
Abstract
Acute kidney injury (AKI) is an inflammatory kidney disease that may lead to kidney failure if untreated. AKI is a widespread disease with limited treatments against it after being diagnosed. AKI is often caused by elevated levels of Reactive Oxygen Species (ROS), which damages the kidney cells, especially their mitochondria. Damaged mitochondria causes several negative impacts to the cell, one of which is an increased level of ROS, causing necrosis and apoptosis. Thus, mitochondria are one of the factors which a treatment can be applied to reduce cell damage in AKI. Specifically, we believe that mitochondrial transplantation may be a treatment towards AKI since it provides healthy mitochondria to the damaged cells, replacing the damaged mitochondria and halting further damage. Nevertheless, mitochondrial transplantation is considered a novel treatment towards multiple inflammatory-related diseases, and there are no direct studies showing its treatment against AKI; therefore, our study aims to discover the effect of mitochondrial transplantation. In this study, we use H2O2—a type of ROS—to induce AKI in NRK52E cells in vitro. Then, with isolated healthy mitochondria, we treat a group of cells and compare its result. The result presents a significant treatment by mitochondria towards cells damaged by H2O2. Specifically, Hypoxia-inducible factor-1α reduces in cells treated with mitochondria. Moreover, ROS reduced in production and generation; antioxidant-related proteins such as nuclear factor erythroid 2–related factor 2 (Nrf2), silent information regulator 1 (Sirt1), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PCG-1α), and superoxide dismutase 2 (SOD2) increased; and apoptosis-related proteins such as Caspase3 reduced in the group of cells treated with mitochondria. In conclusion, this study demonstrates mitochondrial transplantation as a promising future treatment towards AKI.
Introduction
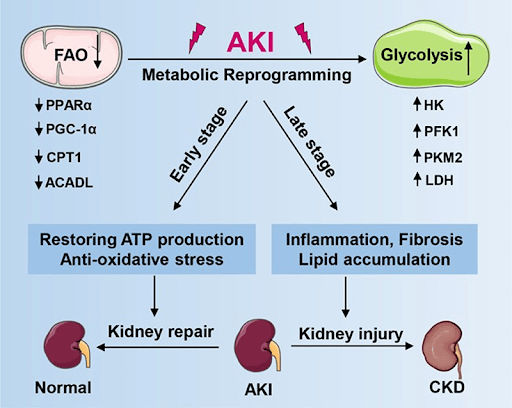
Acute kidney injury (AKI) is a pressing public health concern which affects approximately 13.3 million patients each year and leads to 1.7 million deaths1. The condition poses a considerable burden on healthcare systems and patients alike. AKI not only results from various factors such as dehydration, and medication toxicity, but also a systemic inflammatory response syndrome with severe tubular cell injury (Figure 1)2,3.
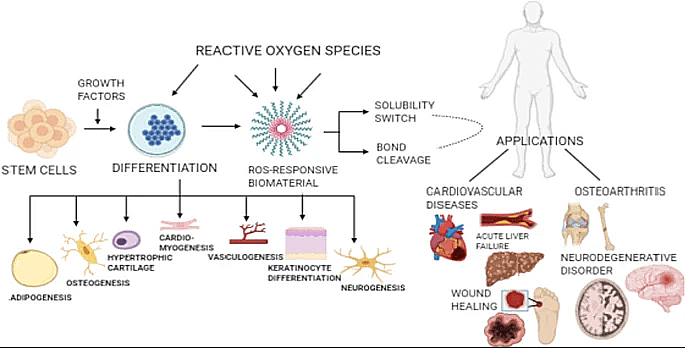
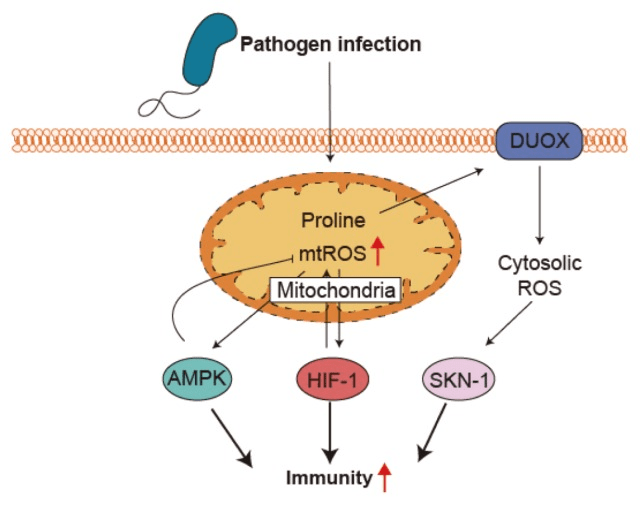
Reactive oxygen species (ROS) is one of the main factors that cause tubular cell injury during systemic inflammatory response syndrome (Figure 2)5,6. ROS production is largely ascribed to inflammation since ROS is one of the mechanisms used by the immune system to eliminate pathogens (Figure 3)7.
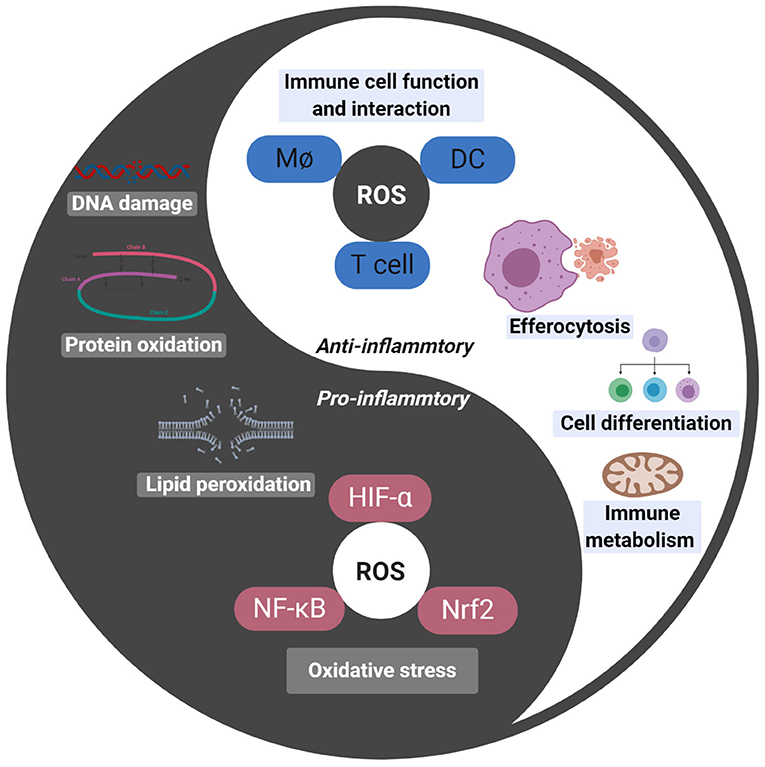
ROS damages both pathogenic and non-pathogenic cells by breaking DNA strands in cells, a process that induces apoptosis9. While ROS eliminates pathogens, it also damages the DNA of the organisms’ cells. Among ROS, H2O2 is one of the most stable; therefore, H2O2 is considered to contribute the most ROS damage10.
In order to balance the oxidative stress caused by ROS, cells produce proteins involved in countering the oxidative stress, one of which is silent information regulator 1 (Sirt1)12. Some findings suggest the correlation between the Sirt1 and the resistance to oxidative stress of cultured renal medullary interstitial cells (Figure 4)13. The findings also mentioned that in mice with AKI, the lack of Sirt1 exacerbates renal apoptosis and fibrosis14,15. Sirt1 is also related to several other proteins, some of them are nuclear factor erythroid 2–related factor 2 (Nrf2) and heme oxygenase-1 (HO-1). Nrf2 is involved in detoxifying ROS and exhibits elevated levels during high oxidative stress in active cells (Figure 5)16. Therefore, the Nrf2/HO-1 pathway can be used as an index for the amount of active cells.

Figure 4. Sirt1 Plays an Important Role in ROS17
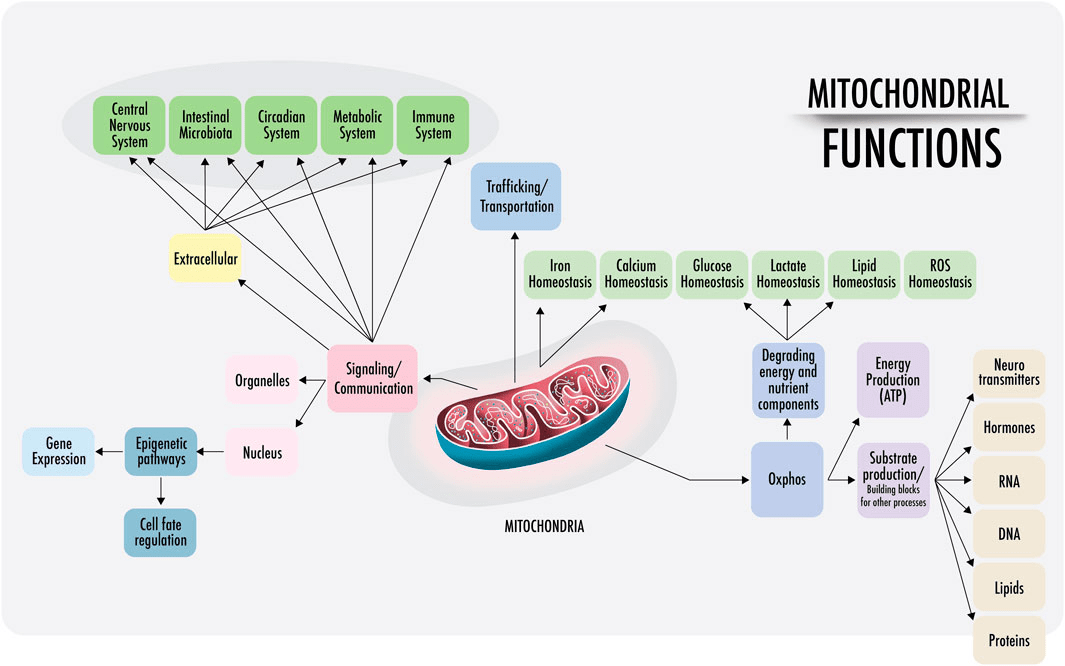
Mitochondria are especially susceptible to ROS. Mitochondrial damage can cause insufficient production of ATP and cell damage from built-up oxygen, sugar, and fat molecules in the cell (Figure 6)19. Furthermore, damaged mitochondria also produce ROS, which ultimately leads to cell necrosis and apoptosis.

