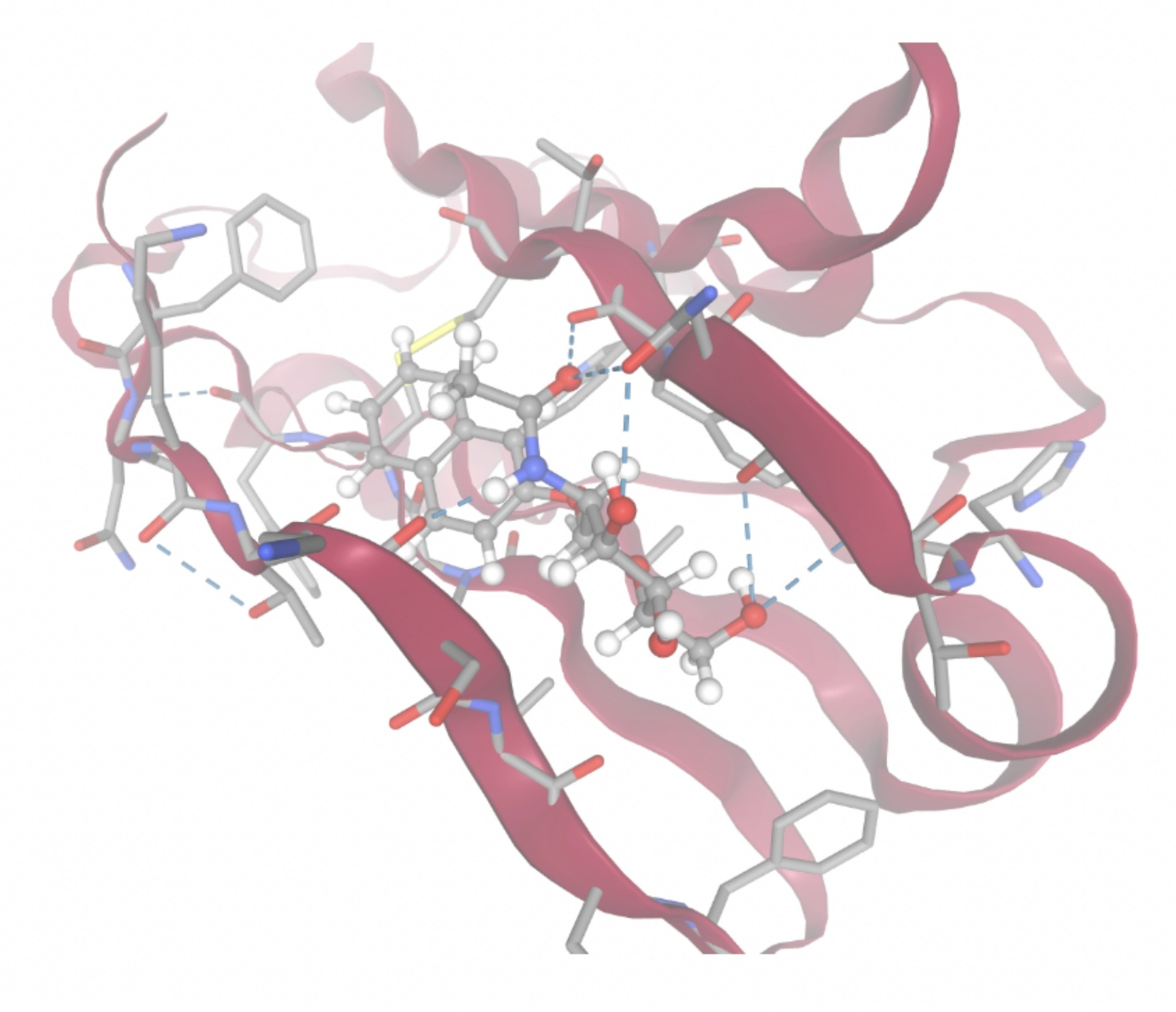 | 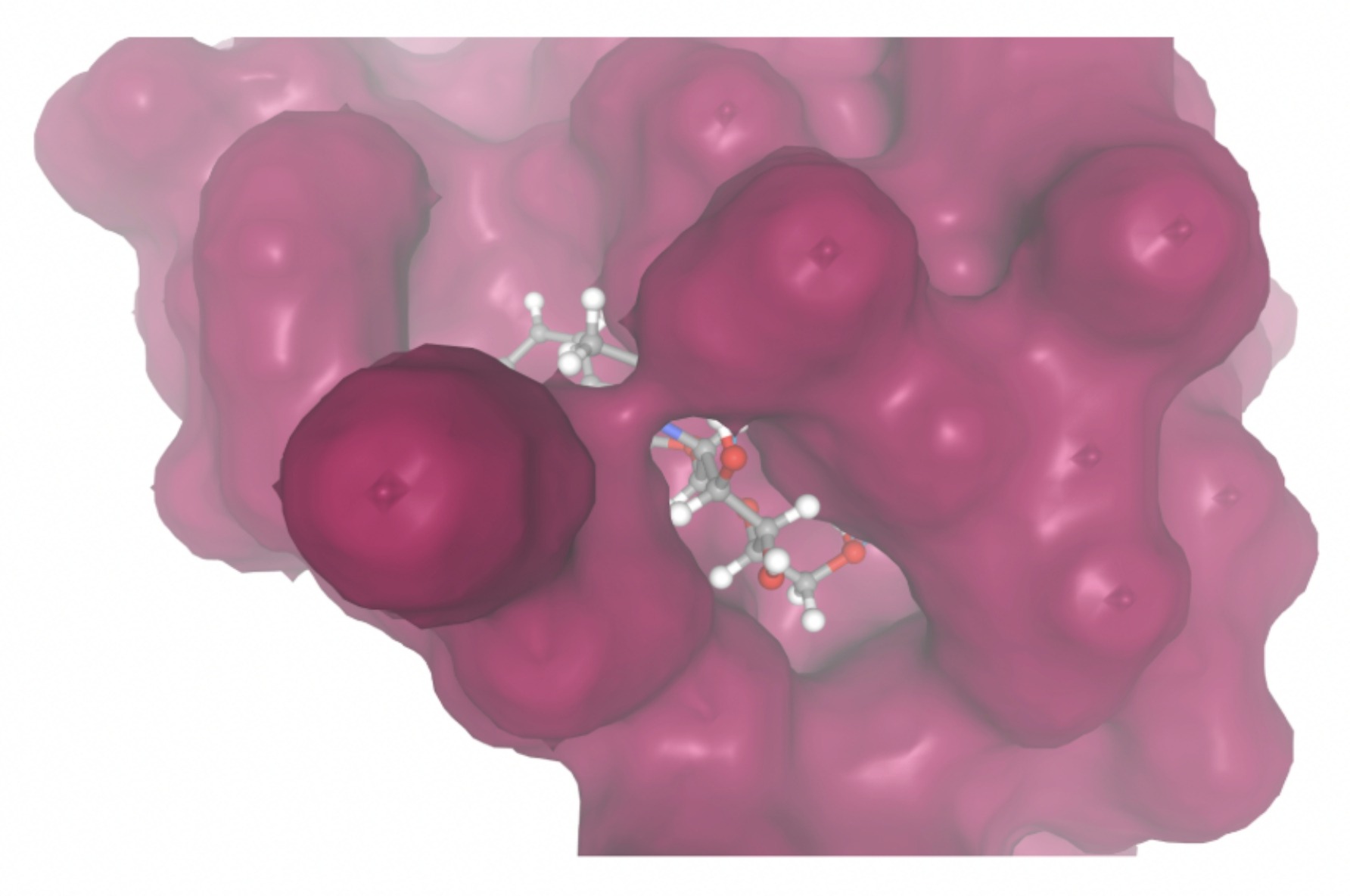 |
| MolPort-023-223-232 | -8.3501 | 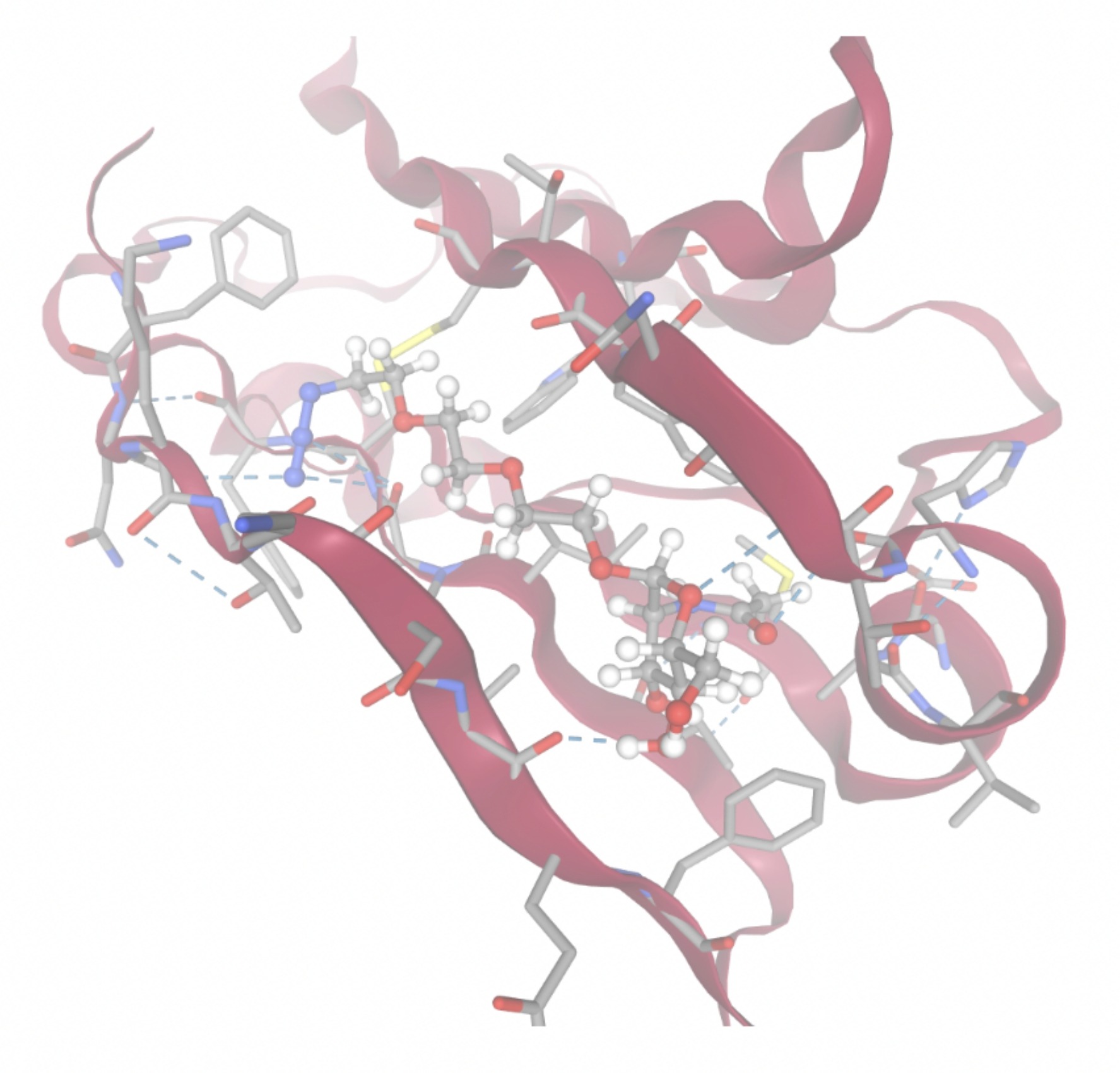 | 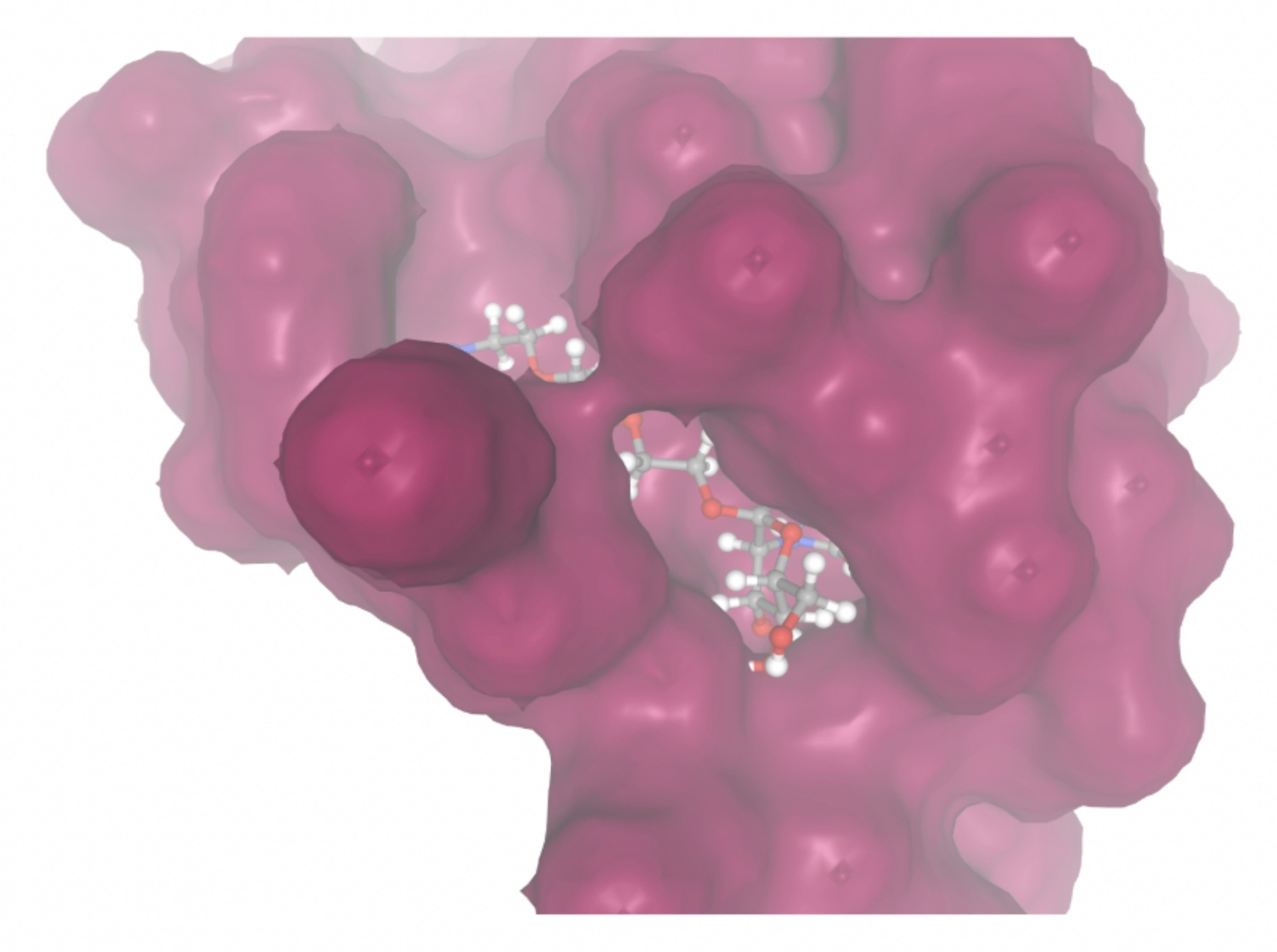 |
| MolPort-000-416-985 | -7.8677 | 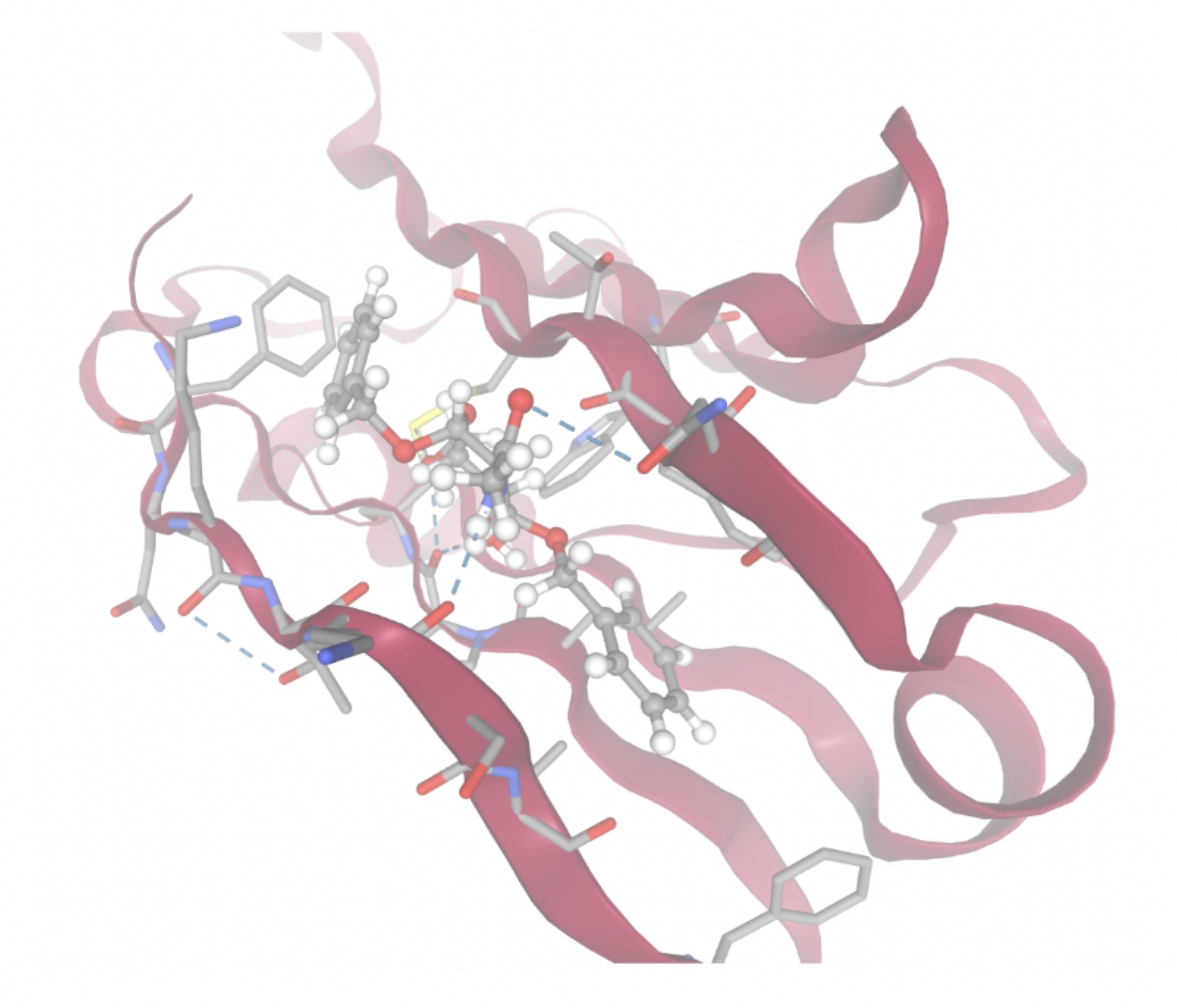 | 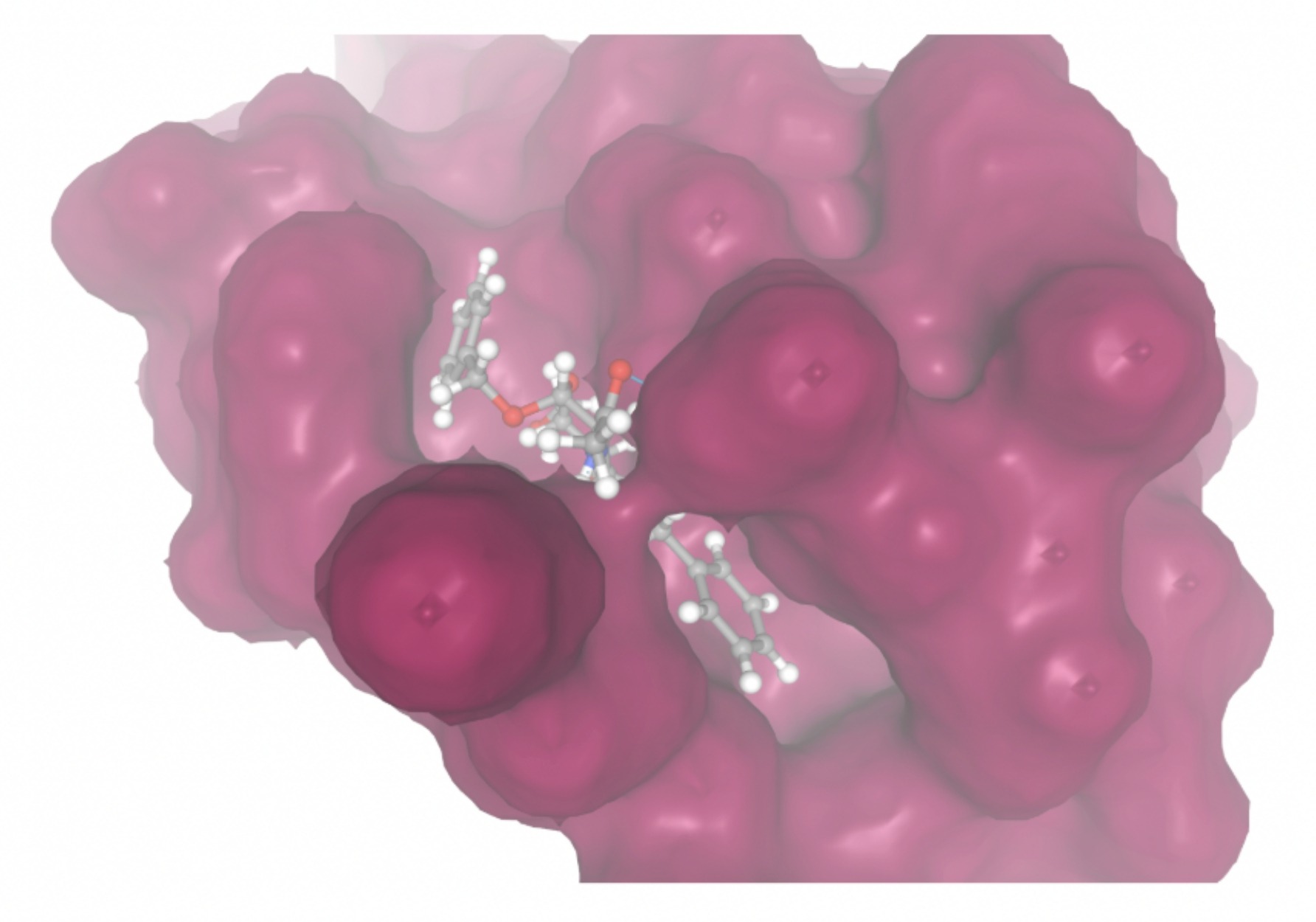 |
| MolPort-047-134-715 | -8.1448 | 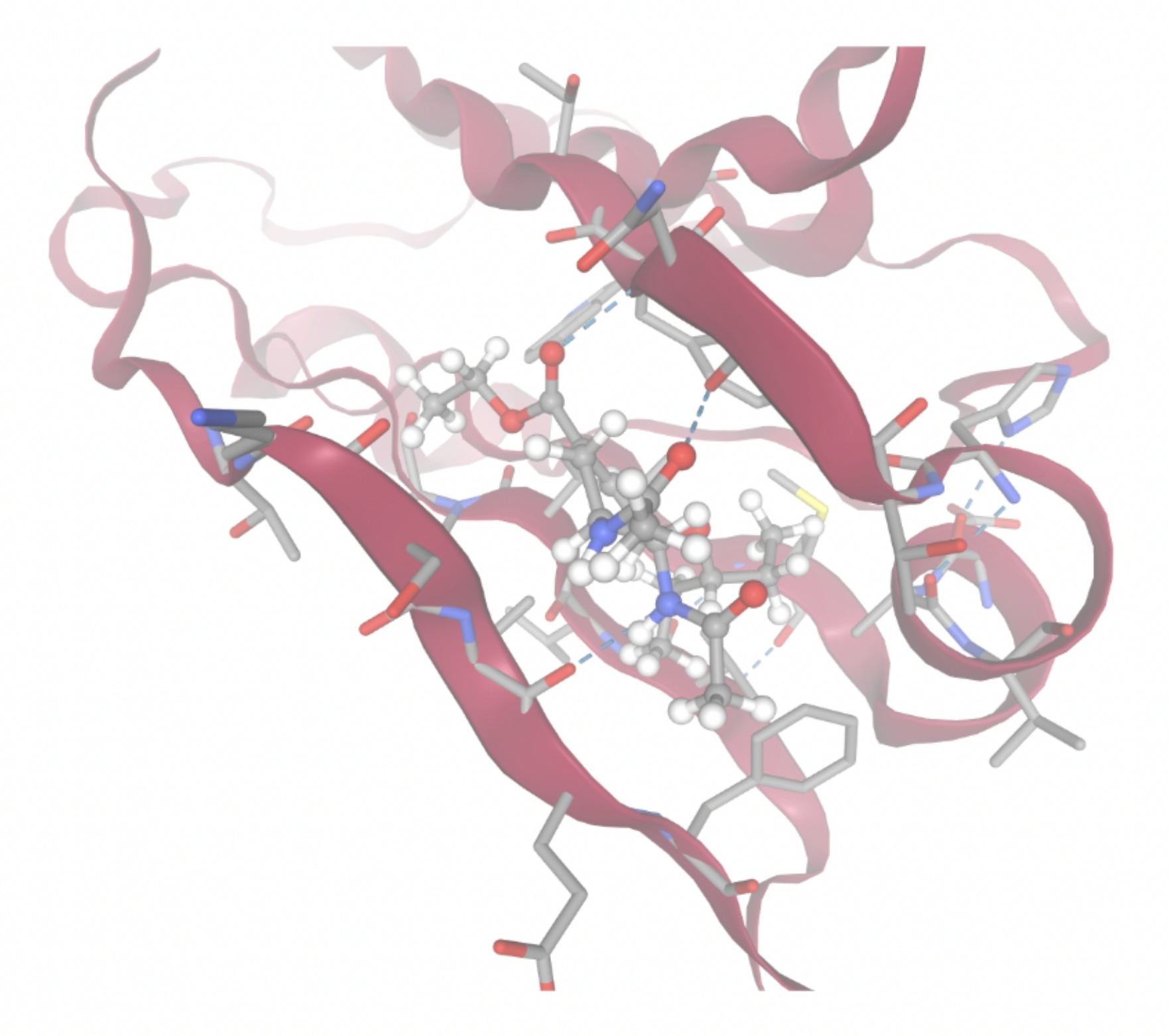 | 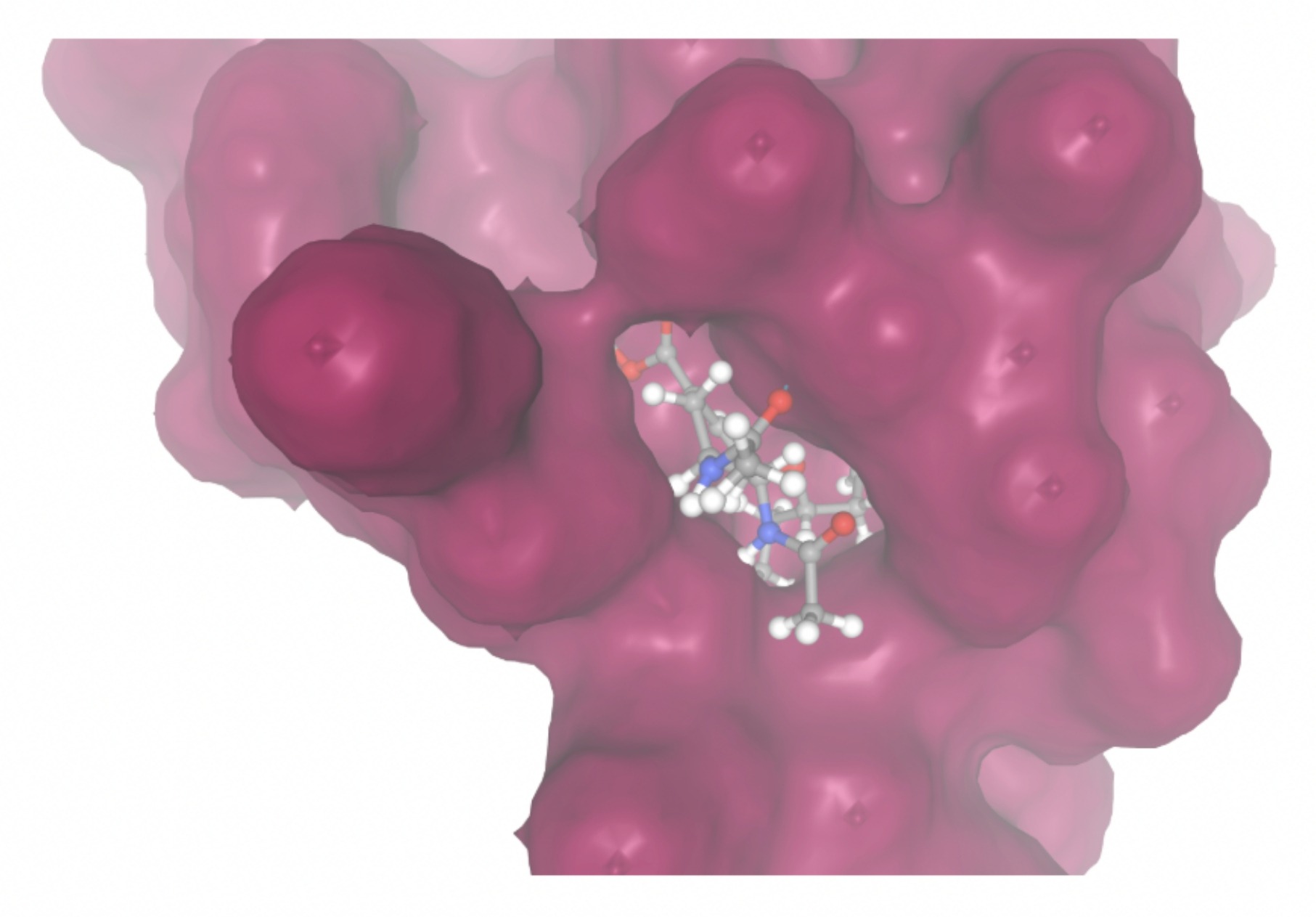 |
| MolPort-051-860-991 | -7.9923 | 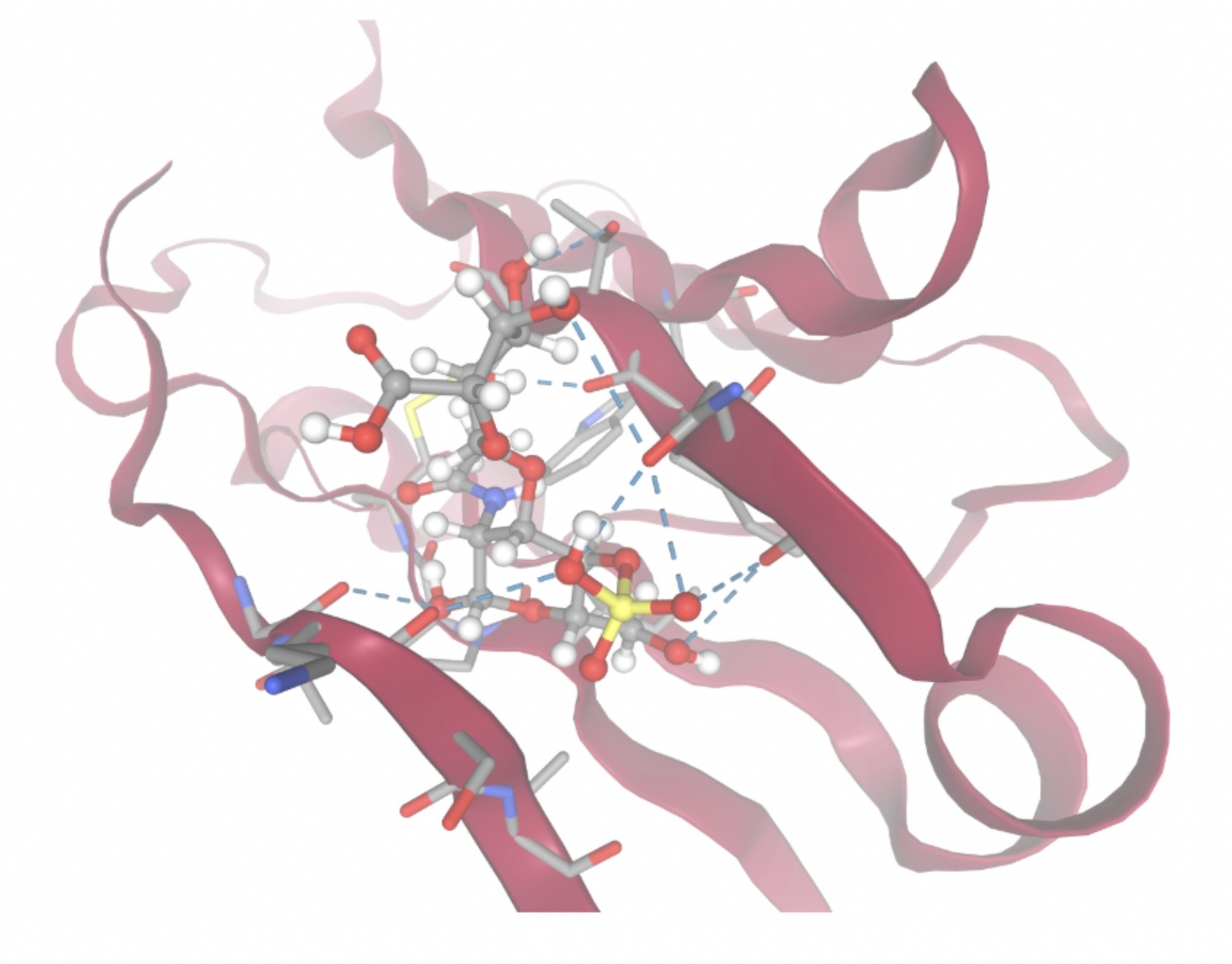 | 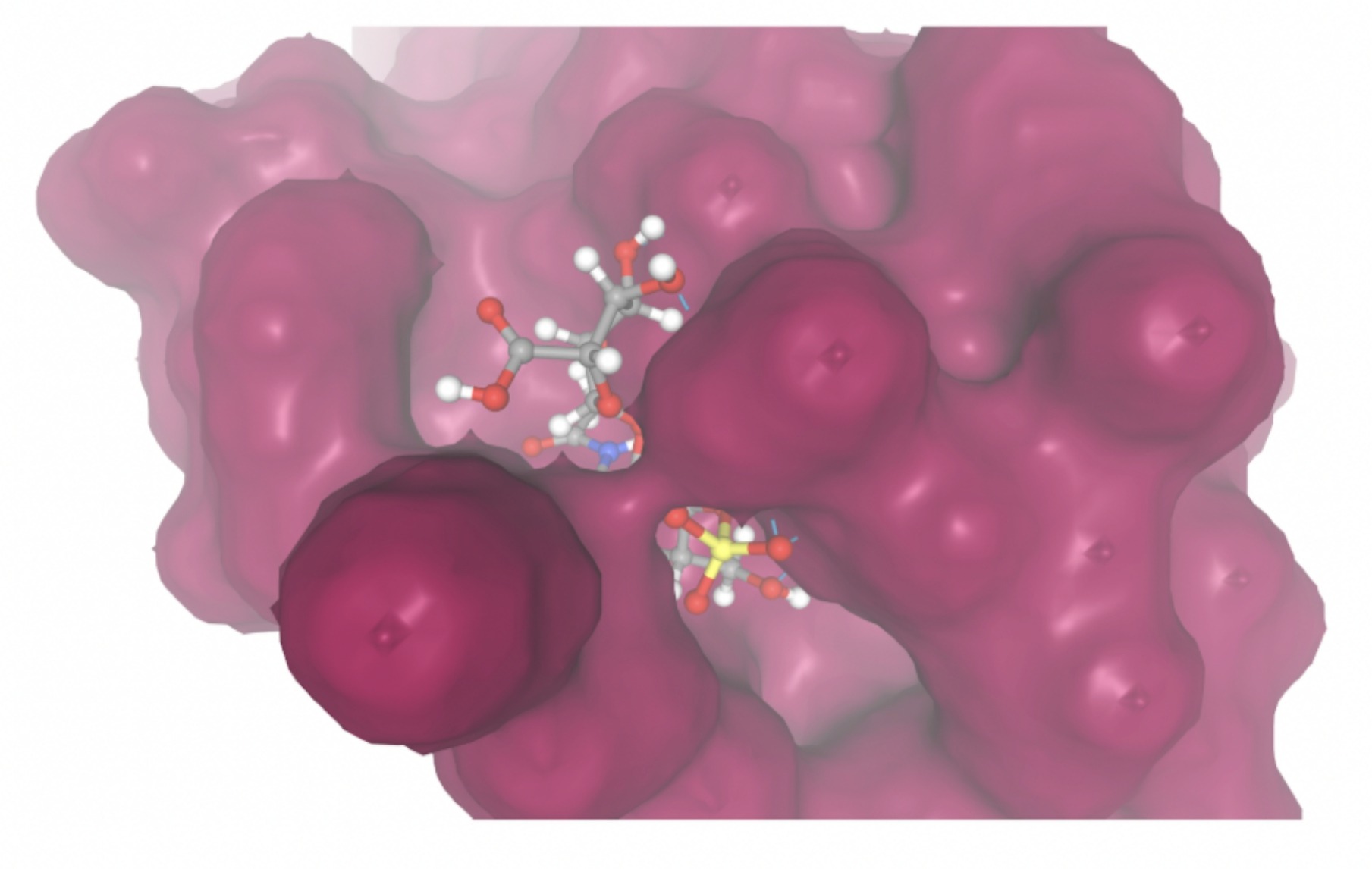 |
| MolPort-006-167-538 | -9.5669 | 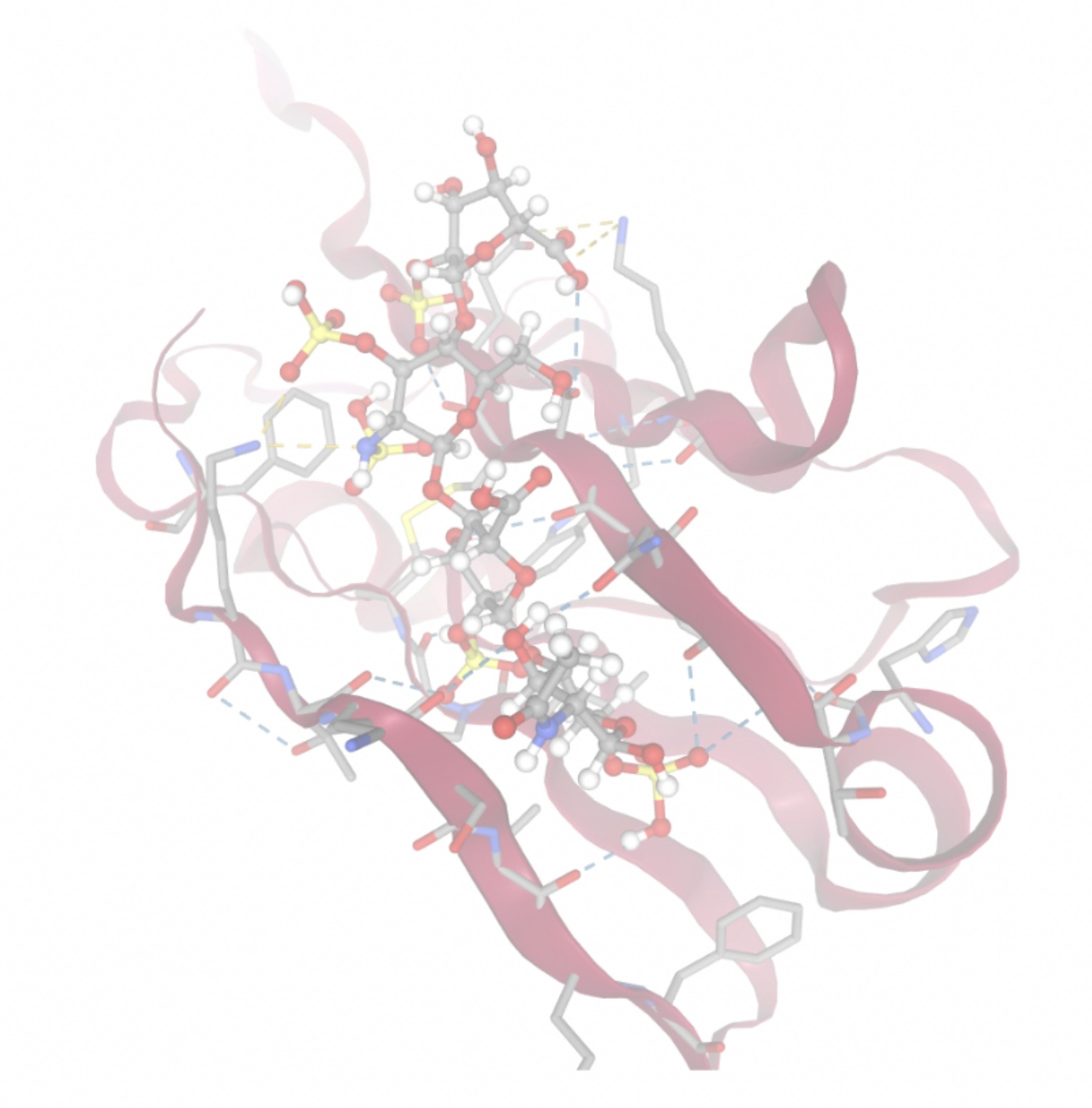 | 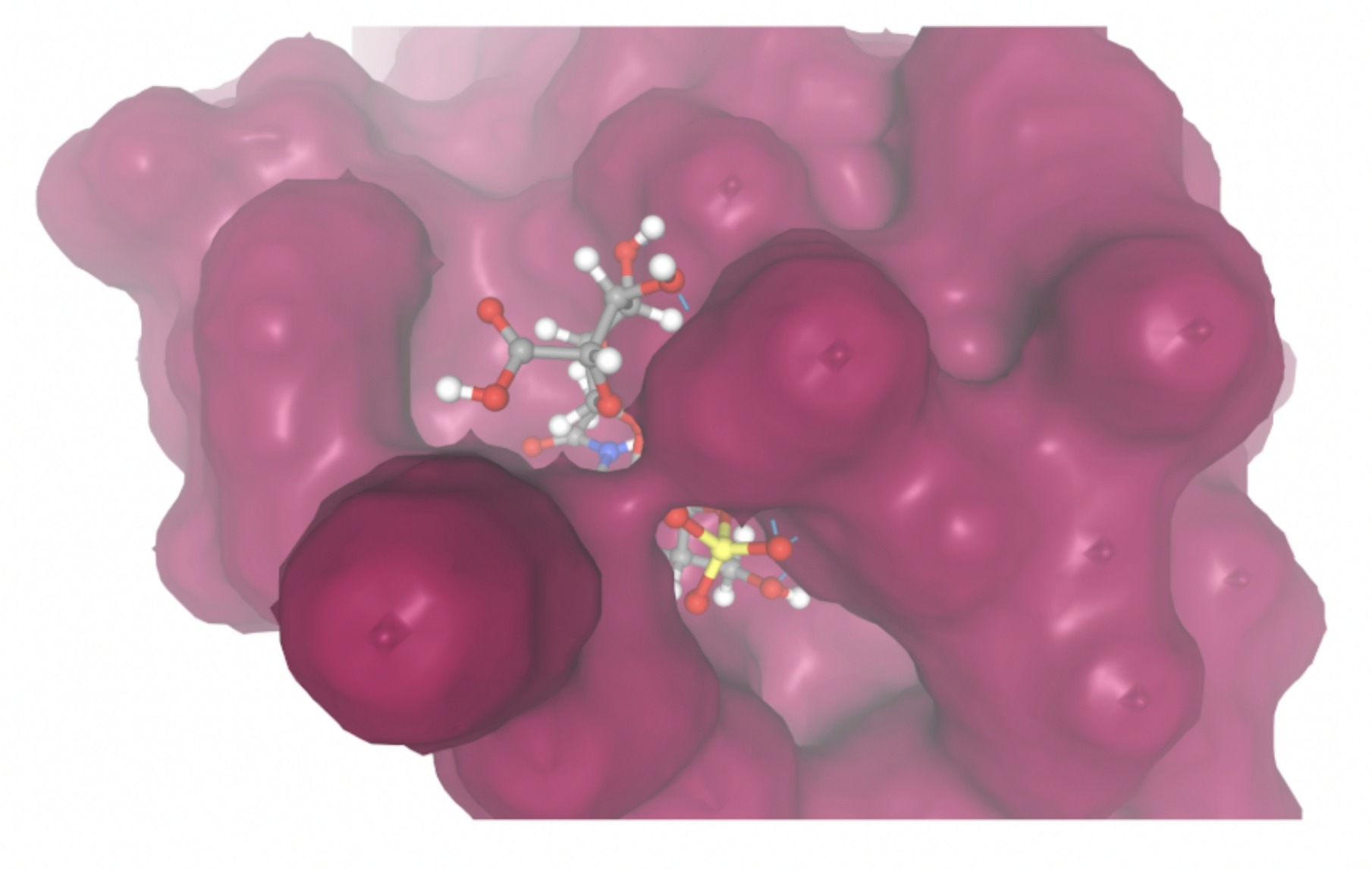 |
| MolPort-004-849-816 | -8.3624 | 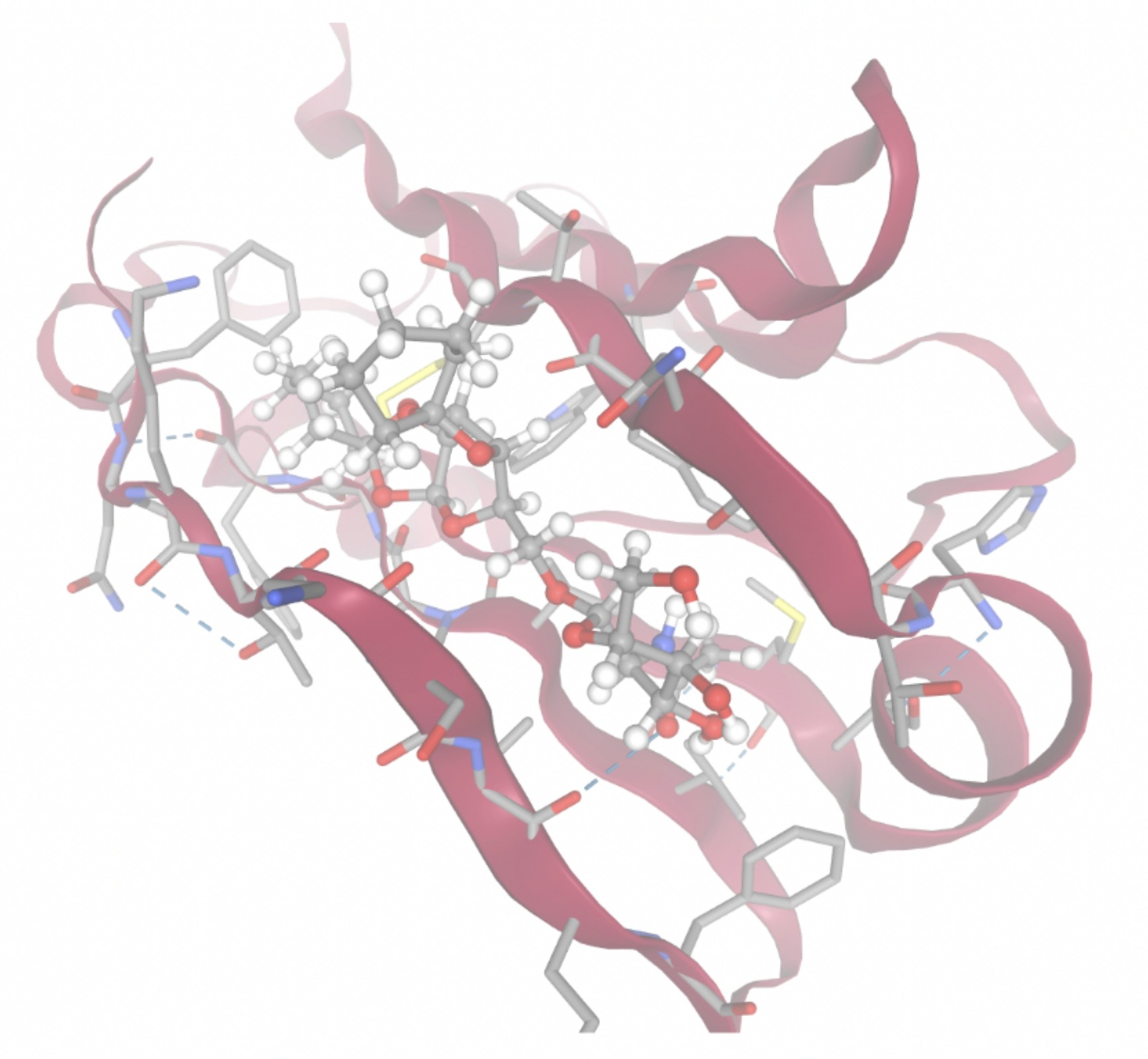 | 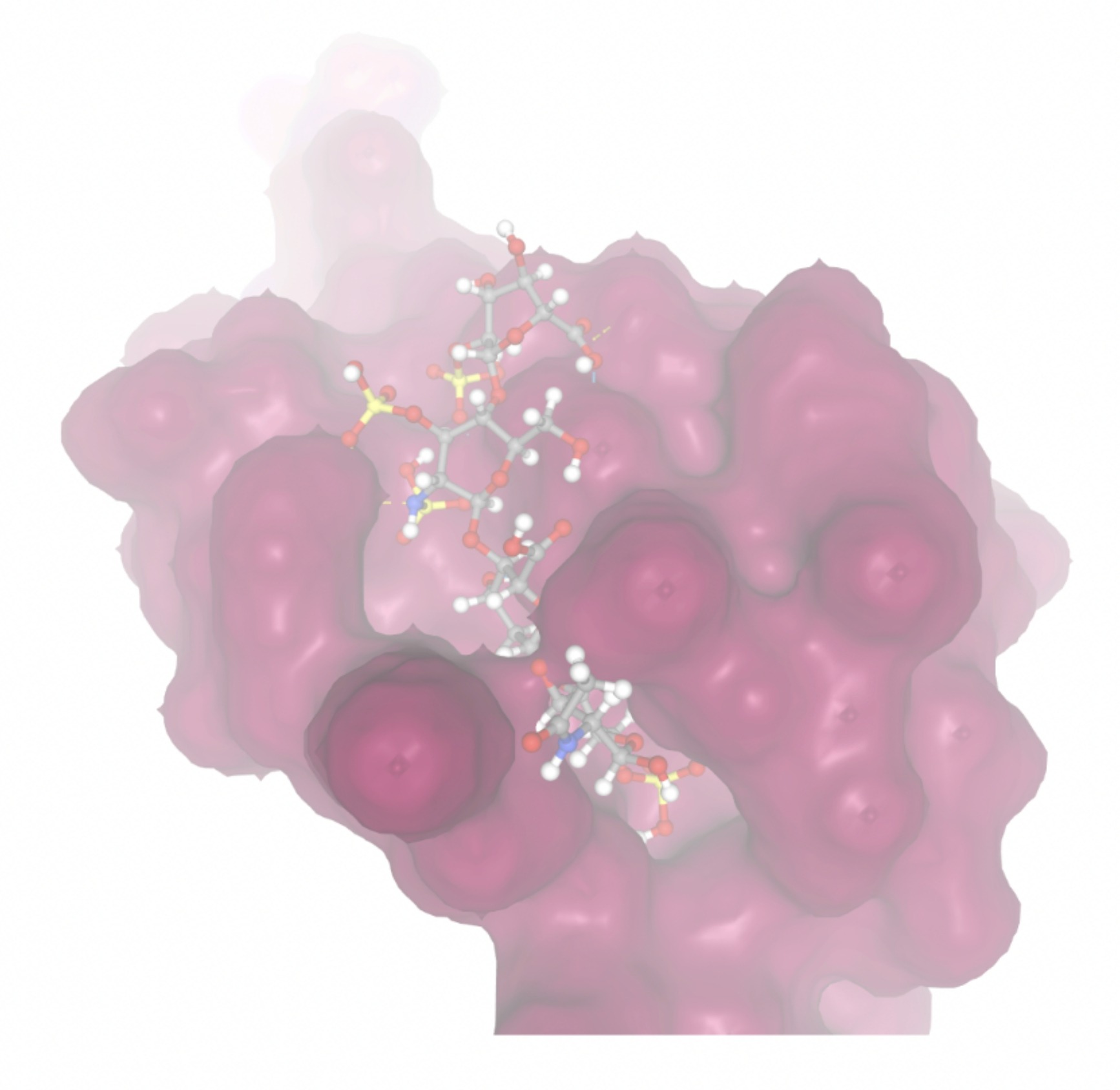 |
| MolPort-008-267-878 | -6.8695 | 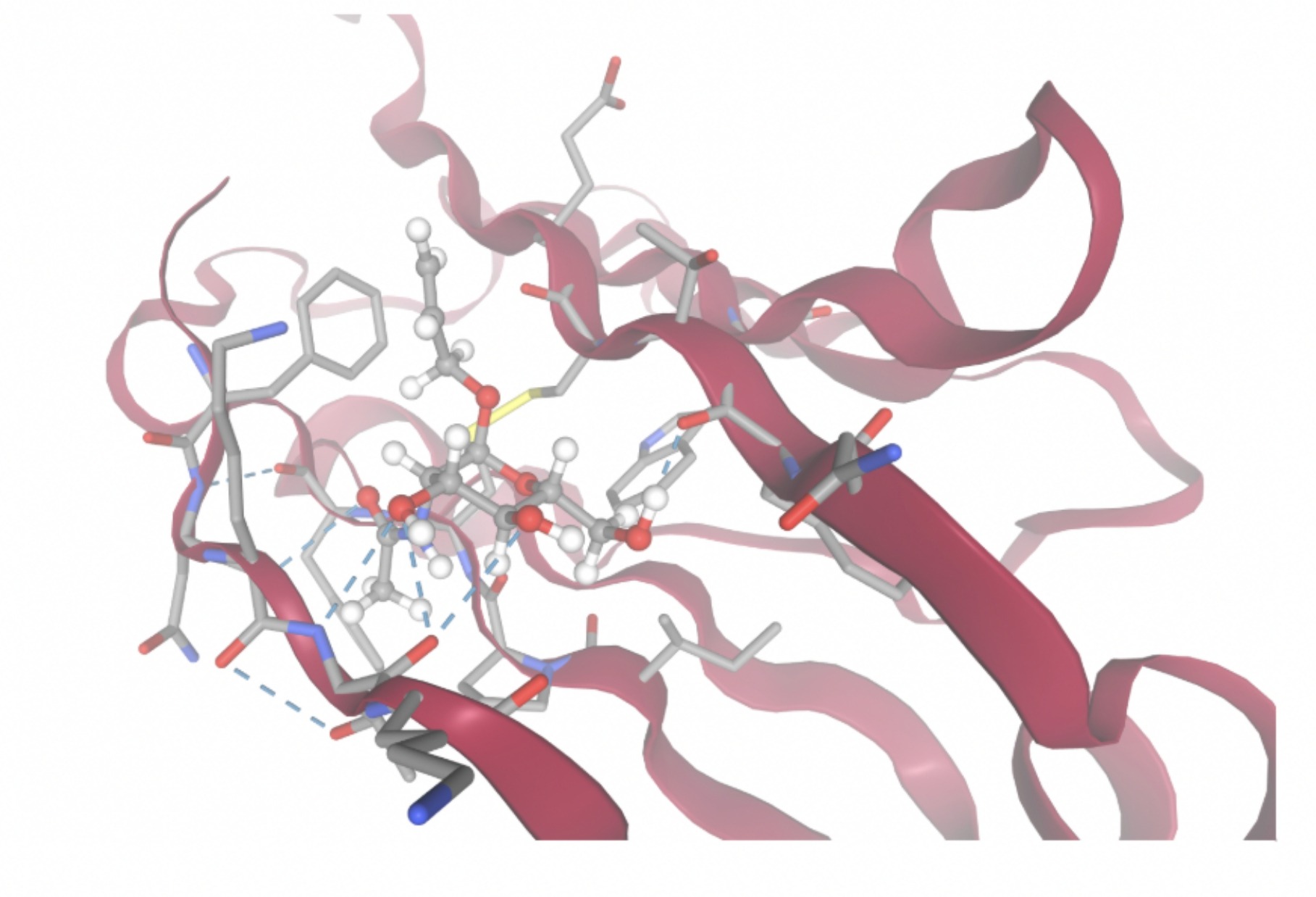 | 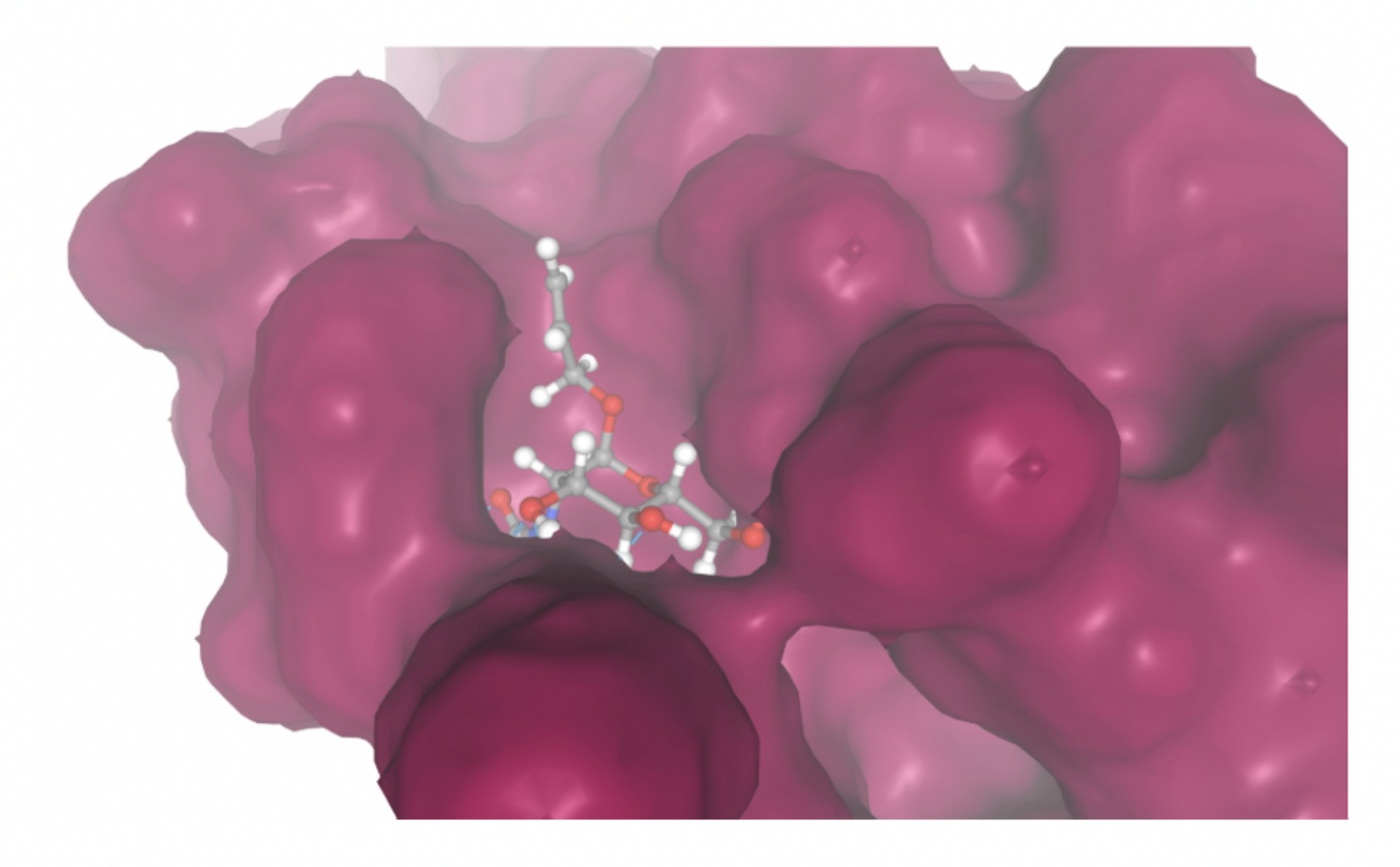 |
| MolPort-002-511-046 | -8.1479 | 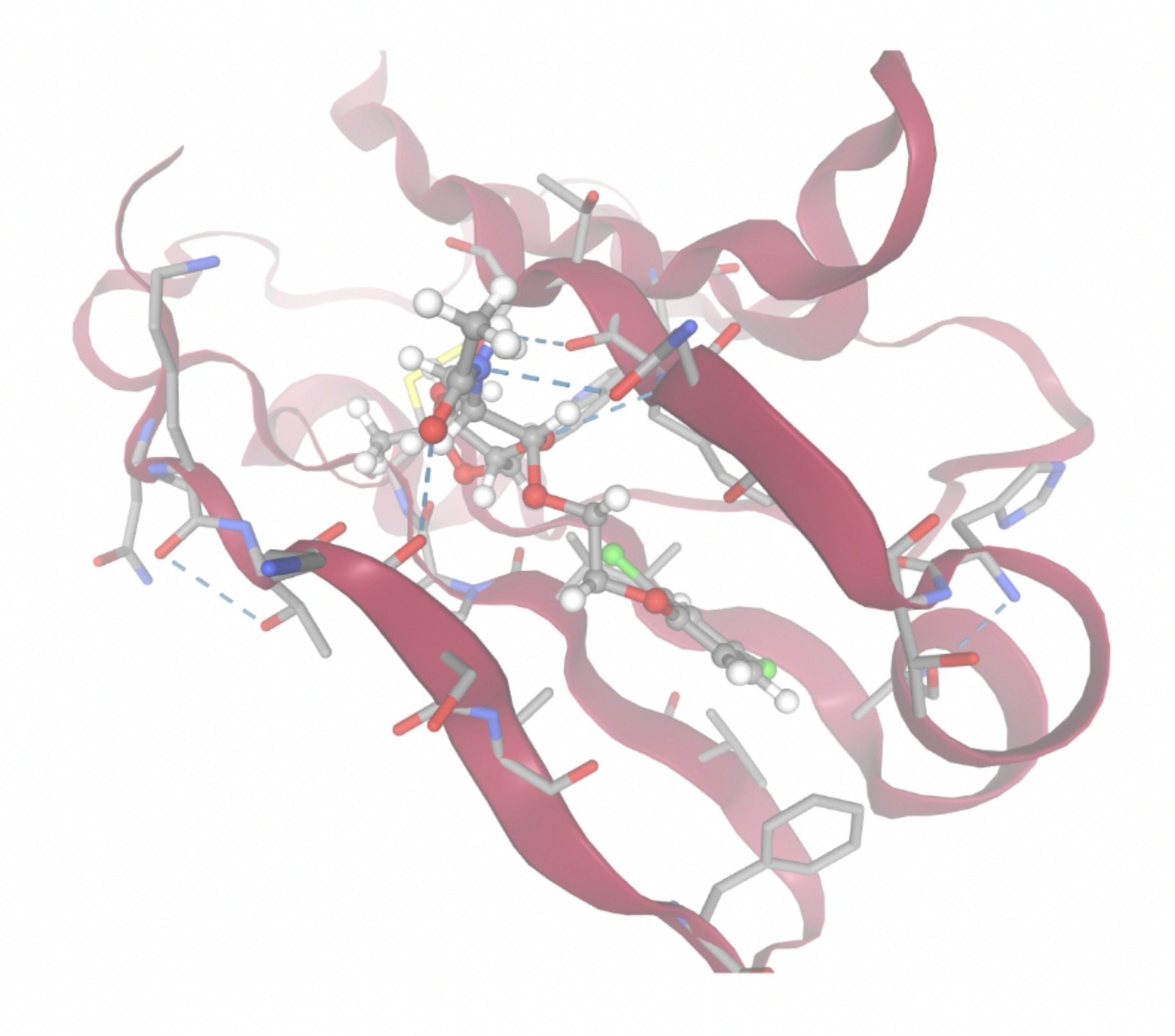 | 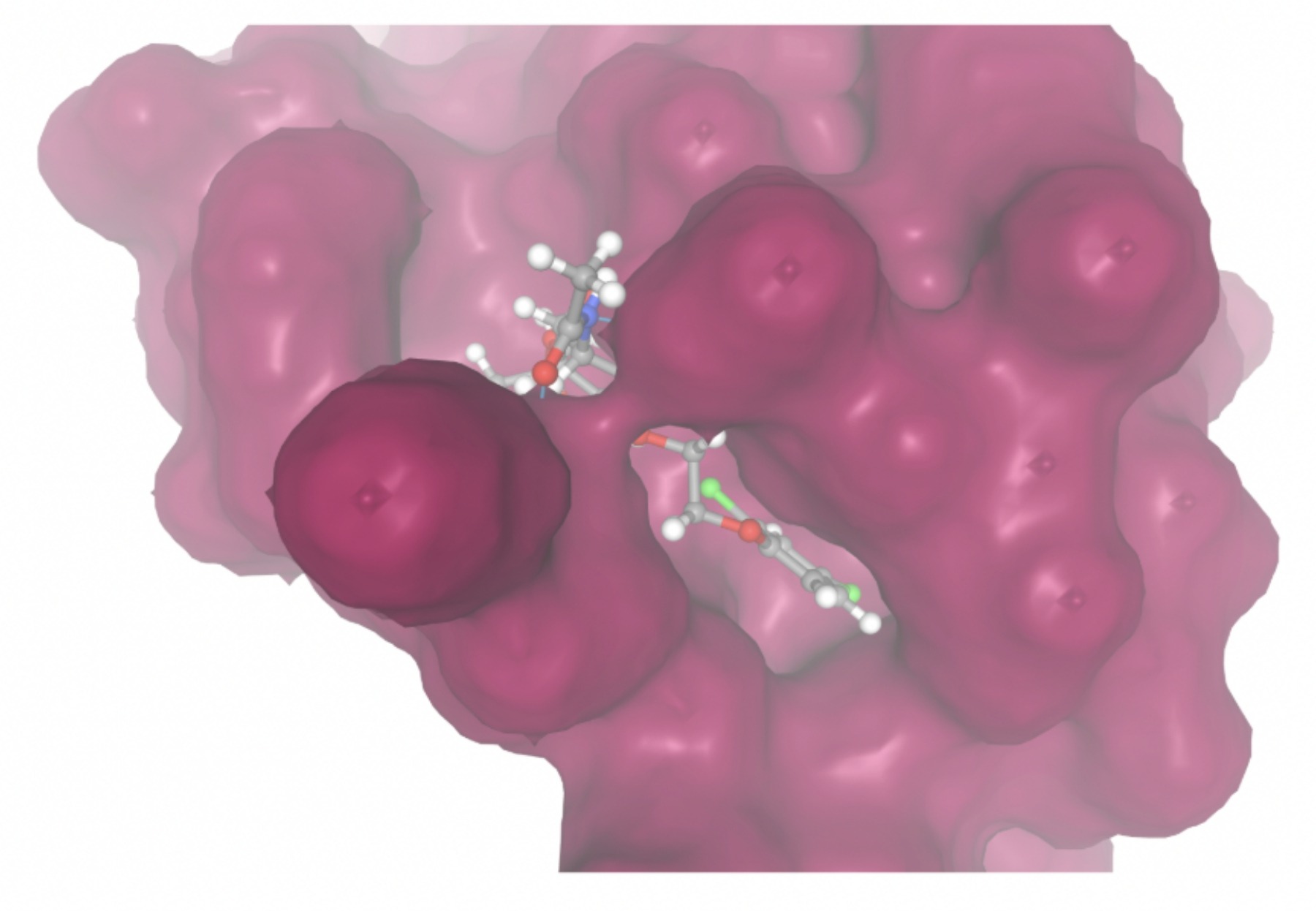 |
| MolPort-003-011-602 | -9.1922 | 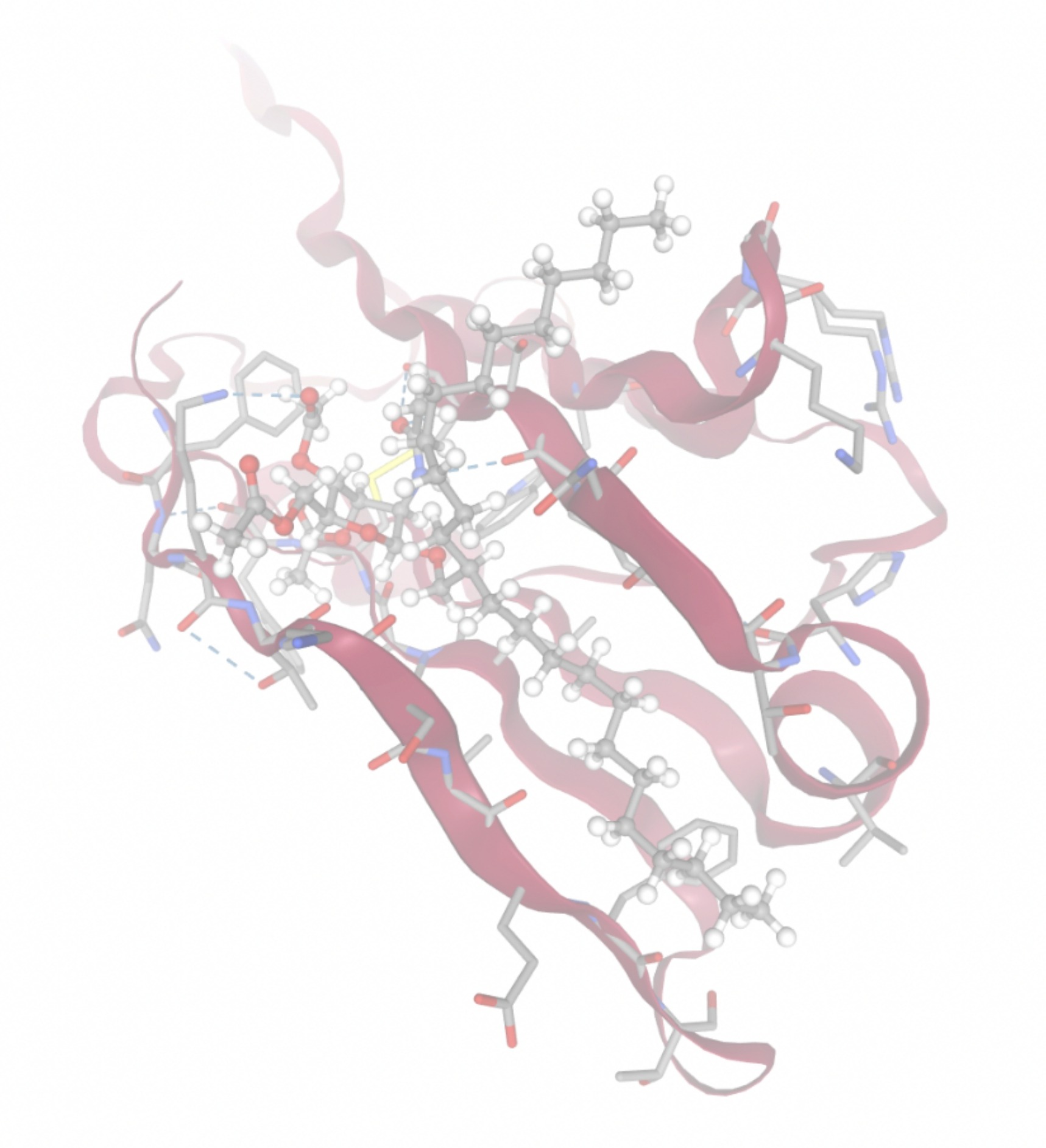 | 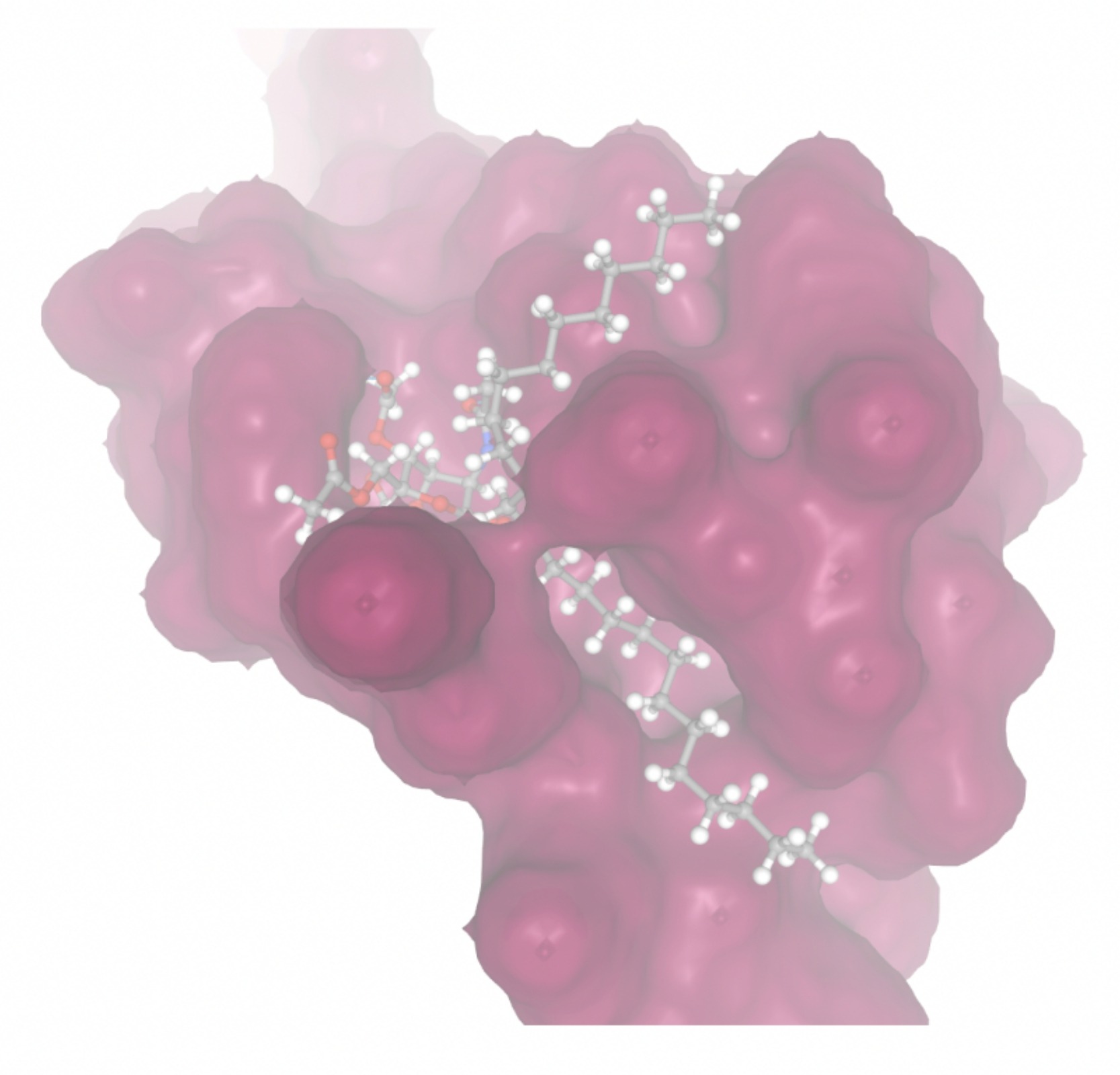 |
| MolPort-038-394-503 | -6.9777 | 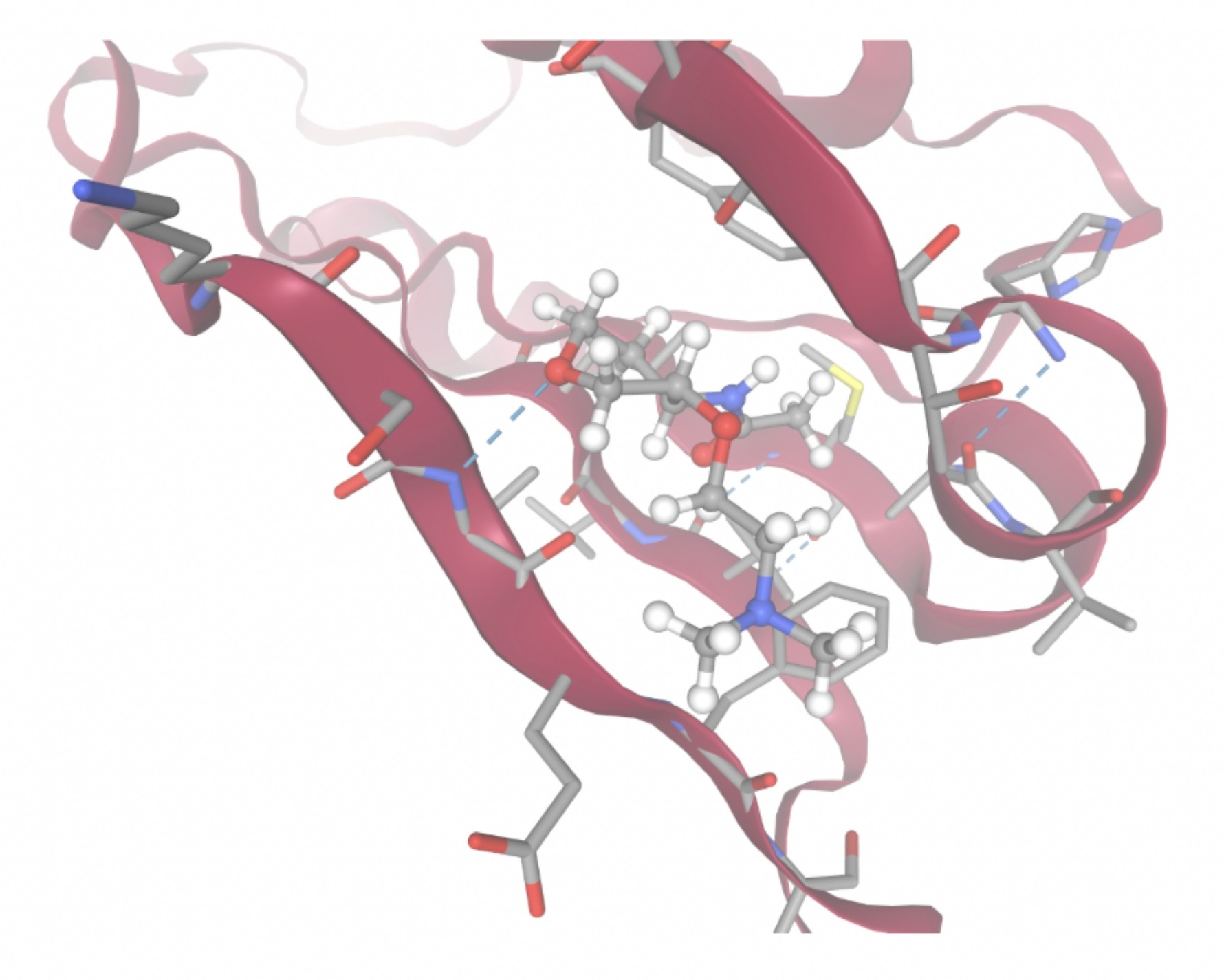 | 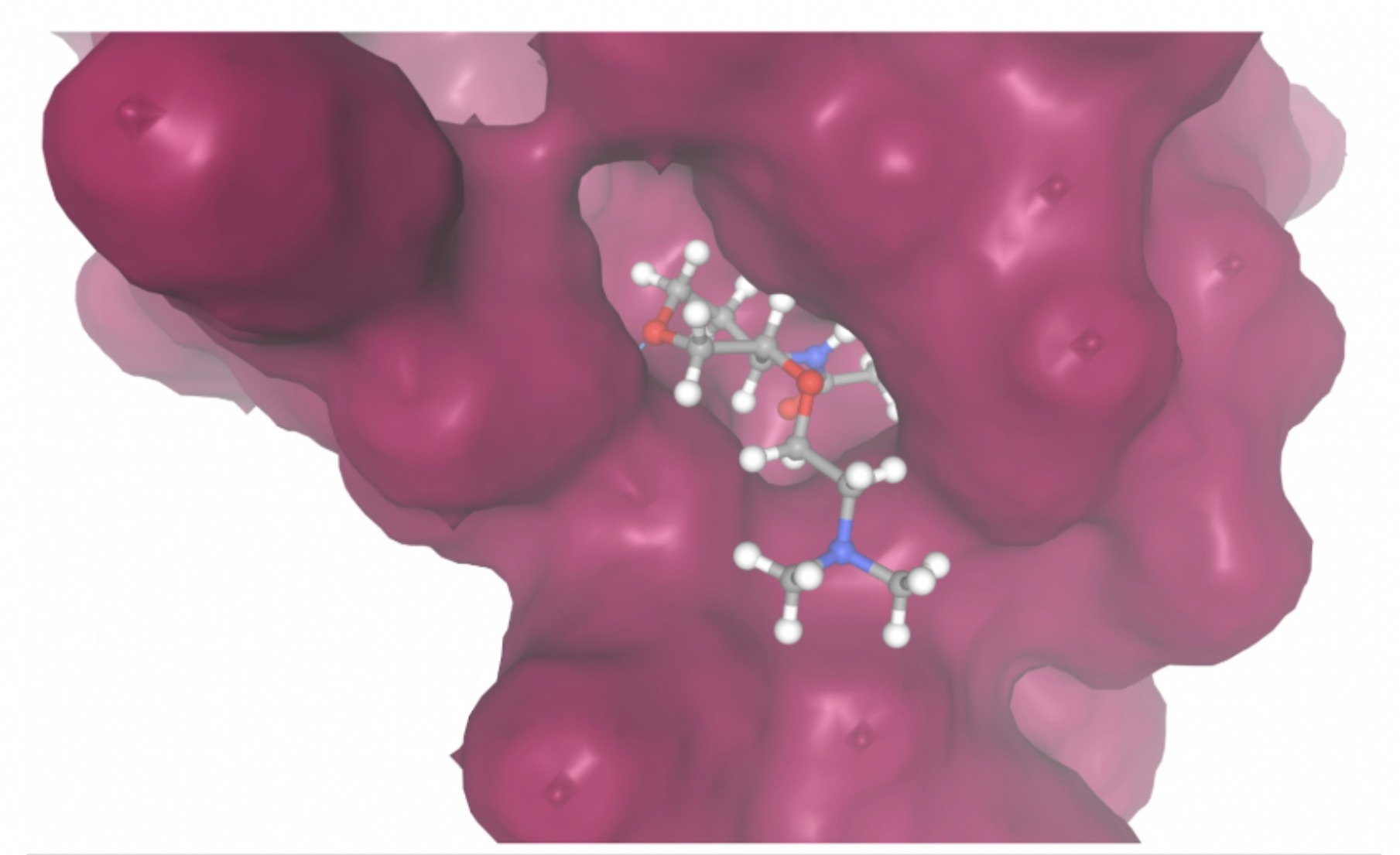 |
| MolPort-052-043-087 | -8.7609 | 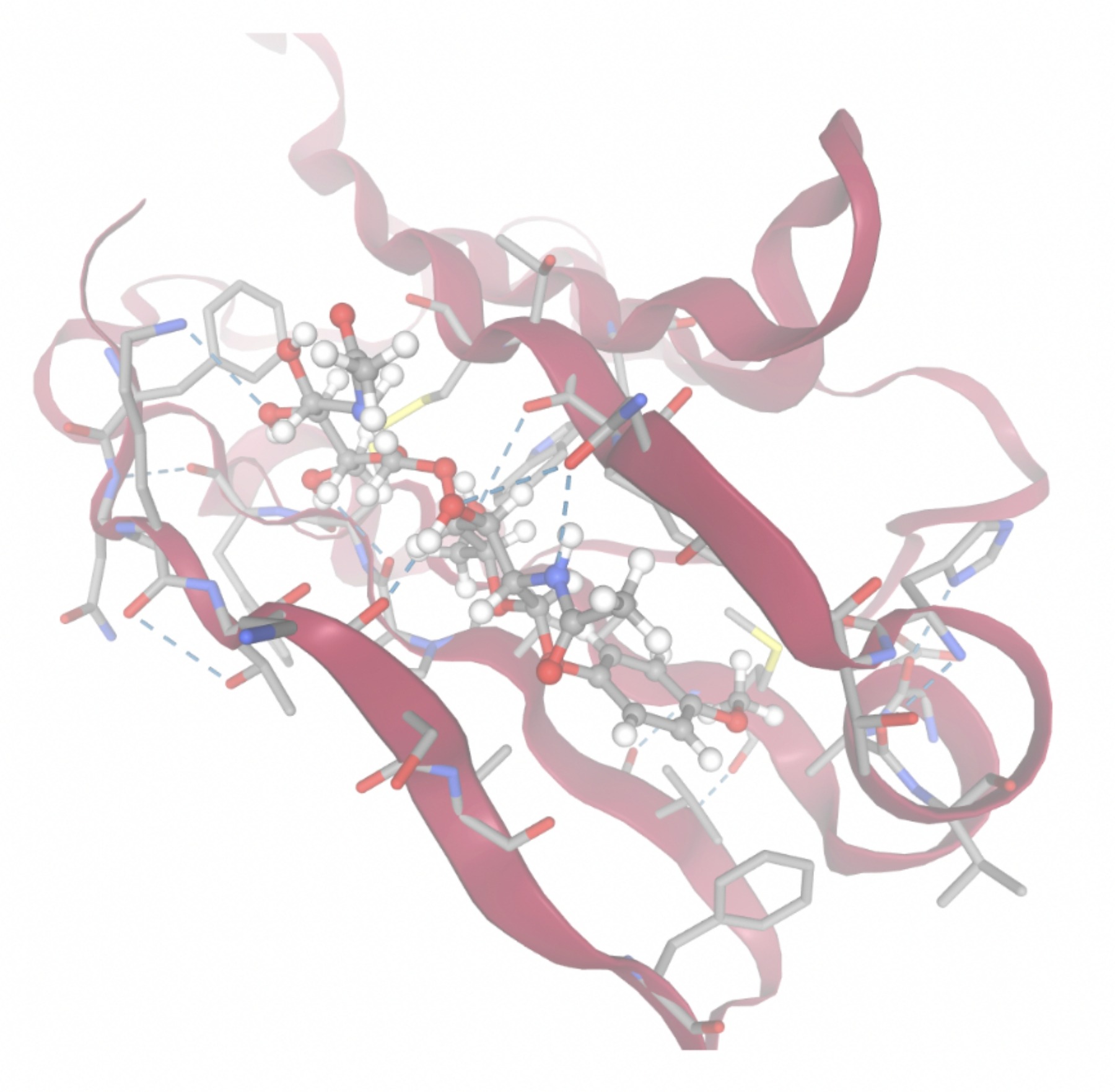 | 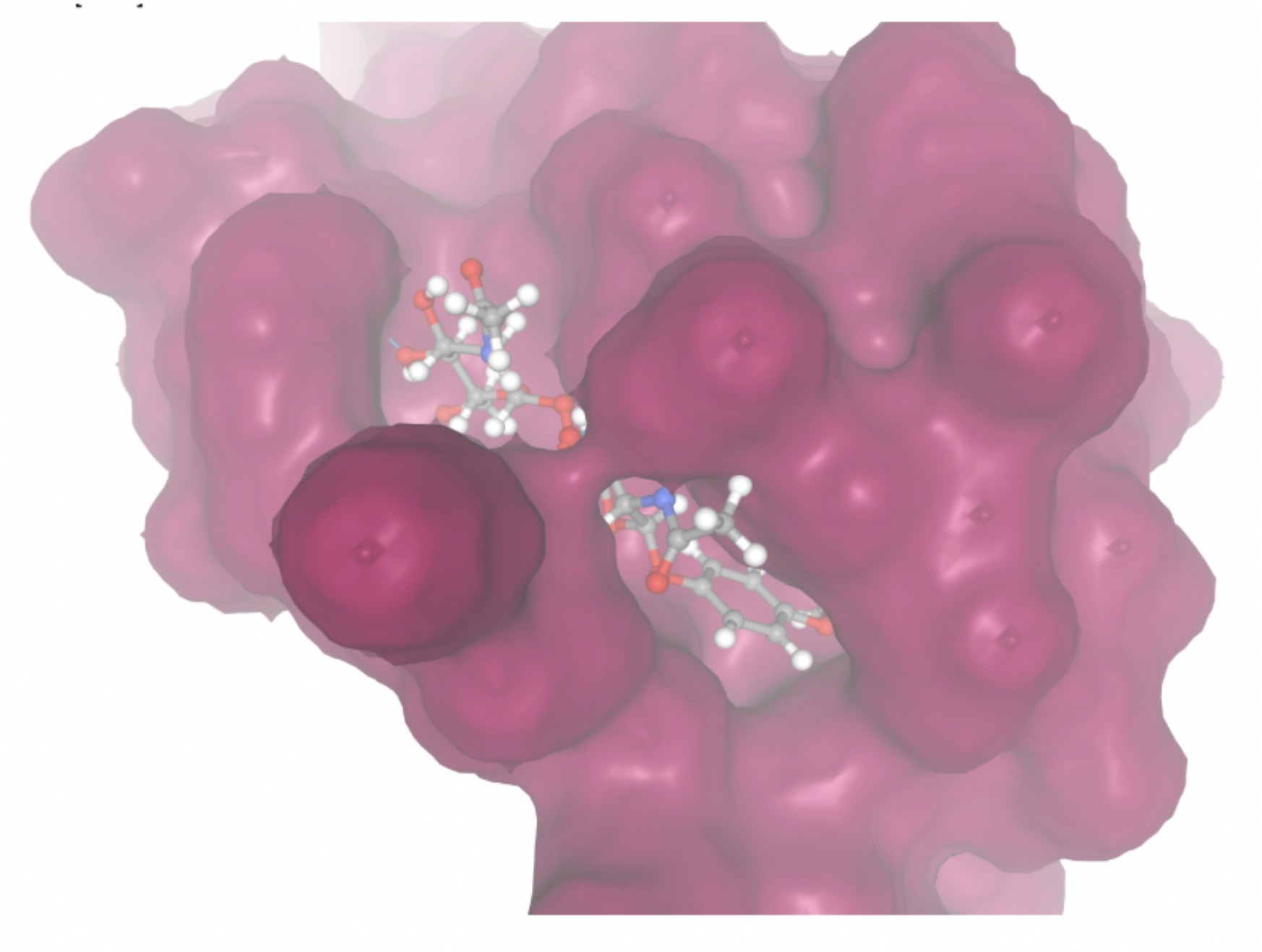 |
| MolPort-005-289-910 | -8.3285 | 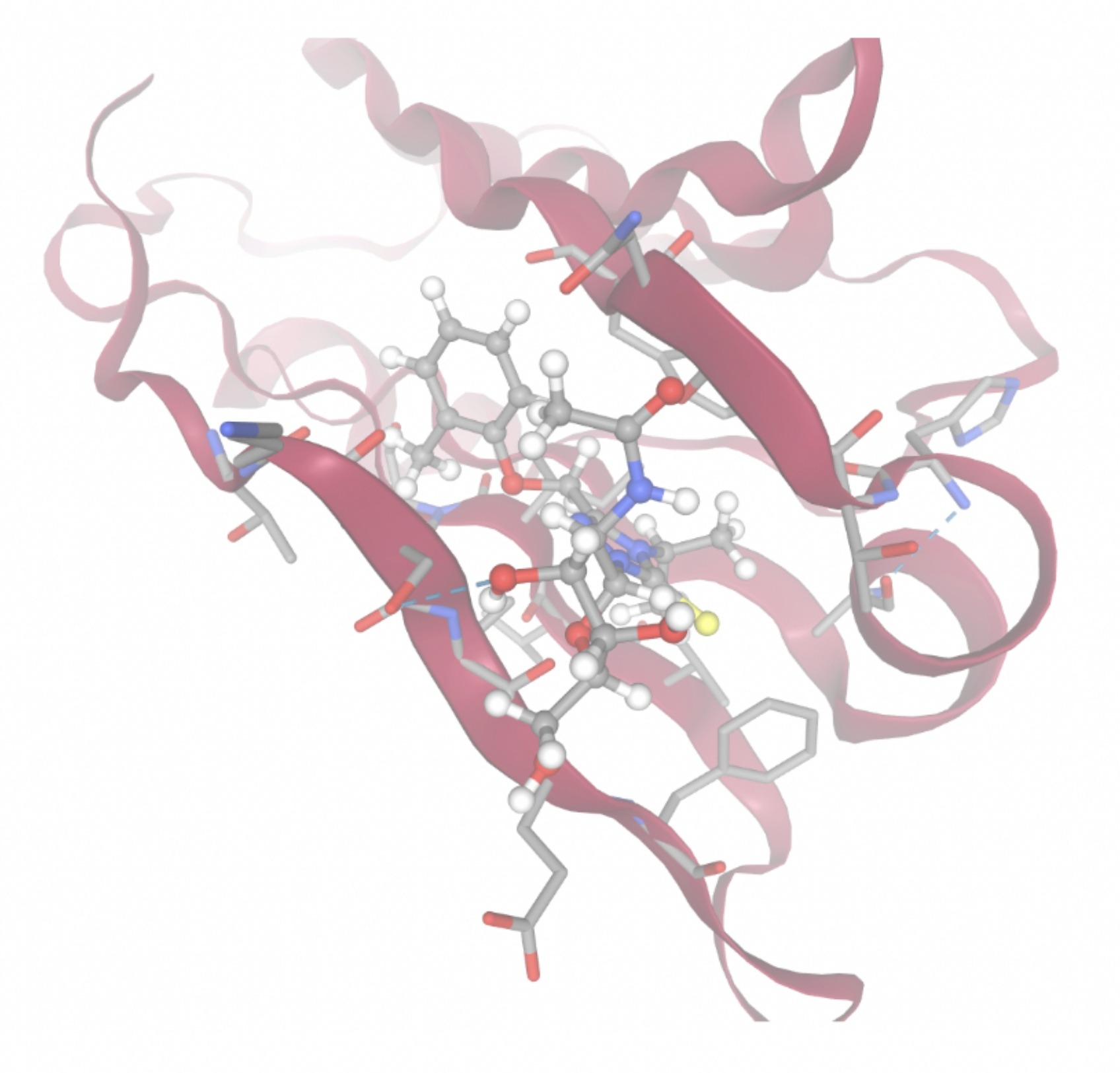 | 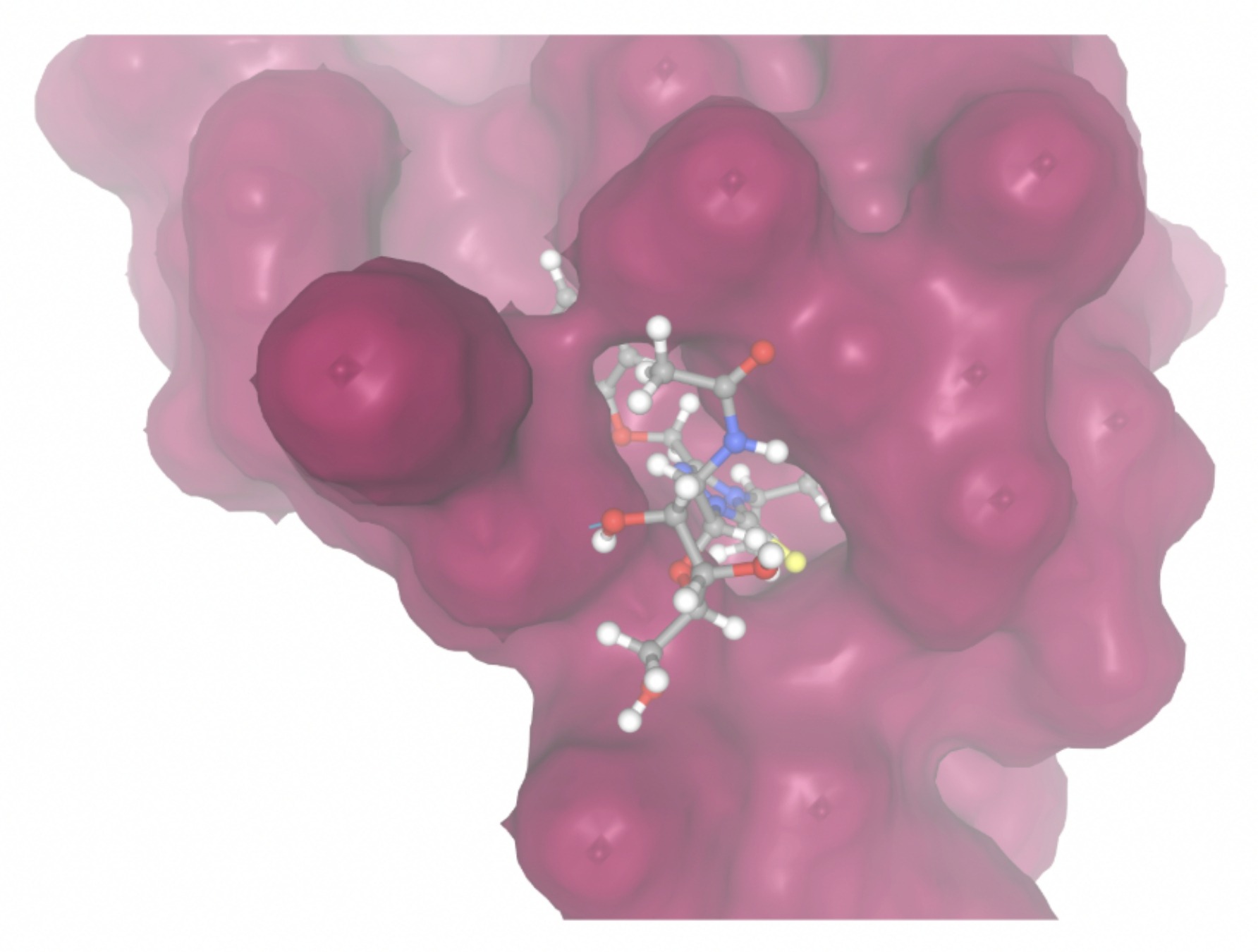 |
| MolPort-051-856-633 | -8.5876 | 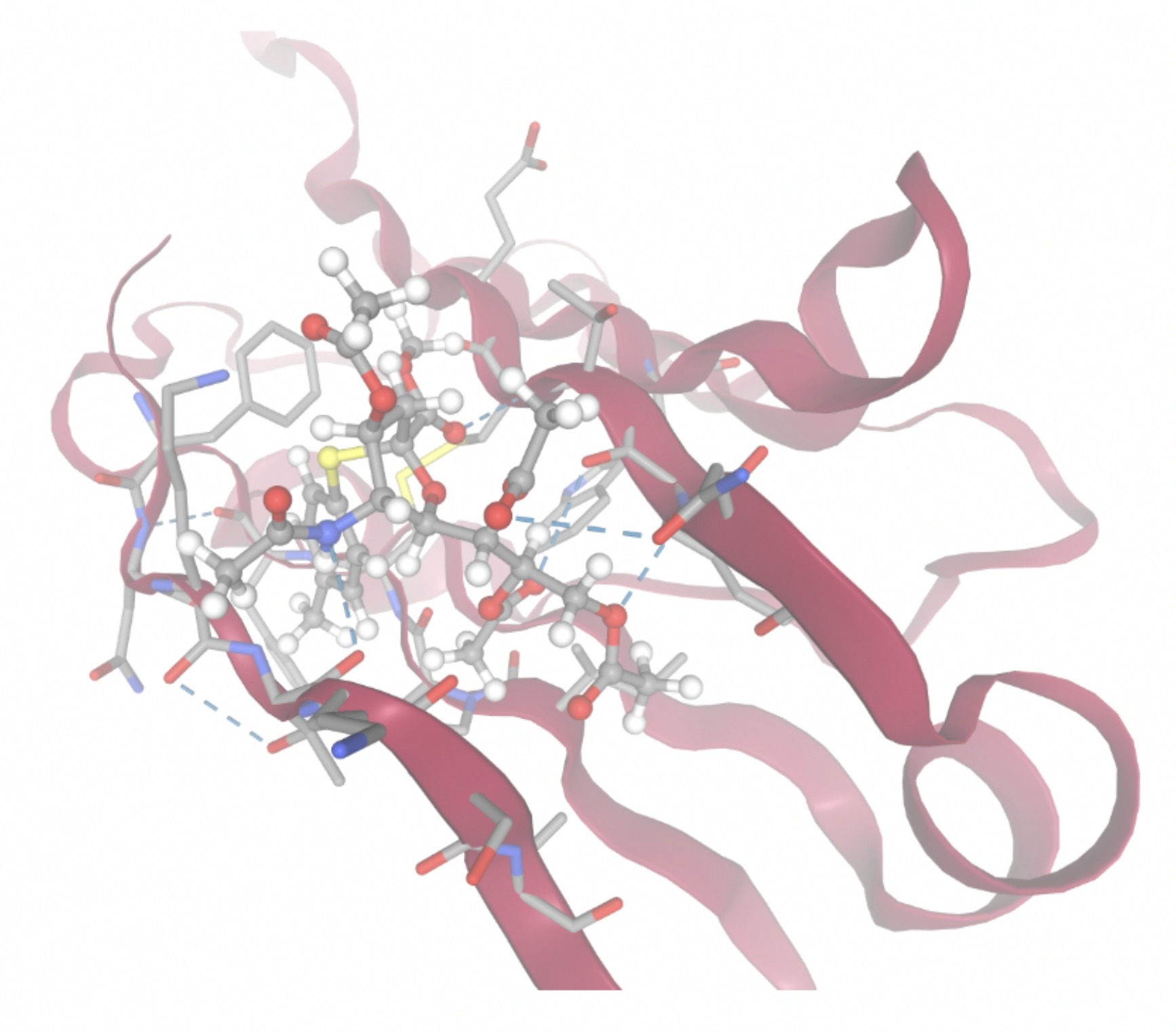 | 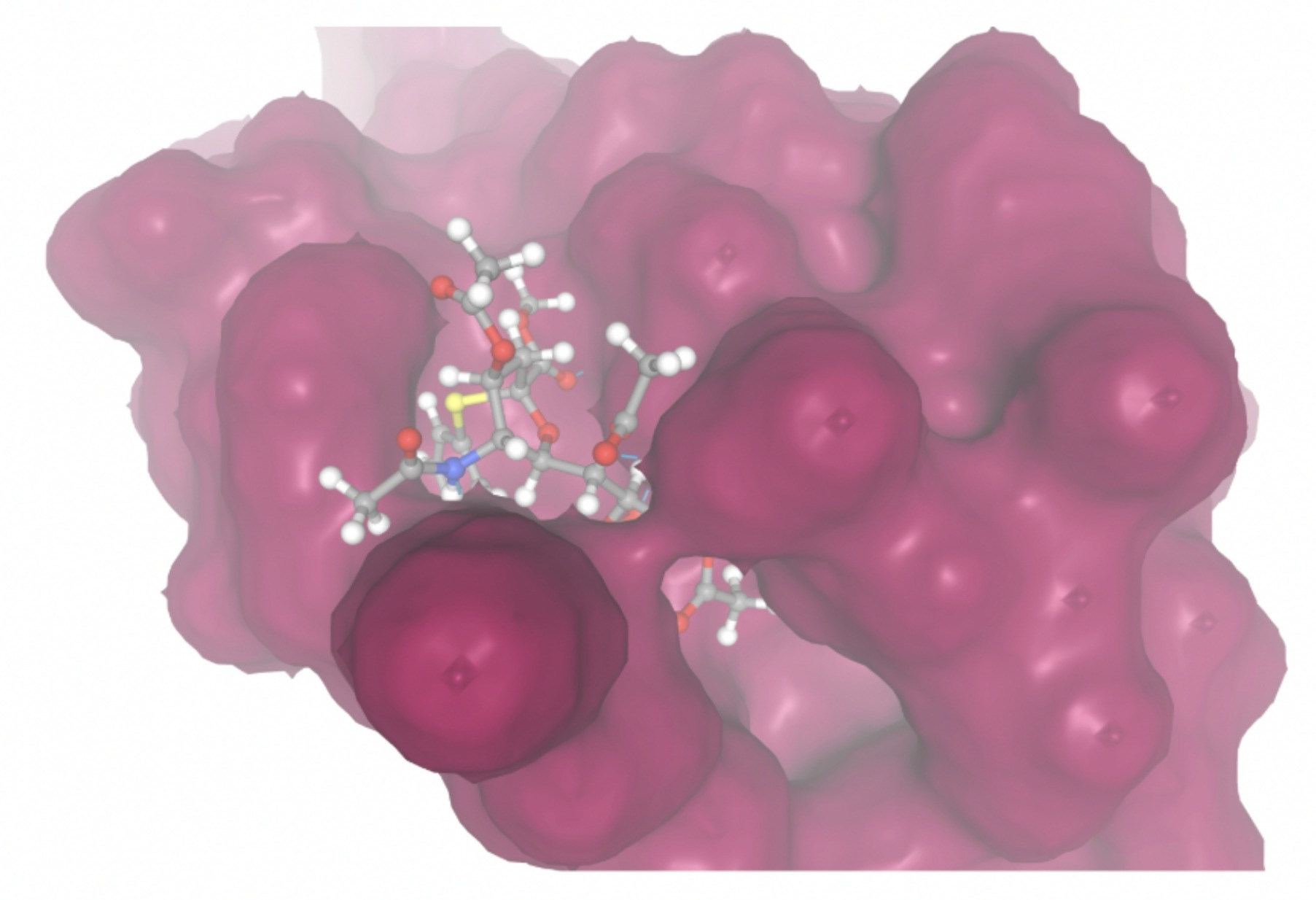 |
| MolPort-047-590-656 | -8.4094 | 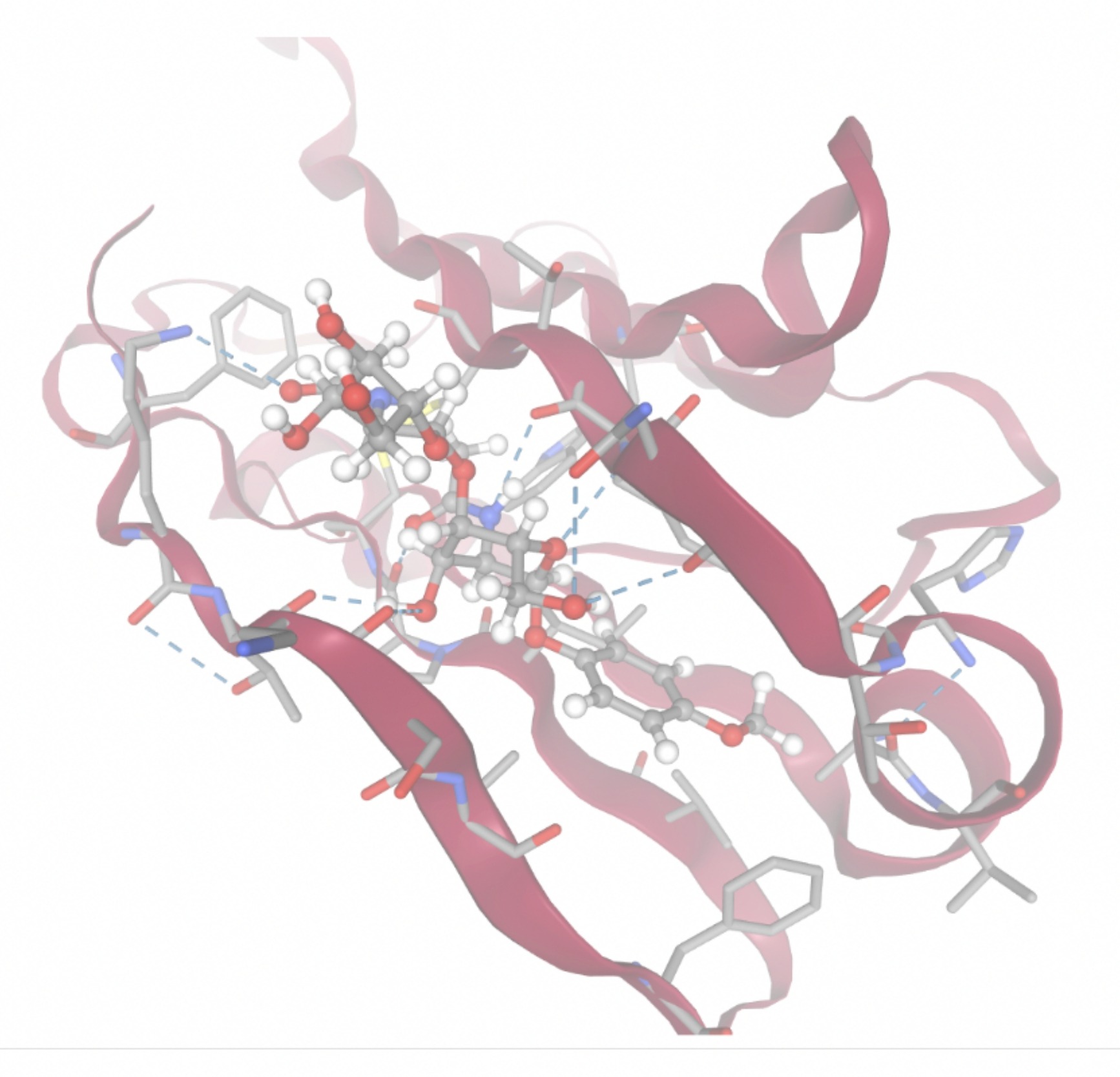 | 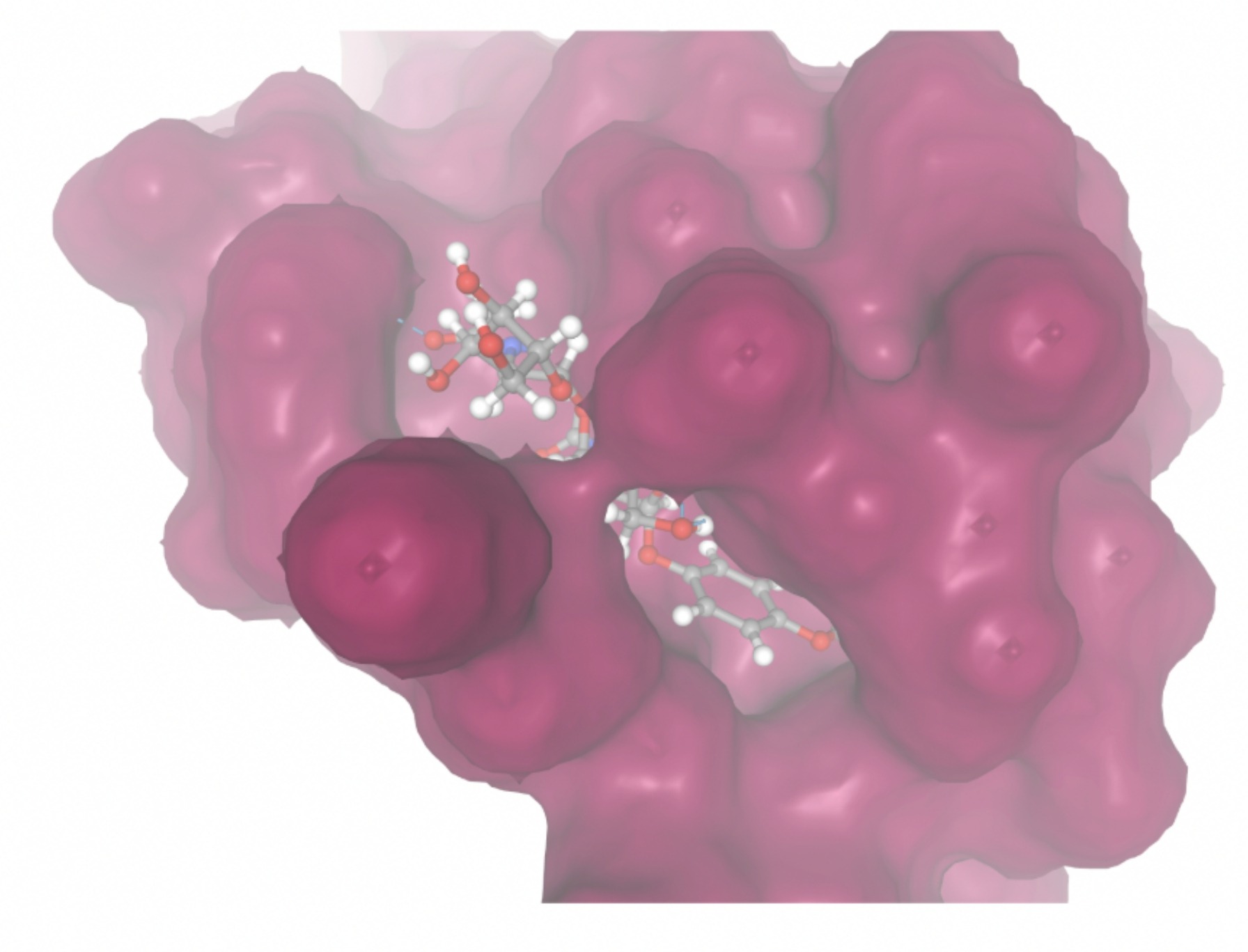 |
| MolPort-046-763-086 | -7.7984 | 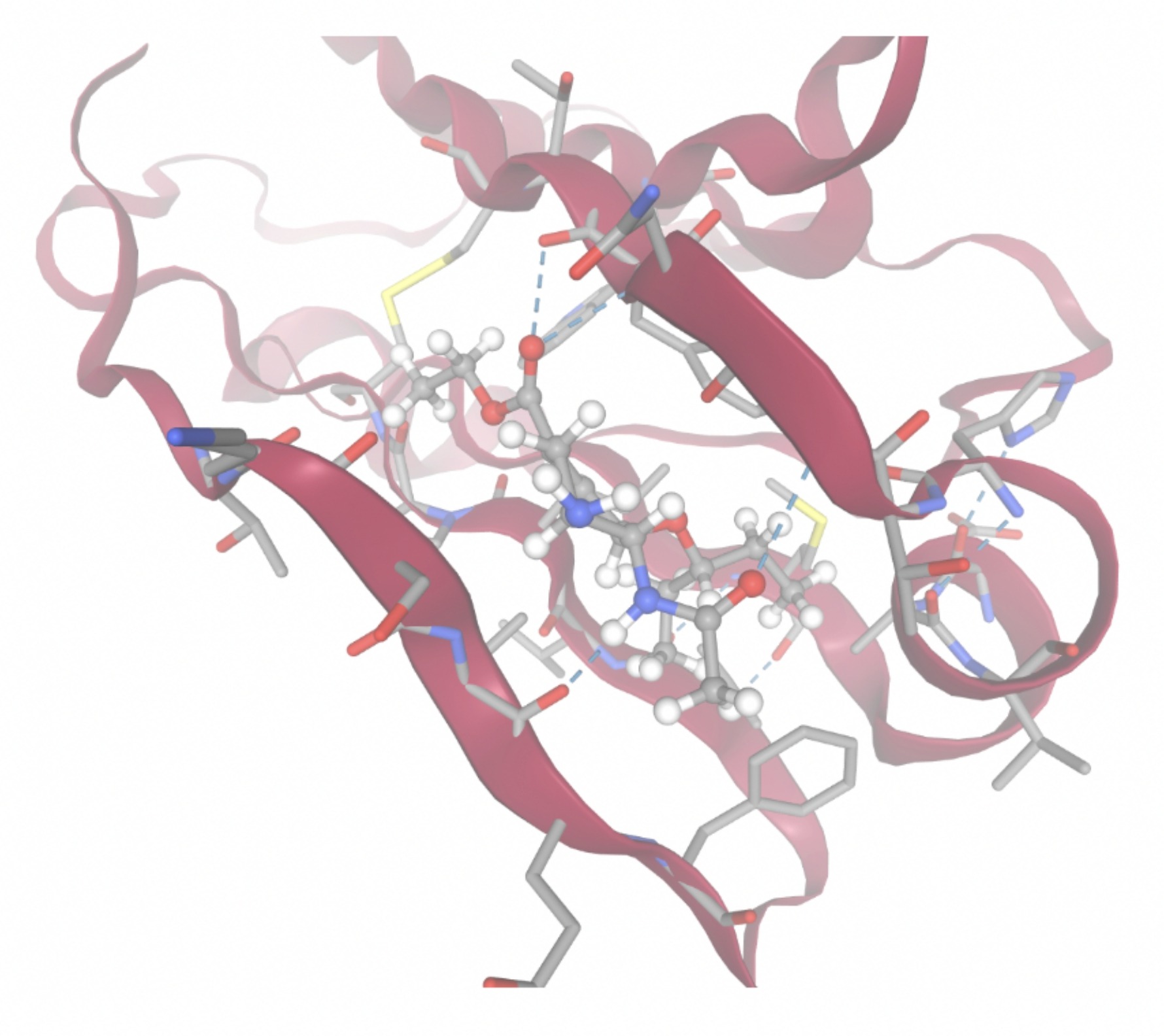 | 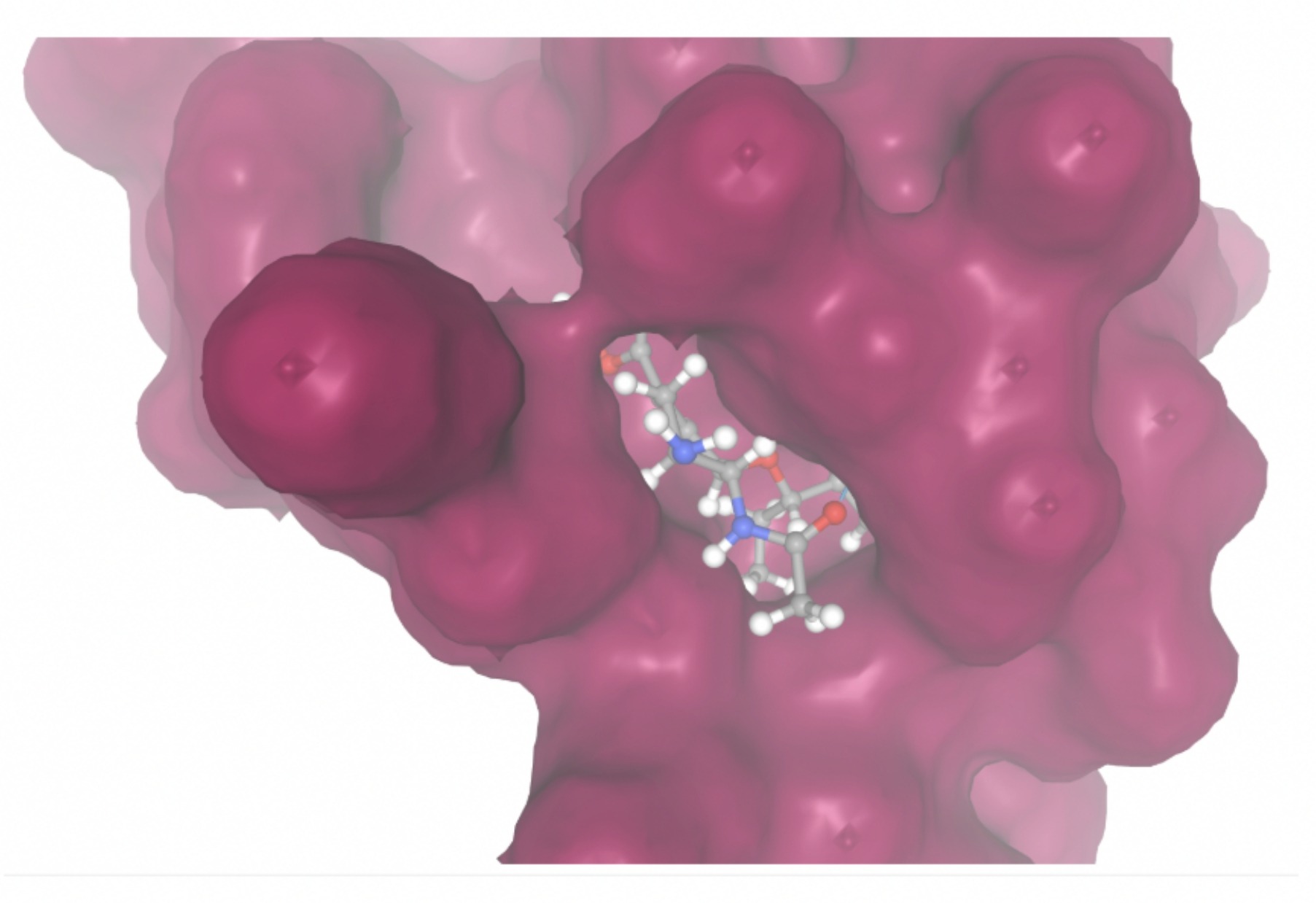 |
| MolPort-004-849-824 | -7.8716 | 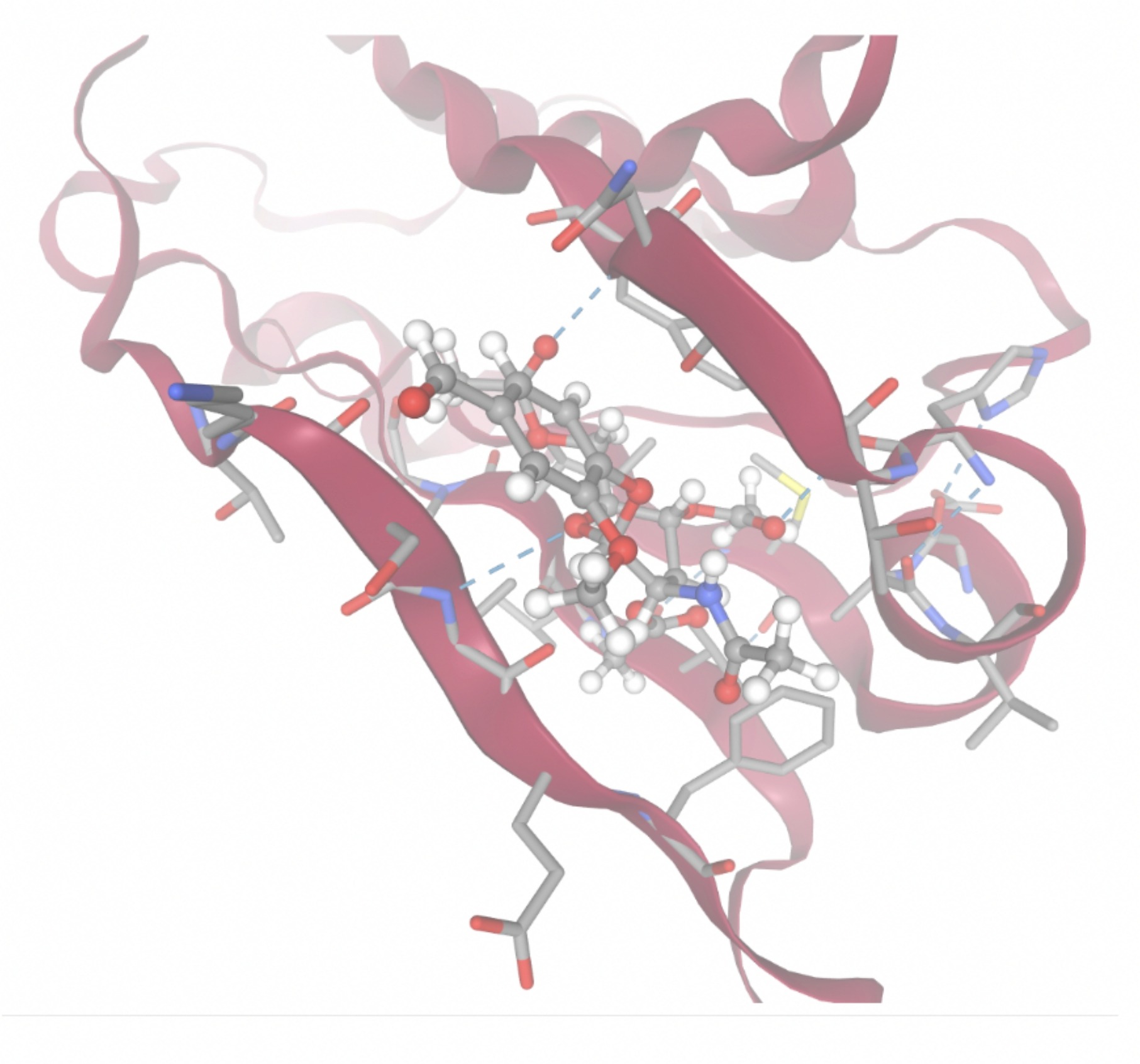 | 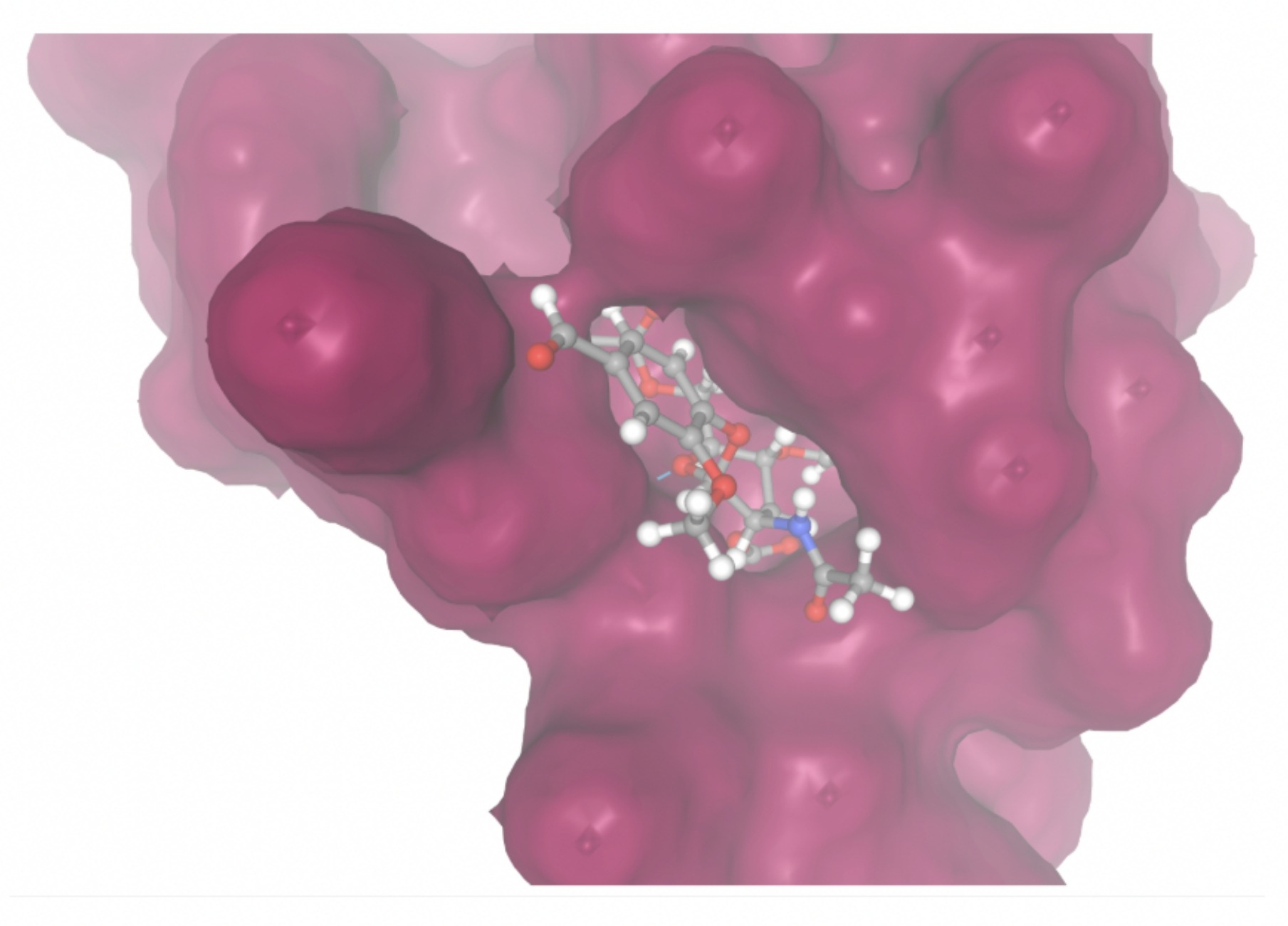 |
| MolPort-003-060-590 | -7.9330 | 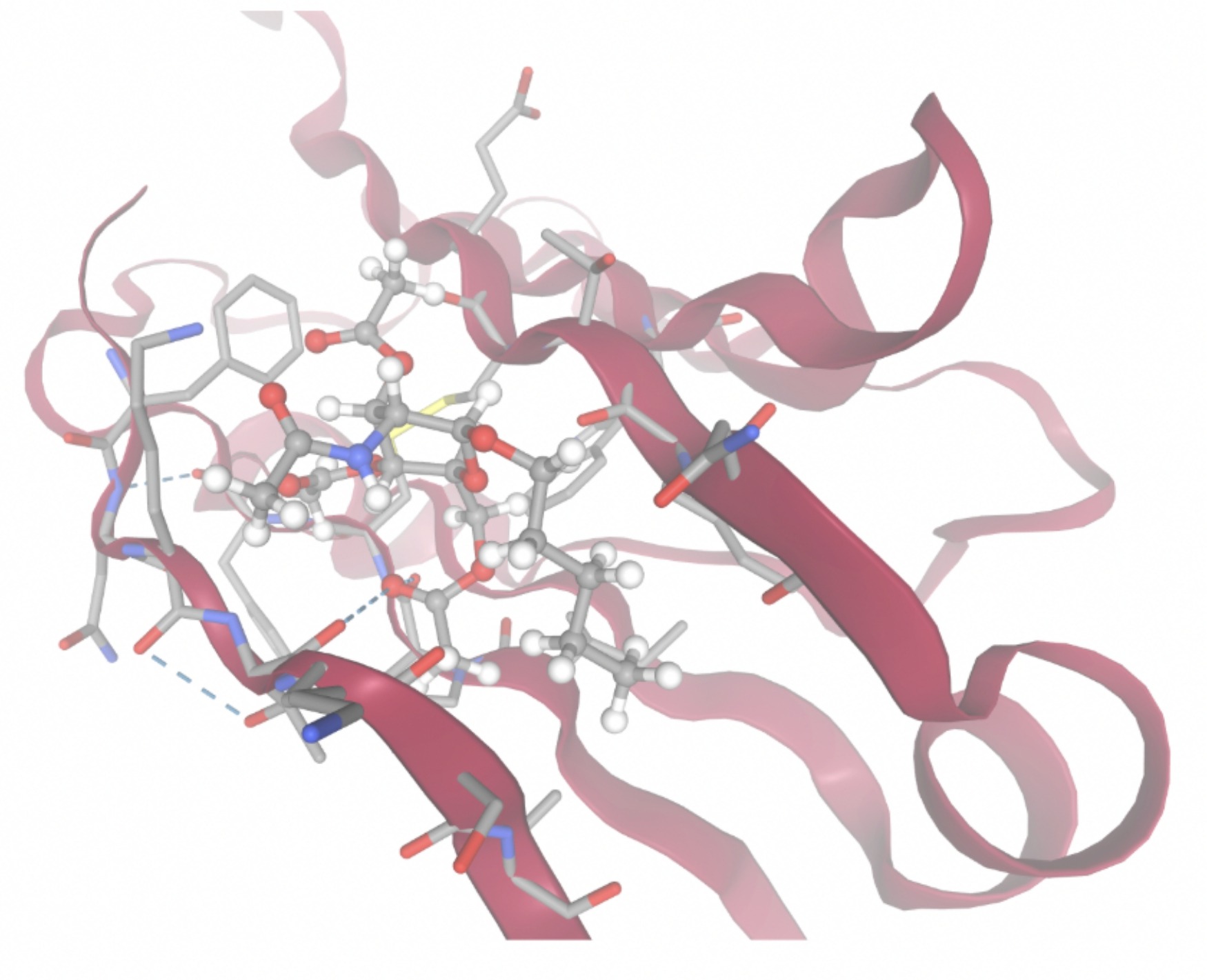 | 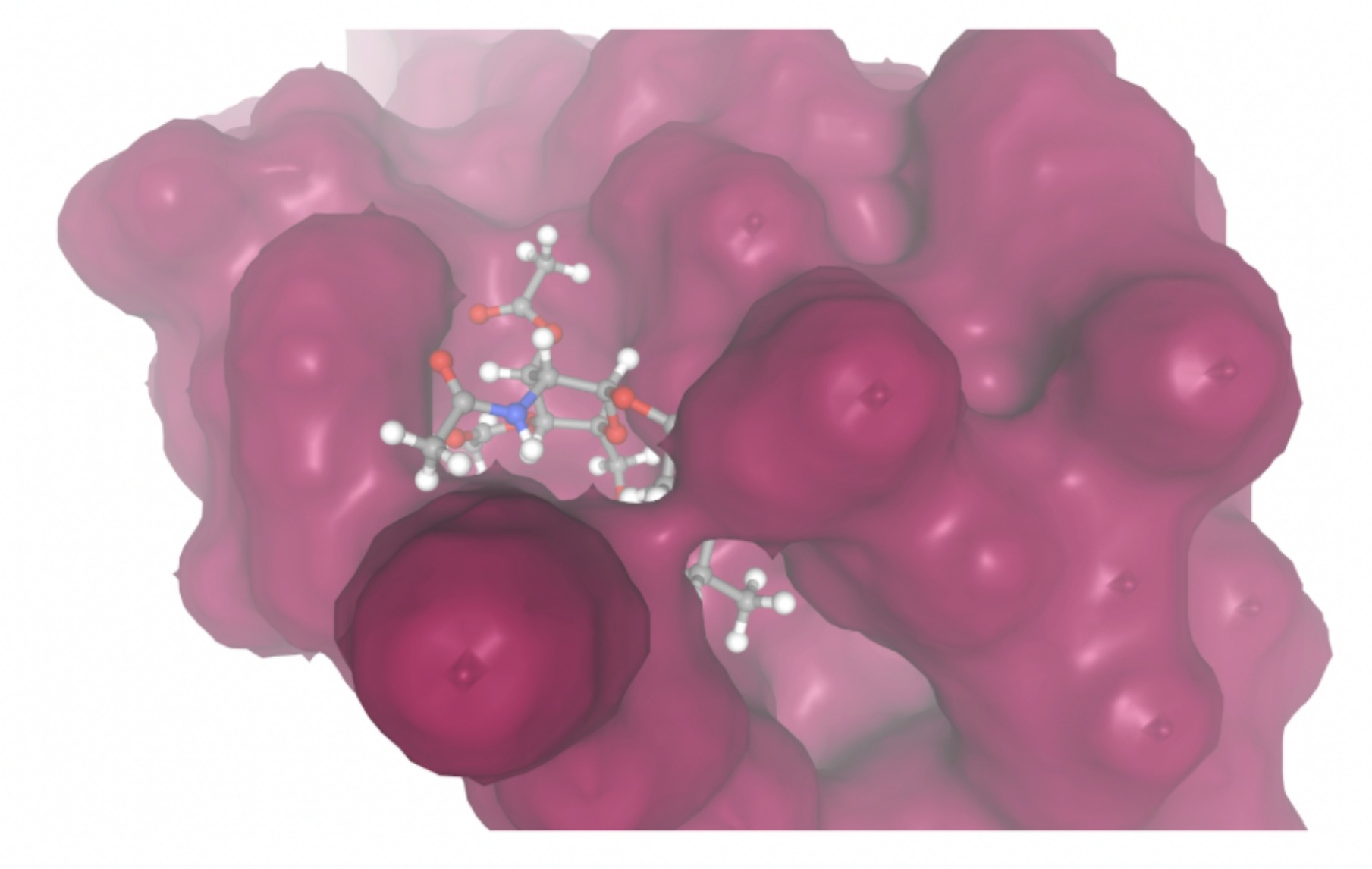 |
ADME Profile Evaluation
SwissADME analysis was used to evaluate the top compounds with the lowest SwissParam docking scores. Each compound was assessed according to Lipinski’s rule to predict a potential favorable physicochemical profile based on hydrogen bonding capacity, molecular weight, and lipophilicity. The results of the assessment are given in Table 6.
| MolPort ID | # of Hydrogen Bond Donors | # of Hydrogen Bond Acceptors | Molecular Weight (Daltons) | Log Po/w (iLOGP) | # of Rule Violations |
| MolPort-006-167-538 | 15 | 38 | 1134.93 | -1.66 | 4 |
| MolPort-005-280-798 | 1 | 11 | 519.50 | 3.25 | 2 |
| MolPort-004-849-816 | 4 | 11 | 543.60 | 3.43 | 2 |
| MolPort-023-223-232 | 4 | 11 | 378.38 | 1.98 | 1 |
| MolPort-002-511-046 | 2 | 7 | 450.31 | 3.83 | 0 |
| MolPort-047-134-715 | 2 | 5 | 354.44 | 3.12 | 0 |
| MolPort-008-326-608 | 4 | 7 | 427.45 | 0.35 | 0 |
| MolPort-051-860-991 | 8 | 15 | 477.40 | -0.99 | 3 |
| MolPort-000-416-985 | 3 | 6 | 401.45 | 2.56 | 0 |
| MolPort-051-689-380 | 4 | 6 | 347.36 | 2.14 | 0 |
| MolPort-006-391-514 | 6 | 7 | 333.38 | 1.64 | 1 |
| MolPort-002-512-231 | 4 | 8 | 342.30 | 0.75 | 0 |
| MolPort-003-011-602 | 1 | 9 | 712.01 | 7.86 | 2 |
