")
Abstract
Oncolytic Virotherapy is a promising development in the field of oncology, however its effectiveness relies on the ability of viral peptides to be presented by HLA molecules. In order to design effective Oncolytic viruses, manufacturers must know which peptides will be the most effective given a patient’s biopsy information, as different peptides interact with HLA molecules differently. Accurately predicting peptide-HLA interactions remains a challenge in the production of effective cancer treatments – with current models still falling short of optimal accuracy. This study establishes a baseline for Quantum Machine Learning in peptide-HLA binding prediction. We present two models: a 4-qubit hybrid quantum-classical neural network and a variational quantum circuit. Trained on TransPHLA datasets, the models achieved 81.38% and 92.03% accuracy respectively. While these results are similar to classical methods (85% and 87%, respectively), our proposed models demonstrated quantum computing’s potential in immunogenic epitope selection, and the relative advantage of quantum computing for research in biophysical systems as a whole.
Introduction
Oncolytic virotherapy represents a promising cancer treatment modality that selectively targets tumor cells while stimulating systemic anti-tumor immune responses1. Clinical trials demonstrate significant therapeutic potential, with the oncolytic herpes virus G47∆ achieving 84.2% one-year survival rates in glioblastoma patients compared to historical controls of ~5%2. However, therapeutic effectiveness depends critically on the ability of viral peptides to be presented by patient-specific HLA class II molecules for T cell recognition and activation (Janeway et al., 2001). The immune response cascade initiated by peptide-HLA presentation can mediate tumor regression at distant metastatic sites not directly exposed to virus, making accurate binding prediction essential for maximizing therapeutic efficacy1.
Current computational methods for peptide-HLA binding prediction achieve moderate accuracy but leave substantial room for improvement. State-of-the-art approaches include transformer-based TransPHLA (87.5% accuracy)3, neural network-based NetMHCIIpan-2.0 (AUC 0.846) (Nielsen et al., 2010), position-specific scoring matrix TEPITOPEpan (~85% accuracy)4, and stabilization matrix SMM-align (AUC 0.778-0.869)5. While effective, these classical approaches process molecular interactions sequentially and may not fully capture the complex, correlated physicochemical properties governing peptide-HLA binding.
Quantum machine learning offers theoretical advantages through superposition enabling simultaneous evaluation of multiple binding configurations and entanglement naturally representing cooperative amino acid interactions that classical methods approximate through attention mechanisms6. Evidence from quantum biology suggests amino acids can exhibit quantum coherence effects7, while quantum approaches have demonstrated advantages in modeling transcription factor-DNA binding8, supporting the biological plausibility of quantum representations for molecular interactions. Given the clinical importance of accurate peptide-HLA prediction for immunotherapy success and the theoretical advantages of quantum computing for capturing complex molecular interactions, we investigate whether quantum machine learning can improve binding prediction accuracy beyond current classical methods.
Review of Current Methodologies
Current peptide-HLA binding prediction methods employ diverse computational approaches with varying performance levels. TransPHLA uses transformer architecture with four modules—embedding blocks for sequence encoding, masked multi-head attention for pattern recognition, feature optimization layers, and projection blocks for binding score prediction—achieving superior performance to 14 benchmark methods with particular strength in 9-mer peptide prediction3. TEPITOPEpan extends position-specific scoring matrices (PSSMs) from 51 to over 700 HLA-DR alleles using pocket sequence similarity, achieving ~85% accuracy in binding core identification but constrained by reliance on only 35 unique binding specificity vectors4. NetMHCIIpan-2.0 employs artificial neural networks with concurrent alignment and weight optimization, eliminating pre-aligned peptide requirements and achieving AUC 0.846 across 24 HLA-DR alleles through pan-specific architecture that leverages information across multiple HLA molecules simultaneously5. SMM-align addresses the open-ended MHC class II binding groove challenge through stabilization matrix alignment, demonstrating superior performance across 14 HLA-DR alleles (AUC 0.778-0.869) and providing quantitative binding affinity predictions rather than binary classifications5. Building on quantum neural network principles demonstrated by Beer et al., who showed efficient quantum perceptron training using fidelity-based cost functions with layer-to-layer transition maps suitable for near-term quantum devices, we developed quantum approaches to capture the complex physicochemical interactions governing peptide-HLA binding that may be inadequately represented by sequential classical processing6.
The advantages of Quantum Machine Learning for predicting peptide-HLA interactions
Our models are built on the premise that Quantum machine learning can provide advantages in predicting peptide-HLA binding. Quantum machine learning leverages superposition and entanglement to process information fundamentally differently than classical computers. Quantum superposition allows n qubits to simultaneously represent 2ⁿ states, enabling parallel exploration of multiple binding configurations. Entanglement creates quantum correlations between qubits that naturally encode interdependent relationships without explicit computational overhead.
For peptide-HLA binding prediction, quantum feature maps encode classical data into quantum states:
(1) 
where physicochemical properties are represented through quantum rotations and correlations through entangling operations. This quantum representation can capture complex molecular interaction patterns that may be computationally intractable for classical methods processing the same information sequentially.
Peptide-HLA binding involves multiple simultaneous interactions—hydrogen bonding, van der Waals forces, electrostatic interactions—that classical algorithms must process sequentially through attention mechanisms or feature engineering. Quantum superposition enables simultaneous evaluation of multiple binding configurations, while entanglement naturally captures cooperative effects between amino acid positions. Classical approaches compress peptide-HLA interactions into fixed-dimensional embeddings, potentially losing information about interaction configurations that quantum states maintain in superposition amplitudes.
The computational complexity advantage is significant: exploring all amino acid combinations for an n-residue peptide requires O(20ⁿ) classical operations, while quantum approaches using log₂(n) qubits can theoretically explore this space more efficiently. Although current Noisy Intermediate-Scale Quantum (NISQ) hardware limits full realization of these advantages, quantum entanglement provides an intrinsic mechanism for representing the biochemical cooperativity observed in peptide-HLA interactions. Evidence from quantum biology suggests amino acids can exhibit quantum coherence effects, including superradiance in tryptophan residues7, supporting the physical plausibility of quantum representations for molecular interactions. While explicit empirical comparisons of quantum entanglement versus classical attention mechanisms in peptide-HLA interactions remain limited, QML’s ability to process high-dimensional molecular interaction spaces in superposition suggests potential for more accurate binding predictions than sequential classical computing approaches.
