
Abstracts
Background: Oral squamous cell carcinoma (OSCC) is a growing global health concern, yet its early detection remains challenging due to the lack of reliable, non-invasive biomarkers. Recent studies suggest that the oral microbiome may play a pivotal role in tumor development and immune modulation.
Objective: This study aimed to investigate alterations in the salivary microbiome of OSCC patients compared to healthy individuals, identify cancer-enriched bacterial taxa, and evaluate their functional roles in driving inflammation, with the ultimate goal of developing a qPCR-based microbial biomarker panel for non-invasive oral cancer diagnosis.
Methods: Unstimulated saliva samples were collected from five healthy controls and five OSCC patients under IRB approval. Microbial composition was analyzed via 16S rRNA gene sequencing (V3–V4 region), followed by alpha and beta diversity analyses, LEfSe for differential abundance, and ROC curve modeling. Fusobacterium nucleatum was further validated via qPCR and functionally tested by co-culturing with human oral keratinocytes (HOKs), measuring cytokine expression (IL-6, IL-8, TNF-α) through qRT-PCR and Western blot.
Results: OSCC patients exhibited increased microbial diversity and distinct microbial community structures compared to controls. Four bacterial species— Fusobacterium nucleatum (Fn), Prevotella intermedia (Pi), Porphyromonas gingivalis (Pg), and Treponema denticola (Td) —were significantly enriched in the cancer group. qPCR confirmed the elevated abundance of F. nucleatum in OSCC saliva (Ct < 24). Co-culture of F. nucleatum with HOKs induced marked upregulation of pro-inflammatory cytokines at both mRNA and protein levels. A Ct-based decision tree model using four taxa achieved high discriminatory performance for classifying oral cancer versus healthy individuals.
Conclusion: Our findings suggest that oral cancer is associated with specific microbial signatures and a pro-inflammatory shift in the host–microbiome interaction. The identified bacterial panel, particularly F. nucleatum, shows promise as a non-invasive, saliva-based biomarker for early detection of OSCC and may offer novel insights into tumor–microbiome crosstalk.
Introduction
Oral cancer is one of the most common malignancies worldwide, with over 370,000 new cases reported in 20201, and its incidence continues to rise. While established risk factors such as tobacco use, alcohol consumption, human papillomavirus (HPV) infection, and ultraviolet exposure are well recognized2‘3‘4, these alone do not fully explain the complexity of oral carcinogenesis. Recently, increasing attention has been paid to the potential role of the oral microbiome in the development and progression of oral cancer5‘6.
The oral cavity is a complex ecosystem composed of hard and soft tissues and harbors the second-largest microbial community in the human body after the gastrointestinal tract7. More than 700 bacterial species coexist in a healthy oral environment, where they contribute to homeostasis through vitamin and amino acid synthesis, immune regulation, and inhibition of pathogenic colonization8‘9‘10. However, when this microbial balance is disrupted—a condition known as oral microbial dysbiosis—it can lead to chronic inflammation, immune suppression, and alterations in cell signaling pathways that are closely linked to various diseases, including cancer11.
Certain bacteria such as Streptococcus species and Fusobacterium nucleatum have been implicated in promoting a tumor-permissive microenvironment by inducing pro-inflammatory and immunosuppressive responses12‘13. In patients with oral cancer, shifts in microbial composition have been associated with changes in fatty acid metabolism, pro-inflammatory cytokine expression, and immune cell function14.
Given this emerging evidence, the oral microbiome is gaining recognition not only as an ecological indicator but also as a promising source of diagnostic biomarkers, early detection tools, and eventherapeutic targets that influence metabolic and immune pathways15. The oral cavity’s accessibility and the non-invasive nature of saliva sampling further enhance the translational potential of microbiome-based diagnostics16.
However, a clear, functionally validated, and clinically translatable microbial signature for OSCC diagnosis is still lacking. Therefore, this study aimed to address this gap by identifying cancer-enriched salivary bacteria, experimentally validating their inflammatory potential, and evaluating their utility as a non-invasive qPCR-based biomarker panel.