| MolPort-038-394-503 | 1 | 4 | 230.30 | 2.57 | 0 |
| MolPort-052-043-087 | 7 | 12 | 530.52 | 1.03 | 3 |
| MolPort-005-289-910 | 4 | 7 | 466.55 | 2.87 | 0 |
| MolPort-051-856-633 | 1 | 12 | 597.63 | 4.19 | 2 |
| MolPort-047-590-656 | 7 | 12 | 530.52 | 1.47 | 3 |
| MolPort-046-763-086 | 5 | 9 | 413.42 | 1.24 | 0 |
| MolPort-004-849-824 | 1 | 11 | 481.45 | 2.60 | 1 |
| MolPort-003-060-590 | 1 | 9 | 417.45 | 3.49 | 0 |
Toxicity Prediction
A toxicity screening of the top candidate drugs was performed using the Protox server. Three acceptance criteria were applied: LD50 ≥ 400mg/kg, toxicity class ≥ 3, and the predicted toxicity in each system must not greatly exceed the average toxicity observed for approved drugs. The results for the screening are shown in Table 7.
To summarize how docking performance was integrated with downstream ADME and toxicity considerations, Table 8 consolidates the quantitative and qualitative criteria implicitly used during hit prioritization.
| Compound ID | Predicted LD50 (mg/kg) | Predicted Toxicity Class | Toxic Systems | Comparison Between Toxicity of Compound and That of the Average Drug |
| MolPort-002-511-046 | 5000 | 5 | Nephrotoxicity, Respiratory toxicity, Cardiotoxicity, Immunotoxicity, Clinical toxicity, Thyroid hormone receptor beta | 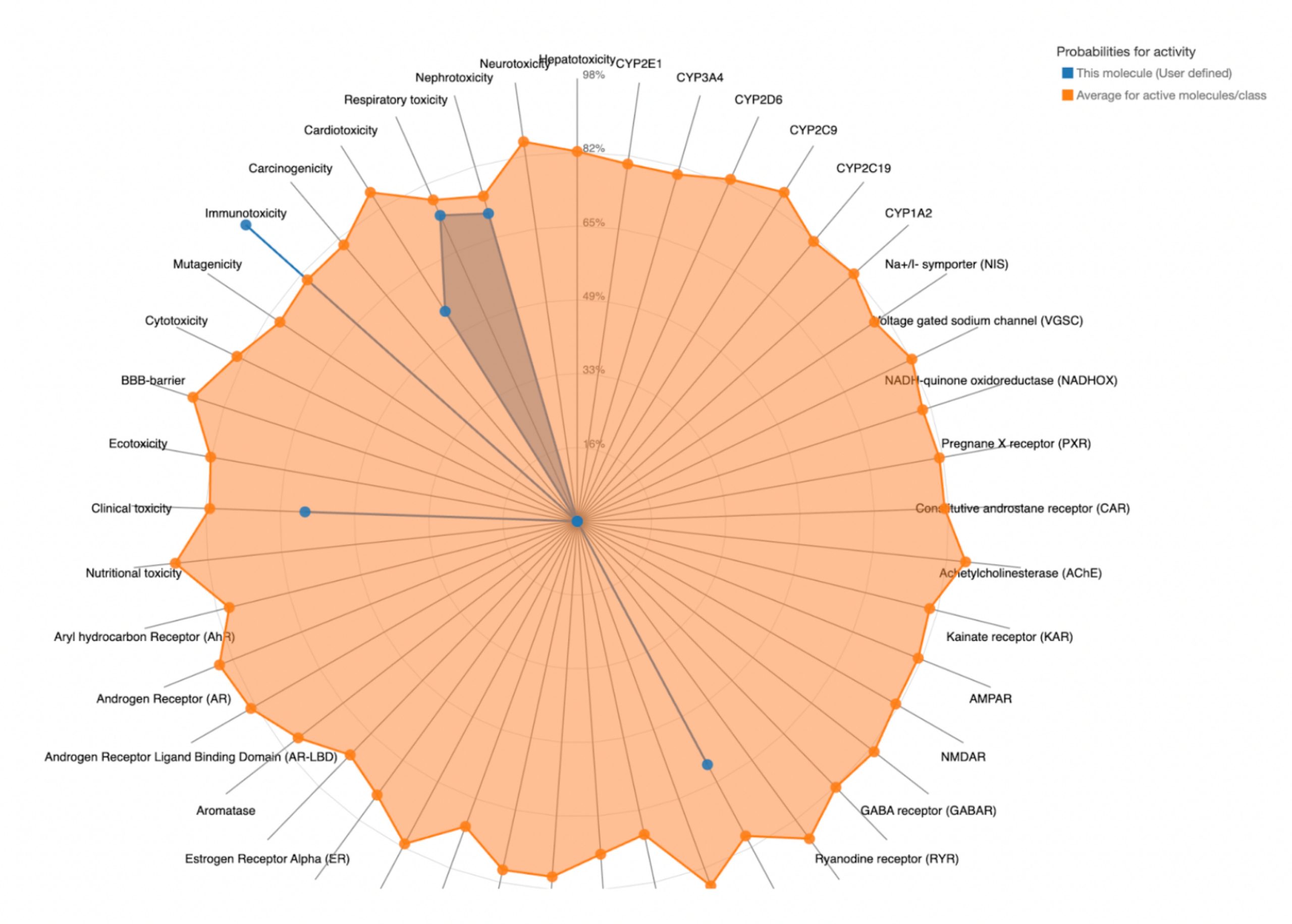 |
| MolPort-047-134-715 | 260 | 3 | Nephrotoxicity, Respiratory toxicity, Cardiotoxicity, Clinical toxicity | 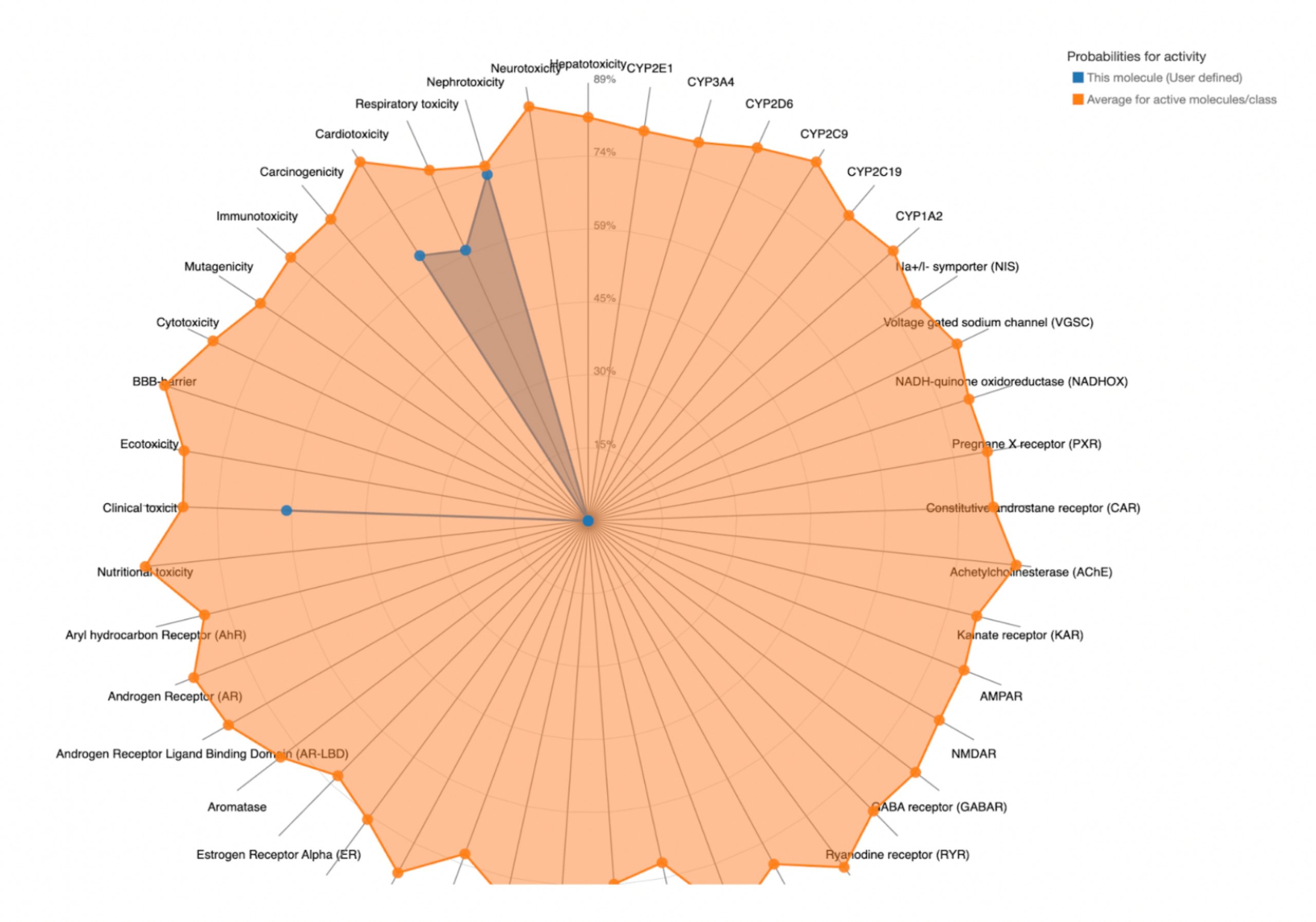 |
| MolPort-008-326-608 | 2500 | 5 | Nephrotoxicity, Respiratory toxicity, Cardiotoxicity, Immunotoxicity, Clinical toxicity | 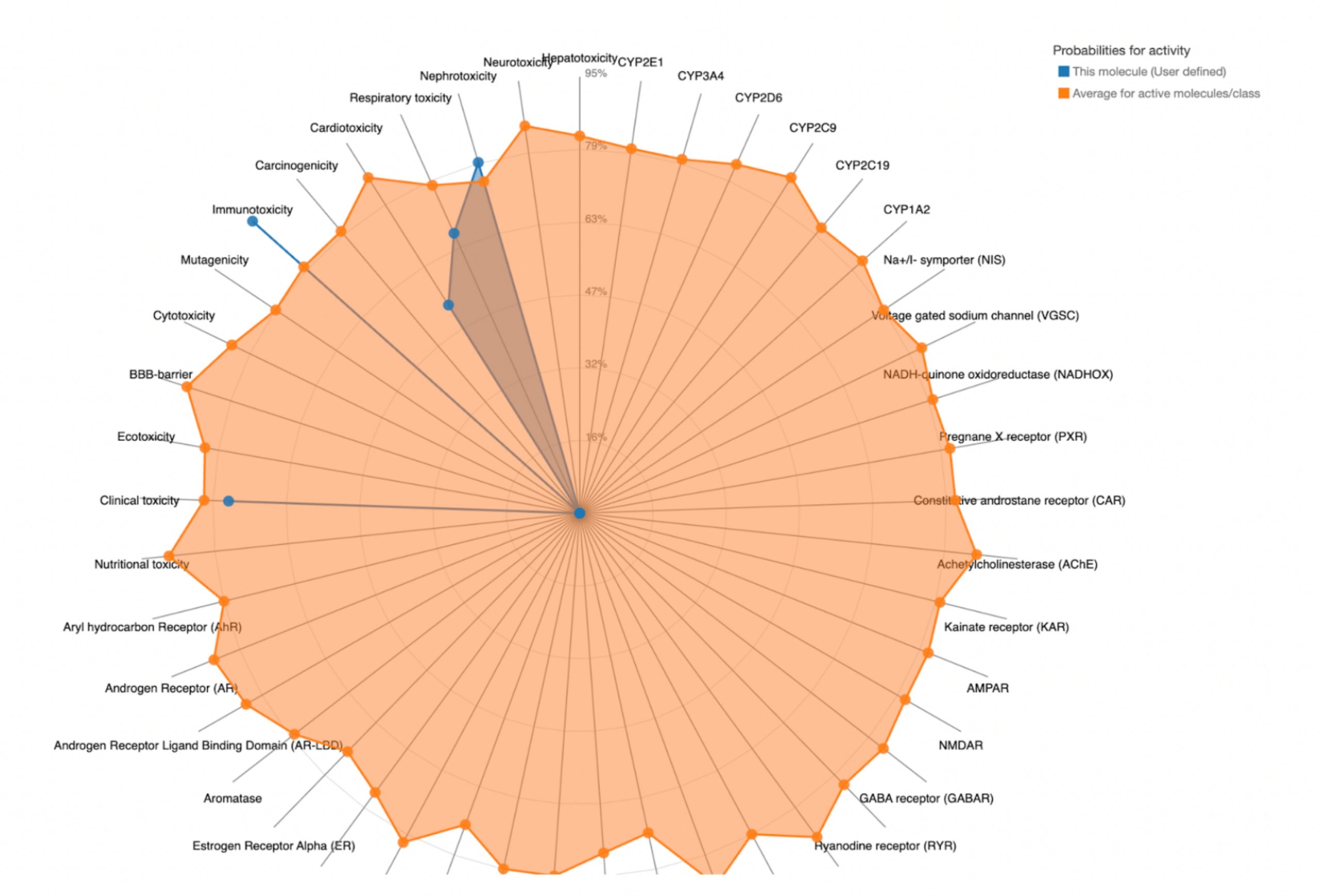 |
| MolPort-000-416-985 | 5000 | 5 | Nephrotoxicity, Respiratory toxicity, Cardiotoxicity, Clinical toxicity | 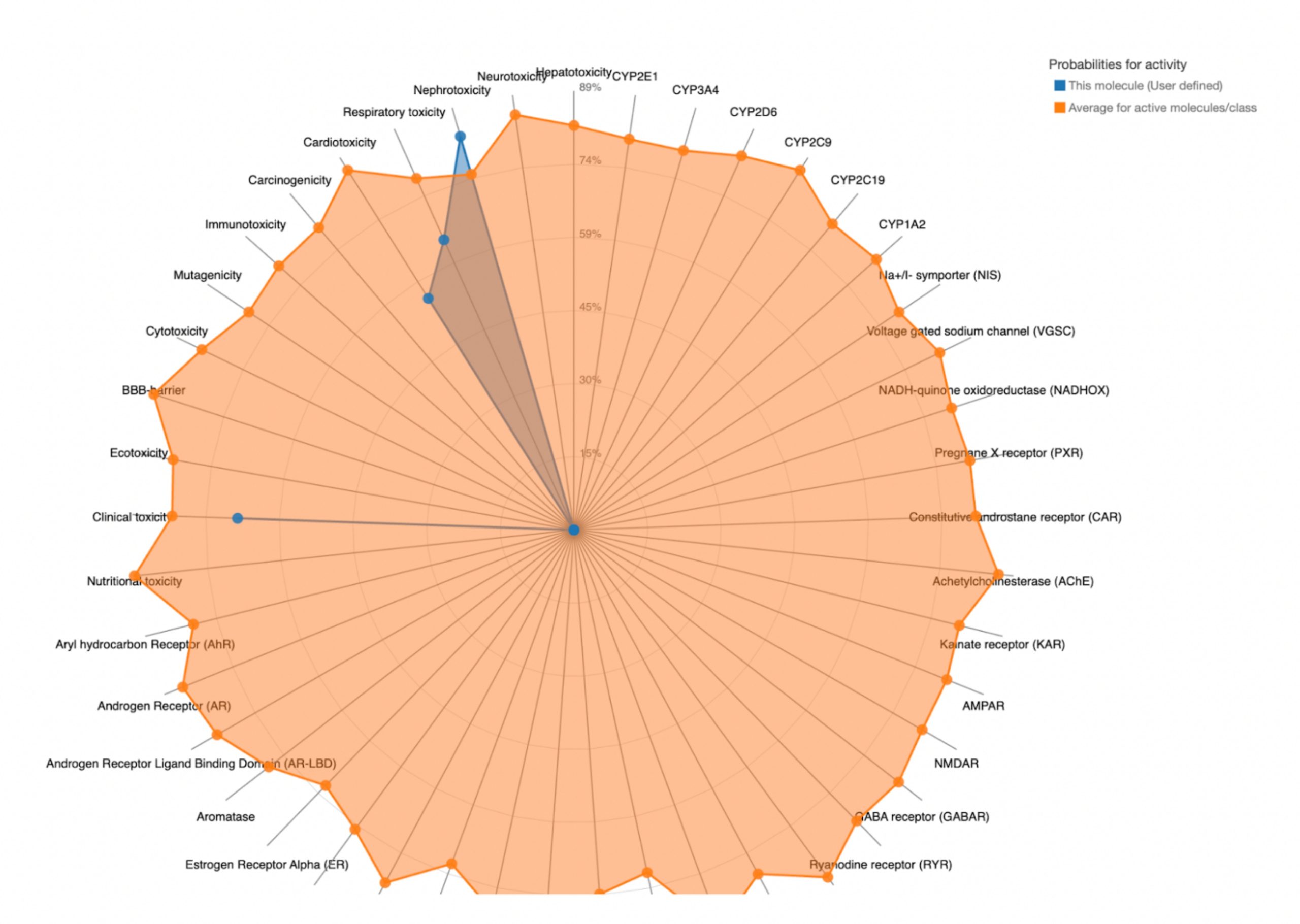 |
| MolPort-051-689-380 | 5000 | 5 | Nephrotoxicity, Respiratory toxicity, Cardiotoxicity, Clinical toxicity | 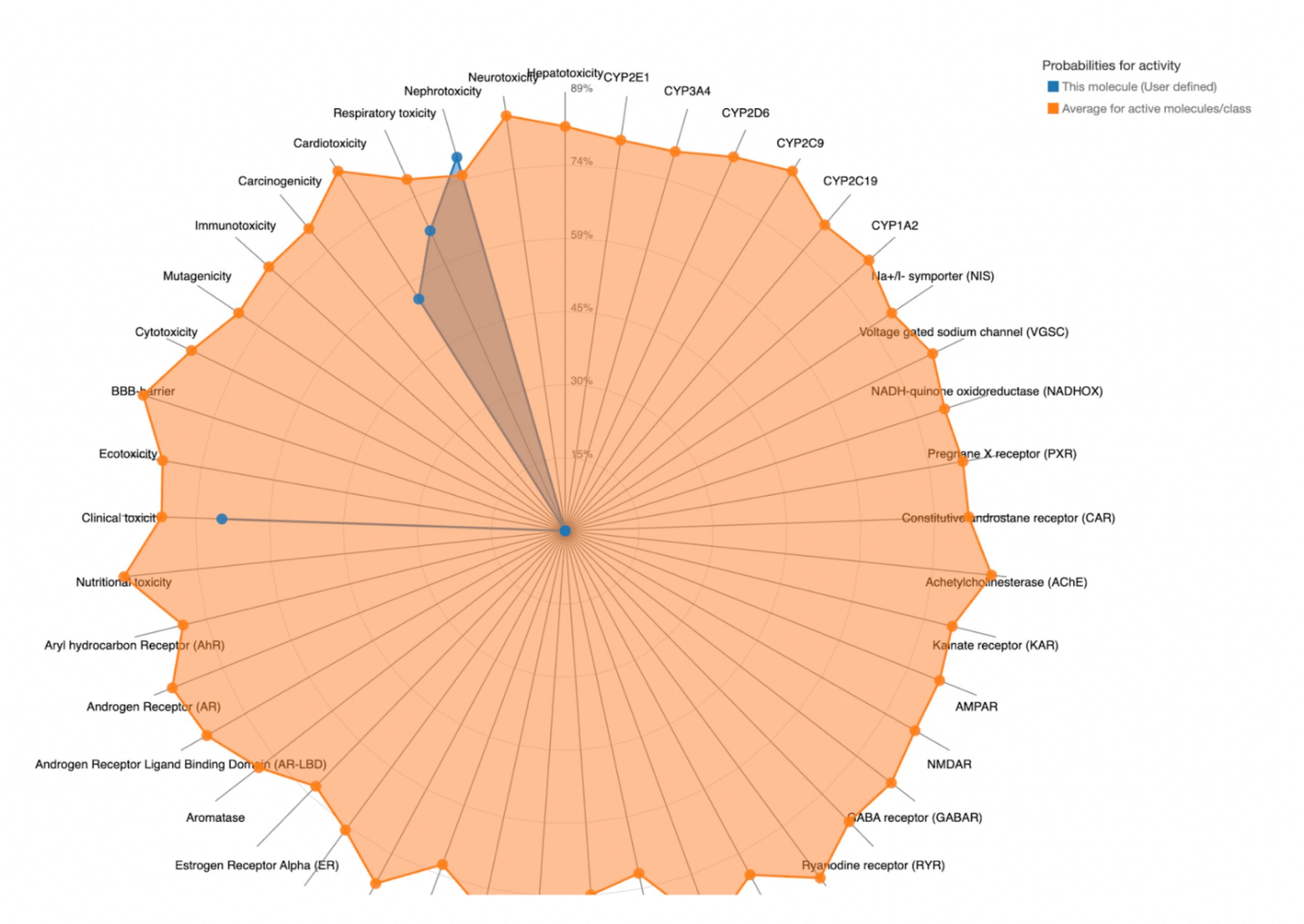 |
| MolPort-002-512-231 | 5000 | 5 | Nephrotoxicity, Respiratory toxicity, Mutagenicity | 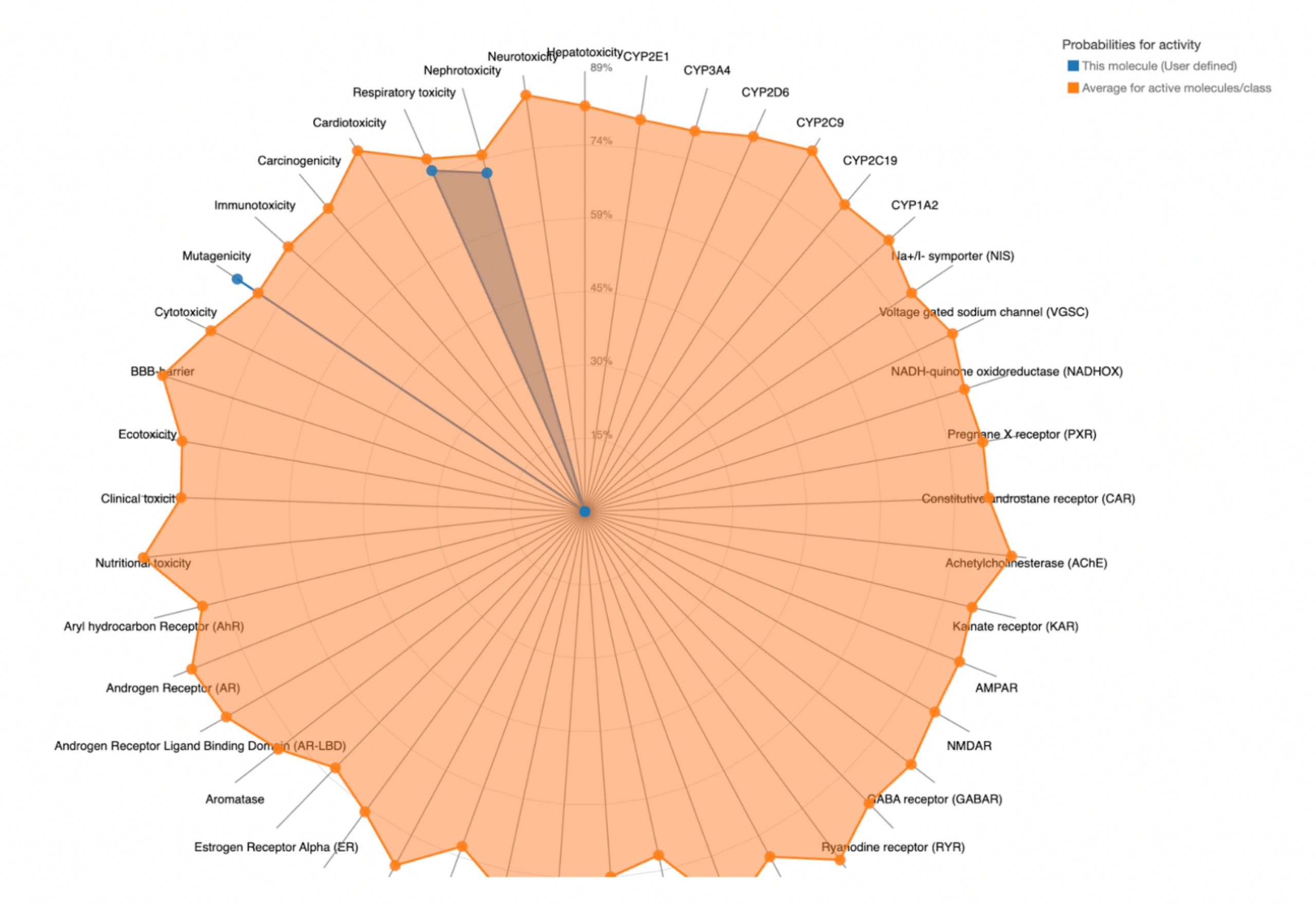 |
| MolPort-038-394-503 | 700 | 4 | Neurotoxicity, Nephrotoxicity, Respiratory toxicity, BBB-barrier, Ecotoxicity, Clinical toxicity | 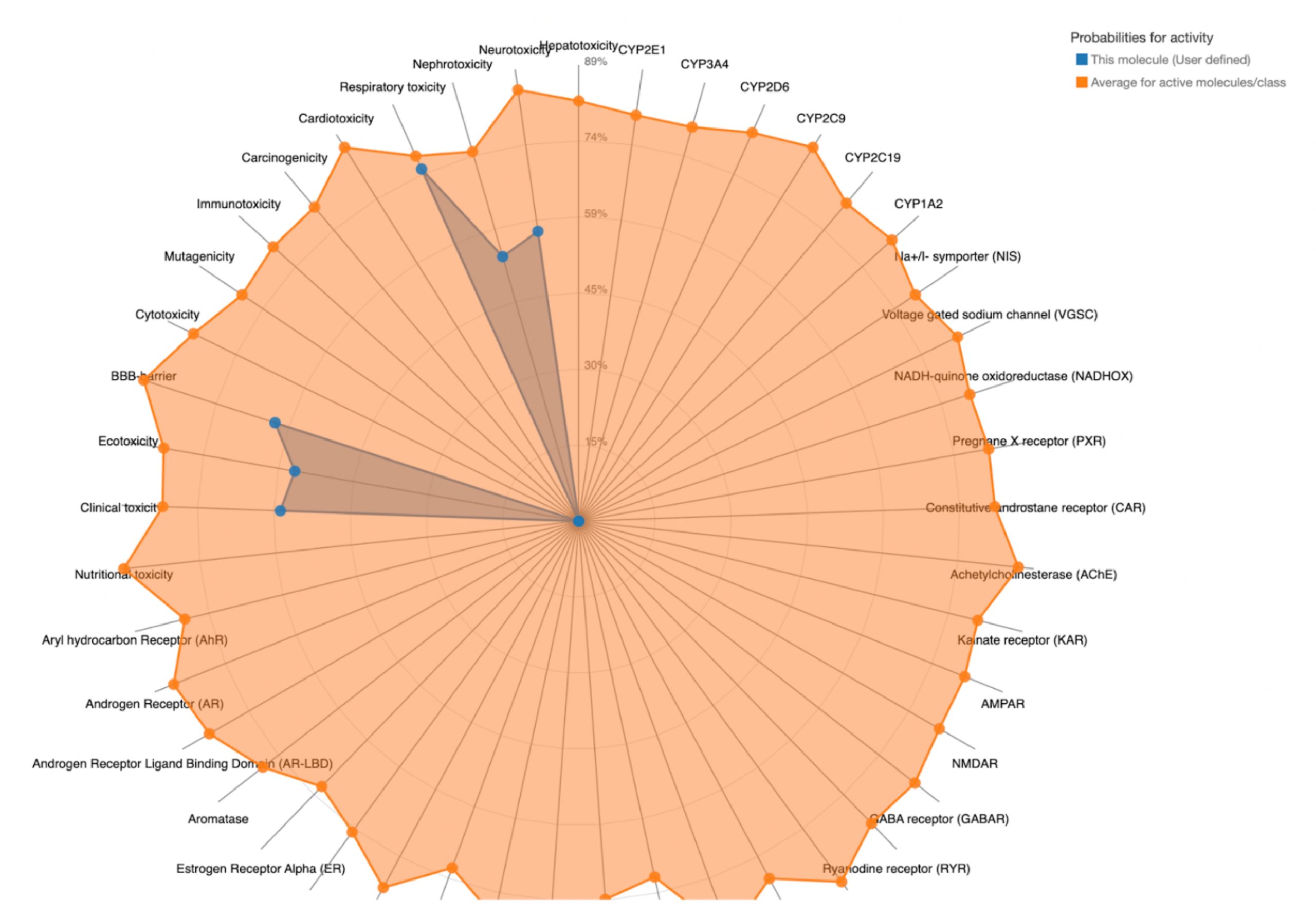 |
| MolPort-005-289-910 | 300 | 3 | Hepatotoxicity, Neurotoxicity, Nephrotoxicity, Respiratory toxicity | 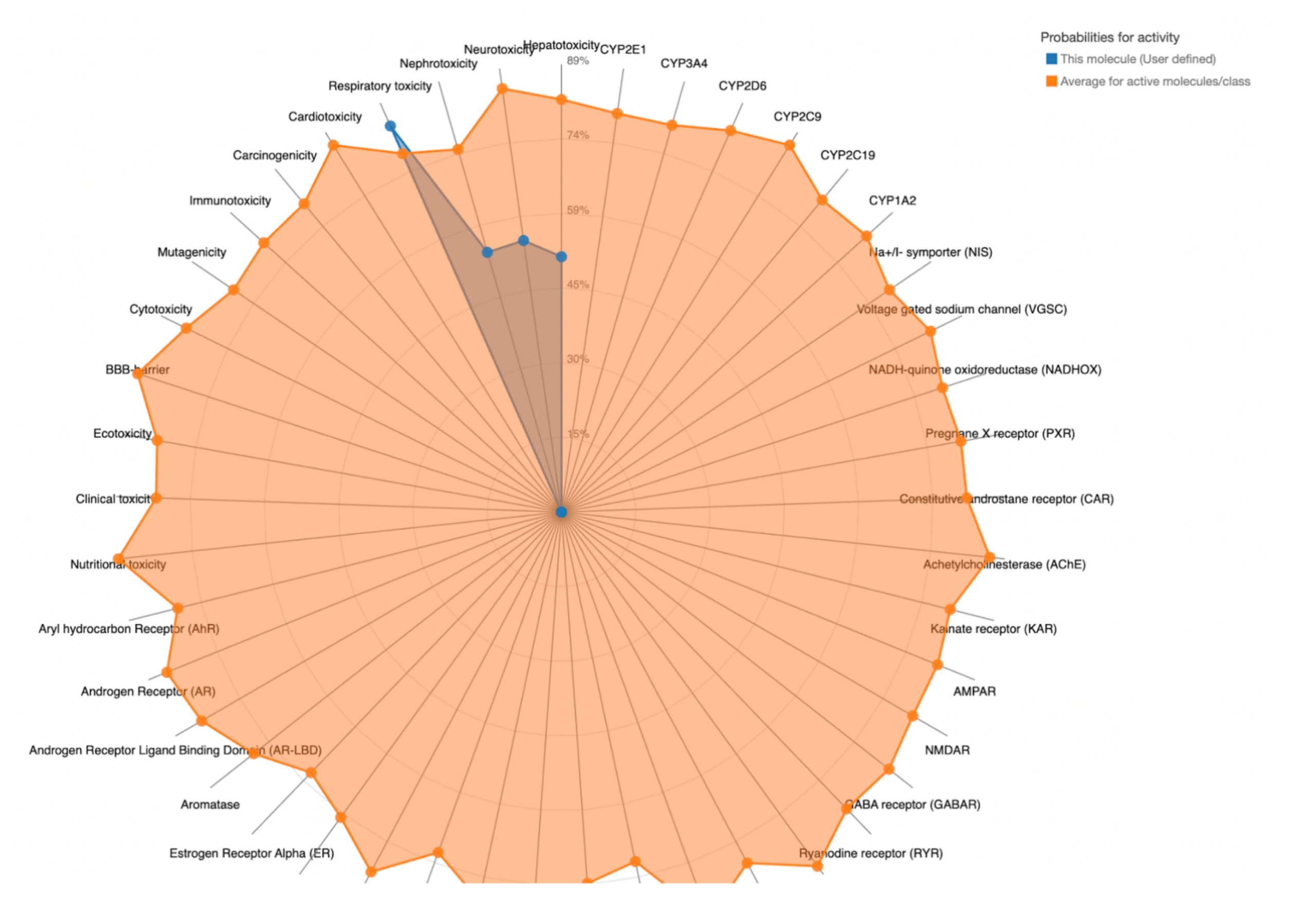 |
| MolPort-046-763-086 | 260 | 3 | Nephrotoxicity, Respiratory toxicity, Cardiotoxicity, Clinical toxicity | 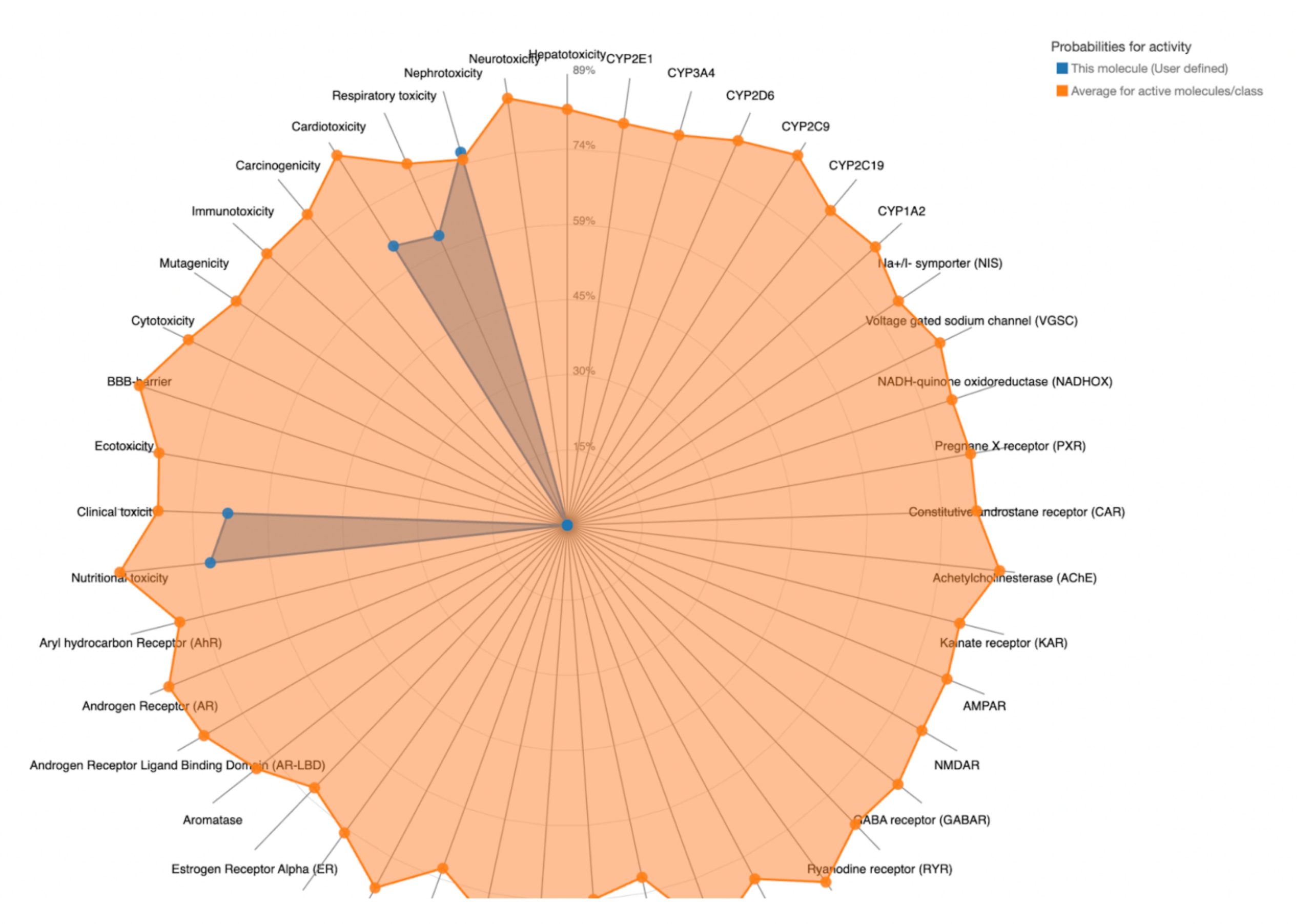 |
| MolPort-003-060-590 | 5000 | 5 | Nephrotoxicity, Respiratory toxicity, Cardiotoxicity, BBB-barrier | 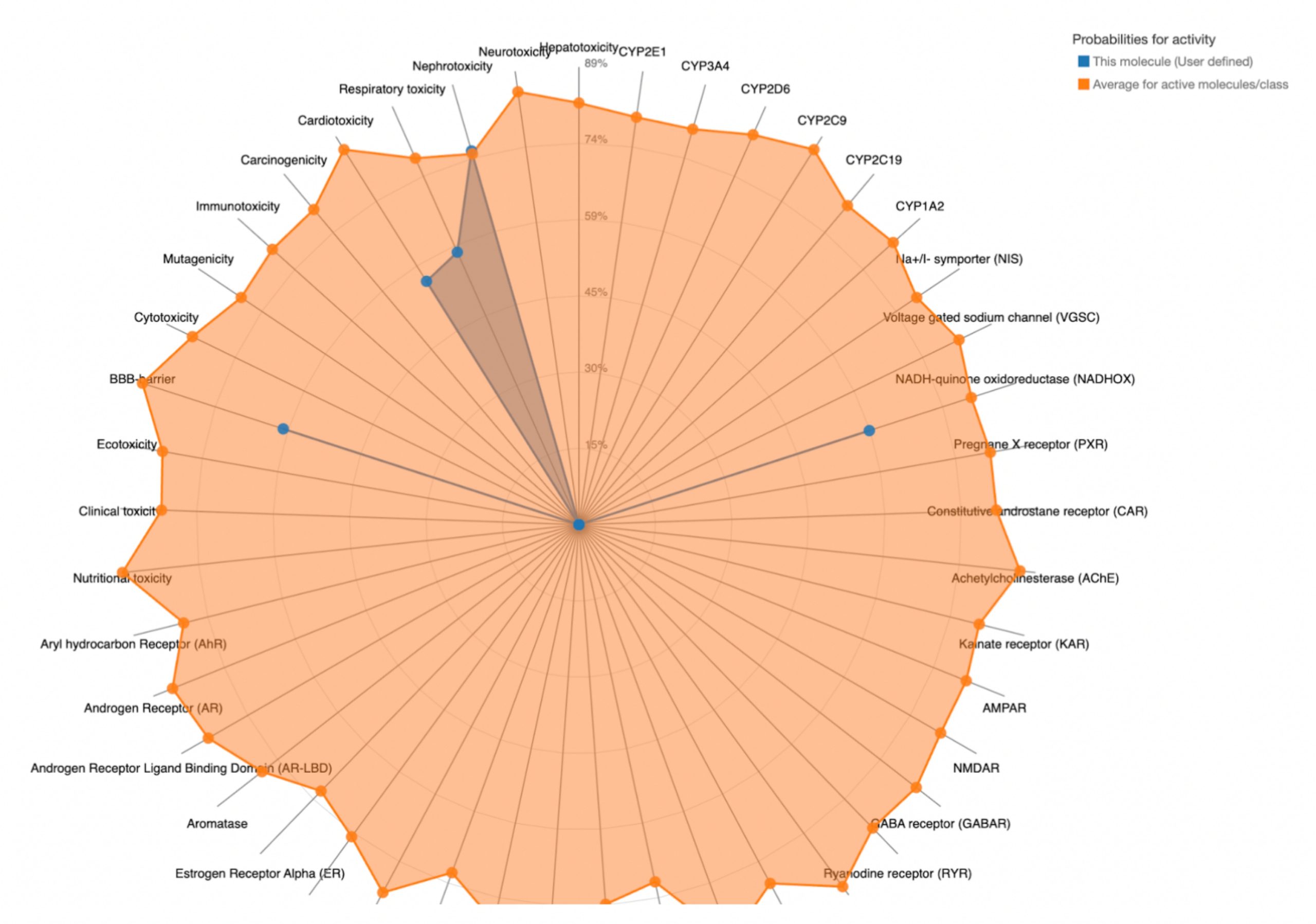 |
| Compound ID | Supplier ID (SMILES Code) | Lowest Observed Estimated ∆G From SwissDock (Kcal/mol) | ADME Summary (# of Lipinski’s Rule Violations) | Ligand Efficiency (∆G / Heavy Atoms) | Toxicity Summary |
| MolPort-008-326-608 | CC(=O)NC1C(O)C(O)C(CO)OC1Oc1ccccc1\C=C\C(=O)c1ccccc1 | -8.1251 | 0 | -0.2621 | Predicted Toxicity Class 5; LD50 ≥ 400 |
| MolPort-005-280-798 | CCN1C(=O)\C(=N\OC2OC(COC(C)=O)C(OC(C)=O)C(OC(C)=O)C2NC(C)=O)c2ccccc12 | -8.7131 | 2 | -0.2355 | Not Evaluated |
| MolPort-002-512-231 | CC(=O)N[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@@H]1Oc1ccc(cc1)[N+]([O-])=O | -7.6195 | 0 | -0.3175 | Predicted Toxicity Class 5; LD50 ≥ 400 |