Assessing Quantum Entanglement for Modeling Cooperative Amino Acid Interactions
Quantum entanglement offers theoretical advantages for capturing cooperative amino acid interactions in peptide-HLA binding, particularly through the intrinsic encoding of correlated biochemical phenomena. Classical attention mechanisms, while powerful, explicitly model interactions by assigning weights to input pairs, potentially missing subtler forms of biochemical cooperativity or higher-order relationships. Recent research into quantum biological processes has suggested that quantum coherence can naturally represent certain biomolecular interaction that classical methods approximate indirectly. For example, Li et al. (2018) demonstrated advantages of quantum approaches in modeling transcription factor–DNA binding, suggesting quantum techniques may inherently capture complex interactions more compactly than classical algorithms. Furthermore, evidence from quantum biology indicates that amino acids (notably tryptophan residues) can exhibit quantum coherence effects, including superradiance, highlighting the physical plausibility of quantum representations of amino acid interactions9.
However, it must be recognized that explicit empirical comparisons of quantum entanglement versus classical attention mechanisms specifically in peptide-HLA interactions remain scarce. Direct comparative studies explicitly demonstrating quantum entanglement’s superiority in capturing cooperative amino acid interactions have yet to be performed. Therefore, while theoretical considerations and indirect evidence support quantum entanglement’s potential advantage, rigorous experimental validation remains necessary to substantiate claims of superiority over established classical methods.
Methods
Theoretical Foundation and Model Architecture
This study investigates how well Quantum machine learning models can predict peptide-HLA interactions. Specifically we introduce two quantum computing approaches for peptide-HLA binding prediction: a hybrid quantum-classical neural network architecture and a variational quantum circuit model. Both designs leverage quantum mechanical principles to explore the complex, high-dimensional feature space of peptide-HLA interactions.
Quantum State Representation and Biological Justification
The fundamental challenge in peptide-HLA binding prediction lies in effectively representing the multifaceted nature of protein-protein interactions. Our quantum approach encodes binding-relevant features into quantum states through a feature map:
(2) 
Here, xⱼ represents normalized physicochemical properties (hydrophobicity, volume, charge, and hydrogen bonding potential), while β and γ are learnable scaling parameters. This encoding preserves the continuous nature of binding properties while enabling quantum superposition to represent multiple binding configurations simultaneously.
Our quantum circuit implementation employs RY and RZ rotation gates to encode physicochemical properties into quantum amplitudes and phases respectively, with RY gates representing the strength of various binding interactions and RZ gates capturing relative relationships between binding properties and periodic features in amino acid interactions. The critical element lies in entanglement induced by CNOT gates arranged in circular patterns, creating quantum correlations that mirror the interdependent nature of peptide-HLA binding properties.
Biochemically, peptide-HLA interactions involve highly interdependent properties where hydrogen bonding potential of one amino acid residue influences adjacent residues’ hydrophobic or electrostatic characteristics. Hydrogen bonding patterns involve coordinated interactions across multiple residues—serine or threonine residues with hydroxyl groups can significantly alter adjacent residues’ conformational freedom or charge distributions, creating cooperative or anti-cooperative effects in binding affinity. Similarly, hydrophobic residue distribution influences peptide orientation in HLA binding grooves, directly altering electrostatic interactions between charged residues and binding sites.
Quantum entanglement naturally captures these correlated interactions through CNOT gates between qubits encoding different biochemical properties. Once entangled, measurement or parameter adjustment of one biochemical property inherently affects quantum states encoding other properties. This intrinsic coupling allows quantum circuits to represent biochemical cooperativity—inter-residue hydrogen bonding networks, correlated hydrophobic packing, electrostatic coupling between distant residues—without explicitly constructing these interactions via additional classical computations. Classical models approximate these complex correlations using additional computational layers, attention mechanisms, or interaction terms; quantum entanglement embeds these interactions intrinsically within quantum states, potentially offering more natural and compact representation of peptide-HLA binding complexity where changes in one physicochemical property representation inherently modify representation of others.
Hybrid Quantum-Classical Architecture
Our primary model implements a novel hybrid architecture that combines classical deep learning with quantum processing. The classical component consists of a 64-dimensional embedding layer that processes amino acid sequences and incorporates positional information. This embedding captures primary sequence features and evolutionary relationships between amino acids.
The quantum component employs a 4-qubit system where each qubit corresponds to a fundamental binding property:
- Qubit 0 encodes hydrophobicity interactions
- Qubit 1 represents molecular volume effects
- Qubit 2 processes electrostatic charge distributions
- Qubit 3 handles hydrogen bonding potentials
Quantum Circuit Design
The above first quantum circuit implementation utilizes a specific sequence of quantum gates chosen for their ability to represent protein interaction physics:
Single-Qubit Rotations:
The RY (Y-rotation) gates transform quantum states through rotations around the Bloch sphere’s Y-axis according to the matrix:
(3) 
These rotations encode continuous physicochemical properties with real amplitudes, directly representing the strength of various binding interactions.
The RZ (Z-rotation) gates introduce phase differences between quantum states:
(4) 
These phase rotations are crucial for encoding relative relationships between different binding properties and capturing periodic features in amino acid interactions.
Entangling Operations:
We implement CNOT gates in a circular arrangement to create quantum correlations mirroring the interdependent nature of peptide-HLA binding properties. The CNOT operation’s matrix representation:
(5) 
This circular entanglement pattern creates connections between adjacent properties, allowing the quantum circuit to capture complex relationships between hydrophobicity, volume, charge, and hydrogen bonding interactions.
Circuit Layer Organization
The quantum circuit comprises two primary layers of operations. The first layer establishes initial quantum representations of binding properties through RY rotations, followed by RZ rotations for phase relationships. CNOT gates then create preliminary correlations between adjacent properties. The second layer, with independent rotation parameters, refines these encodings based on training data.