In this study, we aimed to compare the salivary microbiome profiles of healthy individuals and patients with oral cancer, identify cancer-enriched microbial taxa, and investigate their functional roles in modulating host inflammatory and immune responses. Through this approach, we sought to gain insights into the microbial mechanisms underlying oral carcinogenesis and evaluate the potential of microbiome-based biomarkers for early detection.
Materials and Methods
Study Subjects and Sample Collection
Unstimulated saliva samples were collected from five healthy individuals and five patients diagnosed with oral squamous cell carcinoma (OSCC), following approval by the Institutional Review Board (IRB no. P01-202506-02-009). All participants provided written informed consent. Participants were asked to refrain from eating, drinking, and oral hygiene for at least 1 hour before sample collection. All samples were frozen at -80°C within 30 minutes of collection. Samples were frozen at −80°C within 30 minutes.
DNA Extraction and 16S rRNA Gene Sequencing
Genomic DNA was extracted from saliva samples using a commercial microbial DNA extraction kit (QIAamp DNA Mini Kit), following the manufacturer’s protocol. The V3–V4 hypervariable region of the bacterial 16S rRNA gene was amplified using universal primers (341F/806R). PCR amplification of the V3–V4 hypervariable region was performed using primer pair 341F/806R and the following cycling parameters: 95°C for 3 min; 30 cycles of 95°C for 30 sec, 55°C for 30 sec, 72°C for 30 sec; final extension at 72°C for 5 min. And paired-end sequencing was performed on the Illumina MiSeq platform. The mean sequencing depth was approximately 4 × 10⁴ reads per sample.
Raw reads were processed using QIIME2. Denoising and chimera removal were performed with DADA2. Taxonomic assignment was carried out using the SILVA reference database. Alpha diversity (Shannon index) and beta diversity (Bray–Curtis dissimilarity) analyses were conducted using the phyloseq and vegan packages in R. Principal Coordinates Analysis (PCoA) was used to visualize group differences. Differentially abundant taxa were identified using Linear Discriminant Analysis Effect Size (LEfSe)17. Taxonomic assignments were reported at the genus level unless species-level confidence scores were sufficiently high. Given the known limitations of 16S V3–V4 resolution, species-level labels are interpreted cautiously.
Quantitative PCR (qPCR) Validation
qPCR was performed to validate the abundance of Fusobacterium nucleatum using species-specific primers. Reactions were carried out with SYBR Green master mix on a StepOnePlus Real-Time PCR System. qPCR reactions were performed in a total volume of 20 µL, consisting of 10 µL SYBR Green Master Mix (Bio-Rad), 0.5 µM forward and reverse primers, 2 µL of template DNA, and nuclease-free water.
Cycling conditions were as follows: 95°C for 3 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 30 sec, with a melt-curve analysis from 65°C to 95°C to confirm amplicon specificity. All reactions were performed in triplicate. Each reaction was performed in triplicate.
Ct values were compared between groups and used to develop a rule-based diagnostic algorithm based on threshold values for , P. intermedia, P. gingivalis, and T. denticola. All saliva samples were collected and processed using identical input volumes to reduce variability related to sample quantity. For qPCR normalization, relative abundance was calculated using ΔCt = Ct(target) – Ct(universal 16S), with forward primer 5′ CAACCATTACTTTAACTCTACCATGTTCA and reverse primer 5′-GTTGACTTTACAGAAGGAGATTATGTAAAAATC
Cell Culture and Co-culture with F. nucleatum
Human oral keratinocytes (HOKs) were cultured in keratinocyte growth medium (KGM, Lonza) at 37 °C with 5% CO₂. For co-culture experiments, live (KCTC 2640) was added to confluent HOK monolayers at a multiplicity of infection (MOI) of ~100. F. nucleatum (KCTC 2640) was cultured in an anaerobic chamber under 85% N₂/10% H₂/5% CO₂ using CDC anaerobe medium. MOI was calculated based on OD600 and confirmed by CFU plating. After 24 hours of incubation, cells were harvested for RNA and protein extraction.
