
Abstract
Vemurafenib, as a first-generation BRAF inhibitor, has received approval for use in cases of BRAF V600E-mutated melanoma. The clinical utility of Vemurafenib is limited due to low target specificity, off-target toxicity, and improper delivery pathways. The study proposes a dual-targeted delivery system using gold nanoparticles (AuNPs) that target the Alpha-V Beta-3 ( ) integrin receptor, potentially promoting increased binding specificity, enhanced target release, and intracellular availability. Molecular docking simulations and structural validations were completed to collect binding affinity data, structural validation, and analysis. We compared two different nanoparticles, Polyamidoamine (PAMAM) and AuNP, and selected the optimal one to conduct docking affinity evaluation for the integrin receptor. Upon molecular docking analysis, PAMAM dendrimers exhibited a weak bonding affinity with Vemurafenib. On the contrary, gold nanoparticles (AuNPs) were associated with direct bonds to both the drug and the integrin receptor, revealing promising prospects in target-specific delivery. The subsequent exercise of molecular simulations was done to reconfirm the validity of the structural compatibility and favorable orientation of the nanoparticle in receptor binding. This method of dual-ligand targeting is potentially beneficial to the treatment of melanoma, as it relates to the process of recognition of growth factors influenced by the local binding of the tumor receptor and the subsequent tumor cell proliferation. These results are highly advisable for the field of nanomedicine and locations where local interventions for melanoma control are of urgent importance, particularly in high UV-exposed areas.
) integrin receptor, potentially promoting increased binding specificity, enhanced target release, and intracellular availability. Molecular docking simulations and structural validations were completed to collect binding affinity data, structural validation, and analysis. We compared two different nanoparticles, Polyamidoamine (PAMAM) and AuNP, and selected the optimal one to conduct docking affinity evaluation for the integrin receptor. Upon molecular docking analysis, PAMAM dendrimers exhibited a weak bonding affinity with Vemurafenib. On the contrary, gold nanoparticles (AuNPs) were associated with direct bonds to both the drug and the integrin receptor, revealing promising prospects in target-specific delivery. The subsequent exercise of molecular simulations was done to reconfirm the validity of the structural compatibility and favorable orientation of the nanoparticle in receptor binding. This method of dual-ligand targeting is potentially beneficial to the treatment of melanoma, as it relates to the process of recognition of growth factors influenced by the local binding of the tumor receptor and the subsequent tumor cell proliferation. These results are highly advisable for the field of nanomedicine and locations where local interventions for melanoma control are of urgent importance, particularly in high UV-exposed areas.
Keywords: Vemurafenib, Gold Nanoparticles, Targeted Drug Delivery, Melanoma, Molecular Docking, , Integrin, BRAF, Nanomedicine
Introduction
Melanoma, from the Greek word for “black tumor,” is the most dangerous form of skin cancer due to its aggressive growth potential and high potential for metastasis1. Melanoma accounts for approximately 90% of cases related to ultraviolet (UV) radiation2, most intense in equatorial regions and high-altitude regions. Ecuador, located directly along the equator and with the high-altitude capital city of Quito, offers a distinctive and pressing environment to study melanoma. Studies in Ecuador are motivated by the geographical location of the region in combination with the need for better and more precise therapeutic options. Skin cancer can develop into aggressive melanoma, which is characterized by a high rate of mutations and both metastatic behavior and resistance to traditional chemotherapy treatments3. BRAF is a gene located on chromosome seven that produces both the BRAF gene and the protein. The protein helps cells grow through internal signaling mechanisms, which support cell division as one of its primary functions. Vemurafenib belongs to a category of targeted cancer drugs called cancer growth blockers. Vemurafenib is a BRAF V600E inhibitor, and has the ability to treat unresectable melanoma skin cancer and advanced metastasis4. Targeted melanoma therapy utilizes Vemurafenib as its primary therapeutic component; however, systemically administered Vemurafenib often leads to non-specific distribution, off-target toxicity, and future drug resistance development5. Recent advances in nanomedicine have opened possibilities for artificially engineered nanoparticles capable of delivering medication precisely to tumor cells without harming healthy tissues, minimizing collateral damage6.
Even though ligand-targeted nanoparticles have showcased their potential, single-ligand models are impractical due to the fact that solid tumors are heterogeneous, which makes it difficult for the selective reaction to occur7. The presence of competitive inhibitors at binding sites weakens the strength of the drug, especially when both the carrier nanoparticle and the drug target at the same proteins. In the same way, nanoparticles and Vemurafenib both compete for the BRAF protein binding site, which may have limitation for Vemurafenib’s binding to BRAF. Therefore, the study illustrates a two-ligand approach that is presented by the idea of attaching nanoparticles to the surface receptor of a protein, so that it allows Vemurafenib to bind to BRAF unimpeded, thus achieving complete drug efficiency.
This research brings a new delivery system via dual-ligand methods using Polyamidoamine (PAMAM) dendrimers8 and gold nanoparticles (AuNP)9 as model carriers, testing its binding affinity to both the drug and the surface receptor and finding the optimal nanoparticle; thus improving drug targeting effects and drug release simulation with enhanced specificity levels. The innovation enables both the enhancement of current nanomedicine processes and provides a computational platform for future experimental validation.
integrin is an important transmembrane receptor that has been shown to play a critical role in tumor angiogenesis, invasion, and metastasis, and researchers previously documented overexpression in several aggressive melanomas10. These properties provide a good rationale for receptor-mediated drug delivery using as a targeted strategy, specifically by taking advantage of active targeting and the preferential accumulation of particles through enhanced permeability. However, it is also essential to acknowledge that is not purely tumor-specific, and while highly expressed in malignant cells, it is also expressed at lower levels in some normal endothelial cells, osteoclasts, and smooth muscle cells, which raises concerns about off-target binding and toxicity. Therefore, while this study uses docking in a proof-of-concept study for targeted drug delivery, future work should assess the differences in receptor density between melanoma and healthy tissue, provide comparative docking to other integrin subtypes, and better elucidate the ligand specificity to reduce off-target effects.
The AuNP–VG16KRKP construct was chosen over unmodified AuNPs in this study. VG16KRKP is an artificial antimicrobial peptide that was originally created for targeting intracellular bacterial infections, especially those induced by S. Typhi11. The peptide contains seven amino acids with a sequence of Val-Gly-Lys-Arg-Lys-Arg-Pro. Although the conventional use of AuNP-VG16KRKP lies in antimicrobial therapy, in this study, the potential of the AuNP–VG16KRKP complex as a novel drug delivery system for melanoma treatment is demonstrated. The hypothesis that motivates the current research is that VG16KRKP’s (lysine and arginine) positively charged residues may increase electrostatic interactions with Vemurafenib and thus improve cellular uptake12. For this design, the gold nanoparticle is the carrier while the peptide is the targeting ligand that can be used theoretically for delivery and internalization of the therapeutic agent13.
This choice of using AuNP–VG16KRKP was due to several key reasons. To begin with, VG16KRKP excerpts cell membrane penetration, thus greatly promoting the internalization of nanoparticles into the target cells14. Furthermore, although VG16KRKP was initially designed for antimicrobial use, it is cationic and amphipathic as well—two features that make it easy to interact with cell membranes negatively charged, such as those of cancer cells, and intracellular vesicles like endosomes and lysosomes. These traits are very likely to be the best options for drug delivery in the intracellular space, especially in the acidic microenvironment of the tumor tissues. Likewise, VG16KRKP can help Vemurafenib to disrupt the endosomes and membranes15; hence, it can better access its intracellular targets, in this case, the BRAF V600E in the melanoma cells. Thus, enabling the transportation and the intracellular concentration of Vemurafenib, the AuNP–VG16KRKP platform, sees a potentially effective way of theoretically elevating the therapeutic efficacy in melanoma treatment. A scoring matrix system based on available literature will be used below to visualize the effectiveness of AuNP and AuNP–VG16KRKP complex. An in-depth molecular docking analysis and the evaluation of the binding affinity will be given in the discussion section in order to provide further substantiation of these findings.
Originally characterized for antimicrobial activity, VG16KRKP works via membrane disruption in bacterial cells. Antimicrobial peptides (AMPs), like anticancer peptides (ACPs), are cationic, amphipathic, and membrane-active and penetrate cells, however, their target membranes differ in lipid composition, charge density, and receptor expression. VG16KRKP was chosen for this study because its availability aligns with the objective of this study and due to the potential that VG16KRKP will augment electrostatic association with melanoma cell membranes, and given it is membrane-active and has cell penetrating properties, we hypothesized VG16KRKP would enhance nanoparticle association with negatively charged cancer cell membranes and facilitate internalization16. Cationic residues (lysine, arginine) increase the electrostatic attraction to negatively charged membrane components, and encourage endosomal uptake and membrane translocation17. Previous work has shown the successful binding of peptides to AuNP scaffolds, and has demonstrated the ability to enhance cell uptake, indicating that this strategy is chemically feasible18. While VG16KRKP was not selected because it is a known integrin binder, this study models targeting with peptide-conjugated AuNPs in the presence of affixed docking studies to evaluate the nanoparticle-integrin interface. We acknowledge that VG16KRKP’s melanoma specific targeting ability is yet to be established. Experimental validation such as peptide-integrin binding assays, competitive binding studies with known ligands, and in vitro cell uptake studies using melanoma lines, will be necessary to confirm whether VG16KRKP provides selective targeting, or if a different peptide (such as RGD derivative would be improved).
| Score | Performance Description |
| 0 | No capability or significant disadvantage |
| 1–2 | Minimal effect |
| 3 | Moderate effect but with limitations |
| 4 | Good performance, well-documented in literature |
| 5 | Optimal effect, strong evidence |
Description: A scoring matrix system based on available literature is used to visualize the effectiveness of AuNP and AuNP–VG16KRKP complex. An in-depth molecular docking analysis and the evaluation of the binding affinity will be given in the discussion section in order to provide further substantiation of these findings. Each feature was assigned a score from 0 (poor performance) to 5 (excellent performance) based on available experimental and literature-based evidence.
| Criterion | Importance for Vemurafenib Delivery |
| Targeting Capability | Determines how specifically the nanoparticle reaches tumor cells (e.g., melanoma), affecting drug accumulation and side effects. |
| Cellular Uptake | Affects how efficiently Vemurafenib is taken up into cancer cells, where BRAF V600E is located. |
| Intracellular Delivery | Directly impacts the ability of the nanoparticle to release the drug in the cytoplas, where the target (BRAF) is located. |
| Cytotoxicity Profile | Affects safety and therapeutic index, essential for ensuring normal cells are not harmed. |
Description: The comparison is based on five core features that are critical to the success of nanoparticle-based drug delivery systems. These features were selected for their direct relevance to Vemurafenib delivery efficiency, intracellular targeting, safety, and broader therapeutic potential.