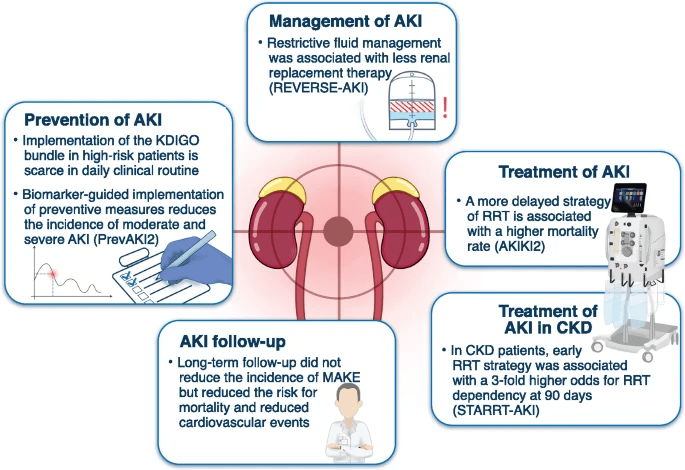
Current clinical therapies are typically supportive in terms of retarding the damage caused by AKI, such therapies include providing fluids challenge and dialysis (Figure 7). Nevertheless, these therapies have no substantial impact on damaged cells. Previous studies frequently observed Mitochondrial damage in AKI.
Mitochondrial damage causes insufficient production of ATP and damage from built-up oxygen, sugar, and fat molecules in the cell (Figure 8)20. Furthermore, damaged mitochondria also produce ROS21. ROS ultimately leads to cell necrosis and apoptosis22.
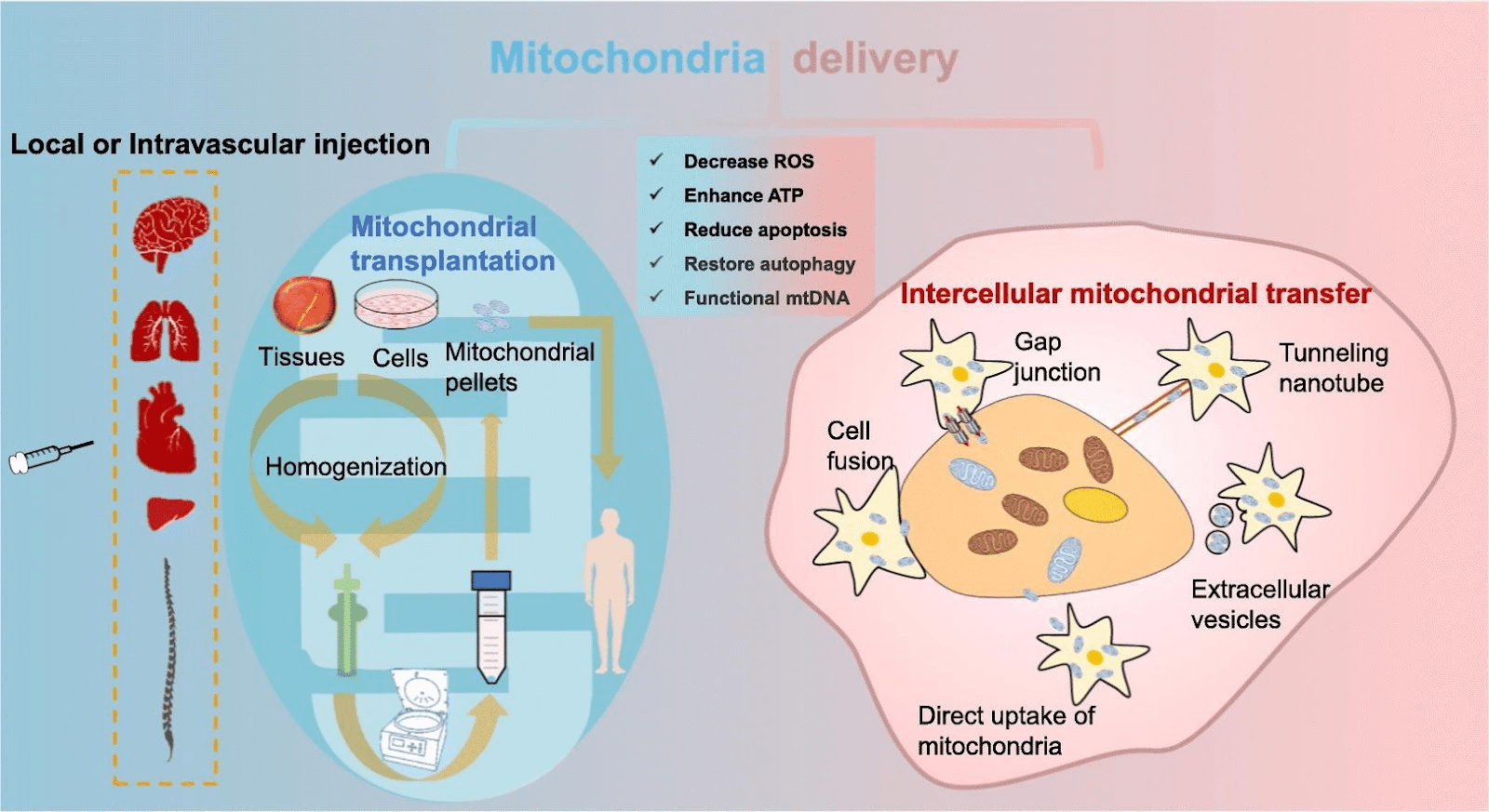
Increasingly, studies have pointed out that mitochondrial transplantation therapy can use the phenomenon of mitochondrial transfer to replace damaged mitochondria in cells and further rescue cells (Figure 9)23,24,25.
Mitochondrial transplantation is studied in different disease models, Hayashida, K et al. discovered that mitochondrial transplantation recovers neural function26. McCully et al. implemented mitochondrial transplantation for the rescue of cell viability and cell function following ischemia-reperfusion injury27,28. Emani et al. implemented mitochondrial transplantation in clinical trials on patients with ischemia-reperfusion injury29. In addition, studies implemented mitochondrial transplantation on curing cancer, Tsai et al. shows that mitochondrial transplantation reduces gastric cancer tumor growth in vivo30. Furthermore, according to the study of Andrea Rossi et. al., mitochondrial transplantation mitigates damage in an in vitro model of renal tubular injury and in an ex vivo model of DCD renal transplantation, proving the ability of mitochondrial transplantation to reduce kidney damage in clinical conditions31.
However, there is currently no research related to the effect of mitochondrial transplantation on AKI. Thus, we hypothesise that mitochondrial transplantation is able to recover the function of mitochondria and suppress the ROS level in cells to reduce cell damage. We used a cell model in our study to prove our hypothesis. According to the study of Kenji Kasuno et. al., H2O2 can be used for a factor to induce AKI in cell models33. Therefore, we used a similar method to conduct our study. Our experiment design uses H2O2 to stimulate cells and observe whether providing healthy mitochondria to cells reduces ROS and restores mitochondrial activity in cells, hence saving the damaged cells (Figure 10).

Goals of the Study
- To demonstrate whether H2O2 induces cell-death
- The cells are treated with different concentrations of H2O2 and CCK8 assay is used to analyze cell viability.
- To demonstrate whether mitochondrial transplantation recover the oxidative stress by H2O2 treatment
- After mitochondrial transplantation, the anti-oxidative capacity of the cell is analyzed by flow cytometry to observe oxidation-related markers (mitoSOX, H2DCFDA, DHE).
- The level of oxidative stress after mitochondrial transplantation is analyzed by western blot to observe HIF-1α expression.
- After mitochondrial transplantation, the anti-oxidative capacity of the cell is analyzed by western blot to observe oxidation-related markers (Nrf2, Sirt1, PCG-1α, SOD2).
- To demonstrate whether mitochondrial transplantation recover the damage by H2O2 treatment
- After mitochondrial transplantation, the cell-death is analyzed by flow cytometry to observe apoptosis markers (Caspase3 activity).
- After mitochondrial transplantation, the cell-death is analyzed by western blot to observe apoptosis markers (Bax, Bcl2, Caspase3).
- After mitochondrial transplantation, the cell viability is analyzed by CCK8 assay.
Result
H2O2 Induces Cell-death
In our study, we use H2O2 to simulate the AKI in vitro model. Our results suggest H2O2 induced cell damage in different concentrations (Figure 11). Our data show the concentration of IC50 is 20 µM. Thus, the experiment is conducted under 20 µM of H2O2 treatment.