Bioinformatic Analysis
To ensure equal sequencing depth across samples, the feature table was rarefied to 2.0 × 10⁴ high-quality reads per sample, which corresponded to the minimum post-filtering read count among all samples. Rarefaction was performed prior to alpha diversity (Shannon index) and beta diversity (Bray–Curtis dissimilarity) analyses to avoid sequencing-depth–related bias.
RNA Extraction and qRT-PCR
Total RNA was extracted using TRIzol reagent, and cDNA synthesis was performed using a reverse transcription kit (iScript, Bio-Rad). Quantitative reverse transcription PCR (qRT-PCR) was performed to assess the expression of IL-6, IL-8, and TNF-α, normalized to GAPDH. Relative expression was calculated using the ΔΔCt method.
Western Blot Analysis
Total protein was extracted from co-cultured HOKs using RIPA buffer supplemented with protease inhibitors. Protein concentrations were measured using a BCA assay (Thermo Fisher Scientific). For SDS-PAGE, 20 µg of total protein per sample was loaded onto 10–12% polyacrylamide gels, transferred to PVDF membranes, and blocked with 5% skim milk in TBST for 1 hour. Blots were probed with antibodies against IL-6 (Cell Signaling Technology, #12153), IL-8 (Cell Signaling Technology, #94407), and GAPDH (Santa Cruz, sc-59540), followed by HRP-conjugated secondary antibodies. Signal was detected using ECL substrate and quantified via densitometry.
Diagnostic Model and ROC Analysis
A rule-based decision tree model was developed using Ct threshold cutoffs: < 24, P. intermedia < 25, P. gingivalis < 26, and T. denticola < 27. Samples satisfying ≥3 of the 4 criteria were classified as “Likely Oral Cancer.” Ct cutoffs were determined from the 90th percentile of healthy-control ΔCt values. ROC analysis and AUC calculations were performed using the pROC package in R.
Results
Schematic representation of the oral microbiome composition in healthy individuals and oral cancer patients
This diagram illustrates the conceptual framework of the study, comparing the salivary microbiome between five healthy individuals and five patients diagnosed with oral squamous cell carcinoma (OSCC) (Fig. 1A). Saliva samples were collected from each participant to serve as a non-invasive and clinically accessible source of oral microbial DNA.
The illustration highlights bacterial species commonly associated with either oral health or dysbiosis observed in cancer patients.
Key pathogenic taxa such as Fusobacterium nucleatum, Prevotella intermedia, Porphyromonas gingivalis, and Treponema denticola have been previously implicated in chronic inflammation and tumorigenesis, while commensal species associated with healthy oral flora are also indicated.
This visual framework serves to emphasize the potential of specific microbial signatures in distinguishing between cancerous and non-cancerous oral environments (Fig. 1B), forming the rationale for further metagenomic and functional analyses in this study.
Oral cancer is associated with altered salivary microbiome composition and increased microbial diversity
| Participant ID | Group | Age (years) | Sex | Smoking status | Alcohol consumption |
| H1 | Healthy | 42 | Female | Non-smoker | No regular alcohol use |
| H2 | Healthy | 39 | Male | Non-smoker | No regular alcohol use |
| H3 | Healthy | 47 | Female | Non-smoker | No regular alcohol use |
| H4 | Healthy | 51 | Male | Non-smoker | No regular alcohol use |
| H5 | Healthy | 44 | Female | Non-smoker | No regular alcohol use |
| O1 | OSCC | 55 | Male | Non-smoker | No regular alcohol use |
| O2 | OSCC | 52 | Female | Non-smoker | No regular alcohol use |
| O3 | OSCC | 58 | Male | Non-smoker | No regular alcohol use |
| O4 | OSCC | 49 | Male | Non-smoker | No regular alcohol use |
| O5 | OSCC | 61 | Female | Non-smoker | No regular alcohol use |

(A) Top 10 most abundant bacterial taxa in saliva from 5 healthy controls and 5 OSCC patients. Cancer-enriched species (, P. intermedia, P. gingivalis) and health-associated taxa are shown.