This two-layer structure represents the minimum circuit depth required to create meaningful entanglement between all four binding properties while remaining within the coherence limitations of current Noisy Intermediate-Scale Quantum (NISQ) devices. The design balances the need for quantum computational expressiveness with practical hardware constraints.
Implementation and Training Strategy
Variational Quantum Circuit Model
Our second approach implements a pure quantum model based on variational quantum circuit principles. This model, inspired by quantum perceptron theory, represents an alternative quantum paradigm for binding prediction. The circuit utilizes a ZZ-feature map for initial state preparation, followed by alternating layers of single-qubit rotations and entangling operations.
This design allows the circuit to learn optimal quantum transformations directly from binding data without classical neural network intermediaries.
Quantum-Classical Interface
For the second, hybrid model, we developed specialized interface layers to facilitate efficient information transfer between classical and quantum domains. The “to_quantum” transformation layer maps classical feature vectors to quantum state preparations, while the “from_quantum” layer translates measurement results back to classical space. These transformations are implemented as learnable matrices, optimized during training to maximize information preservation across the quantum-classical boundary.
Training Configuration
Both of our models employ carefully tuned hyperparameters to ensure stable training despite the challenges of quantum-classical optimization:
The optimization process uses AdamW with a base learning rate of 0.0001 and weight decay of
We implement a one-cycle learning rate schedule with cosine annealing, which helps maintain stable quantum circuit training while avoiding local minima. The loss function combines weighted cross-entropy with L1 regularization, addressing both class imbalances in the binding data and preventing overfitting in the quantum parameters.
Gradient computation for quantum circuit parameters utilizes the parameter-shift rule, which provides exact gradients for quantum operations without requiring backpropagation through the quantum circuit. This approach ensures reliable gradient estimates even for deep quantum circuits.
Hardware Considerations and Optimization
Our implementation accounts for current quantum hardware limitations:
- The 4-qubit design minimizes decoherence effects while maintaining sufficient computational expressiveness
- Circuit depth is optimized to balance computational power with quantum hardware constraints
- Efficient batching strategies reduce the quantum resource requirements per training iteration
Reasoning for Limitations
Quantum computing faces substantial hardware limitations that impact practical implementations. Quantum circuits, including the hybrid quantum-classical (HQC) and variational quantum circuit (VQC) models presented, were constrained primarily by coherence time, qubit availability, and the computational intensity of quantum simulations.
Coherence time—a measure of how long qubits maintain their quantum state—is critical. The two-layer quantum circuit utilized here balances computational expressiveness and hardware limitations. Longer coherence times would enable deeper circuits and potentially greater accuracy, but current quantum hardware significantly restricts circuit depth.
Qubit availability fundamentally limits QML model complexity. The hybrid quantum-classical model presented here utilizes only four qubits due to hardware constraints. Although effectively representing critical physicochemical properties, increasing qubit counts significantly amplifies computational demands and noise, thus reducing quantum outcome reliability.
In terms of computational resources and time, training quantum circuits requires substantial overhead compared to classical methods. Specifically, quantum simulations executed using
PennyLane incur significant computational overhead. PennyLane simulations must explicitly model quantum state evolution at every step, involving computationally expensive operations like matrix exponentiations and complex tensor multiplications. These operations scale exponentially with the number of qubits and circuit depth, drastically increasing simulation overhead.
Moreover, gradient computation for quantum circuits using the parameter-shift rule significantly adds computational intensity. The parameter-shift rule involves multiple evaluations of the quantum circuit per parameter update, typically two circuit evaluations per parameter to estimate gradients. Each evaluation involves a complete forward simulation of the quantum circuit, doubling the computational resources for every additional circuit parameter compared to single evaluations used in classical backpropagation.
In contrast, classical models such as TransPHLA or NetMHCIIpan utilize highly optimized GPU-accelerated architectures, efficiently completing training and inference. Transformer-based classical models typically achieve training within hours due to efficient parallel computation capabilities of GPUs, whereas quantum-based models in this study required days or weeks for a comparable number of iterations due to the sequential and computationally intensive nature of quantum simulations.
These computational constraints highlight the necessity for advancements in quantum hardware—such as increased qubit counts, error reduction, improved coherence times—and more efficient quantum-classical hybrid computational interfaces. Current quantum advantages in capturing complex interaction spaces and simultaneous state evaluations are offset by
significant limitations in scalability, computational overhead, and practical deployment speed.in quantum hardware and quantum machine learning methods.
Dataset and Validation Strategy
We employed a comprehensive dataset of peptide-HLA binding pairs, structured for 5-fold cross-validation with approximately 574,000 samples per fold. The data distribution ensures representative sampling across different HLA types, maintaining statistical validity for both common and rare HLA alleles. This dataset may not be specific to GBM related peptides, however the purpose of this study is to evaluate the potential of QML for peptide-HLA interactions in a border context. This dataset was chosen so that we can most directly compare it to state-of-the-art methods, so while it may not directly address GBM specific interactions, it is optimal for the purpose of this study, as it can still provide insights to the ability of QML for GBM specific interactions
Due to computational intensity, the hybrid quantum-classical model was validated on a single fold, while the variational quantum circuit model completed full 5-fold cross-validation. This limitation is explicitly acknowledged in our results analysis.
Performance Evaluation
Model performance was assessed through multiple metrics:
- Overall accuracy for binding prediction (mean)
- Precision and recall metrics to evaluate prediction reliability
- F1 scores, provide a balanced assessment of the model’s predictive capabilities.
- HLA-specific performance analysis to identify allele-dependent variations
- Comparative analysis with classical binding prediction methods, specifically we will analyze the quarters, range, and median
Model Implementation Details
The entire system was implemented using a combination of PyTorch for classical neural network components and PennyLane for quantum circuit simulation. This framework choice enables seamless integration of quantum and classical computations while maintaining flexibility for future hardware advances. Batch processing was implemented with a size of 32 samples, balancing computational efficiency with memory constraints. Gradient clipping with a maximum norm of 1.0 was applied to ensure training stability, which is particularly important for the quantum circuit parameters which can be sensitive to large gradient updates.