Mitochondrial Transplantation Effectively Recovers ROS on NRK52E Cells
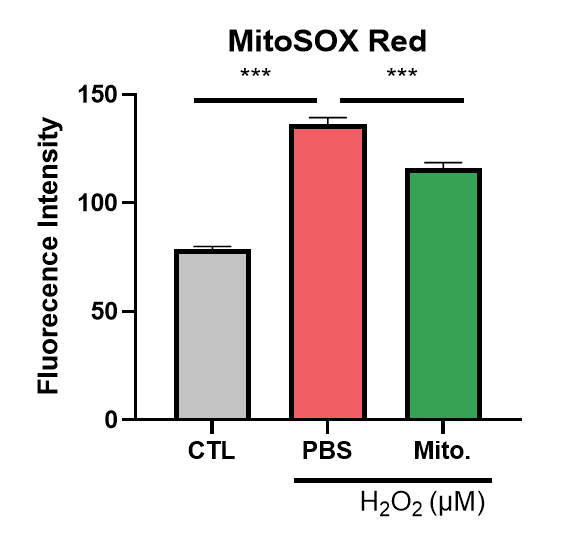
Present study shows that AKI is an inflammatory disease. Hence, our study uses mitochondria transportation to observe if it recovers the oxidative stress by H2O2 treatment. First, we observe if mitochondrial transplantation reduces mitochondrial superoxide production. The cell is co-cultured with or without mitochondria under H2O2 treatment for 24 hours. We used mitoSOX Red to analyze the superoxide production in mitochondria. We observed accumulation of superoxide in the group treated with H2O2 and decreased superoxide level in the group with both mitochondria and H2O2. The data show mitochondrial transplantation reduces superoxide production effectively (Figure 12). Next, we used H2DCFDA and DHE dye to reveal that H2O2 increases ROS level and mitochondria reduces ROS production (Figure 13 and 14). The results above show that mitochondrial transplantation has a significant effect on decreasing cell oxidative stress.

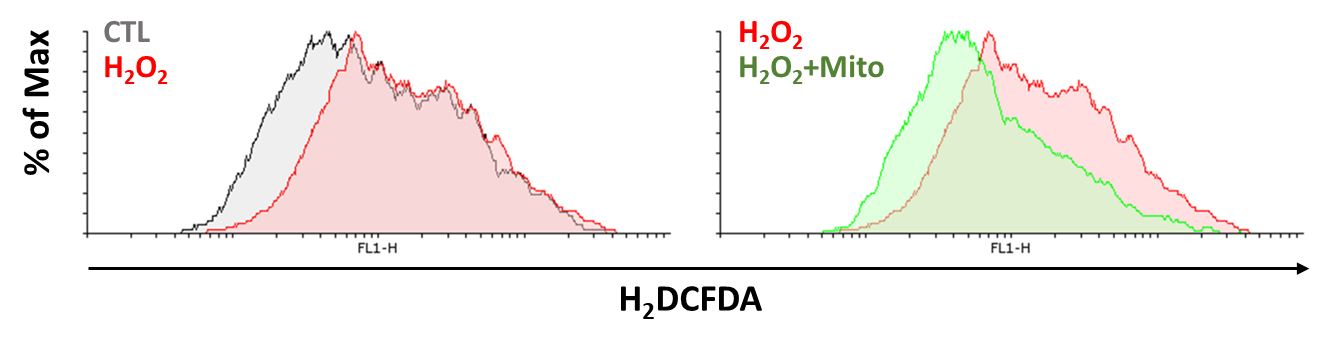



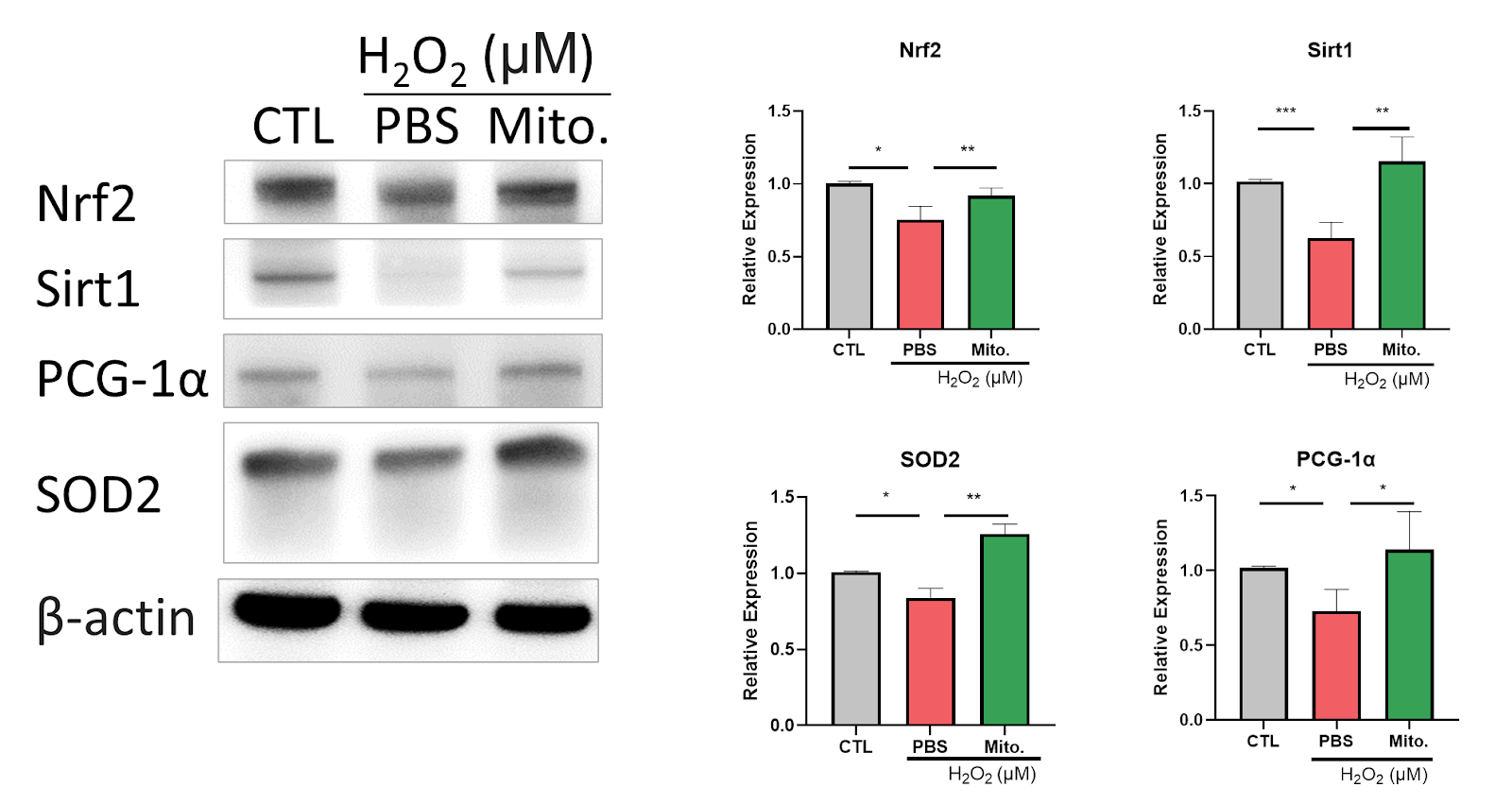
Due to the correlation between AKI and hypoxia, We observe hypoxia-inducible factor 1-alpha (HIF-1α) expression after mitochondrial transplantation. Our data show H2O2 induces HIF-1α expression, which is reduced after mitochondrial transplantation (Figure 15). The result shows the potential for mitochondrial transplantation to recover the injury caused by AKI. Furthermore, we observed the antioxidative ability of mitochondrial transplantation. Our data show antioxidant-related proteins such as Nrf2, Sirt1, PGC-1α, Superoxide dismutase 2 (SOD2) are inhibited by H2O2 treatment. Those proteins increased in expression after mitochondrial transplantation (Figure 16). According to Figure 15 and 16, the groups with mitochondria show that mitochondrial transplantation effectively recovers the increase in inflammatory factors induced by H2O2 and defences the inflammatory response by upregulating the antioxidant related protein, proving that mitochondrial transplantation has the ability to counteract the oxidative stress. In summary, our study proved the potential of AKI treatment by mitochondrial transplantation.

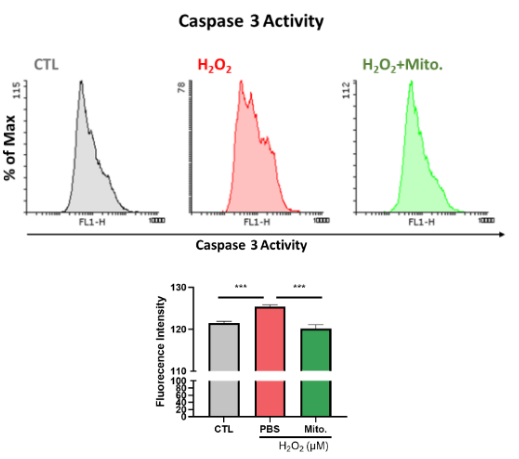
Mitochondrial Transplantation Recovers the Cell Death of NRK52E Cells
Furthermore, our research aims to know whether mitochondria are able to recover the cells damaged by H2O2. We utilized flow cytometry to analyze the activity of Caspase3 (Figure. 17). Figure 16 shows that the fluorescence density of the H2O2 group increases compared to the control group and the group with mitochondria decreases in fluorescence density compared to the H2O2 group. The result demonstrates the ability for mitochondria to reduce the Caspase3 activity effectively. In addition, we utilize western blot to analyze the apoptosis-related protein expression. The result shows a significant increase in Bax expression after H2O2 treatment, and mitochondrial transplantation inhibited Bax expression. On the contrary, the result shows decrease in BCL2 expression after H2O2 treatment, and mitochondrial transplantation increased Bax expression. Bax and BCL2 are known to play important roles in the process of apoptosis. We prove that mitochondria inhibits H2O2 induced cell apoptosis. Similarly, mitochondria inhibits Cleaved Caspase-3 (c-Caspase3) expression, which aligns with the result of flow cytometry (Figure 18). According to Figure 17 and 18, mitochondrial transplantation prevents cell apoptosis in acute inflammation, rescuing the cell from death. Finally, the result of CCK8 shows that mitochondrial transplantation increases the cell viability under H2O2 treatment (Figure 19). In summary, our study proves that mitochondrial transplantation may recover the cell damage from AKI.