| Feature | Bare AuNPs (score 1–5) | AuNP–VG16KRKP (score 1–5) |
| Targeting Capability | 3 (Passive accumulation relies on EPR effect for tumor targeting, leading to non-specific distribution19) | 4 (Facilitates specific interactions with membranes, enhancing targeting of infected cells14) |
| Cellular Uptake | 3 (Can bypass endocytosis; however, requires a large charge density20) | 5 (Promotes efficient internalization into host cells, improving intracellular delivery11) |
| Intracellular Delivery | 3 (Able to penetrate cellular membranes, however, causing disorders in the surrounding lipid20) | 5 (Demonstrated capability to potentially deliver therapeutic agents into infected cells, effectively targeting intracellular bacteria11) |
| Cytotoxicity Profile | 3 (Exhibit cytotoxicity when AuNPs penetrate the cell membrane, disrupting its structure by disordering the lipids20) | 5 (Significantly reduced cytotoxicity, preserving biocompatibility21) |
| Total score | 12/20 | 19/20 |
Description: With the evidence presented above, we can visualize the justification for the creation of the hypothesis of the increased effectiveness of AuNP–VG16KRKP. Literature references and sources used in this table are adapted from Chowdhury et al., 2017, Datta et al., 2016, Ioanna-Aglaia Vagena et al., 2025, Lin et al., 2010, Mohid et al., 2022
The primary aim of the research is to create an in silico nanoparticle-mediated dual-ligand targeting approach to deliver Vemurafenib to melanoma cells. This is achieved through molecular docking in AutoDock Vina to evaluate ligand-receptor and drug-target docking affinities, and then analyze docking poses in Chimera. Finally, show the complete process of a nanoparticle-drug device performing receptor-mediated release of the drug and BRAF inhibition within cancer cells. It is hypothesized that AuNPs will be better than PAMAM dendrimers in the roles of not only facilitating drug delivery but also performing the receptor-mediated targeting and releasing of Vemurafenib22, because the former could possess a more favorable structure compatibility with Vemurafenib and better interaction with the melanoma-specific surface receptors23.
This study focuses solely on in silico simulations, which include conducting molecular docking and structural analysis to predict the feasibility of the existence of a dual ligand system24. It is confined to the scope of computational modeling and predictive simulation. The complexity of in vivo conditions, including pharmacokinetics, immune responses, systemic toxicity, and biodistribution, is not considered in this phase. Likewise, no chemical synthesis, biological assays, or experimental validation are included, due to limited access to laboratory resources and constraints on material availability. As such, the study selects only two accessible nanoparticles to construct a conceptual framework for dual-ligand targeting. The findings are intended to serve as a theoretical basis for future experimental work in targeted melanoma therapy. Additionally, the final model is intended as a conceptual demonstration of the theoretical sequence needed to achieve a theoretically precise binding of Vemurafenib to the intracellular BRAF target. It isolates each step as an independent docking event, focusing on structural feasibility rather than simulating the dynamic, biochemical processes that would occur in vivo. The research leverages principles of receptor-ligand binding kinetics25, surface plasmon chemistry of AuNPs26, and nanoparticle encapsulation techniques27. Computational drug discovery methodologies, especially docking algorithms and molecular surface modeling, underpin the predictive aspects of this study. Ligand-receptor docking scores were compared to assess targeting potential, while spatial visualization supported hypotheses about nanoparticle-mediated delivery.
Results
Systematic Review
Books, articles, and websites were searched extensively in the databases of PubMed, NIH, and Web of Science using the search terms: “nanoparticle drug delivery”, “melanoma”, “dual ligand targeting”, “nanoparticles”, “gold nanoparticles”, and “Vemurafenib.” Articles discussing the mechanism of action of Vemurafenib as a melanoma treatment, and relevant articles about recent advancements in nanoparticle treatments were included. Compilations that used computational modeling involved multiple structure selections blasted from 3D PDB structures and obtained from RCSB PDB and PubChem. Relevant data from each selected article were extracted, including the type of nanoparticle, the ligand used, the targeted type of cancer and receptor, the method’s specificity for docking, the software used for docking, the key findings including binding affinities, and delivery efficacies. Findings not only included thematic analysis of each relevant finding, but they were also analyzed for comparisons between studies with nanoparticles based on type, and some studies concurred with drug-binding performance with delivery specificity. They then analyzed further trends between methodological strengths and innovative approaches to ligand conjugation. The methodological quality of each study was also assessed using the PRIMSA checklist. The basis of quality assessment was based on transparency of methodology, consistency of methodology, reproducibility, validation of software, and interpretation of coupled docking results.
Molecular Docking and Analysis
Vemurafenib–BRAF V600E Docking (CB-Dock2)
BRAF is a serine/threonine-protein kinase in the MAPK/ERK signaling cascade, and it contains an ATP-binding cleft formed between its N-terminal and C-terminal lobes. The V600E mutation replaces valine with glutamic acid at residue 600, causing the kinase to be stabilized in its active conformation, resulting in constitutive downstream signaling that drives proliferation of melanoma cells. Vemurafenib (e.g., PDB ID: 4RZV) binds in a type I½ binding mode, interacting with BRAF and occupying the ATP-binding pocket of BRAF V600E28. The types of stabilizing interactions made by vemurafenib include a hydrogen bond from the sulfonamide group of vemurafenib to hinge residue, Cys532;  stacking interactions between the two aromatic moieties of vemurafenib and Phe595; and hydrophobic interactions with Val471, Ala481, and Ile527. In the docking simulations presented here, vemurafenib had a predicted binding affinity of –5.4 kcal/mol with BRAF V600E, reproducing the hinge-binding hydrogen bond and hydrophobic interactions seen in crystallographic complexes. The predicted binding pose showed an RMSD of 0.715 Å from the experimental reference structure, suggesting the docking protocol is reliable, and validating the interaction model.
stacking interactions between the two aromatic moieties of vemurafenib and Phe595; and hydrophobic interactions with Val471, Ala481, and Ile527. In the docking simulations presented here, vemurafenib had a predicted binding affinity of –5.4 kcal/mol with BRAF V600E, reproducing the hinge-binding hydrogen bond and hydrophobic interactions seen in crystallographic complexes. The predicted binding pose showed an RMSD of 0.715 Å from the experimental reference structure, suggesting the docking protocol is reliable, and validating the interaction model.
To evaluate Vemurafenib – BRAF V600E binding, molecular docking was conducted using CB-Dock2, which integrates automated cavity detection with AutoDock Vina scoring. Five candidate binding pockets were predicted, with binding energies ranging from –5.8 to –12.4 kcal/mol:
| CurPocket ID | Vina Score | Cavity Volume (ų) | Center (x, y, z) | Docking Size (x, y, z) |
| C1 | -12.4 | 3799 | 58, 20, 48 | 22, 35, 22 |
| C2 | -10.9 | 2536 | 85, 11, 15 | 22, 22, 22 |
| C4 | -8.6 | 1095 | 91, 9, 33 | 22, 22, 22 |
| C3 | -7.4 | 1604 | 70, 15, 31 | 22, 31, 22 |
| C5 | -5.8 | 853 | 80, 24, 63 | 22, 22, 22 |
Description: All data in this table were generated in this study.
Residues involved in the C1 pocket include: ILE463, GLY464, SER465, SER467, VAL471, TYR472, ALA481, VAL482, LYS483, LEU505, LEU514, ILE527, THR529, GLN530, TRP531, CYS532, SER535, SER536, HIS539, ASN580, ASN581, PHE583, ILE592, GLY593, ASP594, PHE595, GLY596, and LEU597—all located in Chain B.
The C1 pocket was identified to have the highest binding affinity of -12.4 kcal/mol; therefore, the most worthy of examination of its interactions. Furthermore, its location, nearby to the ATP-binding pocket of BRAF V600E, indicates that the pocket is completely within the area of the known kinase inhibitor Vemurafenib. The most critical parts of the structure are arguably the hinge area that joins the ligand with the amino acid residue Cys532, and together with a DFG motif at position Asp594. Several hydrophobic regions were noted, such as Leu514, Ile527, and Val471, while pi-stacking was found at Phe583. The representation of the amino acids—residues, is located in some drawings—Ile463, Gly464, Ser465, Ser467, Tyr472, Val482, Leu505, Thr529, Ser535, Ser536, His539, Asn580, Asn581, Ile592, and Leu597— that are positioned around the ATP-binding pocket as secondary stabilizers and also participate in numerous interactions.
According to the above observations, it is anticipated that Vemurafenib will form a hydrogen bond in the hinge region by its sulfonamide group to Cys532 and also Asp594 within the DFG motif. The binding of the aromatic rings of Vemurafenib with the interacting amino acid residues Leu514, Ile527, and Val471 is via hydrophobic contacts and is also estimated. Furthermore, the Pi-stacking will be one of the interactions of Phe583 with the fluorophenyl ring of Vemurafenib.