| MolPort-006-391-514 | CC(=O)NC1C(O)C(O)C(CO)OC1NC(=O)CCCCCN | -7.5954 | 1 | -0.3302 | Not Evaluated |
| MolPort-051-689-380 | CC(=O)N[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@@H]1Oc1ccc2ccccc2c | -7.5941 | 0 | -0.3038 | Predicted Toxicity Class 5; LD50 ≥ 400 |
| MolPort-023-223-232 | CC(=O)NC1C(O)C(O)C(CO)OC1OCCOCCOCCN=[N+]=[N-] | -8.3501 | 1 | -0.3212 | Not Evaluated |
| MolPort-000-416-985 | CC(=O)NC1C(OCc2ccccc2)OC(CO)C(O)C1OCc1ccccc1 | -7.8677 | 0 | -0.2713 | Predicted Toxicity Class 5; LD50 ≥ 400 |
| MolPort-047-134-715 | CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@H](C1)NC(C)=O | -8.1448 | 0 | -0.3258 | Predicted Toxicity Class 3; LD50 ≤ 400 |
| MolPort-051-860-991 | CC(=O)NC1C(O)OC(CO)C(OS(O)(=O)=O)C1OC1OC(C(O)C(O)C1O)C(O)=O | -7.9923 | 3 | -0.2578 | Not Evaluated |
| MolPort-006-167-538 | CC(=O)NC1C(O)OC(COS(O)(=O)=O)C(OC2OC(C(OC3OC(CO)C(OC4OC(C(O)C(O)C4OS(O)(=O)=O)C(O)=O)C(OS(O)(=O)=O)C3NS(O)(=O)=O)C(O)C2OS(O)(=O)=O)C(O)=O)C1O | -9.5669 | 4 | -0.1367 | Not Evaluated |
| MolPort-004-849-816 | CC(=O)NC1C(O)C(O)C(CO)OC1OCC1OC2OC3(CCCCC3)OC2C2OC3(CCCCC3)OC12 | -8.3624 | 2 | -0.2201 | Not Evaluated |
| MolPort-008-267-878 | CC(=O)NC1C(O)C(O)C(CO)OC1OCC=C | -6.8695 | Not Evaluated | -0.3816 | Not Evaluated |
| MolPort-002-511-046 | CC(=O)NC1C(O)C2OC(C)(C)OCC2OC1OCCOc1ccc(Cl)cc1Cl | -8.1479 | 0 | -0.2810 | Predicted Toxicity Class 5; LD50 ≥ 400 |
| MolPort-003-011-602 | CCCCCCCCCCCCC(CCCCCCCCCCCC)COC1OC(COC(C)=O)C(OC(C)=O)C(OC(C)=O)C1NC(C)=O | -9.1922 | 2 | -0.1838 | Not Evaluated |
| MolPort-038-394-503 | CN(C)CCO[C@@H]1COCC[C@H]1NC(C)=O | -6.9777 | 0 | -0.4361 | Predicted Toxicity Class 4; LD50 ≥ 400 |
| MolPort-052-043-087 | COc1ccc(OC2OC(CO)C(OC3OC(CO)C(O)C(O)C3NC(C)=O)C(O)C2NC(C)=O)cc1 | -8.7609 | 3 | -0.2368 | Not Evaluated |
| MolPort-005-289-910 | CC(C)n1c(COc2ccccc2C)nn(C2OC(CO)C(O)C(O)C2NC(C)=O)c1=S | -8.3285 | 0 | -0.2603 | Predicted Toxicity Class 3; LD50 ≤ 400 |
| MolPort-051-856-633 | COC(=O)[C@@]1(C[C@H](OC(C)=O)[C@@H](NC(C)=O)[C@@H](O1)[C@H](OC(C)=O)[C@@H](COC(C)=O)OC(C)=O)Sc1ccc(C)cc1 | -8.5876 | 2 | -0.2095 | Not Evaluated |
| MolPort-047-590-656 | COc1ccc(O[C@@H]2O[C@H](CO)[C@@H](O[C@@H]3O[C@H](CO)[C@H](O)[C@H](O)[C@H]3NC(C)=O)[C@H](O)[C@H]2NC(C)=O)cc1 | -8.4094 | 3 | -0.2273 | Not Evaluated |
| MolPort-046-763-086 | OP(O)(O)=O.[2H]C([2H])([2H])C(=O)N[C@@H]1[C@@H](N)CC(=C[C@H]1OC(CC)CC)C(=O)OCC | -7.7984 | 0 | -0.2888 | Predicted Toxicity Class 3; LD50 ≤ 400 |
| MolPort-004-849-824 | COc1cc(C=O)ccc1OC1OC(COC(C)=O)C(OC(C)=O)C(OC(C)=O)C1NC(C)=O | -7.8716 | 1 | -0.2315 | Not Evaluated |
| MolPort-003-060-590 | CCCCCOC1OC(COC(C)=O)C(OC(C)=O)C(OC(C)=O)C1NC(C)=O | -7.9330 | 0 | -0.2736 | Predicted Toxicity Class 5; LD50 ≥ 400 |
Discussion
Analysis of Binding Sites in CD47