Mitochondrial Transplantation Increase the ATP Concentration on NRK52E Cells
Finally, our study analyses the ATP concentration in cells with mitochondrial transplantation. The data shows that the cells treated with H2O2 are significantly lower than control and there is a significant increase in ATP concentration in cells with mitochondria transplantation, proving mitochondrial transplantation recovers the quantity of ATP decreased by H2O2. Since mitochondrial activity directly affects the production of ATP in cells, the result proves that mitochondrial transplantation increases mitochondrial activity, presenting the method as having a rescuing effect towards mitochondrial disability in AKI and decrease in ATP quantity.

Discussion
Mitochondrial transplantation for the prospective treatment of AKI
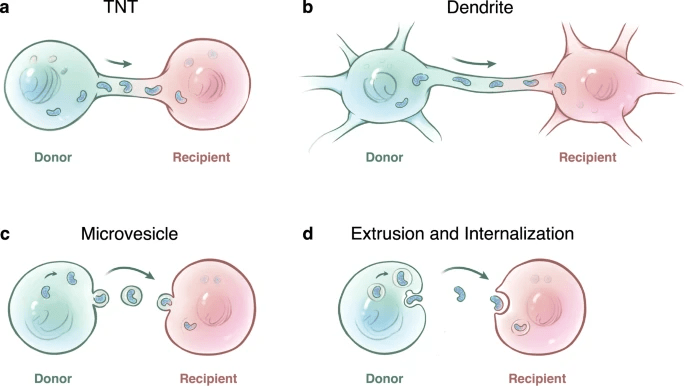
Acute kidney injury (AKI) is an inflammation related disease. AKI refers to rapid decline in renal function with various causes. Patients worldwide experience AKI in hospitals34, and the long-term prognosis is poor35. Therefore, elucidating the repair mechanism is necessary36. Currently, the clinical therapy for AKI is typically supportive37, and there is no effective method to quickly rescue kidney injury Therefore, it is urgent to find suitable treatment options. Increasingly, studies have identified the phenomenon of mitochondrial transfer via tunneling nanotubes, dendrites, microvesicles, and extrusion and internalization (Figure 21)38.
Many studies point out that by treating the damaged cells with mitochondria, the cell viability increased and cell damage decreased40,28. Related studies demonstrate methods to increase the efficiency in mitochondrial treatment in vitro, in addition to directly extracting mitochondria and co-culture with cells (Figure 22)41.
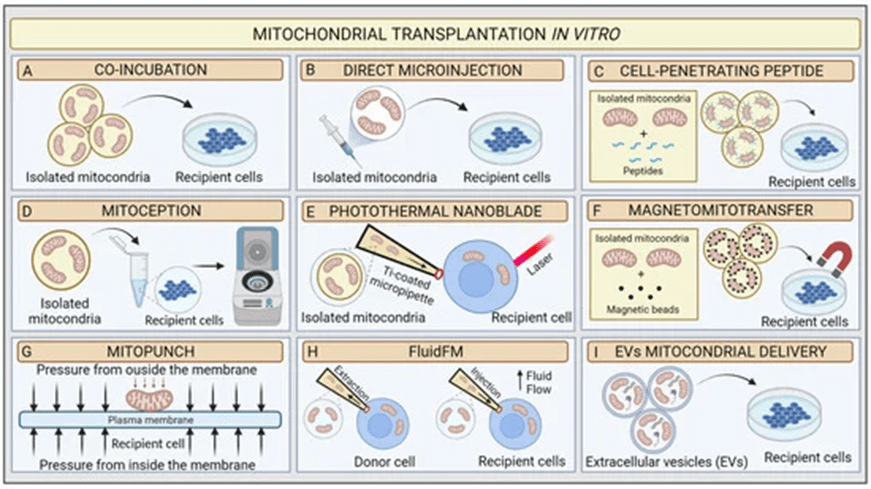
Since no current research is related to the effect of mitochondrial transplantation on AKI, the purpose of our study is to observe if mitochondrial transplantation reduces ROS and restores mitochondrial activity in cells, hence rescuing the damaged cells. To achieve our research goal, this study conducted a series of experiments, the result of which will be discussed in the following section.
H2O2 simulates the cell damage caused by AKI
Our study is based on the study of Ishihara et al.43, which utilized H2O2 as the medicine to induce AKI. The result of our cell viability experiment presents the significance of H2O2’s effect in decreasing NRK52E cell survival rate. The result proves the reliability of our experiment. To demonstrate the effect of mitochondria treatment, we choose IC50 of the medicine as the concentration in the following experiments.
Mitochondrial transplantation regulates HIF-1α to alleviate the inflammatory reaction caused by AKI
Hypoxia has long been recognized as an important factor in the pathogenesis of acute kidney injury44. Hypoxia-inducible factor (HIF) is the master transcription factor that regulates adaptive responses against hypoxia. Many studies prove that HIF-1α is highly expressed in the early stage after AKI36. In addition, its activation protects the kidney against AKI via various mechanisms, such as apoptosis suppression45, autophagy activation46, and interstitial macrophage-dependent inflammatory response modulation47. Therefore, our study analyzes HIF-1α, whose expression increased in the cell treated by H2O2 and decreased in the cell transplanted with mitochondria in our study. The result illustrates how mitochondrial transplantation can recover the inflammation caused by AKI.
Mitochondrial transplantation increases the cell’s resistance against oxidative stress
Oxidative stress plays a crucial role in the complex web of processes behind AKI as it is evident in its circuitous and interdependent pathways, which both provoke and harm the organ’s delicate responses48. Mitochondrial dysfunction is one of the important indicators of AKI. Our research analyzed how mitochondrial transplantation affected the accumulation of superoxides and the generation of ROS. Our result proved the ability for mitochondrial transplantation to reduce the accumulation of superoxides and the generation of ROS induced by H2O2, illustrating the potential for mitochondrial transplantation to treat AKI. Hypoxia is another crucial factor that contributes to AKI. Hypoxia is not only related to Ischemia-Reperfusion injury, but also related to toxins, sepsis, and ureteral obstruction36,49.
Mitochondrial transplantation treats AKI though Sirt1-Nrf2 mediated pathway
Nuclear factor erythroid 2 related factor 2 (Nrf2) is a regulator of redox balance showing improvement to kidney disease by reducing ROS. Past research found the use of compounds activated by Nrf-2 reduces ROS effectively, thereby halting the progression of various types of AKI (Figure 23)50. Our study proves the ability of mitochondrial transplantation to restore the reduced Nrf2 expression caused by H2O2, displaying the ability of mitochondrial transplantation to protect NRK52E.
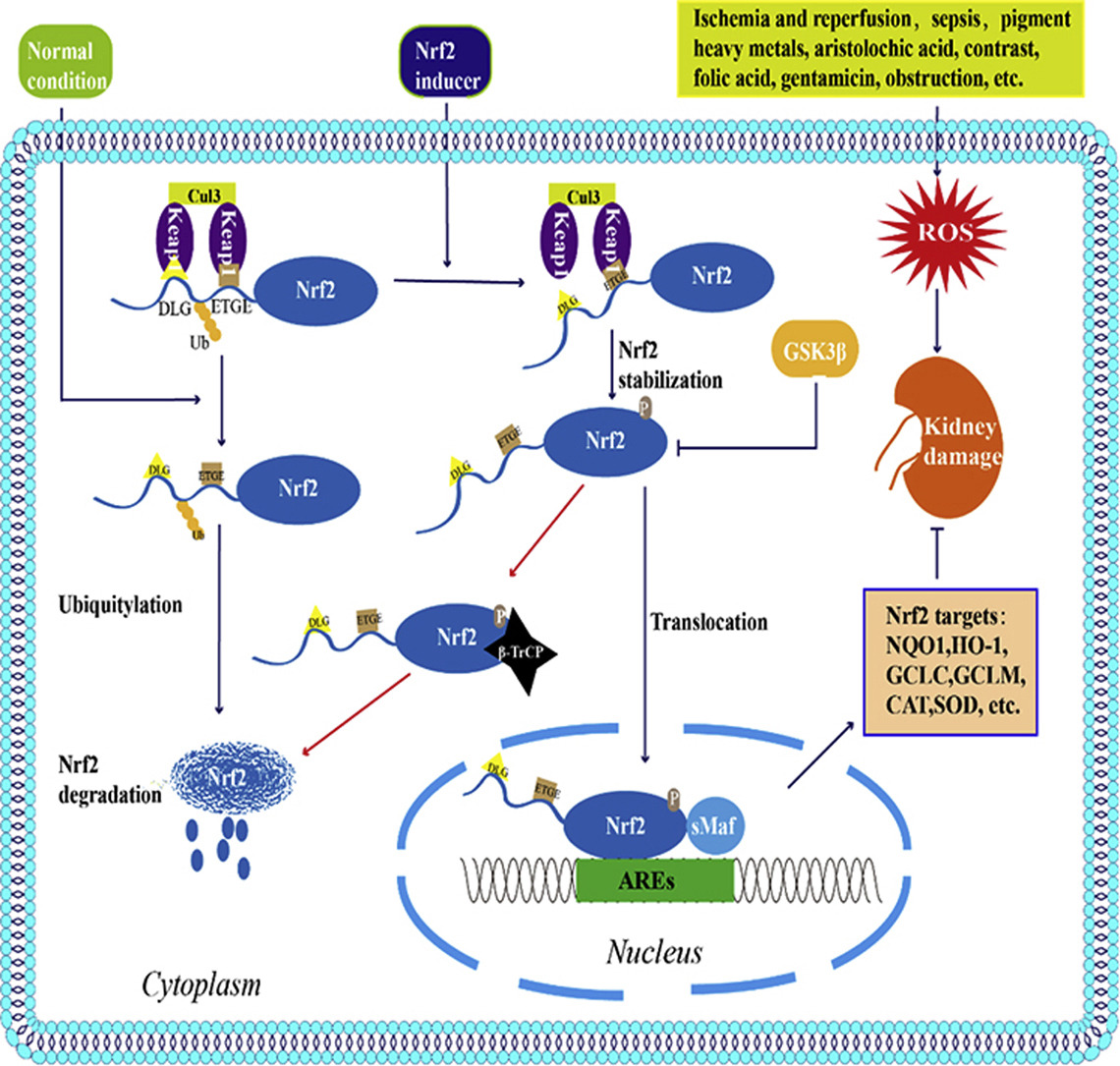
Sirt1 is a member of Class III HDACs, and it is a protein encoded by the SIRT1 gene in humans. Multiple results show that increased Sirt1 expression reduces the ROS and damage of renal cells52),53,54. The study of Shi S et al. points out Melatonin can attenuate oxidative stress in AKI by activating the SIRT1/Nrf2/HO-1 pathway55,56, proving Sirt1 is an important biomarker of treatment of AKI (Figure 24) .Our study observed the restoration of Sirt1 expression after mitochondrial transplantation, demonstrating mitochondrial transplantation may recover cell damage caused by AKI though Sirt1-Nrf2 mediated pathway.