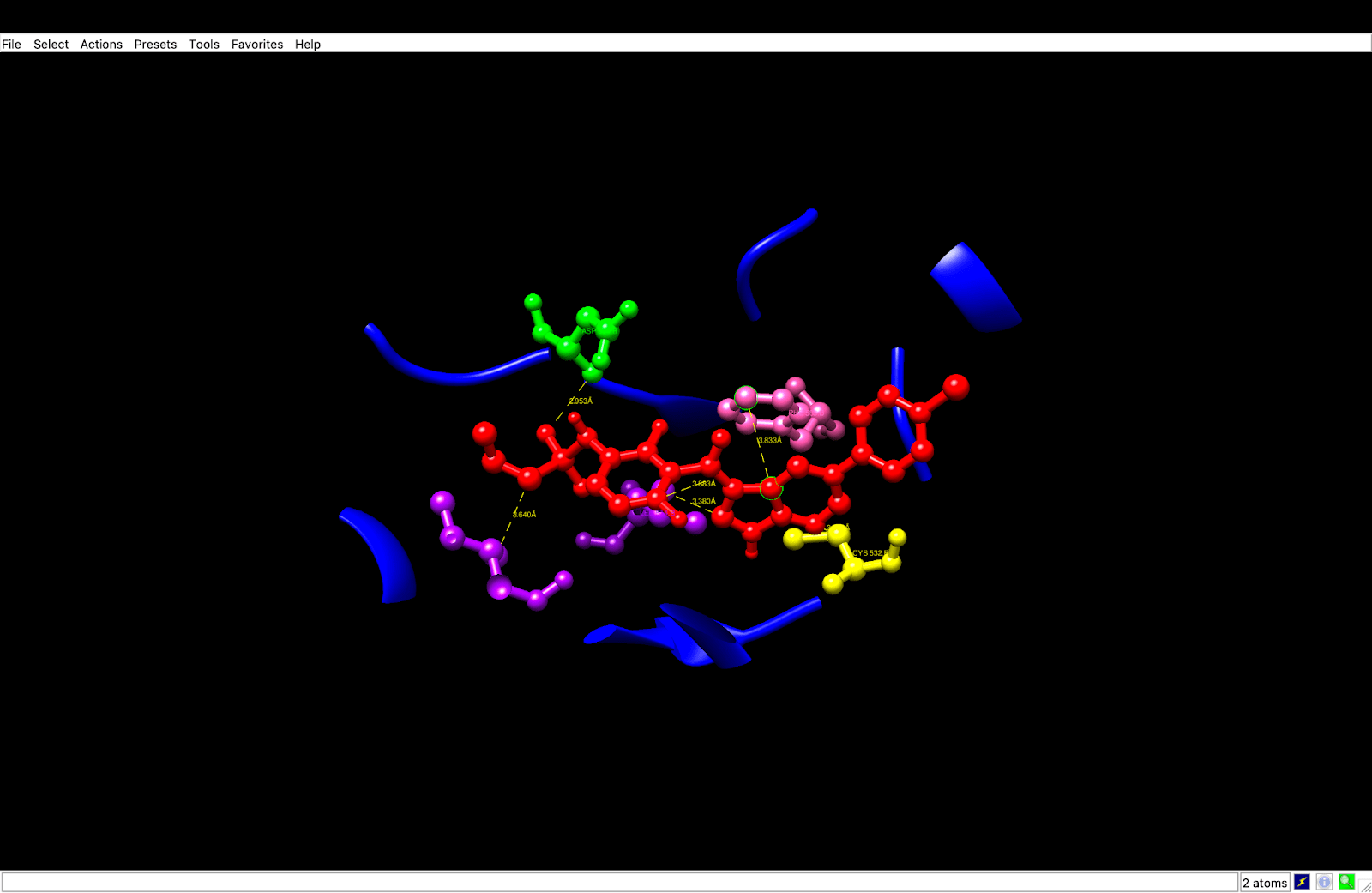
To verify the above-mentioned interactions, the C1 pose was isolated and visualized in UCSF Chimera. According to the analysis, the C1 pose was manually identified as hydrogen bonds, so it was possible to identify the hydrogen bond between the sulfonamide oxygen of Vemurafenib and the backbone nitrogen of Cys532 at a distance of 3.351 Å, and the inhibitor binding was confirmed within the hinge region and a similar process like ATP’s interaction was performed. Additionally, the distance of 2.953 Å between Vemurafenib’s polar group and the side-chain oxygen of Asp594 confirmed the inactivation DFG-out conformation. Both bonds were found to be of the most common donor-acceptor distance (< 3.5 Å), consistent with the literature, thereby confirming the role of Vemurafenib in kinase inhibition through the essential interactions.

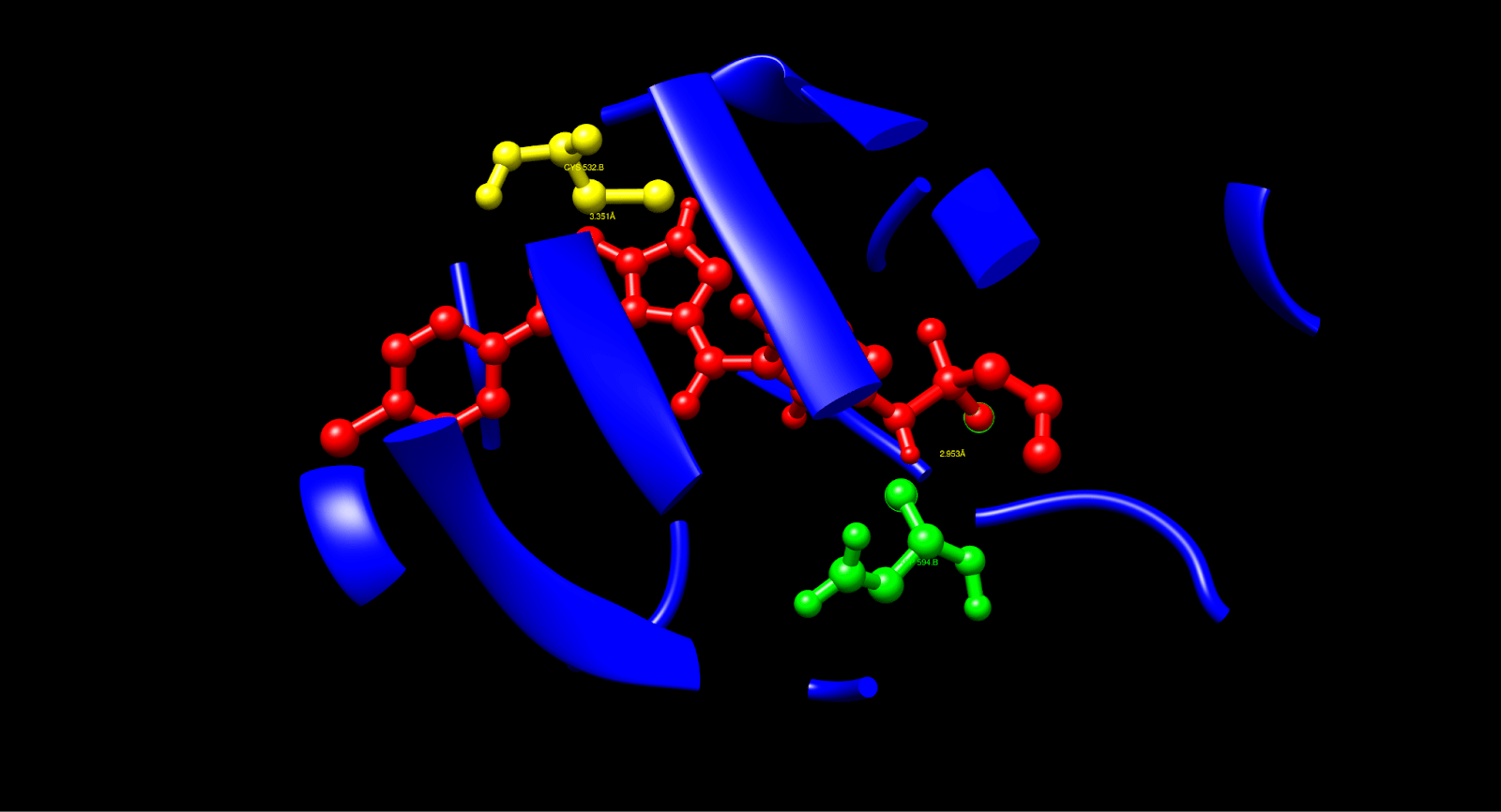
After a manual identification process, the hydrophobic interactions were recognized: Leu514 had a distance of 3.380 Å, which was the interaction of the aromatic carbons of Vemurafenib and the side chain carbons of Leu514, e.g., CD1, CD2. Val471 had a distance of 3.883 Å, which was achieved in contact with the side chain CG1, CG2. Ile527 had a distance of 3.640 Å, which was also in contact with the side chain CD1, CG2. These interactions display that hydrophobic contacts (< 4.0 Å) cause Vemurafenib to be stable in the ATP pocket, in line with its known binding mode, thus increasing binding affinity and specificity.

The process also identified Pi-Stacking interaction: Phe583 determined a 3.833 Å distance between the fluorophenyl ring of Vemurafenib and the phenyl ring of Phe583, nearly parallel arrangement. Pi-stacking also offers extra stability, which is an often-seen trait in kinase inhibitors such as Vemurafenib.

The fully analyzed structure is shown below:

| Interaction Type | Residue | Distance (Å) | Functional Role |
| Hydrogen Bond | Asp594 (green) | 2.953 | Stabilizes inactive DFG-out conformation |
| Hydrogen Bond | Cys532 (yellow) | 3.351 | Anchors the ligand at the hinge region |
| Hydrophobic Contact | Leu514 (purple) | 3.380 | Enhances pocket stability |
| Hydrophobic Contact | Val471 (purple) | 3.883 | Stabilizes aromatic core |
| Hydrophobic Contact | Ile527 (purple) | 3.640 | Adds additional hydrophobic interactions |
| Pi-Stacking | Phe583 (pink) | 3.833 | Aromatic stabilization |
Description: All data in this table were generated in this study.
Docking Validation and Structural Accuracy Against 3OG7
To further evaluate the accuracy of the docked pose, the ligand conformation from the C1 complex was aligned with the Vemurafenib molecule from PDB 3OG7 based on the match command in UCSF Chimera. The root-mean-square deviation (RMSD) between the two structures was 0.715 Å, indicating a high level of structural alignment. This low RMSD value (< 2 Å) also further indicates that the C1 pose is very accurate, nearly identical, in terms of structure and orientation to the structure determined experimentally in 3OG7, indicating compelling support for the docking and its associated results.

The descriptive interaction analyses from above and validation against the experimental data in 3OG7 (RMSD = 0.715 Å) demonstrate that the docked pose of Vemurafenib in BRAF V600E is accurate and realistic, as the H-bond interactions, hydrophobic interactions, and pi-stacking interactions are consistent with Vemurafenib’s known mechanism of anticancer activity.
Gefitinib was also used to validate the docking protocol as a negative control, given that it is a drug-like structure but has no measurable inhibition of BRAF. Using the same docking software and methods, Gefitinib gave a docking score of -8.8 kcal/mol which was 3.6 kcal/mol less favorable than the best scoring active ligand, vemurafenib (-12.4 kcal/mol). This separation illustrates that the docking protocol was able to distinguish between a known BRAF inhibitor and a non-binding molecule.
| CurPocket ID | Vina Score | Cavity Volume (ų) | Center (x, y, z) | Docking Size (x, y, z) |
| C2 | -8.8 | 2121 | 1, -12, 41 | 22, 22, 22 |
| C1 | -8.6 | 2436 | 4, -9, 15 | 22, 29, 22 |
| C3 | -8.6 | 1855 | 3, -19, 27 | 22, 22, 22 |
| C4 | -7.9 | 1065 | 3, -5, 6 | 22, 22, 22 |
| C5 | -5.4 | 309 | 23, -16, 57 | 22, 22, 22 |
Description: All data in this table were generated in this study.