As shown in Table 2, multiple binding sites exhibited drug scores greater than 0.7, indicating potential for a small molecule interaction. Among these binding sites, P_1 shows to be a particularly promising candidate. It exhibited a high drug score of 0.81, due to its volume (just under 1000 Å3) and its surface area (slightly over 1000 Å2). In addition to P_1, binding sites P_0, P_2, and P_5 show potential, each with drug scores greater than 0.7. FTSite then identified several binding sites on CD47 where small molecules would interact in an energetically favorable way. PrankWeb later found 5 binding sites that met both criteria: an energetically favorable reaction and a suitable volume for small molecules. These results serve as strong evidence that CD47 has several binding sites that are volumetrically suitable and energetically favorable for small molecule drugs. This finding supports the targeting of CD47 with small molecule drugs as a promising cancer therapy strategy.
Virtual Screening for Identifying Small Molecules
The goal of this experiment was to identify potential small molecule drugs that bind to CD47 through virtual screening using the Pharmit website. Pharmit allows users to match candidate compounds to a pharmacophore model, which represents the chemical features needed for molecule recognition. Using pharmacophore efficiently allows for the narrowing of large chemical libraries to a list of compounds with high potential affinity to the target protein.
Though three binding pockets were generated by Pharmit, only one was viable for virtual screening. One pocket lacked the complexity needed to construct a pharmacophore model. The other pocket, though it contained more features, produced compounds with RMSD scores greater than 0.1, indicating a poor alignment with the binding site. In contrast, the binding site used had a balance of features, making it most suitable for screening. As a result, all compounds in Table 4 came from the third binding pocket with 4 interactions. Still, this binding pocket interacted favorably with thousands of compounds in the MolPort library, allowing for further testing.
Molecular Docking Tests
To determine the binding potential of the small molecule drugs identified in section 3.2, molecular docking was performed through SwissDock. Each compound was evaluated based on the resulting estimated binding free energies. The lower energies, which are more negative, indicate a more favorable and spontaneous interaction between the drug and target protein.
Docking-derived ∆G values were interpreted comparatively within this dataset, and the threshold -7 kcal/mol was applied as a practical prioritization. Across all docked compounds, MolPort-006-167-538 exhibited the most favorable interaction with a binding free energy of -9.5669 kcal/mol. Its low score indicates a high potential binding affinity for CD47 among the docked molecules. The visualization for this molecule shows a well defined pocket and accurate insertion. Other promising compounds include MolPort-005-280-798 (-8.7131 kcal/mol), Molport-023-223-232 (-8.3501 kcal/mol), and Molport-004-849-816 (-8.3624 kacl/mol). The compounds listed also exhibited favorable binding affinities. In these visualizations, the ligands are encompassed in the proteins binding site, indicating a likely stable bind. It can also be seen that all compounds are binding at the same site which is a favorable result. In summary, several compounds indicated promising binding with CD47, mainly those with binding free energies -8.0 kcal/mol. Accordingly, the docking results are intended to guide compound prioritization within the screening funnel rather than to serve as definitive evidence of binding.