Endogenous antioxidant enzymes substantially reduced ROS through catalysis ROS through catalysis. SODs, divided into cytosolic CuZn-SOD (SOD1), mitochondrial Mn-SOD (SOD2), and extracellular (SOD3) SOD enzymes, seem to be the first line of defense in ROS elimination. SODs can be rapidly activated in some situations and catalyze superoxide into molecular oxygen and H2O2 (Figure 25). The catalyzation converts ROS into less reactive species58,59.
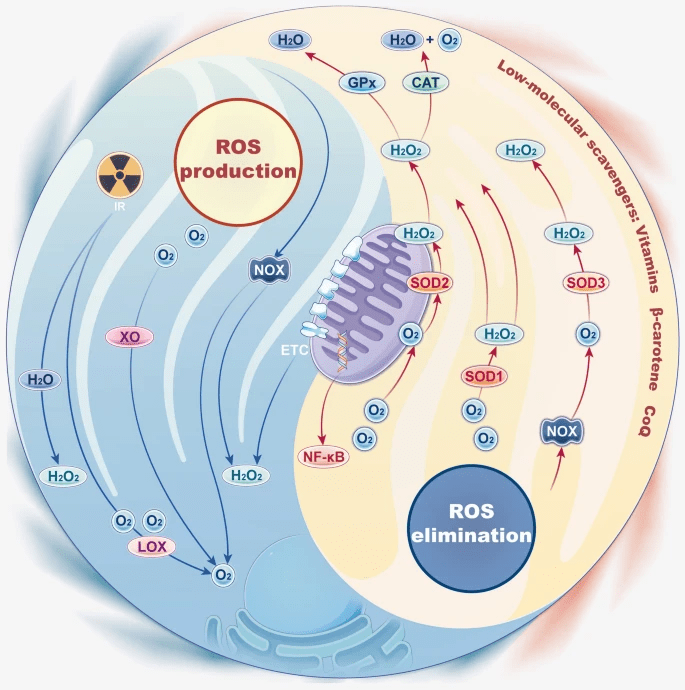
According to the study, we discovered that SOD2 expression increases after mitochondrial transplantation. We conclude that mitochondrial transplantation reduces H2O2 induced ROS and recovers cell damage caused by the inflammation.
Mitochondrial transplantation inhibits the apoptosis caused by AKI
AKI is often caused by transient ischemia from hypovolemia, hypotension, or heart failure, which are considered to be the cause of nearly one-third of patients requiring acute renal replacement therapy. Necrosis is considered as an important cause of ischemic AKI61. Modern research believes that cell apoptosis causes ischemic renal dysfunction62. Apoptosis is a type of programmed cell death, meaning the cell death is an ordered process, which, once the process has started, continues automatically according to the established program, mainly occurring in development and cell renewal. Pothana Saikumar and his colleagues found that cell death may be caused by apoptosis and necrosis, which are caused by ischemia in experimental models of renal injury both in vivo and in vitro. They prove that in hypoxic renal cells, Bax and Bak work together to permeabilize proteins such as cytochrome c. When caspase is inhibited, inflammation is reduced, along with the cascading parenchymal injury that is associated with inflammation (Figure 26)63,64. In our study, we proved that mitochondrial transplantation effectively decreased H2O2 induced Caspase3 activation. Furthermore, The result of western blot also presents a decrease in the ratio of Bax/Bcl2 and c-Caspase3. Moreover, we used CCK8 analysis to analyze cell viability, and the result shows mitochondrial transplantation is able to effectively recover the decrease in cell viability caused by H2O2. Finally, many studies show that mitochondrial disability and the decrease in ATP production is related to AKI65,66,67. These studies see mitochondrial dysfunction as a target to treating AKI. Our study shows that mitochondrial transplantation significantly increases the ATP concentration after cell damage, further proving mitochondrial transplantation being a new strategy in treating AKI. All results above prove that mitochondrial transplantation has the ability to protect the cell from cell death caused by oxidative stress.

Summary
Since ROS production is associated with apoptosis and is observed in several AKI models, it may be reasonable to explore drugs that inhibit or scavenge ROS as potential strategies for managing organ injury. Various antioxidants have been successfully used to prevent ischemic, septic, or toxic AKI in animal models61. However, these interventions are currently limited to prevention, rather than treatment of renal injury. Mitochondrial transplantation is one of the main topics that is being researched around the globe in many inflammation related diseases and has proven the ability to prevent cell and tissue damage, despite this, there was no AKI-related studies that implement mitochondrial transplantation as the treatment. Our study discovered that mitochondrial transplantation is a promising candidate for clinical use in AKI treatment. In the future, we hope to find a suitable animal model to conduct mitochondrial transplantation and conduct a more in-depth discovery in the protein regulatory pathway in the cell to achieve more verification before clinical application.
Conclusion
Our study provides insights into the protective potential of mitochondrial transplantation. mitochondrial transplantation exhibits antioxidant, anti-inflammatory, and antiapoptotic effects both in vitro in the context of AKI. In addition, mitochondrial transplantation recovers the ATP quantity reduced by cell damage. These beneficial effects can be attributed to activation of the antioxidative protein such as Nrf2, Sirt1, PCG-1ɑ, and SOD2. They indicate that mitochondrial transplantation may represent a promising therapeutic option for treatment of AKI (Figure 26).
The results of our study demonstrate that mitochondrial transplantation is a promising therapeutic approach for treating AKI, potentially overcoming the limitations of conventional supportive treatments, whose effectiveness often varies based on the patient’s condition. Specifically, the protective effect of mitochondrial transplantation on cell damage makes an important contribution to slowing down the course of the disease and improving the survival rate of patients. To achieve this, in vivo trials will be an important future work. In addition to animal models, several transcription factors in this study can also serve as important research targets. In-depth exploration of the molecular mechanisms induced by mitochondrial transplantation is also one of the important future work.
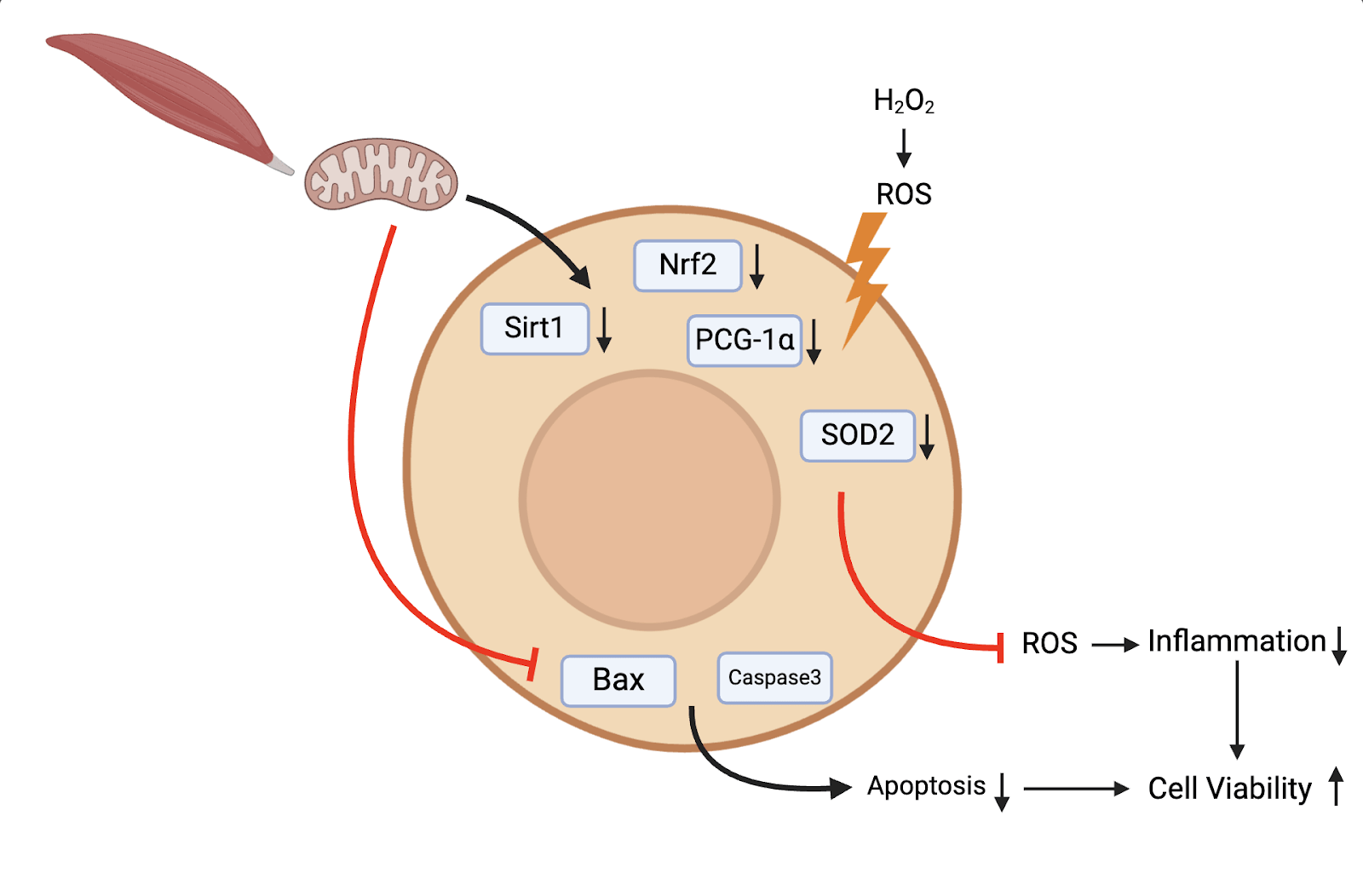
Limitations
In this research, we investigated the importance of mitochondrial transplantation in treating AKI. However, some limitations occur in the quantification of mitochondrial activity and the storage of mitochondria.