(B) Shannon index showing higher alpha diversity in oral cancer samples (**p < 0.01).
(C) PCoA plot based on Bray–Curtis dissimilarity, indicating distinct microbial community structures between the two groups
To compare the salivary microbiome composition between 5 healthy individuals and 5 oral cancer patients (Fig. 1B), 16S rRNA gene sequencing targeting the V3–V4 regions was performed on saliva samples from five healthy controls and five oral squamous cell carcinoma (OSCC) patients. The top 10 most abundant bacterial taxa at the species level are shown (Fig. 2A). Notably, Fusobacterium nucleatum, Prevotella intermedia, and Porphyromonas gingivalis were markedly enriched in the cancer group, while Streptococcus mitis, Actinomyces odontolyticus, and Rothia mucilaginosa were more prevalent in the healthy group. These taxonomic shifts suggest a dysbiotic microbial signature associated with oral cancer.
Microbial alpha diversity was assessed using the Shannon index (Fig. 2B). Interestingly, oral cancer samples exhibited significantly higher diversity compared to healthy controls (p < 0.01), indicating an expansion of microbial richness and evenness in the cancer-associated oral environment. This increase in diversity may reflect the establishment of a more complex and potentially pathogenic microbial community.
Principal Coordinates Analysis (PCoA) based on Bray–Curtis dissimilarity (Fig. 2C) further revealed distinct clustering of samples according to disease status. Samples from oral cancer patients clustered separately from those of healthy individuals, demonstrating a clear separation in microbial beta diversity between the two groups. These results collectively suggest that oral cancer is associated with specific taxonomic alterations, increased microbial diversity, and an overall shift in community structure that may contribute to the tumor microenvironment.
Differentially abundant bacteria in oral cancer and molecular validation of Fusobacterium nucleatum
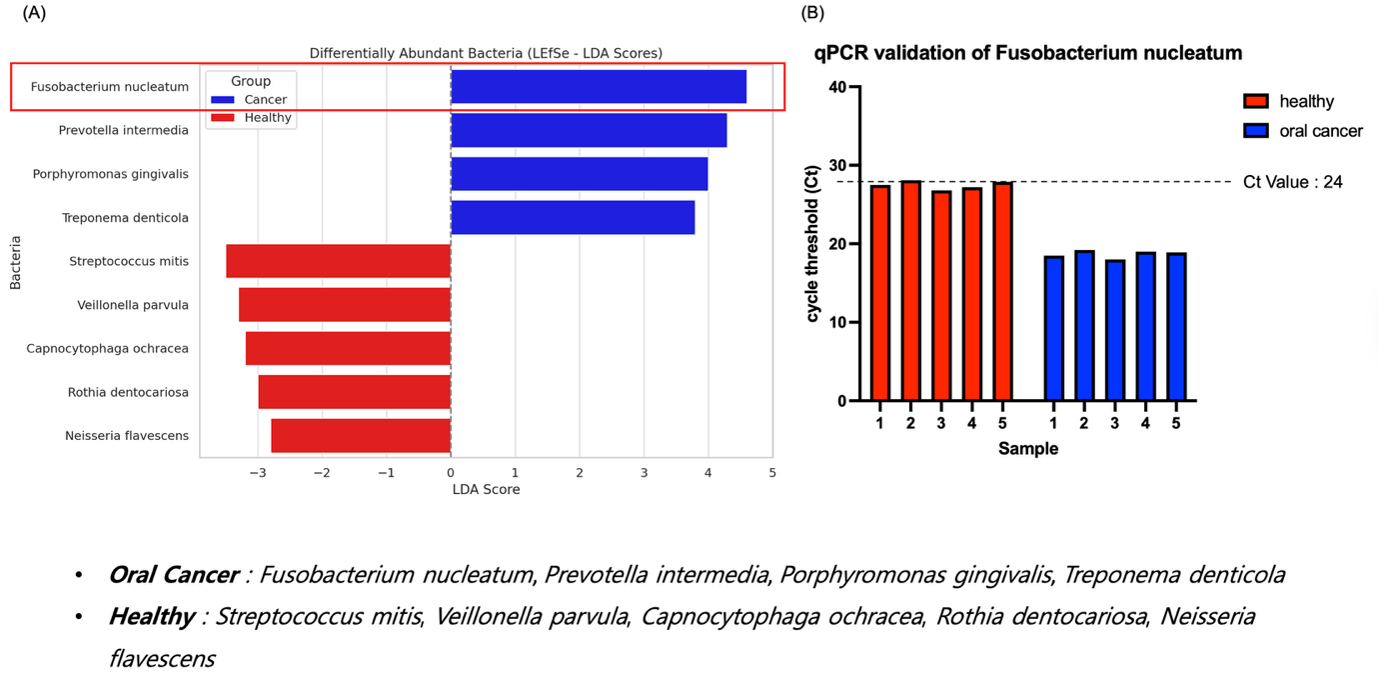
(A) LEfSe analysis showing differentially abundant taxa between OSCC patients and healthy controls. Fusobacterium nucleatum, Prevotella intermedia, Porphyromonas gingivalis, and Treponema denticola were enriched in cancer, while S. mitis and others were more abundant in healthy individuals.
(B) qPCR validation of shows lower Ct values in all OSCC samples (Ct < 24), confirming higher abundance.