As shown in Figure 7, the SIRPα-contacting epitope from the CD47-SIRPα complex was mapped onto the CD47 structure used for docking to provide structural context for the docking results. This mapping supports that the docking search region corresponds to the SIRPα-contacting surface on CD47.
While docking suggests that several candidates can bind CD47 in silico, docking to CD47 alone does not establish whether binding occurs directly at the CD47-SIRPα interface versus an interface adjacent or allosteric site. Therefore, this study identifies computationally prioritized CD47 binding candidates rather than demonstrating competitive inhibition of the CD47-SIRPα interaction. Confirming interface competition would require explicit pose mapping to the CD47-SIRPα complex and/or in-silico steric clash checks with SIRPα present, followed by experimental tests.
It is also important to consider the heavily glycosylated nature of CD47 when interpreting these results. Glycan chains on the extracellular immunoglobulin variable (IgV) domain can dynamically modulate surface topology and partially shield shallow surface features, potentially limiting continuous access to interface-adjacent regions37. As a result, small molecule binders are more likely to engage compact or transiently accessible pockets rather than directly occluding the full CD47-SIRPα interface reinforcing the interpretation of docking results as hypothesis generating rather than definitive evidence of binding37.
ADME Profile Evaluation
As shown in Table 6, several of the compounds exceeded the limits for one or more parameters. For example, MolPort-006-167-538 had 15 hydrogen bond donors, 38 hydrogen bond acceptors, a molecular weight exceeding 1100 Da, and a logP value less than 0, resulting in four total violations.
In contrast, MolPort-002-511-046 exhibited drug-like properties, with 0 rule violations. It had 2 hydrogen bond donors. 7 acceptors, a molecular weight of 450.31 Da, and a logP value of 2.21, all within optimal range. This suggests a favorable balance between solubility and membrane permeability, making it a promising candidate for oral delivery. Importantly, compliance with Lipinski’s rule and SwissADME metrics reflects drug-likeness and pharmacokinetic feasibility rather than biological safety.
Toxicity Prediction
MolPort-038-394-503 satisfied all criteria for the toxicity screening with a predicted LD50 value of 700mg/kg and a predicted toxicity class of 4. The drug also demonstrated favorable organ-specific toxicity profiles in their radar charts. When the blue dot (predicted toxicity for the tested compounds) was positioned closer to the center than the orange dot (average toxicity of approved drugs), the toxicity in that system did not exceed that of the average drug. For MolPort-038-394-503, this was true.
Additionally, MolPort-003-060-590 satisfied the criteria with a predicted LD50 value of 5000mg/kg and a predicted toxicity class of 5. Although its predicted nephrotoxicity is slightly greater than that of the average drug, because of its proximity to the average value the drug is deemed acceptable.
In contrast, the compounds that failed to meet the criteria displayed blue dots extending far beyond the orange dots in one or more organ systems, indicating a predicted toxicity exceeding the average drug. These results suggest reduced safety and lower suitability for drug development.