First of all, each cell has a different number of mitochondria, each with a different mitochondrial activity. Although some current methods such as flow cytometry can give some data, the data is limited in the accuracy and consistency due to differences in individual mitochondrion. We used protein quantification to the mitochondria used in this experiment to resolve the inconsistency in mitochondria used in this experiment. The mitochondria are extracted directly from the living muscle tissue to exclude the inconsistency in mitochondrial activity.
On the other hand, freezing and chemical preservation of mitochondria are unable to sustain its activity and function because freezing preservation of mitochondria damages the structure of mitochondria and chemical preservation of mitochondria decreases its activity. In this experiment, the mitochondria are extracted directly from the living muscle tissue to negate the limitation in its storage. Despite the limitation in storage of mitochondria may be resolved in the future, currently, these limitations exist in the methods used in this research.
Method
Cell Culture and Reagent
Rattus norvegicus (rat) Cell type kidney epithelial-like cells (NRK-52E) was cultured using Dulbecco’s Modified Eagle Medium (Gibco, Waltham, MA, USA) medium with 5% fetal bovine serum (Gibco, Waltham, MA, USA) under 5% CO2 at 37℃. The cell line is maintained using trypLE reagent (Gibco, Waltham, MA, USA) under 5% CO2 at 37℃ for 5 minutes, and the reaction is terminated by medium. Stock solutions of Hydrogen Peroxide (H2O2) 10M solution (Sigma-Aldrich, St. Louis, MO, USA) are prepared and dissolved in culture medium before treatment. Control group: culture medium with PBS, H2O2 group: culture medium with H2O2 solution, H2O2+mito group: culture medium with H2O2 solution and mitochondria in PBS.
Mitochondria Isolation
The muscle tissue of the rat is obtained while the rat is under anesthesia via Isoflurane (Sigma-Aldrich, St. Louis, MO, USA). The muscle tissue is washed with PBS and disrupted with a dounce homogenizer while on ice. The tissue is centrifuged at 1,000 rcf for 3 minutes at 4°C. The supernatant is discarded. The pellet is suspended in 800 µL of BSA/Reagent A Solution. The tube is vortexed at medium speed for 5 seconds and incubated on ice for exactly 2 minutes. 10 µL of Mitochondria Isolation Reagent B is added to the sample, which is vortexed at maximum speed for 5 seconds. The tube is incubated on ice for 5 minutes and vortexed at maximum speed every minute. 800 µL of Mitochondria Isolation Reagent C is added to the tube, which is inverted several times to mix and centrifuged at 700 rcf for 10 minutes at 4°C. The supernatant is transferred to a new tube and centrifuged at 3,000 rcf for 15 minutes at 4°C. The supernatant is discarded, and 500 µL of Wash Buffer is added to the mitochondrial pellet. The tube is centrifuged at 12,000 rcf for 5 minutes, and the supernatant is discarded. Mitochondria are maintained on ice before treatment. Fresh mitochondria are isolated for every experiment at a protein concentration of 25 micrograms per milliliter.
Cell viability analysis
NRK-52E cells are seeded in 96-well dishes in quadruplicate at 8000 cells/well and cultured for 24 hours before treatment. For dose-dependent tests, cells are treated with different concentrations of H2O2 (100, 80, 50, 20, 10, 5 µM) for 24 hours. For mitochondria treatment, cells are treated under 20 µM of H2O2 with or without mitochondria for 24 hours. After treatment, 10 µL of Cell Counting Kit-8 (Sigma–Aldrich, St. Louis, MO, USA) reagent is added to each well. The reagent reacted for 90 minutes, and the absorbance of the reagent is measured at 450 nm using a microplate reader. The cell viability is calculated with the following formula.
![\[\text{Cell viability (\%)} = \frac{OD_{\text{sample}} - OD_{\text{Blank}}}{OD_{\text{Control}} - OD_{\text{Blank}}} \times 100\%\]](https://nhsjs.com/wp-content/ql-cache/quicklatex.com-37dcb6921e818371f8e145c1238cb610_l3.png "Rendered by QuickLaTeX.com")
Mitochondrial Superoxide (MitoSOX) Analysis
NRK-52E cells are seeded in 6-well dishes in quadruplicate at 1×105 cells/well for 24 hours before treatment. Cells are treated under 20 µM of H2O2 with or without mitochondria for 24 hours. After the treatment, the cells are harvested with triPLE and washed with cold PBS. Cells are stained using 5 μM of MitoSOX™ Mitochondrial Superoxide Indicators (Invitrogen, Waltham, MA, USA) under 37℃ for 15 minutes. The sample is centrifuged at 2000 rpm for 10 minutes. The sample is washed with cold PBS and analyzed through flow cytometry.
Intracellular Reactive Oxygen Species Analysis
NRK-52E cells are seeded in 6-well dishes in quadruplicate at 1×105 cells/well for 24 hours before treatment. Cells are treated under 20 µM of H2O2 with or without mitochondria for 24 hours. After the treatment, the cells are harvested with triPLE and washed with cold PBS. Cells are stained using 10 μM of H2DCFDA (MedChemExpress, Monmouth Junction, NJ, USA) under 37℃ for 25 minutes. The sample is centrifuged at 2000 rpm for 10 minutes. The sample is washed with cold PBS and analyzed through flow cytometry.
Superoxide and Hydrogen Peroxide Analysis
NRK-52E cells are seeded in 6-well dishes in quadruplicate at 1×105 cells/well for 24 hours before treatment. Cells are treated under 20 µM of H2O2 with or without mitochondria for 24 hours. After the treatment, the cells are harvested with triPLE and washed with cold PBS. Cells are stained using 10 μM of Dihydroethidium (Hydroethidine) (Invitrogen, Waltham, MA, USA) under 37℃ for 15 minutes. The sample is centrifuged at 2000 rpm for 10 minutes. The sample is washed with cold PBS and analyzed through flow cytometry.
Caspase Activity Analysis
NRK-52E cells are seeded in 6-well dishes in quadruplicate at 1×105 cells/well for 24 hours before treatment. Cells are treated under 20 µM of H2O2 with or without mitochondria for 24 hours. After the treatment, the cells are harvested with triPLE and washed with cold PBS. Cells are suspended in 300 µL of PBS, stained with 1 µL of FITC-DEVD-FMK, and mixed. Cells are incubated in darkness under 37℃ for 30 minutes. The sample is centrifuged at 3000 rpm for 5 minutes, and the supernatant is discarded. The sample is washed with Wash Buffer and analyzed through flow cytometry.
Western Blot Analysis
- Protein Preparation
NRK-52E cells are seeded in 10 cm dishes in quadruplicate at 1×106 cells/well for 24 hours before treatment. Cells are treated under 20 µM of H2O2 with or without mitochondria for 24 hours. Cells are harvested with Cell Scraper and collected into a tube. Cells are washed and dispersed with PBS. Cells are centrifuged at 1500 rpm for 5 minutes, and the supernatant is discarded. The tube containing the cells are added and washed with a solution of lysis buffer containing 1% protease inhibitor and 1% phosphatase inhibitor. The sample is left at rest at 4℃ for 5 minutes. The sample is centrifuged at 15,300 rpm for 30 minutes, and the supernatant is collected as the protein sample.
- Protein Quantification
The standard is set up using stage-diluted BSA (500, 250, 125, 62.5, and 31.25 µg/mL). The quantification is done with Bradford protein assay and ELISA reader. The quantity is determined with the absorbance of the blue complex measured at a wavelength of 595 nm.

- Protein Electrophoresis
20 µg of protein sample is isolated for electrophoresis. The sample is mixed with 4x Laemmli Sample Buffer at 3 to 1 ratio and heated in water at 95℃ for 5 minutes. The SDS page is inserted into the electrophoresis tank, which is filled with a MES buffer. The protein marker is loaded into the wells of the SDS page, which is electrophoresed at 165 volts for 50 minutes.