These results ssupport asa potential salivary biomarker for oral cancer.
To identify specific bacterial taxa differentially enriched between oral cancer patients and healthy individuals, LEfSe (Linear Discriminant Analysis Effect Size) analysis was performed on the 16S rRNA sequencing data. An exploratory LEfSe analysis suggested potential differences in several taxa. Notably, Fusobacterium nucleatum, Prevotella intermedia, Porphyromonas gingivalis, and Treponema denticola exhibited markedly higher abundance in the oral cancer group, while Streptococcus mitis, Veillonella parvula, Capnocytophaga ochracea, Rothia dentocariosa, and Neisseria flavescens were more prevalent in healthy individuals (Fig. 3A). Among these, F. nucleatum displayed the highest LDA score in the cancer group, suggesting its potential as a discriminatory biomarker.
To validate the sequencing results at the molecular level, we performed quantitative PCR (qPCR) analysis targeting using species-specific primers. Oral cancer patients’ samples exhibited significantly lower Ct values compared to healthy controls, indicating a higher abundance of DNA. All five cancer samples fell below the diagnostic threshold of Ct = 24, while none of the healthy samples met this criterion, supporting the discriminatory power of this taxon.
Together, these results confirm as a candidate diagnostic biomarker for oral cancer and demonstrate the consistency between sequencing-based and qPCR-based microbiome profiling.
Inflammatory activation of oral keratinocytes by Fusobacterium nucleatum

(A) Schematic of co-culture system using human oral keratinocytes (HOKs) and
(B) qRT-PCR analysis shows significant upregulation of IL-6, IL-8, and TNF-α mRNA after 24 h co-culture with Fn (2.5–4.5 fold).
(C) Western blot confirms increased IL-6 and IL-8 protein levels in F. nucleatum-treated HOKs. These findings suggest that it promotes inflammation in the oral epithelial microenvironment, potentially contributing to tumor development.
(D) LDH release assay according to FN MOI
To investigate the functional impact of on host epithelial immune responses, a co-culture system was established using human oral keratinocytes (HOKs) and live (Fig. 4A). Following 24-hour co-incubation, total RNA and protein were extracted for gene expression and immunoblot analyses, respectively.