Beyond safety-related considerations, it is also important to interpret these computationally prioritized candidates within the broader landscape of CD47-targeting strategies. Engineered SIRPα variants and peptide based CD47-SIRPα inhibitors have demonstrated that direct disruption of this interface can elicit antitumor responses in experimental models18. In contrast to these protein and peptide based approaches, the present work explores the feasibility of identifying small molecule candidates that may engage CD47 through a complementary mechanism, with the potential advantages of oral bioavailability and scalable synthesis.
Statement of Limitation
This study is limited by various factors. First, the toxicity evaluation was conducted at the end of the workflow rather than earlier screening stages. Incorporating the toxicity prediction earlier would reduce the number of compounds eliminated late in the process and improve efficiency. Second, only 22 compounds were tested, which is significantly fewer than the hundreds to thousands typically screened in comparable studies. This reduced the likelihood of identifying the most effective candidates. An additional limitation of this study is the absence of formal docking validation. No redocking of a reference ligand or peptide was performed. As a result, the docking results presented should be interpreted as a relative prioritization tool rather than a validated predictor of binding. Also, while virtual screening provides valuable insight, physical screening remains necessary for validation, and its absence in this study constrains the strength of the drawn conclusion. Importantly, the findings presented reflect computational predictions intended to guide experimental follow-up rather than direct evidence of biological efficacy. A final limitation of this study is the absence of counter-screening against unrelated IgV domain proteins. As a result, the specificity of the identified compounds for CD47 relative to other IgV-containing receptors cannot be assessed from the present analysis.
Conclusion
This study computationally identifies small molecule candidates predicted to interact favorably with CD47 and provides a structured workflow for prioritizing compounds for experimental testing. MolPort-038-394-503 achieved a favorable docking score of -6.9777 kcal/mol, followed by an LD50 of 700 mg/kg and a toxicity class of 4, placing it within a moderate toxicity range consistent with approved drugs, MolPort-003-060-590 demonstrated an even more promising profile, with a docking score of -7.9330 kcal/mol, an LD50 of 5000mg/kg, and a toxicity class of 5, indicating low toxicity and excellent tolerability. Both compounds also satisfied ADME requirements, supporting their potential oral delivery and physiological stability. Together, these results demonstrate that the computational workflow not only narrowed candidates effectively but also yielded two viable small drug candidates that represent testable candidates prioritized based on predicted binding, drug-likeness, and toxicity profiles. This outcome validates the approach as a successful strategy for identifying testable small molecule inhibitors of CD47, and establishes a foundation for applying the workflow to other immune checkpoint proteins in future studies.