PAMAM–BRAF V600E Docking
Initially, a PAMAM dendrimer was tested as a direct nanoparticle binder to BRAF for the delivery of Vemurafenib. Unfortunately, it was not effective as a drug delivery vehicle. Molecular docking identified that PAMAM cannot directly bind to the ATP-binding pocket of BRAF. Through CB-Dock2, the strongest binding interaction was a ΔG of -6.9 kcal/mol in the C1 pocket of BRAF, which indicated a modest interaction that could inhibit Vemurafenib’s targeting ability. The same docking was conducted in DockThor which resulted in a similar ΔG of -7.167 as well for the purpose of cross-validation which is later used to compare the affinity score against AuNP– in order to eliminate methodological bias. In order for Vemurafenib to inhibit BRAF’s kinase activity, the pocket must be completely available to its binding site. Since PAMAM occupies the space of the ATP-binding pocket, steric hindrance blocks Vemurafenib from binding and thus could potentially decreases the therapeutic effect. In addition, PAMAM dendrimers have a highly branched structure with positively charged amine groups that enhance non-specific electrostatic and hydrogen-bonding interactions with residues near the ATP pocket (e.g. Asp594 & Lys483) that stabilize PAMAM in the pocket and prevent Vemurafenib from binding effectively. PAMAM dendrimers also have a significant steric hindrance associated with their high molecular weight. For example, PAMAM dendrimers are typically used with an overall diameter of about 4-5 nm at G4-G5. Being a large dendrimer only increases steric hindrance and, with a partial resistance to displacing PAMAM during binding, only complicates binding to BRAF. Moreover, the intracellular PAMAM’s strong binding to BRAF inhibits PAMAM’s potential to act as a delivery vehicle.
To mitigate these issues, an alternative targeted approach has been suggested. For example, nanoparticles can be functionalized with targeting melanoma cell surface markers, such as Alpha-V Beta-3 () Integrin. Nanoparticles can bind to the melanoma cell surfaces selectively, thus delivering Vemurafenib intracellularly. Once bound, Vemurafenib could be released from the nanoparticle, thus giving Vemurafenib access to BRAF’s ATP pocket without having to compete with the nanoparticle, improving bioavailability and inhibition potential. This may also decrease cytotoxicity through lowering the intracellular exposure to nanoparticles. By targeting the surface of a cell and releasing Vemurafenib either on or shortly after endocytosis, the drug will have less time to cause off-target effects. This method will take advantage of the great potential of nanoparticles for targeted delivery without aiming to interact directly with BRAF, therefore could improve the effectiveness of Vemurafenib treatments.
PAMAM–Vemurafenib
PAMAM dendrimers are tested as a targeted drug delivery system for melanoma treatment to deliver BRAF inhibitor Vemurafenib, but the study had severe limitations that demonstrated its utility in the alternative delivery platform. Vemurafenib was not able to bind sufficiently to PAMAM, as evidenced by the molecular docking study that found a binding affinity (ΔG) of -3.2 kcal/mol; well below the targeted -4 to -6 kcal/mol range that is preferred for sustained drug loading and release. The non-specific electrostatic interactions between the positively charged amine groups of PAMAM and the polar regions of Vemurafenib (i.e, sulfonamide group) were enough to suggest the drug could dissociate before reaching the target melanoma cells.
Apart from the suboptimal binding affinity, PAMAM dendrimers did not promote sufficient stabilization of Vemurafenib. Although PAMAM dendrimers can encapsulate drugs via hydrophobic interactions and electrostatic interactions, the weak ΔG value (-3.2 kcal/mol) indicates there are no strong hydrogen bonds or hydrophobic contacts to retain the drug while in circulation. The risk of instability meant little drug retention and therefore, a decreased effective concentration of Vemurafenib delivered to melanoma cells and ultimately insufficient BRAF signaling inhibition. Additionally, binding affinity also made regulated release a challenge. Although PAMAM’s amine groups are further pH-responsive, they simply did not form enough specific interactions with Vemurafenib that would successfully provide good release in the acidic tumor microenvironment or intracellular environment, which can further inhibiting therapeutic efficacy.
To overcome these disadvantages, the delivery system was redesigned to be administered with gold nanoparticles (AuNPs). AuNP-based systems provide a multitude of additional benefits over the PAMAM-based approach. The gold nanoparticle acts like a stable scaffold, with VG16KRKP attached via thiol-gold chemistry. In addition, VG16KRKP has specific interactions with Vemurafenib, leading to better drug loading. The surface properties of AuNPs allow for controlled release mechanisms through the tumor microenvironment conditions that can benefit drug release from the AuNP-derived release systems. Ultimately, this opens the door to providing a buttress of additional treatment for Vemurafenib delivery through localized hyperthermia heating, which leads to increased cell membrane permeability, facilitating drug uptake and producing synergy of therapies along with BRAF inhibition. The AuNP-VG16KRKP system has a superior biocompatibility compared to PAMAM dendrimers. Gold nanoparticles can be easily functionalized with peptides such as VG16KRKP, which also provide a great flexibility for targeted delivery and potentially lower the cytotoxicity associated with delivering anticancer agents.
AuNP-VG16KRKP–Vemurafenib
| CurPocket ID | Vina Score | Cavity Volume (ų) | Center (x, y, z) | Docking Size (x, y, z) |
| C1 | -5.4 | 43 | -2, -8, 2 | 22, 22, 22 |
| C2 | -5.2 | 2 | -7, -7, -3 | 22, 22, 22 |
| C4 | -4.8 | 0 | -6, -6, -6 | 22, 22, 22 |
| C5 | -4.4 | 0 | 0, -6, -1 | 22, 22, 22 |
| C3 | -4.2 | 0 | -1, -7, -1 | 22, 22, 22 |
Description: All data in this table were generated in this study.
The efficacy of drug delivery systems is vital to improve the stability, cellular uptake, and the potential therapeutic efficacy of Vemurafenib for melanoma while minimizing off-target effects. With some of the disadvantages in mind, we conducted computational docking to assess the binding of Vemurafenib to a novel delivery system consisting of an AuNP-VG16KRKP. We wanted to transition from the PAMAM dendrimer system, which had a binding affinity (ΔG) of -3.426 kcal/mol, to a more effective target range of -4 to -6 kcal/mol.
Using computational analysis, it was determined that five different binding pockets existed, with the C1 pocket having the largest cavity volume (43 Å) and the most favorable energetics. The best docking pose in the C1 pocket had a Vina score of -5.4 kcal/mol, which is in the range of target affinity obtained and represents an improvement of approximately 2 kcal/mol better the previous work based on PAMAM. The molecular interactions were subsequently analyzed in UCSF Chimera, and contact residues on the VG16KRKP peptide (VAL1, ARG3, GLY4, LYS5, ARG6, LYS3) showed specific interactions with Vemurafenib.
Multiple interactions were identified as providing a molecular basis for the binding affinity. The most important of these involved the confirmation of a hydrogen bond between the NH3⁺ side chain of LYS5 and a sulfonamide oxygen in Vemurafenib at a distance of 3.12 Å (3.5 Å = max threshold for a stable hydrogen bond donor-acceptor distance). There are likely additional hydrogen bonds formed by LYS3, ARG3, and ARG6. The two non-polar residues, VAL1 and GLY4, exhibit complementary hydrophobic interactions with the aromatic rings of Vemurafenib. In the complex under study, the AuNP functions chiefly as a structural scaffold for stabilizing the bioactive conformation and likely forms only weak van der Waals contacts with the aromatic portions of the Vemurafenib molecule. The VG16KRKP peptide performs the bulk of mediating binding through its hydrogen bonds and electrostatic interactions with the drug molecule.
In addition to the molecular recognition and binding aspects provided by the AuNP component, there is also a host for peptide attachment via thiol-gold chemistry and inherent photothermal properties that can be utilized for near-infrared (NIR) laser therapy. The ability of these two features simultaneously increases the possibilities of a combined therapeutic effectiveness: Vemurafenib with its targeted BRAF inhibition, coupled with localized hyperthermia that could likely improve Vemurafenib penetration and efficacy. The Vemurafenib complex of AuNP-VG16KRKP-Vemurafenib demonstrated a reasonable binding affinity for the above chemistry to advance to experimental verification with a melanoma cell model. The computer docking data suggest that the AuNP-VG16KRKP-Vemurafenib complex can effectively transport Vemurafenib and release it directly at the melanoma site, while also potentially maximizing its effect on BRAF-mutant pathway inhibition and minimizing undesired, adverse systemic toxicity. Integrating the photothermal potential with Vemurafenib likely sets this platform apart from the traditional delivery system by potentially allowing for multiple therapeutic modes of delivery in a single nanoscale construct.