Quantitative real-time PCR revealed that co-culture with significantly upregulated the mRNA expression of key pro-inflammatory cytokines including IL-6, IL-8, and TNF-α compared to HOK monoculture controls (Fig. 4B). The fold changes ranged from approximately 2.5-fold to 4.5-fold, indicating a robust transcriptional inflammatory response.
These findings were further supported by Western blot analysis, which showed a marked increase in IL-6 and IL-8 protein levels in HOKs exposed to (Fig. 4C), confirming that the transcriptional upregulation translated into increased protein production. LDH release remained below 20% even at MOI 100 (Fig. 4D), indicating that F. nucleatum did not induce substantial cytotoxicity under our co-culture conditions. Thus, the observed increase in pro-inflammatory cytokines likely reflects an active host response rather than nonspecific cell lysis.
Together, these results demonstrate that induces a pro-inflammatory phenotype in oral epithelial cells, suggesting a potential mechanistic link between microbiome dysbiosis and chronic inflammation in the tumor microenvironment of oral cancer.
Rule-based Ct model predicts oral cancer using four key bacteria
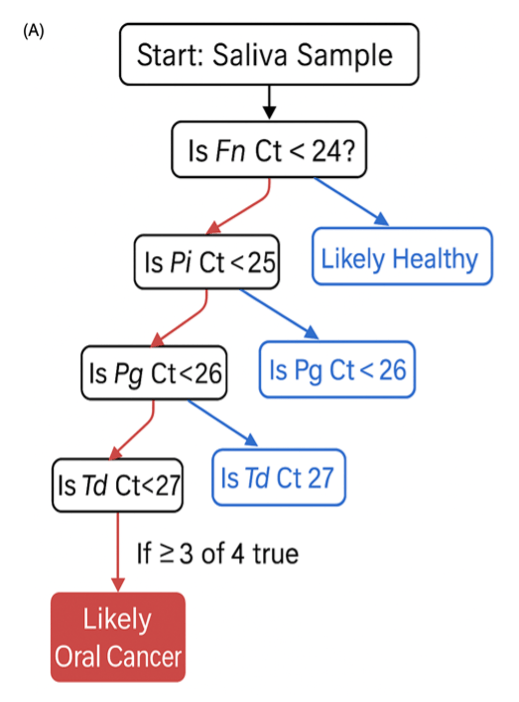
(A) Diagnostic decision tree based on Ct values for four cancer-enriched taxa (, P. intermedia, P. gingivalis, T. denticola).
To assess the potential of microbiome-derived signals as a diagnostic tool for oral cancer, we developed a decision tree model based on species-specific qPCR cycle threshold (Ct) values (Fig. 5A). The model incorporates four bacterial taxa— F. nucleatum, P. intermedia, P. gingivalis, and T.denticola —that were previously identified as significantly enriched in the oral cancer group.
Using optimized diagnostic thresholds derived from ROC analysis (FN < 24, Pi < 25, Pg < 26, Td < 27), the algorithm stratifies individuals based on the number of positive hits. Samples satisfying at least three of the four criteria were classified as “Likely Oral Cancer,” while those with fewer than three were categorized as “Likely Healthy.”
This rule-based classifier offers a simple yet effective framework for early detection of oral cancer using non-invasive saliva samples and cost-effective molecular assays. The model highlights the potential clinical utility of microbiome biomarkers for risk stratification and screening in at-risk populations.
Discussion
This study provides a multi-layered analysis of the oral microbiome in oral squamous cell carcinoma (OSCC), integrating taxonomic, functional, and diagnostic perspectives using both sequencing and molecular validation approaches. Through 16S rRNA sequencing of saliva samples, we identified distinct microbial shifts in OSCC patients compared to healthy controls, characterized by increased microbial diversity and enrichment of specific pathogenic taxa such as Fusobacterium nucleatum, Prevotella intermedia, Porphyromonas gingivalis, and Treponema denticola. These findings align with previous studies suggesting a link between microbial dysbiosis and tumor-associated inflammation in the oral cavity.