References
- National Cancer Institute. Cancer Statistics. https://www.cancer.gov/about-cancer/understanding/statistics (n.d.). [↩]
- National Cancer Institute. What Is Cancer?. https://www.cancer.gov/about-cancer/understanding/what-is-cancer (n.d.). [↩]
- F. Whyte. Metastasis: the deadly part of cancer. British Journal of Nursing. Vol. 5, 1996, https://doi.org/10.12968/bjon.1996.5.9.535. [↩]
- C. Rébé, F. Ghiringhelli. Cytotoxic effects of chemotherapy on cancer and immune cells: how can it be modulated to generate novel therapeutic strategies?. Future oncology. Vol. 11, pg. 2645-2654, 2015, https://doi.org/10.2217/fon.15.198. [↩]
- C. Sordo-Bahamonde, S. Lorenzo-Herrero, A. P. Gonzalez-Rodriguez, A. Martínez-Pérez, J. P. Rodrigo, J. M. García-Pedrero, S. Gonzalez. Chemo-immunotherapy: a new trend in cancer treatment. Cancers. Vol 15, pg. 2912, 2023, https://doi.org/10.3390/cancers15112912. [↩] [↩]
- H. Chen, H. Yang, L. Guo, Q. Sun. The role of immune checkpoint inhibitors in cancer therapy: mechanism and therapeutic advances. MedComm, Vol 6, 2020, https://doi.org/10.1002/mco2.70412. [↩]
- M. A. Hossain. A comprehensive review of immune checkpoint inhibitors for cancer treatment. International immunopharmacology. Vol 143, 2024, https://doi.org/10.1016/j.intimp.2024.113365. [↩] [↩]
- S. S. Said, W. N. Ibrahim. Cancer resistance to immunotherapy: comprehensive insights with future perspectives. Pharmaceutics. Vol 15, pg. 1143, 2023, https://doi.org/10.3390/pharmaceutics15041143. [↩]
- Y. Liu, L. Weng, Y. Wang, J. Zhang, Q. Wu, P. Zhao, Y. Shi, P. Wang, L. Fang. Deciphering the role of CD47 in cancer immunotherapy. Journal of Advanced Research. Vol. 63, pg. 129–158, 2024, https://doi.org/10.1016/j.jare.2023.10.009. [↩]
- M. E. W. Logtenberg, F. A. Scheeren, T. N. Schumacher. The CD47-SIRPα immune checkpoint. Immunity. Vol 52, pg. 742–752, 2020, https://doi.org/10.1016/j.immuni.2020.04.011. [↩] [↩] [↩] [↩]
- Q. Chen, X. Guo, W. Ma. Opportunities and challenges of CD47-targeted therapy in cancer immunotherapy. Oncology Research. Vol 32, pg. 49–60, 2023, https://doi.org/10.32604/or.2023.042383. [↩] [↩]
- X. Luo, Y. Shen, W. Huang, Y. Bao, J. Mo, L. Yao, L. Yuan. Blocking CD47-SIRPα signal axis as promising immunotherapy in ovarian cancer. Cancer Control. Vol 30, 2023, https://doi.org/10.1177/10732748231159706. [↩]
- Q. Li, C. Kang. Mechanisms of action for small molecules revealed by structural biology in drug discovery. International Journal of Molecular Sciences. Vol 21, pg. 5262, 2020, https://doi.org/10.3390/ijms21155262. [↩]
- S. Y. van der Zanden, J. J. Luimstra, J. Neefjes, J. Borst, H. Ovaa. Opportunities for small molecules in cancer immunotherapy. Trends Immunol. Vol 41, pg. 493-511, 2020, https://doi.org/10.1016/j.it.2020.04.004. [↩]
- M. J. Monroy-Iglesias, S. Crescioli, K. Beckmann, N. Le, S. N. Karagiannis, M. V. Hemelrijck, A. Santaolalla. Antibodies as biomarkers for cancer risk: a systematic review. Clinical and Experimental Immunology. Vol 209, pg. 46-63, 2020, https://doi.org/10.1093/cei/uxac030. [↩]