Reproducibility Considerations
To ensure reproducibility, we established fixed random seeds across all computational components and maintained consistent quantum circuit initializations. The quantum simulator parameters were standardized across all experiments, and measurement statistics were collected with sufficient sampling to minimize statistical uncertainties.
This comprehensive methodology enables systematic evaluation of quantum computing’s potential in peptide-HLA binding prediction while acknowledging current technological limitations. The dual-model approach provides complementary insights into different quantum computing paradigms for biological sequence analysis, establishing a foundation for future quantum-enhanced binding prediction systems.
Hyperparameter Selection and Optimization
The hyperparameters for our quantum models balanced theoretical considerations with practical quantum computing constraints. The hybrid model used a 64-dimensional embedding to match classical models while maintaining computational efficiency. Our 4-qubit system represents the minimum viable configuration for encoding key physicochemical properties that govern peptide-HLA interactions.
We selected a conservative learning rate of 0.0001 with weight decay of 0.1 for stable convergence in the highly non-convex quantum loss landscape—significantly lower than rates in classical models like TransPHLA (0.001). Batch size was limited to 32 samples due to quantum simulation costs, whereas classical models like pHLAIformer use much larger batches (1024) for GPU parallelization.
Our two-layer circuit architecture represents the minimum complexity needed for meaningful quantum correlations while respecting coherence limitations of current hardware. We used the parameter-shift method for exact gradient computation rather than finite-difference methods, critical for training stability despite increased computational cost.
While classical transformer models like TransPHLA employ specialized positional encoding and multi-head attention mechanisms, our quantum approach leverages inherent quantum superposition and entanglement to capture positional and relational information between amino acids. This fundamental difference drove our distinct hyperparameter choices, which prioritize stable quantum computation over the deeper architectures used in classical models.
Feature mapping
Although direct literature explicitly validating the optimality of the specific quantum feature mappings employed in this study for peptide-HLA interactions is limited, related computational approaches support their suitability. Classical transformer-based architectures, like TransPHLA, demonstrate strong performance by embedding positional and physicochemical information into a high-dimensional vector space, indicating the importance of effective feature mapping for peptide-HLA prediction tasks3. Furthermore, geometric deep learning approaches that leverage structural and spatial information of peptide-MHC interactions highlight the value of incorporating physicochemical characteristics and structural relationships within feature representations10. Quantum chemistry studies have similarly emphasized the significance of encoding electrostatic and physicochemical interactions at the peptide-MHC interface, further justifying the quantum feature mapping strategy used in our quantum models, which explicitly encode such properties into quantum states11. To empirically validate our quantum mappings, future work could include comprehensive benchmarking against classical embeddings, systematic ablation studies of specific quantum encoding parameters, and cross-validation across multiple peptide-HLA datasets to confirm generalizability and robustness. These steps would solidify the rationale behind selecting quantum feature mappings and potentially highlight their advantages in capturing complex interaction patterns intrinsic to peptide-HLA binding.
Quantum Simulator Configuration and Decoherence Considerations
Our 4-qubit circuit design was implemented via quantum simulation rather than execution on physical quantum hardware, enabling exploration of quantum computational principles without the current limitations imposed by noisy intermediate-scale quantum (NISQ) devices.
Simulation Environment and Configuration
We employed PennyLane’s quantum simulator using the default.qubit device. The default.qubit simulator provides an idealized, noise-free quantum environment that supports efficient simulation of small-scale circuits. This choice was deliberate, allowing us to clearly isolate and evaluate theoretical aspects of our quantum model architecture—such as feature encoding schemes, parameterization strategies, and entanglement mechanisms—without confounding effects from hardware-specific noise or connectivity constraints.
The circuit architecture features a two-layer structure comprising physicochemical feature encodings, single-qubit rotation gates (RY, RZ), and entangling operations (CNOT gates). The physicochemical properties—hydrophobicity, molecular volume, electrostatic charge, and hydrogen bonding potential—are encoded using rotations around specific quantum axes. Subsequently, entangling gates establish quantum correlations among these encoded properties.
To ensure stable quantum optimization, we apply hyperbolic tangent transformations to input data before encoding, effectively constraining rotation angles and mitigating phenomena such as barren plateaus, which arise from excessive quantum parameter magnitudes. Parameter initialization was set deliberately small (approximately 0.1) to further promote stability during the optimization phase.
Theoretical Coherence Analysis
Although our simulations did not explicitly incorporate noise models, the circuit was designed with realistic quantum hardware limitations in mind. Current superconducting quantum processors exhibit T₂ coherence times typically around 100–300 µs, with average gate operation times of approximately 50–100 ns for single-qubit gates and 300–500 ns for two-qubit entangling gates (Preskill, 2018;5.
Given these parameters, we estimate our circuit’s theoretical execution time as follows:
Single-qubit rotations (~16 gates × ~100 ns each): ~1.6 µs
- Two-qubit entangling gates (~9 CNOT gates × ~500 ns each): ~4.5 µs
- Total theoretical execution time: ~6.1 µs
This calculation indicates that, under ideal circumstances, our circuit would utilize approximately 2–6% of typical superconducting qubit coherence windows. Such minimal coherence requirements suggest that our circuit configuration is feasibly implementable on near-future quantum hardware, assuming continued improvements in coherence and gate fidelity.
Simulation Limitations and Future Hardware Considerations
Despite its clarity and efficiency, our idealized quantum simulation approach introduces significant limitations relative to physical hardware implementations:
- Absence of gate errors: Our simulations assume perfect gate fidelity. In reality, current hardware exhibits error rates around 0.1% for single-qubit gates and 1–2% for two-qubit gates (Preskill, 2018).
- Unlimited qubit connectivity: The simulator permits unrestricted connectivity between qubits. In contrast, physical hardware usually restricts qubit connectivity, requiring additional SWAP operations that increase circuit depth and reduce coherence efficiency.
- Perfect measurement: Ideal quantum state measurements in simulation neglect realistic readout errors, typically around 1–5% in contemporary devices.