In contrast to many cancers where microbial diversity tends to decrease, our data revealed a significant increase in Shannon diversity among OSCC samples. Oral cancer samples showed a trend toward increased microbial diversity compared to healthy controls, although this preliminary observation is based on a small sample size (n = 5 per group) and should be interpreted with caution. This may reflect the breakdown of mucosal barriers and the infiltration of opportunistic pathogens in a tumor-altered microenvironment. While this increase may reflect a loss of microbial control, it could also be due to other factors not measured in our study, such as diet or oral hygiene.
The elevated abundance of and other anaerobes is particularly notable, as these species have been implicated not only in oral carcinogenesis but also in colorectal and esophageal cancers, suggesting a broader oncogenic potential mediated through inflammation, immune evasion, and microbial virulence factors18‘19. While our in vitro data suggests a potential mechanism by which could contribute to a pro-tumor environment, further studies are needed to establish a causal role in vivo.
The presence of F. nucleatum detected by sequencing was confirmed using an orthogonal method (qPCR) in the same set of samples. The initial agreement between sequencing results and qPCR validation for F. nucleatum further supports its utility as a microbial biomarker. The lower Ct values observed exclusively in OSCC samples highlight the diagnostic potential of this species, particularly when used in combination with other taxa. Building upon this, we proposed a rule-based decision model that integrates Ct thresholds from four taxa to stratify cancer risk. However, the diagnostic model was generated and tested on the same cohort and is therefore hypothesis-generating only. Independent cohort validation is required before assessing clinical utility.
Functionally, our co-culture experiments with human oral keratinocytes demonstrated that F. nucleatum directly induces pro-inflammatory cytokine expression, including IL-6, IL-8, and TNF-α. This suggests a mechanistic role for this bacterium in remodeling the immune landscape of the oral epithelium. The observed protein-level upregulation of IL-6 and IL-8 further supports the hypothesis that F. nucleatum contributes to a tumor-permissive, inflamed microenvironment that may enhance tumor initiation or progression.
However, we have limitations. Due to the very small cohort (n=10), we were unable to control for potential confounders such as age, sex, smoking habits, and oral hygiene. In the future, formal power calculations indicate that substantially larger cohorts will be required to achieve adequate statistical power for biomarker validation. We therefore plan to conduct validation in an expanded, independent cohort of ≥30 participants to confirm the diagnostic performance of the identified microbial markers and the qPCR-based classifier.
In addition, saliva, while being an easily accessible and non-invasive biospecimen, may not fully reflect localized microbial interactions at the tumor site or in the adjacent mucosa20. Future research incorporating multi-site sampling may better capture the spatial dynamics of the tumor–microbiome interface. Furthermore, 16S rRNA sequencing, while useful for identifying dominant taxa, lacks the resolution to assess strain-level differences and microbial function. Metagenomic or meta-transcriptomic analyses could overcome these limitations and reveal novel functional pathways.
In conclusion, this study suggests a potential salivary microbiome signature associated with oral cancer, highlights the inflammatory potential of cancer-enriched bacteria, and proposes a novel qPCR-based decision model for non-invasive detection. These findings offer new opportunities for microbiome-based diagnostics and therapeutic targeting in oral oncology.