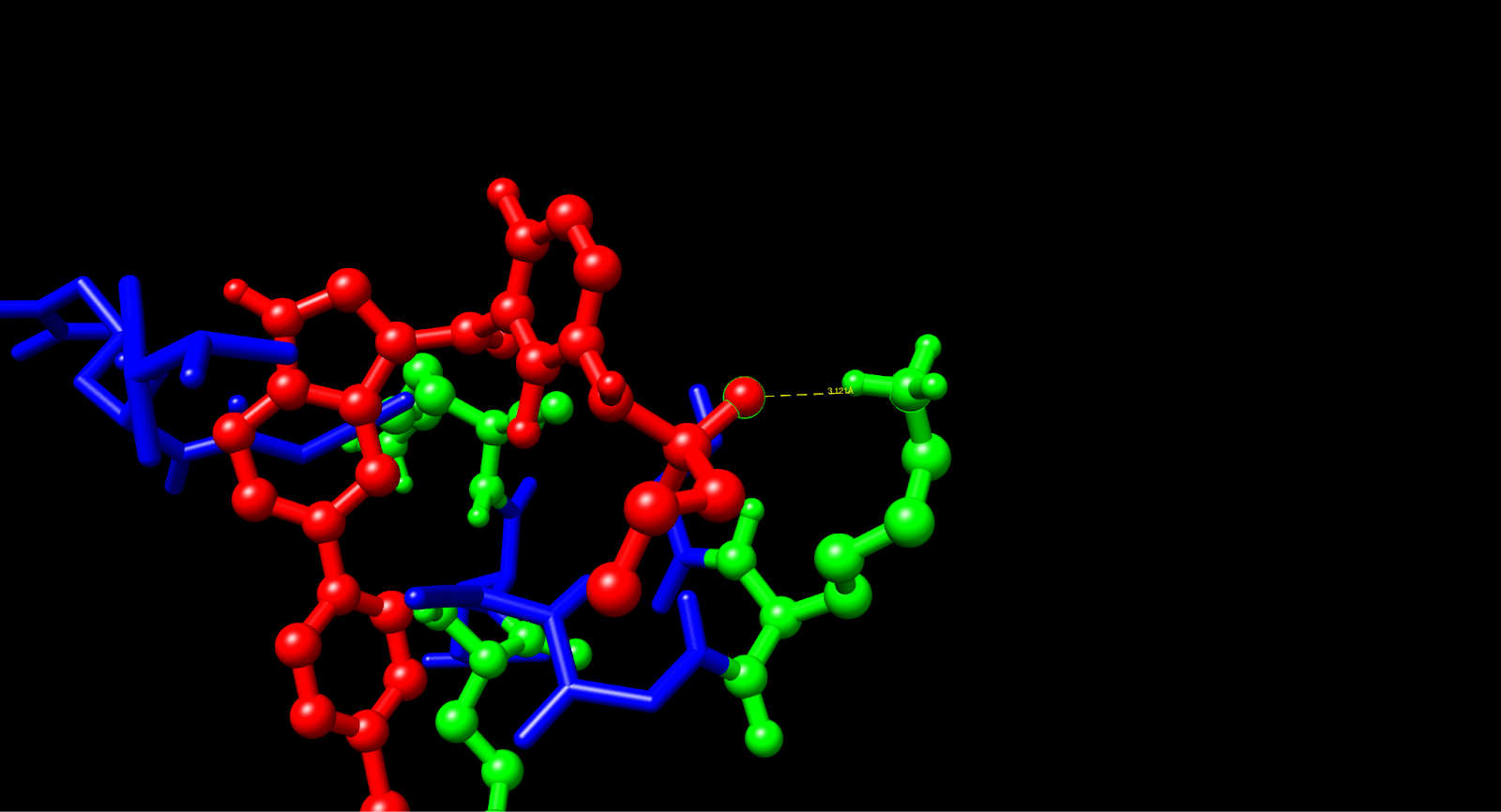
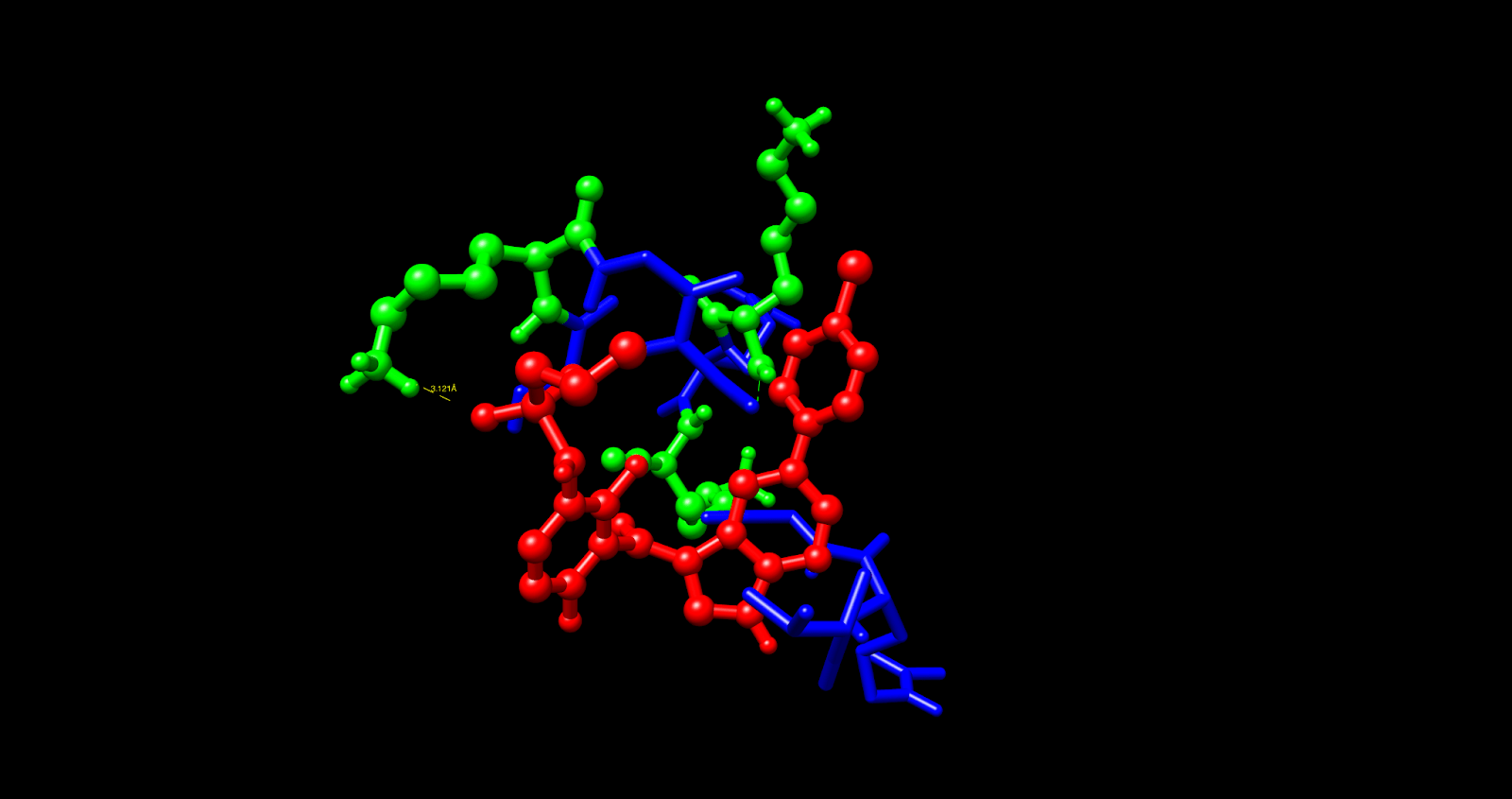
Through an in-depth analysis of the molecular contacts, we found that VAL1 and GLY4 are supporting large contributions to the hydrophobic contacts with the aromatic portions of Vemurafenib, as well as the indole and chlorophenyl groups, creating a stable complex through complementary van der Waals forces. The gold nanoparticle core is a vital part of the structural scaffold that confines the peptide to allow the drug to interact in the proper conformation. A comparison of the two delivery systems showed significant differences in the binding efficacy of the two systems.
The PAMAM-Vemurafenib system had a binding affinity (ΔG) of -3.426 kcal/mol. It was not binding tightly enough to retain the drug due to non-specific electrostatic interactions and large steric hindrance. The AuNP-VG16KRKP system demonstrated further improvements, and binding energy was enhanced by an average of about 2 kcal/mol to a ΔG value of -5.4 kcal/mol. The differences in the binding affinity can be largely explained by the hydrogen bondings of lysine and arginine residues of the VG16KRKP peptide, allowing for a more favorable binding micro-environment to Vemurafenib than the amine functional groups of PAMAM. We also believe the contribution of the gold nanoparticle as a structural scaffold is significant, as it provides a stable structure for peptide conjugation, which is absent in the PAMAM system.
The C1 binding site shows a definitive advantage over other binding sites according to cavity volume analysis because it has 43 ų of volume, while alternative sites C2 – C5 measure from 0-2 ų in volume. Vemurafenib reaches its most optimal fit within the C1 pocket because of its enhanced ability to fit the pharmacological dimensions and shape requirements of the drug. Smaller identified alternative binding sites within the structure suggest how these sites could become important secondary binding modes during changes in physiological characteristics or solution conditions, but these possibilities require further investigation. The AuNP-VG16KRKP-Vemurafenib complex exhibits strong binding capabilities because of peptide-to-drug hydrogen bonds that use both the core structure and the photothermal activities of the AuNP core to support these interactions. Experimental validation of this delivery system as a targeted melanoma therapy should proceed because the computational findings identify it as a promising clinical candidate.
AuNP– integrin
| Metric | Value | Interpretation |
| Affinity | -6.535 kcal/mol | Good binding (lower than -6 is acceptable for drug-like molecules) |
| Total Energy | -136.094 | Overall stability of the ligand-receptor complex |
| vdW Energy | 3.212 | Van der Waals interactions (repulsive or slightly weak here) |
| Electrostatic Energy | -67.343 | Strong electrostatic (charge-based) interactions between Vemurafenib and BRAF |
integrin Docking Results Description: In this case, we decided to use DockThor for our molecular docking platform due to technical limitations: the AuNP file exceeded the 20 kb size limitation for CB-Dock2. All data in this table were generated in this study.
Since the target site for Vemurafenib is in the BRAF ATP pocket, AuNP binding to BRAF could interfere with the drug’s ability to effectively inhibit its target action. Our design philosophy maintained that the AuNP is only a carrier, not another blocker to the therapeutic target. To achieve a melanoma-specific targeting mechanism, we considered integrin, one of its best receptors, since integrin is a melanoma-associated cell-surface marker. Our negative docking affinity values suggest that there is a reasonable binding affinity of AuNP to integrin as a surface-targeting ligand in our delivery system. This targeting mechanism benefits from dual action, as integrin is overexpressed on melanoma cells as well as on angiogenic vasculature, which markedly improves the tumor selectivity of the designed nanocarrier system.
One of the benefits of this method is that it does not interfere with BRAF binding. Previously developed strategies directed the use of nanoparticles to the BRAF V600E ATP-binding pocket. However, our extracellular integrin binding method allows Vemurafenib to reach its intracellular target (after release from the nanoparticle carrier) without being sterically hindered in any way to interfere with binding. Overall, molecular analysis reveals significant electrostatic interactions, such as the energetic potential of negative values of electrostatic energy, which indicate charge-based attraction, important for initial recognition by the receptor and formation of stable ligand-receptor complexes.