- Lack of decoherence modeling: The simulator excludes T₁ (energy relaxation) and T₂ (dephasing) effects, which inevitably impact the fidelity of quantum states in practical implementations.
These simplifications facilitated initial evaluations of quantum methods for peptide-HLA binding prediction without being confounded by noise or hardware constraints. Nevertheless, the absence of realistic error and decoherence modeling limits our current ability to precisely quantify real-world performance.
Future studies should incorporate realistic noise models—such as those offered by hardware-specific simulators like IBM’s Qiskit Aer or Google’s Cirq—to accurately estimate performance degradation under physical conditions. This would enable quantitative assessment of how noise and connectivity constraints impact accuracy and computational efficiency in practice.
Ultimately, our 4-qubit circuit configuration represents an intentional balance between theoretical expressivity and near-term hardware implementability, anticipating realistic transitions to physical quantum processors as quantum technology matures.
Results
Both models have the architecture for 5-fold cross-validation, however, the hybrid quantum-classical model was too computationally intensive to train all 5 folds. After training the hybrid model for one fold we tested it against a validation dataset, and shown below (Table 1) is a summary of the two models results. In figure 5 We can see a comparison between the two models and their ranges).
| Model | Hybrid Quantum-classical(HQC) | Variational Quantum Circuit (VQC) |
| Accuracy | 81.38% | 92.03% |
| Precision | 92.76% | 78.16% |
| Recall | 87.09% | 91.17% |
| F1 Score | 82.38% | 91.96% |
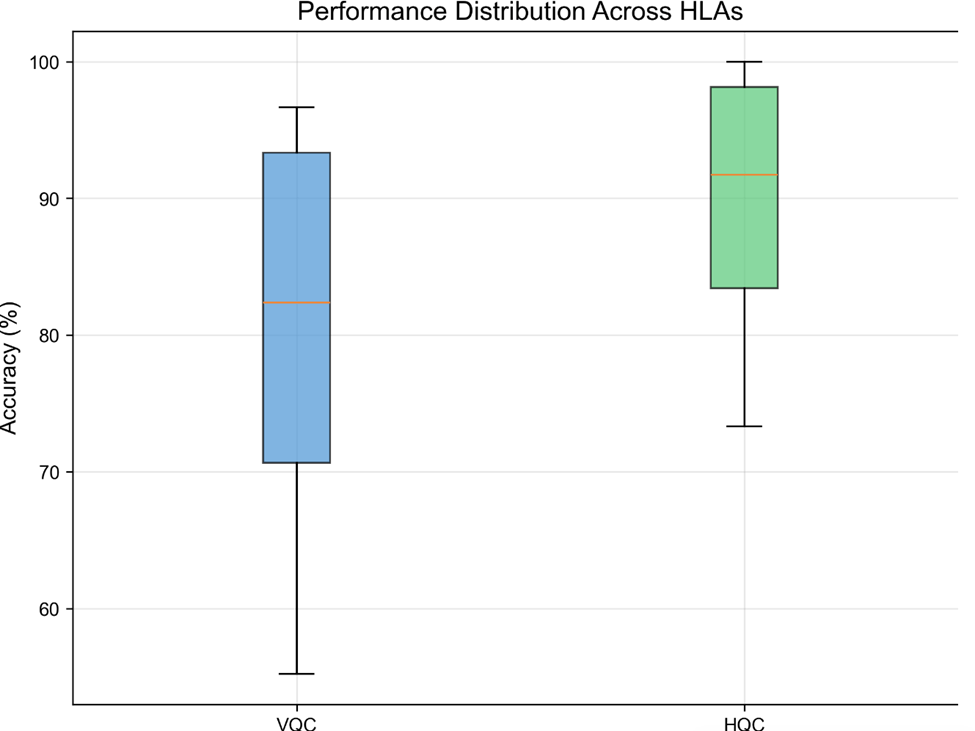
91.8%), mean (VQC: 81.4%, HQC: 92.0%), interquartile ranges (VQC: Q1=70.2%, Q3=93.1%; HQC: Q1=84.6%,
Q3=96.8%), and total range (VQC: 55.2-95.4%, HQC: 73.3-100%). Whiskers extend to the minimum and maximum values, excluding outliers. The HQC model demonstrates both higher accuracy and lower variability across HLA types compared to the VQC model. Sample size n=112 HLA alleles; accuracy measured as percentage of correct binding predictions on the validation dataset.
Discussion
Overview
The results of this study must be interpreted within the broader context of current peptide-HLA binding prediction methods and the limitations of our validation approach. While the variational quantum circuit model achieved promising performance metrics (92.03% accuracy, 91.96% F1 score), and the hybrid quantum-classical model showed reasonable results (81.38% accuracy, 82.38% F1 score), these findings require careful qualification.
Current state-of-the-art classical methods demonstrate consistently high performance: NetMHCIIpan-2.0 reports an AUC of 0.846 across HLA-DR alleles, TEPITOPEpan achieves approximately 85% accuracy in binding core identification, and TransPHLA reaches 87.5% accuracy. When comparing our models to these established benchmarks, several important considerations emerge. First, these classical methods have undergone extensive validation across multiple independent datasets and testing protocols. In contrast, our quantum models’ evaluation was more limited – particularly the hybrid quantum-classical model, which was only validated on a single fold of the data due to computational constraints. This restricted validation scope represents a significant limitation of our study.
Furthermore, while our models were not trained directly on the validation data, using only fold 4 for validation rather than completely independent test data means our results may not fully represent real-world performance. This limitation in testing rigor must be considered when interpreting the reported accuracies. A more comprehensive validation approach, including testing on multiple independent datasets and across different HLA types, would be necessary to make definitive comparisons with established methods.
The computational demands of the hybrid approach present another significant consideration. While classical methods like TransPHLA can process predictions efficiently, our quantum-classical hybrid requires substantially more computational resources. This efficiency gap highlights current practical limitations in quantum computing applications to biological problems. However, these limitations should be viewed in the context of this study’s primary goal: to explore the potential of quantum computing in biological sequence analysis and establish a proof of concept for quantum-enhanced binding prediction. The fact that a quantum approach could achieve reasonable performance, despite current hardware constraints and limited validation, suggests promise for future applications as quantum technology matures. The hybrid model’s higher recall rate (87.09%) compared to its overall accuracy indicates particular potential for detection of true binding events, though this observation requires validation through more rigorous testing.