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71, 209-249,2021. [↩]
- Louredo BV, Vargas PA, Pérez-de-Oliveira ME, Lopes MA, Kowalski LP, Curado MP. Epidemiology and survival outcomes of lip, oral cavity, and oropharyngeal squamous cell carcinoma in a southeast Brazilian population. Med Oral Patol Oral Cir Bucal 27, e274-e284, 2022. [↩]
- Shi L, Yang Y, Li M, Li C, Zhou Z, Tang G, Wu L, Yao Y, Shen X, Hou Z, Jia H. LncRNA IFITM4P promotes immune escape by up-regulating PD-L1 via dual mechanism in oral carcinogenesis. Mol Ther 30, 1564-1577,2022. [↩]
- Petti, S. Lifestyle risk factors for oral cancer. Oral Oncology 45, 340-350, 2009. [↩]
- Bettendorf, O., Piffkò, J. & Bànkfalvi, A. Prognostic and predictive factors in oral squamous cell cancer: important tools for planning individual therapy? Oral Oncol 40, 110-119, 2004. [↩]
- Perera, M., Al-hebshi, N.N., Speicher, D.J., Perera, I. & Johnson, N.W. Emerging role of bacteria in oral carcinogenesis: a review with special reference to perio-pathogenic bacteria. Journal of Oral Microbiology 8, 32762, 2016. [↩]
- Kitamoto, S., Nagao-Kitamoto, H., Hein, R., Schmidt, T.M. & Kamada, N. The Bacterial Connection between the Oral Cavity and the Gut Diseases. Journal of Dental Research 99, 1021-1029, 2020. [↩]
- Stasiewicz, M. & Karpiński, T.M. The oral microbiota and its role in carcinogenesis. Seminars in Cancer Biology 86, 633-642, 2022. [↩]
- Kuramitsu, H.K., He, X., Lux, R., Anderson, M.H. & Shi, W. Interspecies Interactions within Oral Microbial Communities. Microbiology and Molecular Biology Reviews 71, 653-670, 2007. [↩]
- Marsh, P.D. & Zaura, E. Dental biofilm: ecological interactions in health and disease. Journal of Clinical Periodontology 44, S12-S22, 2017. [↩]
- Asoudeh-Fard, A., et al. Lactobacillus plantarum induces apoptosis in oral cancer KB cells through upregulation of PTEN and downregulation of MAPK signalling pathways. Bioimpacts 7, 193-198, 2017). [↩]
- Asoudeh-Fard, A., et al. Lactobacillus plantarum induces apoptosis in oral cancer KB cells through upregulation of PTEN and downregulation of MAPK signalling pathways. Bioimpacts 7, 193-198, 2017. [↩]
- Granato, D.C., et al. Meta-omics analysis indicates the saliva microbiome and its proteins associated with the prognosis of oral cancer patients. Biochimica et Biophysica Acta (BBA) – Proteins and Proteomics 1869, 140659, 2021. [↩]
- Praveen, Z., et al. Oral Microbiome and CPT1A Function in Fatty Acid Metabolism in Oral Cancer. International Journal of Molecular Sciences 25, 10890, 2024. [↩]
- Chang, C., et al. Porphyromonas gingivalis Infection Promoted the Proliferation of Oral Squamous Cell Carcinoma Cells through the miR-21/PDCD4/AP-1 Negative Signaling Pathway. ACS Infectious Diseases 5, 1336-1347, 2019. [↩]
- Kumar, P., Gupta, S. & Das, B.C. Saliva as a potential non-invasive liquid biopsy for early and easy diagnosis/prognosis of head and neck cancer. Transl Oncol 40, 101827, 2024. [↩]
- Hao, Y., et al. The human oral – nasopharynx microbiome as a risk screening tool for nasopharyngeal carcinoma. Frontiers in Cellular and Infection Microbiology 12, 2022. [↩]
- McIlvanna, E., Linden, G.J., Craig, S.G., Lundy, F.T. & James, J.A. Fusobacterium nucleatum and oral cancer: a critical review. BMC Cancer 21, 1212, 2021. [↩]
- Harrandah, A.M., Chukkapalli, S.S., Bhattacharyya, I., Progulske-Fox, A. & Chan, E.K.L. Fusobacteria modulate oral carcinogenesis and promote cancer progression. J Oral Microbiol 13, 1849493, 2020. [↩]
- Gopinath, D., et al. Differences in the bacteriome of swab, saliva, and tissue biopsies in oral cancer. Scientific Reports 11, 1181, 2021. [↩]



{kind=link}