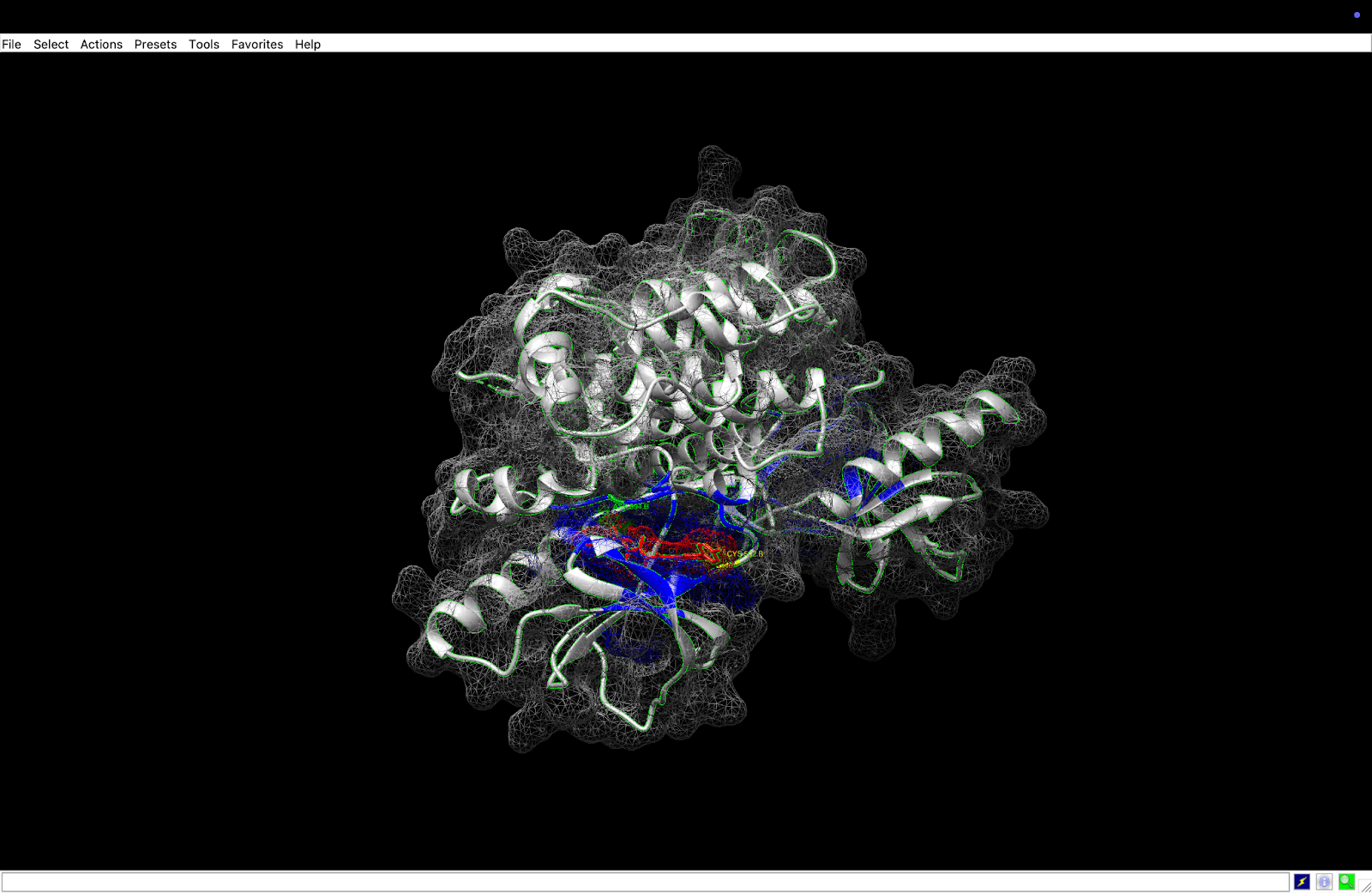
 integrin receptor. The integrin is expressed in light blue, while the AuNP-Verumrafenib complex is expressed in the center in other colors, such as green, red, and blue.
integrin receptor. The integrin is expressed in light blue, while the AuNP-Verumrafenib complex is expressed in the center in other colors, such as green, red, and blue.In the structural representation of the docking findings, the ligand appears to be deeply inserted into the ATP-binding pocket. The ligand orientation indicates the presence of aromatic ring systems associating with the hydrophobic area of the binding pocket, a binding mode mostly expected for kinase inhibitors such as Vemurafenib. These structural data further demonstrate the molecular foundation for our targeted delivery method.
Discussion
As demonstrated by the docking results, the model allows for targeted delivery as a two-step process. In the targeting phase, the AuNP binds to the integrin receptors located on melanoma cells. Once this recognition has occurred via receptor-mediated endocytosis, the delivery phase commences, where the nanoparticle system enables the release of vemurafenib intracellularly for binding to its primary therapeutic target, BRAF V600E.
We believe this two-part model is a noteworthy improvement because of its dual-function ligand design. Rather than providing separate targeting therapeutic agents, common for drug delivery systems, we created a system with vemurafenib as both a targeting ligand and intracellular inhibitor, thereby simplifying the delivery system and reducing formulation complexity, while still providing the dual function of targeting and inhibition. The use of dual-function ligands does offer some potential advantages, particularly with respect to minimizing off-target effects.
In this research, we define “dual-ligand”, as functionalizing the AuNPs with both VG16KRKP (for membrane interaction) and Vemurafenib (for intracellular kinase inhibition). The concepts of potentially combining receptor-mediated uptake with targeted drug action intracellularly is attractive in principle. However, we don’t make actual comparisons to single-ligand systems in this research study and thus, we want the present work to be interpreted merely as a conceptual proof-of-concept for dual-ligand functionalization within limited time and resource constraints rather than proof of better therapeutic performance. Future experimental studies may allow for comparisons of AuNP–VG16KRKP–Vemurafenib to AuNP–VG16KRKP only, AuNP–Vemurafenib only, and perhaps other combinations for measuring any additive or synergistic effects on melanoma targeting and cytotoxicity.
The binding energy of -6.535 kcal/mol indicates a moderate binding; it achieves an optimal compromise for reversible binding of drug targets. The value is in line with many approved kinase inhibitors of the FDA, which is hypothetically beneficial for the pharmacokinetic profile during therapeutic development. The negative total energy value indicates that the ligand is stable in the active site and that Vemurafenib has a good overall conformational fit in the binding pocket. Further consideration of the interaction components shows a strong electrostatic component of the total (-67.343), which suggests that Vemurafenib has multiple hydrogen bonds, salt bridges, or dipole interactions with charged amino acids within the BRAF ATP pocket. In most instances, these electrostatic interactions help to provide both the specificity and stability of orientation that is vitally important for accurate molecular recognition. The van der Waals component also has a slightly positive value (+3.212), which indicates it is likely packing against some areas of repulsion or weak packing with some of the atoms, indicating perhaps narrow clashes or loose fits within the hydrophobic regions. Nevertheless, this value is far less than a value of concern (typically >+10) and should not undermine the overall binding stability or function.
The docking affinity of -6.535 kcal/mol determined from the study indicates that it has potentially comparable binding to existing ligands, such as cilengitide that is a high-affinity -integrin antagonist29. Although this affinity suggests the potential for favourable receptor engagement, the functional targeting efficiency is dependent upon further parameters including receptor density, nanoparticle avidity, and/or in vivo pharmacokinetics. All of which were outside the scope of this computational study.