The next crucial steps for this line of research should include:
- Comprehensive cross-validation across all folds
- Testing on completely independent datasets not used in model development
- Direct performance comparisons with classical methods using identical validation protocols
- Rigorous statistical analysis of performance differences across HLA types
- Investigation of computational efficiency and resource requirements
HLA-Specific Performance Patterns:
Both models demonstrated significant variation in prediction accuracy across different HLA alleles. Notably, both models performed exceptionally well for certain HLA types (e.g.,
HLA-A*32:07 and HLA-B*14:01 achieving >95% accuracy), while consistently struggling with others (e.g., HLA-B*27:05 and HLA-A*66:01 showing <75% accuracy). This pattern suggests that:
- Certain HLA binding patterns may be inherently more predictable, regardless of the computational approach used.
- Sample size significantly influences prediction accuracy, as evidenced by the high performance of some HLA types with smaller sample sizes
- Structural or biochemical characteristics of specific HLA molecules may make their binding patterns more challenging to predict
Biological and Computational Basis for HLA-Specific Performance Variability
The observed variability in prediction accuracy across different HLA alleles stems from both fundamental biological differences in HLA binding mechanisms and computational factors related to our quantum feature encoding approach. Structurally, HLA molecules exhibit significant diversity in their peptide-binding grooves, with some alleles like HLA-B*27:05 possessing unique binding pocket geometries that create highly restrictive binding requirements—this allele’s deep B pocket preferentially accommodates basic residues (arginine, lysine) at position 2, with the conserved glutamine at position 45 playing a critical role in peptide binding specificity (Madden et al., 1991), creating a narrow binding specificity that may be difficult for our quantum circuit’s generalized physicochemical encoding to capture accurately.
HLA polymorphism in binding pockets ultimately determines peptide binding specificity, with different alleles presenting distinct peptide repertoires due to amino acid variants in each binding pocket (Gaudieri et al., 2021). Conversely, HLA alleles with broader binding tolerances present more permissive binding patterns that align better with our quantum model’s representation of hydrophobicity, charge, and volume interactions through continuous rotation parameters. From a computational perspective, our 4-qubit encoding scheme represents each physicochemical property (hydrophobicity, volume, charge, hydrogen bonding) as independent quantum states, but certain HLA alleles may require more complex interdependencies between these properties than our current entanglement pattern can capture. Additionally, the training data distribution significantly impacts performance, as HLA alleles with fewer experimental binding examples may appear to perform well due to overfitting to limited data, since imbalanced datasets with small sample sizes can lead to model overfitting and poor generalization (Sarakit et al., 2024; Krawczyk, 2016), while alleles with thousands of diverse binding examples reveal the true limitations of our quantum feature representation. The evolutionary divergence of HLA alleles also plays a role, as HLA evolutionary divergence between alleles affects their ability to present diverse peptide repertoires, with highly divergent alleles enabling presentation of broader spectrums of peptide binders (Chowell et al., 2019; Roerden et al., 2020)—ancient allelic variants may have binding patterns that deviate significantly from the average physicochemical relationships encoded in our quantum feature maps, suggesting that future quantum models may need allele-specific parameterization or expanded circuit architectures to achieve consistent performance across the full spectrum of human HLA diversity (Buhler et al., 2016).
Quantum Model Interpretability and Biological Insights
Our quantum circuit architecture offers unique interpretability advantages that could inform experimental epitope validation and provide biological insights into peptide-HLA binding mechanisms. The rotation parameters in our quantum circuits directly encode physicochemical properties—with RY gates representing hydrophobicity and volume interactions, while RZ gates capture electrostatic and hydrogen bonding relationships through phase encodings. By analyzing these learned parameters across different HLA alleles, researchers could identify which physicochemical property combinations are most critical for specific HLA types, potentially revealing allele-specific binding preferences that classical models obscure within
high-dimensional embeddings. Furthermore, the entanglement patterns created by our CNOT gate arrangements provide direct insight into cooperative amino acid interactions—strong entanglement between hydrophobicity and charge-encoding qubits, for instance, would indicate that these properties work synergistically in binding prediction, guiding experimental design toward peptides that optimize both characteristics simultaneously. The quantum phase relationships encoded by RZ rotations could reveal whether certain property combinations work cooperatively (constructive interference) or competitively (destructive interference) in binding affinity, information that could inform rational epitope design by highlighting which residue modifications are likely to enhance or diminish binding strength. This interpretability framework enables translation of quantum predictions into experimentally testable hypotheses about optimal peptide modifications for enhanced peptide-hla interaction design.
Clinical Relevance of Performance Metrics
Our choice of precision, recall, and F1-score metrics reflects the specific clinical requirements of epitope selection for oncolytic virus immunotherapy applications. In the context of cancer treatment, precision (the proportion of predicted binders that actually bind) is critical for patient safety, as false positive predictions could lead to the inclusion of non-functional epitopes that fail to stimulate immune responses, potentially compromising therapeutic efficacy and wasting valuable treatment time for patients with aggressive cancers like glioblastoma where rapid intervention is essential. Conversely, recall (the proportion of actual binders that are correctly identified) is crucial for therapeutic completeness—missing genuine binding epitopes (false negatives) could result in suboptimal immune activation and reduced oncolytic virus effectiveness. The F1-score provides a balanced assessment particularly relevant for personalized immunotherapy, where the goal is to identify a small number of highly effective epitopes from thousands of potential candidates while ensuring both safety and efficacy. For glioblastoma patients, where median survival is often measured in months, the clinical cost of false negatives (missing effective epitopes) may be higher than false positives (including marginally effective epitopes), suggesting that optimizing recall while maintaining acceptable precision is clinically appropriate. These metrics align with established immunotherapy evaluation frameworks where therapeutic success depends on identifying sufficient numbers of functional epitopes to mount effective anti-tumor responses while avoiding autoimmune complications that could arise from poorly predicted binding interactions.