- J. L. Messerschmidt, G. C. Prendergast, G. L. Messerschmidt. How cancers escape immune destruction and mechanisms of action for the new significantly active immune therapies: helping nonimmunologists decipher recent advances. The Oncologist. Vol 21, pg. 233-243, 2016, https://doi.org/10.1634/theoncologist.2015-0282. [↩]
- M. Gracia-Hernandez, M. Suresh, A. Villagra. The advances in targeting CD47/SIRPα “do not eat me” axis and their ongoing challenges as an anticancer therapy. Oncotarget. Vol 14, pg 462-465, 2024, https://doi.org/10.18632/oncotarget.28607. [↩]
- K. Weiskopf, A. M. Ring, C. C. Ho, J. P. Volkmer, A. M. Levin, A. K. Volkmer, E. Ozkan, N. B. Fernhoff, M. van de Rijn, I. L. Weissman, K. C. Garcia. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. Vol 341, pg. 88-91, 2013, https://doi.org/10.1126/science.1238856. [↩]
- H. Wang, Y. Sun, X. Zhou, C. Chen, L. Jiao, W. Li, S. Gou, Y. Li, J. Du, G. Chen, W. Zhai, Y. Wu, Y. Qi, Y. Gao. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. Journal for Immunotherapy of Cancer. Vol 8, 2020, https://doi.org/10.1136/jitc-2020-000905. [↩]
- C. Chen, R. Wang, X. Chen, Y. Hou, J. Jiang (2022). Targeting CD47 as a novel immunotherapy for breast cancer. Frontiers in Oncology. Vol 12, 2022, https://doi.org/10.3389/fonc.2022.924740. [↩] [↩] [↩]
- B. Zhang, W. Li, D. Fan, W. Tian, J. Zhou, Z. Ji, Y. Song. Advances in the study of CD47-based bispecific antibody in cancer immunotherapy. Immunology. Vol 167, pg. 15-27, 2022, https://doi.org/10.1111/imm.13498. [↩] [↩] [↩]
- K. Yucebas, S. Ko, J. Zhou, E. M. Hamel, M. G. Hackworth, E. A. Diaz Miranda, H. S. Carpenter, M. I. Hunter, O. M. Khan, I. L. Weissman, S. Jin (2025). Immunotherapy of endometrial cancer via CD47 blockade-mediated macrophage phagocytosis. PNAS Nexus. Vol 4, 2025, https://doi.org/10.1093/pnasnexus/pgaf143. [↩] [↩] [↩] [↩]
- J. Liang, H. Edelsbrunner, C. Woodward. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Science. Vol 7, pg. 1884–1897, 1998, https://doi.org/10.1002/pro.5560070905. [↩]
- R. Aguti, E. Gardini, M. Bertazzo, S. Decherchi, A. Cavalli. Probabilistic pocket druggability prediction via one-class learning. Frontiers in Pharmacology. Vol 13, 2022, https://doi.org/10.3389/fphar.2022.870479. [↩]
- Protein Plus. https://proteins.plus/ (n.d.). [↩]
- RCSB PDB. 2JJT. https://www.rcsb.org/structure/2JJT (n.d.). [↩]
- FTSite. https://ftsite.bu.edu/ (n.d.). [↩]
- PrankWeb. https://prankweb.cz/ (n.d.). [↩]
- Pharmit. https://pharmit.csb.pitt.edu/ (n.d.). [↩]
- X. Y. Meng, H. X. Zhang, M. Mezei, M. Cui. Molecular docking: a powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design. Vol 7, pg. 146–157, 2011, https://doi.org/10.2174/157340911795677602. [↩]
- SwissDock. https://www.swissdock.ch/ (n.d.). [↩]
- RCSB PDB. 2VSC. https://www.rcsb.org/structure/2VSC (n.d.). [↩]
- The PyMOL Molecular Graphics System, Version 3.0 Schrödinger, LLC. [↩]
- L. Z. Benet, C. M. Hosey, O. Ursu, T. I. Oprea. BDDCS, the rule of 5 and drugability. Advanced Drug Delivery Reviews. Vol. 101, pg. 89–98, 2016, https://doi.org/10.1016/j.addr.2016.05.007. [↩]
- SwissADME. https://www.swissadme.ch/ (n.d.). [↩]
- ProTox. https://tox.charite.de/protox3/index.php?site=compound_input (n.d.). [↩]
- T. Zhang, F. Wang, L. Xu, Y. G. Yang. Structural-functional diversity of CD47 proteoforms. Frontiers in Immunology. Vol 15, 2024, https://doi.org/10.3389/fimmu.2024.1329562. [↩] [↩]



{kind=link}