Building the final comprehension Step-by-Step Simulation modeling of Targeted Nanoparticle Drug Delivery System
Step 1: Create the AuNP-VG16KRKP–Vemurafenib complex

Step 2: Simulate the AuNP-VG16KRKP bound to the integrin receptors, simulating membrane localization
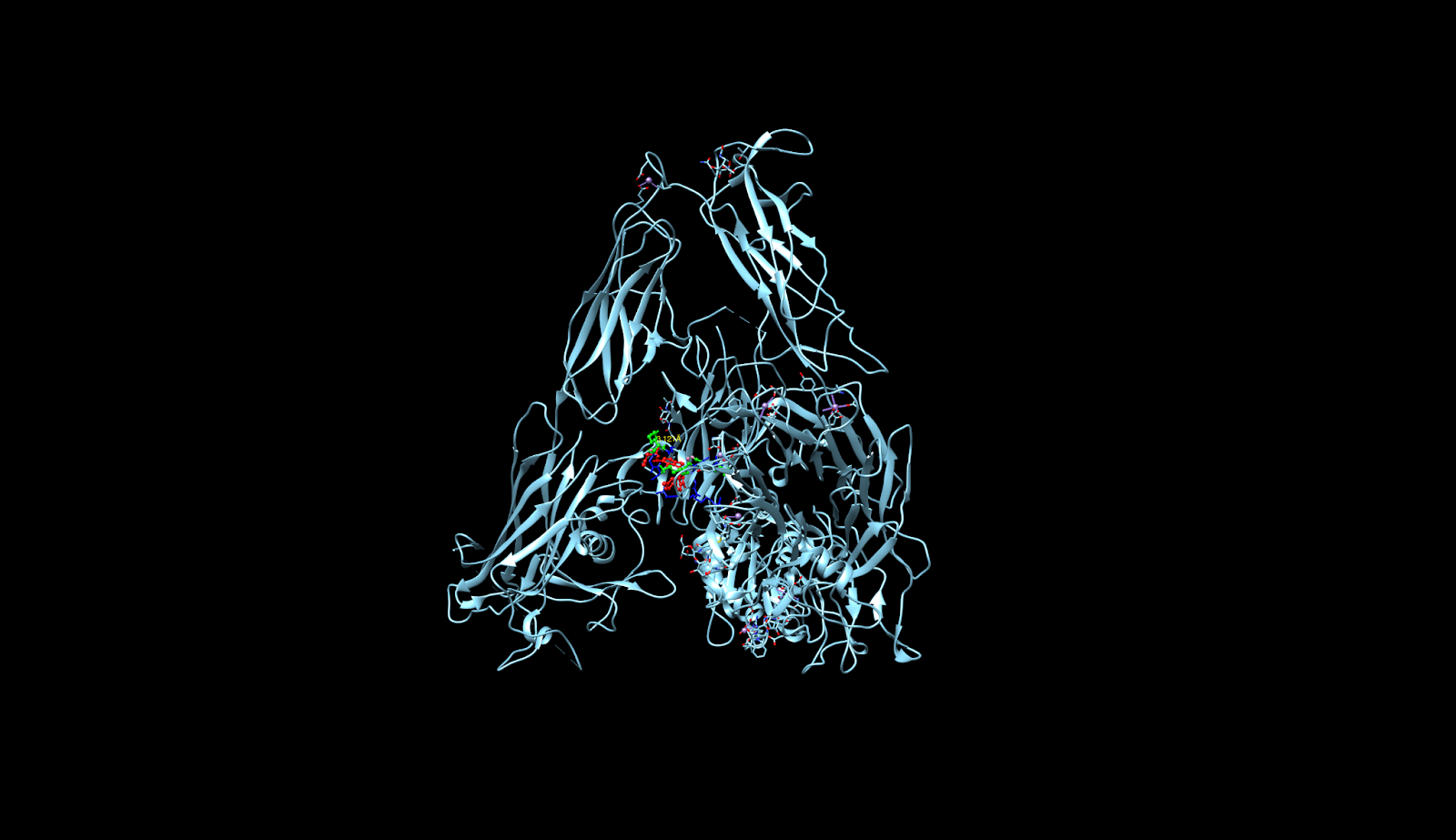
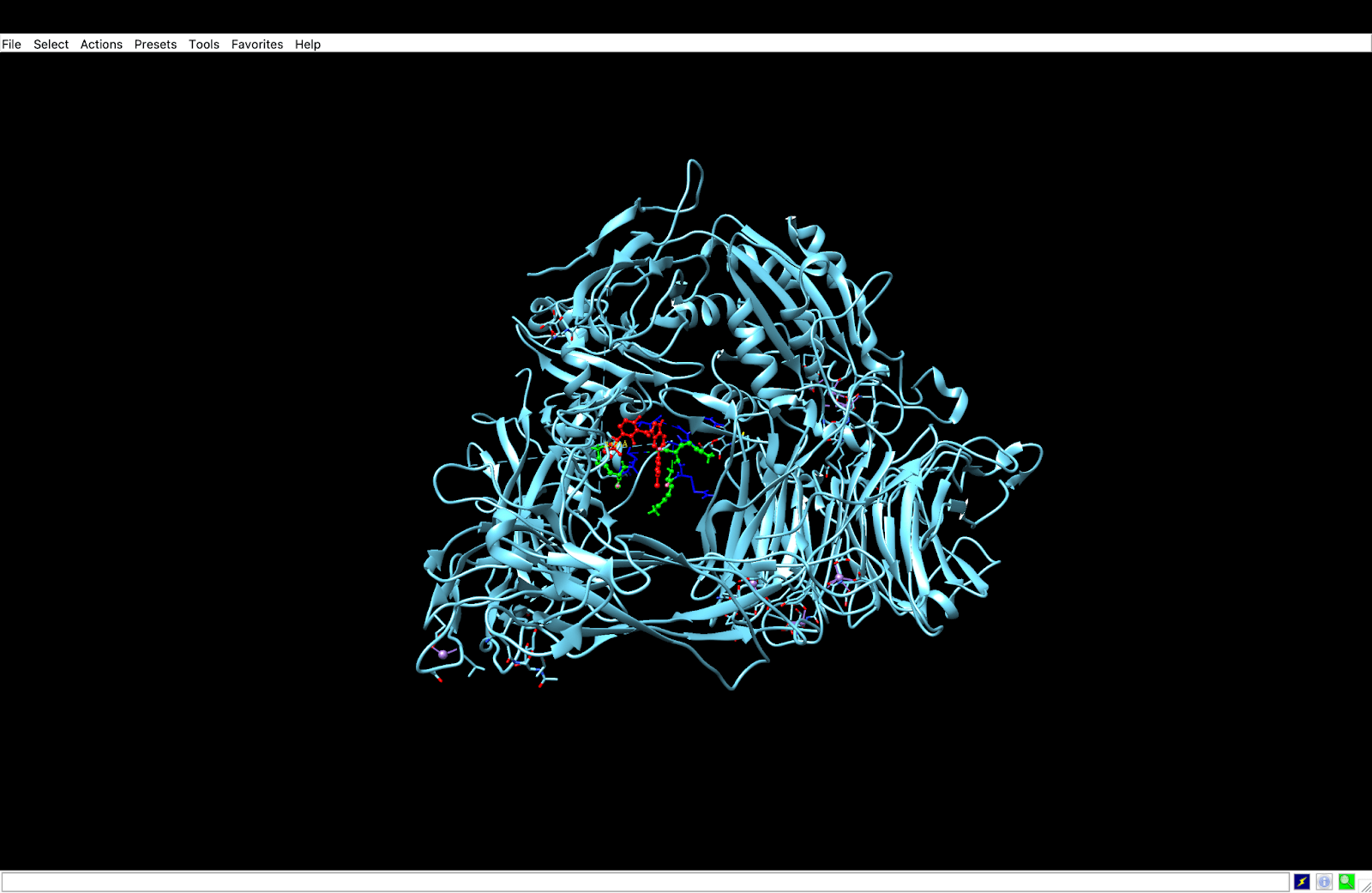

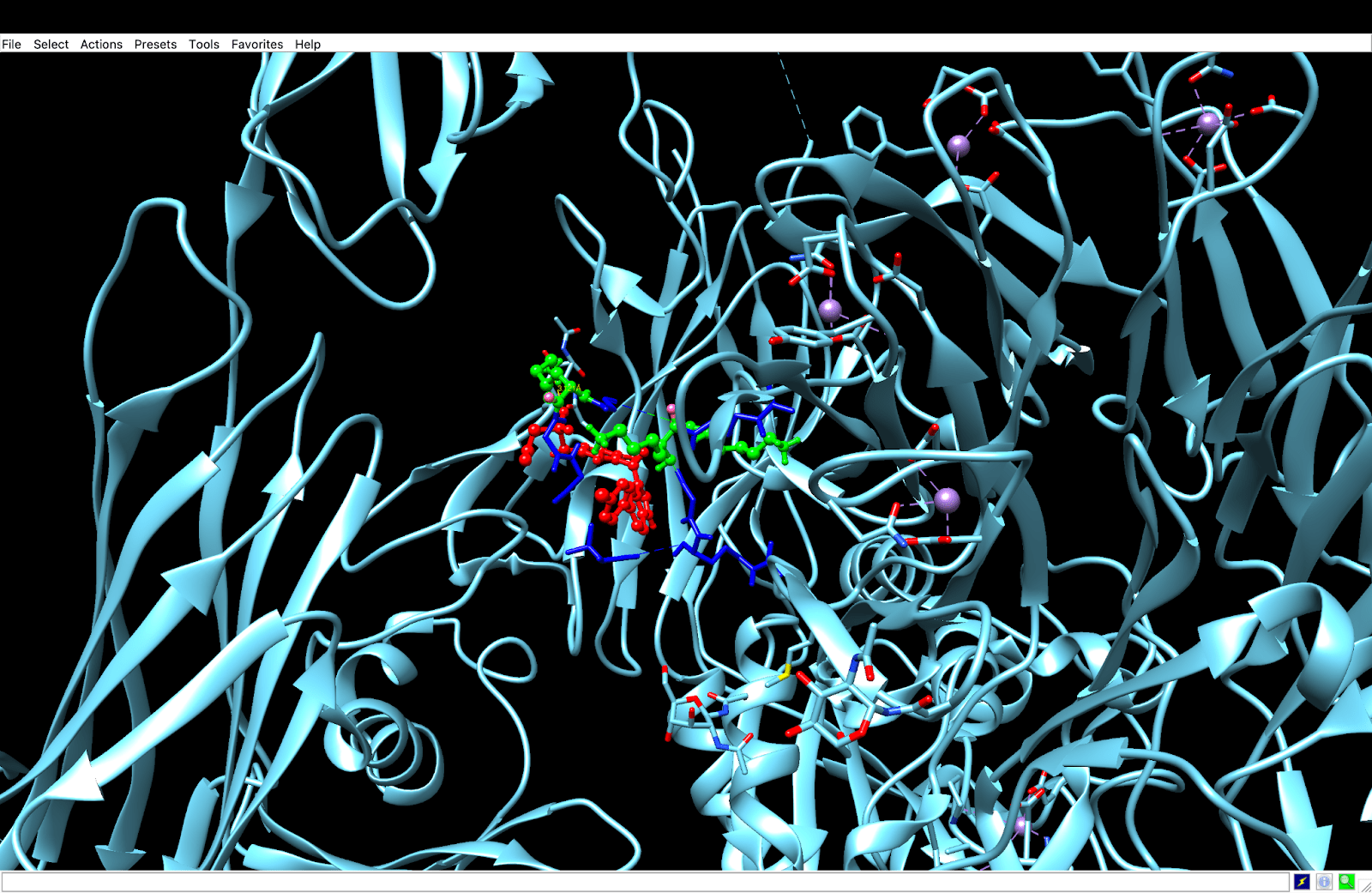 integrin receptor. The integrin is expressed in light blue, while the AuNP-Verumrafenib complex is expressed in the center in other colors, such as green, red, and blue, refer back to Figure 8.
integrin receptor. The integrin is expressed in light blue, while the AuNP-Verumrafenib complex is expressed in the center in other colors, such as green, red, and blue, refer back to Figure 8.Step 3: Upon cellular uptake, AuNP-VG16KRKP releases Vemurafenib inside the cell, then diffuses to and binds to BRAF V600E


| Property | AuNP–VG16KRKP (Score 1–5) | PAMAM Dendrimers (Score 1–5) |
| Vemurafenib Binding | 5 (strong binding affinity: ΔG = -5.4 kcal/mol) | 2 (weaker binding affinity: ΔG = -3.2 kcal/mol) |
| Cancer cell Targeting | 5 (Targets integrin with binding affinity of ΔG = -6.535 kcal/mol) (DockThor) | 3 (Targets BRAF V600E with binding affinity of ΔG = -7.167 kcal/mol, causing competitive inhibition) (DockThor) |
| Hydrophobic/Hydrophilic Balance with Vemurafenib | 5 (exhibits balance at Val1, Gly4, and Lys5) | 2 (weak binding affinity leading to weak interactions) |
| Release Profile | 3 (burst release in the first 10–12 h, and a controlled release thereafter30) | 4 (sustained release31) |
| Biocompatibility | 5 (Literature suggests reduced cytotoxicity, preserving biocompatibility21) | 5 (generally no cytotoxicity32) |
| Total score: | 23/25 | 16/25 |
Description: The final comparison between AuNP–VG16KRKP and PAMAM dendrimers, supported by both this study and existing literature, establishes AuNP–VG16KRKP as the more effective nanoparticle for Vemurafenib binding and delivery. Literature references and sources used in this table are adapted from Mohid et al., 2022, Dehkordi et al., David et al., 2023, Janaszewska et al., 2019
Although Figures 8–10 present the proposed delivery pathway, the figures should be regarded as conceptual schematics and not molecular dynamics simulations. Endosomal escape, trafficking of nanoparticles, and release kinetics were not computationally simulated in this work; future work that includes dynamic simulations, in cognizance of experimental validations, will be imperative to truly characterize these mechanistic steps.