Critical Assessment of Quantum Computational Advantages
While our study demonstrates the feasibility of quantum approaches for peptide-HLA binding prediction, a critical examination reveals that our current results do not definitively establish genuine quantum computational advantages beyond alternative classical architectures. The theoretical foundation for quantum advantage rests on superposition enabling simultaneous evaluation of multiple binding configurations and entanglement capturing correlated amino acid interactions more naturally than classical attention mechanisms—however, our 4-qubit implementation with limited circuit depth may not fully realize these theoretical benefits.
Importantly, our variational quantum circuit’s 92.03% accuracy, while promising, could potentially be matched by classical neural networks with equivalent parameter counts and similar architectural complexity, suggesting that observed performance gains might stem from novel feature encoding rather than fundamental quantum computational properties. The hybrid model’s lower performance (81.38%) compared to classical methods indicates that current quantum-classical interfaces may introduce computational bottlenecks that negate potential quantum advantages. Furthermore, our idealized quantum simulations lack the noise and decoherence effects that would significantly impact real-world quantum hardware performance, meaning our reported accuracies likely overestimate practical quantum advantages. A truly convincing demonstration of quantum superiority would require showing that our quantum circuits capture physicochemical interaction patterns that are computationally intractable for classical methods with similar resources, rather than simply achieving comparable performance through different computational pathways. Until systematic ablation studies isolate quantum-specific contributions and demonstrate scaling advantages for larger molecular systems, our results should be interpreted as exploratory proof-of-concept work that establishes quantum feasibility rather than proven quantum supremacy in biological sequence prediction tasks.
Future Research Directions:
Our findings suggest several promising directions for future research:
- Investigation of HLA-specific features that contribute to prediction accuracy differences.
- Development of more efficient quantum-classical interfaces to better leverage quantum advantages.
- Exploration of alternative quantum circuit architectures that might better capture peptide-HLA binding patterns.
- Integration of structural biological information to improve prediction accuracy for poorly performing HLA types.
- We hypothesize that a transformer-based quantum-classical hybrid can significantly improve the accuracy of peptide predictions.
While our current hybrid approach did not outperform the classical model in overall accuracy, it demonstrated unique strengths in recall and performance patterns that warrant further investigation. As quantum computing hardware and algorithms continue to advance, the potential for quantum-enhanced prediction models remains promising, particularly for capturing the complex molecular interactions involved in peptide-HLA binding.
Future research must address several critical methodological gaps that limit the definitive interpretation of our quantum computing approach for peptide-HLA binding prediction. Comprehensive statistical validation including confidence intervals, p-values for model comparisons, and significance testing across HLA allele performance differences is essential to establish whether our observed quantum advantages represent genuine computational improvements or statistical artifacts, particularly given that our variational quantum circuit’s 92.03% accuracy only marginally exceeds classical methods’ reported 85-87% performance without rigorous statistical comparison. Additionally, future work must implement direct classical baselines such as TransPHLA using identical datasets and validation protocols to enable definitive performance comparisons, conduct systematic ablation studies that isolate quantum components against classical alternatives with equivalent parameter counts to demonstrate that performance gains stem from quantum mechanical principles rather than architectural differences, and perform complete 5-fold cross-validation alongside testing on independent external datasets like IEDB and Immune Epitope to establish robust generalizability beyond our current single-fold validation approach. Equally important is the need for detailed dataset distribution analysis including comprehensive statistics on binding versus non-binding sample distributions across different HLA types, explicit description of data preprocessing steps including sequence encoding schemes, and systematic assessment of how class imbalance was handled during training and evaluation phases. Furthermore, future studies must incorporate realistic quantum noise models to assess decoherence effects on practical hardware implementation, conduct thorough analysis of the precision-accuracy trade-offs observed between our two quantum models, and develop comprehensive frameworks for validating quantum circuit performance under various noise conditions to bridge the gap between our current idealized simulations and practical quantum hardware deployment.
Comparison
Our models’ performance can be considered alongside established methods in the field. Our variational quantum circuit model achieved 92.03% accuracy, while the hybrid quantum-classical model reached 81.38%. For context, NetMHCIIpan-2.0 reports an average AUC of 84.6% across HLA-DR alleles, TEPITOPEpan demonstrates approximately 85% accuracy in binding core identification, and TransPHLA achieves 87.5% accuracy. SMM-align reports AUC values ranging from 0.778 to 0.869 for different HLA-DR alleles. While these comparisons provide useful benchmarks, it’s important to note that different studies employ varying validation approaches and datasets. The established methods offer faster processing times for predictions, while our quantum approaches explore new computational paradigms for this biological prediction task. Our results were tested against a separate folds validation dataset, so we suggest that our models be more rigorously tested with different datasets, to gain more representative descriptions of our models’ abilities.
Experimental Validation for Clinical Translation
Translating our quantum-enhanced peptide-HLA binding predictions into therapeutic applications requires specific biological validation steps that our purely computational study cannot provide. The most critical next step involves direct binding assays using surface plasmon resonance (SPR) or competitive binding experiments to verify that our top-ranked peptide predictions actually bind to their predicted HLA molecules with the expected affinities, as computational accuracy doesn’t guarantee biological accuracy. Beyond binding confirmation, we
must test whether our predicted peptide-HLA complexes trigger appropriate immune responses through primary T cell proliferation assays using patient-derived PBMCs to validate that our predicted epitopes actually activate CD4+ and CD8+ T cells and produce relevant cytokines like IFN-γ and TNF-α. Since our models showed significant variation across HLA alleles (55.2-100% accuracy range), validation studies should focus on glioblastoma patients with the specific HLA types where our models performed best (>95% accuracy) and worst (<75% accuracy) to identify systematic prediction biases. Finally, a small-scale proof-of-concept study should test whether incorporating our quantum-predicted peptides into established oncolytic virus platforms (like HSV or adenovirus) enhances immune activation in glioblastoma cell lines compared to randomly selected peptides or those predicted by classical methods. These targeted validation experiments would provide the essential biological evidence needed to justify larger preclinical studies and eventual clinical translation, addressing the current limitation that our promising computational results lack experimental verification.