- Transfer
The PVDF transfer membrane is activated with methanol then assembled with the electrophoresed SDS page in the transfer buffer. 100 volts of electricity is applied for 90 minutes to transfer the protein onto the PVDF transfer membrane. The membrane is submerged in a solution of blocking buffer for an hour before being submerged into a solution of the primary antibody. The membrane is submerged in the solution of the primary antibody under 4℃ overnight, allowing the primary antibody to attach to the proteins on the membrane.
| Primary antibody | KDa | Source | Recommended Dilutions | Catalog No. |
| BCL-2 | 28 | Rabbit | 1:1000 | #2876 |
| c-Caspase | 17, 19 | Rabbit | 1:1000 | #9661 |
| nrf2 | 110 | Rabbit | 1:1000 | A3577 |
| sirt1 | 90 | Rabbit | 1:1000 | #2310 |
| Bax | 20 | Rabbit | 1:1000 | ab182734 |
| Caspase3 | 17, 19, 35 | Rabbit | 1:1000 | #14220 |
| PCG-1α | 91 | Goat | 1:1000 | NB-100-60955 |
| HIF-1α | 120 | Rabbit | 1:1000 | #14179 |
| SOD2 | 22 | Rabbit | 1:1000 | #13141 |
| β-actin | 46 | Mouse | 1:5000 | #MAB1501 |
PBST is utilized to wash out the unbonded primary antibodies from the PVDF transfer membrane. The washing is repeated 3 times, each wash lasting 10 minutes. The washed PVDF transfer membrane is submerged under the solution of secondary antibodies for an hour under room temperature. PBST is utilized to wash out the unbonded secondary antibodies from the PVDF transfer membrane. The washing is repeated 3 times, each wash lasting 10 minutes. The washed PVDF transfer membrane is dyed with ECL.
| Secondary antibody | Recommended Dilutions | Catalog No. |
| ECL™ Anti-Rabbit IgG antibody, Peroxidase (HRP)-conjugated | 1:5000 | Cytiva NA934-1ML |
| ECL™ Anti-mouse IgG antibody, Peroxidase (HRP)-conjugated | 1:10000 | Cytiva NA931-1ML |
| Peroxidase AffiniPure™ Donkey Anti-Goat IgG (H+L) | 1:10000 | 705-035-003 |
ATPlite™ luminescence assay
- Sample preparation
NRK-52E cells are seeded in 6-well dishes in quadruplicate at 1×105 cells/well for 24 hours before treatment. Cells are treated under 20 µM of H2O2 with or without mitochondria for 24 hours. After the treatment, the cells are harvested with triPLE and washed with cold PBS.
- ATP Standard Solution Preparation
Use M.Q. water to dissolve ATP in the kit to 10 mM. Then, use M.Q. water to stair dilute the sample (200、100、50、25、12.5、6.25、3.125、0 mM).
- Analysis
Add cells into 96 well white plates at the concentration of 1×104 cells/50 µL well (n=8). Add ATP standard solution into cell-free well (n=2). Add 50 µL of mammalian cell lysis solution into every well. Use a shaker to mix the sample at 700 rpm for 5 minutes.
Remove the sample from the shaker and add 10 µL ATP control into the standard solution. Use a shaker to mix the sample at 700 rpm for 5 minutes. Then, add 50 µL of substrate solution to the well. Use a shaker to mix the sample at 700 rpm for 5 minutes.
Remove the sample from the shaker and put it in a dark room for 10 minutes. Then, collect the luminescence data and calculate the concentration of ATP.
Statistical Analysis
All data were analyzed by GraphPad Prism version 10. All graphs in figures were presented as means ± standard deviation (SD). Statistical analysis was performed one-way ANOVA analysis to compare data between each group. The experiment is conducted independently with more than 3 trials. All analyses with statistical significance were P < 0.05. Statistical results were labeled in each figure as *P < 0.05, **P < 0.01, ***P < 0.001.
References
- Mehta RL, Cerdá J, Burdmann EA, Tonelli M, García-García G, Jha V, Susantitaphong P, Rocco M, Vanholder R, Sever MS, Cruz D, Jaber B, Lameire NH, Lombardi R, Lewington A, Feehally J, Finkelstein F, Levin N, Pannu N, Thomas B, Aronoff-Spencer E, Remuzzi G. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. Jun 27;385(9987):2616-43. (2015) [↩]
- Zhu Z, Hu J, Chen Z, Feng J, Yang X, Liang W, Ding G. Transition of acute kidney injury to chronic kidney disease: role of metabolic reprogramming. Metabolism. Jun;131:155194. (2022) [↩]
- Zuk A, Bonventre JV. Acute Kidney Injury. Annu Rev Med. 2016;67:293-307. [↩]
- Zhu Z, Hu J, Chen Z, Feng J, Yang X, Liang W, Ding G. Transition of acute kidney injury to chronic kidney disease: role of metabolic reprogramming. Metabolism. Jun;131:155194. (2022) [↩]
- Tyagi, N., Gambhir, K., Kumar, S. et al. Interplay of reactive oxygen species (ROS) and tissue engineering: a review on clinical aspects of ROS-responsive biomaterials. J Mater Sci 56, 16790–16823. (2021) [↩]
- Jing Zhou, Chao Fang, Chao Rong, Tao Luo, Junjie Liu, Kun Zhang, Reactive oxygen species-sensitive materials: A promising strategy for regulating inflammation and favoring tissue regeneration, Smart Materials in Medicine, Volume 4, Pages 427-446 (2023) [↩]
- Kwon S, Kim EJE, Lee SV. Mitochondria-mediated defense mechanisms against pathogens in Caenorhabditis elegans. BMB Rep. Jun;51(6):274-279. (2018) [↩]
- Tyagi, N., Gambhir, K., Kumar, S. et al. Interplay of reactive oxygen species (ROS) and tissue engineering: a review on clinical aspects of ROS-responsive biomaterials. J Mater Sci 56, 16790–16823. (2021) [↩]
- Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. Dec;1863(12):2977-2992. (2016) [↩]
- Herb M, Schramm M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants (Basel). Feb 19;10(2):313. (2021) [↩]
- Kwon S, Kim EJE, Lee SV. Mitochondria-mediated defense mechanisms against pathogens in Caenorhabditis elegans. BMB Rep. Jun;51(6):274-279. (2018 [↩]
- Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int J Mol Sci. Feb 11;14(2):3834-59. (2013) [↩]
- Alam F, Syed H, Amjad S, Baig M, Khan TA, Rehman R. Interplay between oxidative stress, SIRT1, reproductive and metabolic functions. Curr Res Physiol. Mar 27;4:119-124. (2021) [↩]
- He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, Yang HC, Fogo AB, Zent R, Harris RC, Breyer MD, Hao CM. Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest. Apr;120(4):1056-68. (2010) [↩]
- Guo C, Dong G, Liang X, Dong Z. Epigenetic regulation in AKI and kidney repair: mechanisms and therapeutic implications. Nat Rev Nephrol. Apr;15(4):220-239. (2019) [↩]
- Lin W, Shen P, Song Y, Huang Y, Tu S. Reactive Oxygen Species in Autoimmune Cells: Function, Differentiation, and Metabolism. Front Immunol. Feb 25;12:635021. (2021) [↩]
- Alam F, Syed H, Amjad S, Baig M, Khan TA, Rehman R. Interplay between oxidative stress, SIRT1, reproductive and metabolic functions. Curr Res Physiol. Mar 27;4:119-124. (2021) [↩]
- Lin W, Shen P, Song Y, Huang Y, Tu S. Reactive Oxygen Species in Autoimmune Cells: Function, Differentiation, and Metabolism. Front Immunol. Feb 25;12:635021. (2021) [↩]
- Chen S, Li Q, Shi H, Li F, Duan Y, Guo Q. New insights into the role of mitochondrial dynamics in oxidative stress-induced diseases. Biomed Pharmacother. Sep;178:117084. (2024) [↩]
- Casanova A, Wevers A, Navarro-Ledesma S, Pruimboom L. Mitochondria: It is all about energy. Front Physiol. Apr 25;14:1114231. (2023) [↩] [↩]
- Wen X, Tang L, Zhong R, Liu L, Chen L, Zhang H. Role of Mitophagy in Regulating Intestinal Oxidative Damage. Antioxidants (Basel). Feb 14;12(2):480. (2023) [↩]
- Villalpando-Rodriguez GE, Gibson SB. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid Med Cell Longev. Aug 14;2021:9912436. (2021) [↩]
- Zhao R, Dong C, Liang Q, Gao J, Sun C, Gu Z, Zhu Y. Engineered Mitochondrial Transplantation as An Anti-Aging Therapy. Aging Dis. Jul 19. (2024) [↩]
- Chen MM, Li Y, Deng SL, Zhao Y, Lian ZX, Yu K. Mitochondrial Function and Reactive Oxygen/Nitrogen Species in Skeletal Muscle. Front Cell Dev Biol. Feb 21;10:826981. (2022) [↩]
- Huang T, Zhang T, Gao J. Targeted mitochondrial delivery: A therapeutic new era for disease treatment. J Control Release. Mar;343:89-106. (2022) [↩]
- Hayashida K, Takegawa R, Endo Y, Yin T, Choudhary RC, Aoki T, Nishikimi M, Murao A, Nakamura E, Shoaib M, Kuschner C, Miyara SJ, Kim J, Shinozaki K, Wang P, Becker LB. Exogenous mitochondrial transplantation improves survival and neurological outcomes after resuscitation from cardiac arrest. BMC Med. 2023;21(1):56. [↩]
- McCully JD, Cowan DB, Pacak CA, Toumpoulis IK, Dayalan H, Levitsky S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol. 2009 Jan;296(1):H94-H105. [↩]
- McCully JD, Del Nido PJ, Emani SM. Mitochondrial transplantation: the advance to therapeutic application and molecular modulation. Front Cardiovasc Med. Dec 15;10:1268814. (2023) [↩] [↩]
- Emani SM, Piekarski BL, Harrild D, Del Nido PJ, McCully JD. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J Thorac Cardiovasc Surg. Jul;154(1):286-289. (2017) [↩]
- Tsai HY, Tsai KJ, Wu DC, Huang YB, Lin MW. Transplantation of gastric epithelial mitochondria into human gastric cancer cells inhibits tumor growth and enhances chemosensitivity by reducing cancer stemness and modulating gastric cancer metabolism. Stem Cell Res Ther. 2025 Feb 23;16(1):87. [↩]
- Rossi A, Asthana A, Riganti C, Sedrakyan S, Byers LN, Robertson J, Senger RS, Montali F, Grange C, Dalmasso A, Porporato PE, Palles C, Thornton ME, Da Sacco S, Perin L, Ahn B, McCully J, Orlando G, Bussolati B. Mitochondria Transplantation Mitigates Damage in an In Vitro Model of Renal Tubular Injury and in an Ex Vivo Model of DCD Renal Transplantation. Ann Surg. 2023 Dec 1;278(6):e1313-e1326. [↩]
- Huang T, Zhang T, Gao J. Targeted mitochondrial delivery: A therapeutic new era for disease treatment. J Control Release. Mar;343:89-106. (2022) [↩]
- Kasuno K, Shirakawa K, Yoshida H, Mori K, Kimura H, Takahashi N, Nobukawa Y, Shigemi K, Tanabe S, Yamada N, Koshiji T, Nogaki F, Kusano H, Ono T, Uno K, Nakamura H, Yodoi J, Muso E, Iwano M. Renal redox dysregulation in AKI: application for oxidative stress marker of AKI. Am J Physiol Renal Physiol. 2014 Dec 15;307(12):F1342-51. [↩]
- Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, Jaber BL; Acute Kidney Injury Advisory Group of the American Society of Nephrology. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol 8: 1482–1493. (2013) [↩]
- Noble RA, Lucas BJ, Selby NM. Long-term outcomes in patients with acute kidney injury. Clin J Am Soc Nephrol 15: 423–429. (2020) [↩]
- Xu ZH, Wang C, He YX, Mao XY, Zhang MZ, Hou YP, Li B. Hypoxia-inducible factor protects against acute kidney injury via the Wnt/β-catenin signaling pathway. Am J Physiol Renal Physiol. Jun 1;322(6):F611-F624. (2022) [↩] [↩] [↩]
- Moore PK, Hsu RK, Liu KD. Management of Acute Kidney Injury: Core Curriculum 2018. Am J Kidney Dis. Jul;72(1):136-148. (2018) [↩]
- Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, Shi L, Meloni BP, Zhang C, Zheng M, Gao J. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther. Feb 16;6(1):65. (2021) [↩]
- Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, Shi L, Meloni BP, Zhang C, Zheng M, Gao J. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther. Feb 16;6(1):65. (2021) [↩]
- Emani SM, Piekarski BL, Harrild D, Del Nido PJ, McCully JD. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J Thorac Cardiovasc Surg. Jul;154(1):286-289. (2017) [↩]
- D’Amato M, Morra F, Di Meo I, Tiranti V. Mitochondrial Transplantation in Mitochondrial Medicine: Current Challenges and Future Perspectives. Int J Mol Sci. Jan 19;24(3):1969. (2023) [↩]
- D’Amato M, Morra F, Di Meo I, Tiranti V. Mitochondrial Transplantation in Mitochondrial Medicine: Current Challenges and Future 19;24(3):1969. (2023) [↩]
- Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, Fujimoto S, Terada Y. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. Aug 15;305(4):F495-509. (2013) [↩]
- Liu H, Li Y, Xiong J. The Role of Hypoxia-Inducible Factor-1 Alpha in Renal Disease. Molecules. 2022 Oct 28;27(21):7318. [↩]
- Li X, Chen W, Feng J, Zhao B. The effects of HIF-1α overexpression on renal injury, immune disorders and mitochondrial apoptotic pathways in renal ischemia/reperfusion rats. Transl Androl Urol. Oct;9(5):2157-2165. (2020) [↩]
- Qiu S, Chen X, Pang Y, Zhang Z. Lipocalin-2 protects against renal ischemia/reperfusion injury in mice through autophagy activation mediated by HIF1α and NF-κb crosstalk. Biomed Pharmacother. Dec;108:244-253. (2018) [↩]
- Li ZL, Ji JL, Wen Y, Cao JY, Kharbuja N, Ni WJ, Yin D, Feng ST, Liu H, Lv LL, Liu BC, Wang B. HIF-1α is transcriptionally regulated by NF-κB in acute kidney injury. Am J Physiol Renal Physiol. Aug 1;321(2):F225-F235. (2021) [↩]
- Rashid H, Jali A, Akhter MS, Abdi SAH. Molecular Mechanisms of Oxidative Stress in Acute Kidney Injury: Targeting the Loci by Resveratrol. Int J Mol Sci. Dec 19;25(1):3. (2023) [↩]
- Li X, Chen W, Feng J, Zhao B. The effects of HIF-1α overexpression on renal injury, immune disorders and mitochondrial apoptotic pathways in renal ischemia/reperfusion rats. Transl Androl Urol. Oct;9(5):2157-2165. (2020) [↩]
- Wei W, Ma N, Fan X, Yu Q, Ci X. The role of Nrf2 in acute kidney injury: Novel molecular mechanisms and therapeutic approaches. Free Radic Biol Med. Oct;158:1-12. (2020) [↩]
- Wei W, Ma N, Fan X, Yu Q, Ci X. The role of Nrf2 in acute kidney injury: Novel molecular mechanisms and therapeutic approaches. Free Radic Biol Med. Oct;158:1-12. (2020 [↩]
- Kim JY, Jo J, Kim K, An HJ, Gwon MG, Gu H, Kim HJ, Yang AY, Kim SW, Jeon EJ, Park JH, Leem J, Park KK. Pharmacological Activation of Sirt1 Ameliorates Cisplatin-Induced Acute Kidney Injury by Suppressing Apoptosis, Oxidative Stress, and Inflammation in Mice. Antioxidants (Basel).Aug 19;8(8):322. (2019 [↩]
- Xu S, Gao Y, Zhang Q, Wei S, Chen Z, Dai X, Zeng Z, Zhao KS. SIRT1/3 Activation by Resveratrol Attenuates Acute Kidney Injury in a Septic Rat Model. Oxid Med Cell Longev. 2016:7296092. (2016) [↩]
- Khajevand-Khazaei MR, Mohseni-Moghaddam P, Hosseini M, Gholami L, Baluchnejadmojarad T, Roghani M. Rutin, a quercetin glycoside, alleviates acute endotoxemic kidney injury in C57BL/6 mice via suppression of inflammation and up-regulation of antioxidants and SIRT1. Eur J Pharmacol. Aug 15;833:307-313. (2018) [↩]
- Shi S, Lei S, Tang C, Wang K, Xia Z. Melatonin attenuates acute kidney ischemia/reperfusion injury in diabetic rats by activation of the SIRT1/Nrf2/HO-1 signaling pathway. Biosci Rep.Jan 15;39(1):BSR20181614. (2019) [↩]
- Raji-Amirhasani A, Khaksari M, Darvishzadeh Mahani F, Hajializadeh Z. Activators of SIRT1 in the kidney and protective effects of SIRT1 during acute kidney injury (AKI) (effect of SIRT1 activators on acute kidney injury. Clin Exp Nephrol. Aug;25(8):807-821. (2021) [↩]
- Raji-Amirhasani A, Khaksari M, Darvishzadeh Mahani F, Hajializadeh Z. Activators of SIRT1 in the kidney and protective effects of SIRT1 during acute kidney injury (AKI) (effect of SIRT1 activators on acute kidney injury. Clin Exp Nephrol. Aug;25(8):807-821. (2021 [↩]
- Zhou J, Wang X, Wang M, et al. The lysine catabolite saccharopine impairs development by disrupting mitochondrial homeostasis. J Cell Biol.218(2):580-597.(2019) [↩]
- Liu J, Han X, Zhang T, Tian K, Li Z, Luo F. Reactive oxygen species (ROS) scavenging biomaterials for anti-inflammatory diseases: from mechanism to therapy. J Hematol Oncol. 16(1):116.(2023) [↩]
- Liu J, Han X, Zhang T, Tian K, Li Z, Luo F. Reactive oxygen species (ROS) scavenging biomaterials for anti-inflammatory diseases: from mechanism to therapy. J Hematol Oncol. 16(1):116.(2023) [↩]
- Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int.Jul;80(1):29-40.(2011) [↩] [↩]
- Saikumar P, Venkatachalam MA. Role of apoptosis in hypoxic/ischemic damage in the kidney. Semin Nephrol. Nov;23(6):511-2.(2003) [↩]
- Rui Y, Li S, Luan F, Li D, Liu R, Zeng N. Several Alkaloids in Chinese Herbal Medicine Exert Protection in Acute Kidney Injury: Focus on Mechanism and Target Analysis. Oxid Med Cell Longev. May 13;2022:2427802. (2022) [↩]
- Saikumar P, Venkatachalam MA. Role of apoptosis in hypoxic/ischemic damage in the kidney. Semin Nephrol. Nov;23(6):511-2.(2003) [↩]
- Yao C, Li Z, Sun K, Zhang Y, Shou S, Jin H. Mitochondrial dysfunction in acute kidney injury. Ren Fail. 2024 Dec;46(2):2393262. [↩]
- Hoogstraten CA, Hoenderop JG, de Baaij JHF. Mitochondrial Dysfunction in Kidney Tubulopathies. Annu Rev Physiol. 2024 Feb 12;86:379-403. [↩]
- Chen, Y., Li, Z., Zhang, H. et al. Mitochondrial metabolism and targeted treatment strategies in ischemic-induced acute kidney injury. Cell Death Discov. 10, 69 (2024). [↩]
- Rui Y, Li S, Luan F, Li D, Liu R, Zeng N. Several Alkaloids in Chinese Herbal Medicine Exert Protection in Acute Kidney Injury: Focus on Mechanism and Target Analysis. Oxid Med Cell Longev. May 13;2022:2427802. (2022) [↩]



{kind=link}