Conclusion
This study represents an initial exploration of quantum computing’s potential in biotechnology, specifically for peptide-HLA binding prediction in oncolytic virus design. While our hybrid quantum-classical model (81.38% accuracy) didn’t surpass classical approaches, and our variational model achieved 92.03% accuracy, these results demonstrate the feasibility of integrating quantum computing into biological prediction tasks. While quantum computing in biotechnology remains in its infancy, this framework establishes a foundation for future approaches. Future experiments, particularly ablation studies that selectively disrupt entanglement or comparative analyses with classical models, could provide further empirical evidence of the biochemical fidelity and practical advantage of quantum entanglement in modeling peptide-HLA interaction networks.
Several important factors should be considered when interpreting the results of this study. The computational intensity of the hybrid model limited our ability to perform complete 5-fold cross-validation, which affects the statistical robustness of our performance metrics. Sample sizes across HLA types ranged from 28 to 14,668 samples, introducing potential variability in the reliability of performance metrics across different alleles. Furthermore, current quantum computing hardware constraints may impact the full utilization of quantum resources in our hybrid model. Our validation approach, while showing promising initial results, would benefit from testing on additional independent datasets. These considerations suggest that while our results demonstrate the potential of quantum approaches in peptide-HLA binding prediction, additional validation studies would help establish the broader applicability and reproducibility of these findings.
Another important limitation in our study arises from potential biases introduced by data imbalance and selection within our training and validation datasets. While our dataset includes approximately 574,000 peptide-HLA binding pairs spanning 112 HLA alleles, the distribution across these alleles is significantly uneven. Certain alleles are represented by tens of thousands of samples, whereas others have fewer than a hundred examples. This imbalance may bias model performance towards alleles that are more frequent in the dataset, potentially leading to inflated accuracy estimates for common alleles and poorer generalization for rare or less studied variants.
The dataset primarily consists of experimentally validated binding peptides, which introduces potential selection bias towards peptides and HLA alleles commonly studied or experimentally accessible. As a result, peptides relevant to specific clinical contexts, such as glioblastoma-associated neoantigens or rare HLA alleles more frequently observed in particular patient populations, might be underrepresented. Consequently, our reported model performance may not fully generalize to clinical scenarios involving diverse or underrepresented peptide-HLA interactions.
Future work should explicitly employ strategies such as stratified sampling, oversampling of minority classes, or synthetic data generation to mitigate these biases. Additionally, validation on independent datasets specifically representing clinically relevant or underrepresented HLA alleles and peptides would help ensure robust generalizability and reduce potential biases in
real-world clinical predictions. Future strategies would have to include wet-lab verification of this model’s predictions. The path from these initial steps to clinical applications requires continued innovation, but the potential benefits for cancer patient outcomes, particularly for challenging cases like brain tumors, make this a worthwhile pursuit.
References
- Kaufman, H. L., Kohlhapp, F. J., & Zloza, A. (2015). Oncolytic viruses: a new class of immunotherapy drugs. Nature Reviews Drug Discovery, 14(9), 642–662. https://doi.org/10.1038/nrd4663 [↩] [↩]
- Todo, T., Ito, H., Ino, Y., Ohtsu, H., Ota, Y., Shibahara, J., & Tanaka, M. (2022). Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nature Medicine, 28(8), 1630–1639. https://doi.org/10.1038/s41591-022-01897-x [↩]
- Chu, Y., Zhang, Y., Wang, Q., Zhang, L., Wang, X., Wang, Y., Salahub, D. R., Xu, Q., Wang, J., Jiang, X., Xiong, Y., & Wei, D. (2022). A transformer-based model to predict peptide–HLA class I binding and optimize mutated peptides for vaccine design. Nature Machine Intelligence, 4(3), 300–311. https://doi.org/10.1038/s42256-022-00459-7 [↩] [↩] [↩]
- Zhang, L., Chen, Y., Wong, H., Zhou, S., Mamitsuka, H., & Zhu, S. (2012). TEPITOPEpan: Extending TEPITOPE for Peptide Binding Prediction Covering over 700 HLA-DR Molecules. PLoS ONE, 7(2), e30483. https://doi.org/10.1371/journal.pone.0030483 [↩] [↩]
- Nielsen, M., Justesen, S., Lund, O., Lundegaard, C., & Buus, S. (2010). NetMHCIIpan-2.0 – Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Research, 6(1), 9. https://doi.org/10.1186/1745-7580-6-9 [↩] [↩] [↩] [↩]
- Beer, K., Bondarenko, D., Farrelly, T., Osborne, T. J., Salzmann, R., Scheiermann, D., & Wolf, R. (2020). Training deep quantum neural networks. Nature Communications, 11(1). https://doi.org/10.1038/s41467-020-14454-2 [↩] [↩]
- Montes-Cabrera, A., et al. (2022). Ultraviolet superradiance from tryptophan residues highlights quantum coherence in biological systems. Physical Review Letters, 129(2), 028102. https://doi.org/10.1103/PhysRevLett.129.028102 [↩] [↩]
- Li, R. Y., Di Felice, R., Rohs, R., & Lidar, D. A. (2018). Quantum annealing versus classical machine learning applied to a simplified computational biology problem. npj Quantum Information, 4(1), 14. https://doi.org/10.1038/s41534-018-0060-8 [↩]
- Montes-Cabrera, A., et al. (2022). Ultraviolet superradiance from tryptophan residues highlights quantum coherence in biological systems. Physical Review Letters, 129(2), 028102. https://doi.org/10.1103/PhysRevLett.129.028102 [↩]
- Gainza, P., et al. (2024). Geometric deep learning reveals functional peptide binding patterns across MHC alleles. Communications Biology, 7, 210 [↩]
- Fagerberg, T., et al. (2006). Prediction of peptide binding to MHC class I proteins using artificial neural networks and quantum chemical descriptors. Journal of Molecular Graphics and Modelling, 25(1), 112–12 [↩]



{kind=link}