The research objectives of the study were fully achieved, showing that dual-ligand functionalized gold nanoparticles can enhance targeting efficiency and drug-binding stability. The study clearly places AuNPs as viable drug delivery carriers via targeting, particularly with ligands that target specific receptors that are overexpressed in skin and epithelial cancers. Computational modeling indicated that Vemurafenib is well-positioned and conjugate adequately to AuNPs, while the dual-targeting delivered greater specificity relative to adjacent healthy cells and decreased uncertainty regarding non-target delivery.
Gold nanoparticle (AuNP) biocompatibility is well established, however, factors contributing to safety can be influenced by core size, surface chemistry, dose, and route of administration. After systemic administration, AuNPs are sequestered primarily by macrophages in the liver and the spleen, where they can remain for months as the gold core is non-biodegradable. Cationic surface ligands can improve cellular uptake but can also lead to non-specific binding interactions with healthy cells and serum proteins that can contribute to off-target biodistribution and undesired immune activation. While the current study only uses in silico structural modeling, clear in vitro and in vivo evaluations of toxicity and biodistribution will need to be the next steps of the process before being considered for clinical use.
We used vemurafenib in this study as our representative BRAF inhibitor because of its established clinical significance and rich structural documentation has been generated. The commercially available BRAF inhibitors are numerous, and while other experimental BRAF inhibitors (dabrafenib, encorafenib) are validated, we limited the scope of exploration to the vemurafenib molecule to keep our analysis simplified and manageable. The focus of this project was to explore the nature of the interaction and binding characteristics of vemurafenib with the target protein to develop our proof of concept for the computational process that we have employed. In the future, the opportunities to expand on our study will be to explore a variety of BRAF inhibitors to further substantiation and generalization of our findings.
This paper is primarily concerned with comparing AuNPs and PAMAM dendrimers. Additionally, other nanocarrier platforms such as liposomes and polymeric micelles, which have been in clinical testing or consideration for melanoma drug delivery, deserve mention. Liposomes composed of phosphatidylcholine and cholesterol have been particularly successful in preclinical melanoma models and can encapsulate hydrophobic drugs with high loading capacity, and limit systemic toxicity33. Polymeric micelles generated from amphiphilic block copolymers have demonstrated improved solubility of hydrophobic kinase inhibitors as well as controlled drug release, with several formulations advancing to clinical trials34. Future work should involve direct computational and experimental comparisons with other nanocarrier platforms, such as liposomal and polymeric micelle systems, in order to gain a better understanding of their relative targeting efficiencies.
Additionally, although these platforms may provide benefits for hydrophobic delivery, the present study only investigated gold nanoparticles, so in our future computational screening we will also consider lipid-based and polymeric nanoparticles to evaluate whether their drug loading characteristics and release kinetics result in a more advantageous delivery system for Vemurafenib than AuNP-based systems.
Docking studies showed that the peptide VG16KRKP engages Vemurafenib principally through one hydrogen bond between Lys5 and the sulfonamide (3.12 Å) and additional hydrophobic contacts, namely Val1 and Gly4. Though these methods suggest that the VG16KRKP–Vemurafenib complex is stable in the docking environment, we do not have any residue-specific energy decompositions to verify this, nor do we have any directly relevant data to determine if this stability would hold up under physiological conditions. Serum proteins and blood flow could facilitate either premature drug dissociation or drug leakage into the blood. In the future, molecular dynamics simulations in a simulated plasma environment and experimental binding assays will need to be performed to evaluate if the peptide-drug conjugation, is stable enough to eliminate drug leakage during systemic perfusion of the peptide–drug conjugation.
Peptide-functionalized nanoparticles (VG16KRKP) may nonspecifically interact with other anionic cell membranes or integrins besides (e.g., αvβ5 and α5β1) that expression in some healthy tissues35. We recognize that nonspecific adsorption could result in unintended biodistribution and toxicity. While focused on for docking, we did not model binding with off-target integrins or other membrane receptors, and future computational work should complete comparative docking to a panel of integrins and serum proteins to follow with in vitro uptake assays in melanoma and non-melanoma cell lines to measure targeting selective and avoid off-target effects.
A key limitation of the docking study outlined in this paper is the lack of experimentally validated negative controls for AuNP– integrin binding. In an ideal situation, these controls could include nanoparticles with the same size and surface chemistry that were functionalized with non-binding ligands (e.g., RAD peptide or scrambled RGD), or tested in -negative cell lines. However, negative control systems were not feasible to implement in this study, due to lack of structural information and availability of resources for the nanoparticle–ligand constructs. In the future, the original plan to create property-matched non-binding AuNP conjugates and examine them for docking or binding behaviour will be fully validated, opening the framework for a more rigorous examination of “specificity” and potential for false-positive predictions of binding.
Future research should aim to validate these computational predictions experimentally in vitro and in vivo, to evaluate biocompatibility, pharmacokinetics, and clinical relevance. Combining a computational modeling approach with available AI drug discovery platforms can facilitate applications for drug design via ligand-receptor interactions and broaden the usage of smart delivery nanoparticles beyond the two cancers considered in the study, due to enhanced knowledge of the differences in receptor turnover and expression between receptor subtypes. Additionally, further assessment of optimal compositions of the nanoparticles may yield more favorable stabilization of their biological durability in the biological environment, therefore improving clinical application of energy-conjugated systems.
Important shortcomings of this investigation, including the lack of experimental verification of the utilized computational predictions and the limited two ligands and two receptors that can be modeled in full, need to be addressed in future work. Despite identified limitations, the current work signifies important contributions to a burgeoning field of study in nanomedicine by confirming the optimization of a dual-ligand nanoparticle system being developed for targeted drug delivery. The ability of drug design via computational modeling to integrate with nanotherapeutic design illustrates a new frontier for innovation regarding targeted drug delivery systems and potential future translation into the clinical realm for more efficient melanoma treatment.
While we have focused purely on issues of structural feasibility and receptor binding in silico and therefore will not consider pharmacokinetic (ADME) modelling or experimental release profiles, it should be noted that drug release kinetics, plasma protein binding, metabolic stability, biodistribution and clearance are key factors determining therapeutic efficacy and safety in any nanoparticle delivery system. Future work would be needed to investigate each of these parameters by (1) performing in-silico ADMET predictions of Vemurafenib and the impact of conjugation, (2) performing in vitro release assays under conditions that define physiological relevance, worth examining the measure of rate of release and burst vs sustained release profiles, (3) examining serum stability and protein corona formation, and (4) evaluating biodistribution and clearance in small-animal models. These studies would indicate whether or not the nanoparticle formation will extend circulation half-life, reduce off-target exposure, and even achieve therapeutically relevant intracellular drug concentrations without unacceptable organ accumulation or toxicity.
As an in silico study, we do not provide experimental physicochemical or biodistribution data. However, these parameters are integral to assessing the translational viability of nanoparticle-based delivery systems; thus, we provide broad insight below, and additional work will be required to investigate these parameters.
Surface chemistry/charge has a significant impact on stability in biological media; therefore, it is essential to measure the hydrodynamic size, polydispersity index (PDI), and zeta potential in vitro, using dynamic light scattering (DLS), and zeta potential measurements. Further, stability can be investigated by measuring shifts in UV–Vis absorbance; peptide conjugation efficiency can also potentially be verified by Fourier-transform infrared spectroscopy (FTIR) or X-ray photoelectron spectroscopy (XPS).
Biocompatibility and delivery potential are contingent on nanoparticle circulation half-life, biodistribution, and clearance pathway. Previous in vivo studies of AuNP have determined a considerable increase in the liver, spleen, and gallbladder, with clearance primarily through the hepatobiliary route36. Future work should investigate the AuNP–VG16KRKP–Vemurafenib complex for serum stability, protein corona formation, and cytotoxicity in melanoma and healthy cell lines to confirm targeted delivery and limit off-target effects.
Methods
This study employed a comparative computational modeling approach to evaluate the drug-binding efficiencies and targeting performance of nanoparticle-based drug delivery systems for targeted skin cancer therapy. The biological structures were sourced from the Protein Data Bank (PDB) and PubChem. Nanoparticle models were built based on established chemical structures of gold nanoparticles (AuNPs), and PAMAM dendrimers. The data collection process involved identifying and optimizing all relevant 3D structures:
| Structures | Description | ID |
| Vemurafenib | targeted cancer drug inhibitor molecule for targeting BRAF V600E | PubChem CID 42611257 |
| PAMAM dendrimer | Nanoparticle made of repetitively branched subunits of amide and amine functionality are used for drug delivery | PubChem CID 4140276 |
| BRAF V600E | The most common type of BRAF protein to undergo mutations | PDB ID 4RZV |
| Integrin | Surface receptor for BRAF | PDB ID 1L5G |
| AuNP-VG16KRKP | Improves drug delivery in comparison to only AuNP. The structure, adapted from PDB ID 5WYE, excludes LPS to avoid immune artifacts irrelevant to melanoma. | PDB ID 5WYE |
Description: Five 3D structures were used for this study, found through the databases of PubChem and PDB.
These molecules were selected by their biological relevance to targeted melanoma therapeutics and their availability in computer-aided structural databases and molecular modeling tools. All molecular models were justified and prepared using standard computational chemistry methods before all docking and simulations. In this study, we used a range of biologically relevant molecular constructs in the context of targeted cancer therapy. Vemurafenib is a small-molecule inhibitor found to be the best when targeting the BRAF V600E mutation37, which is one of the most common oncogenic drivers of melanoma and other cancers. The associated mutant BRAF V600E protein is the most prevalent BRAF mutation, with its constitutively active kinase activity38. To model surface binding, we used integrin, which is commonly overexpressed in tumor cells, contributing to invasive behavior and resistance to drugs, as a PKC and PDGF cell surface receptor39. To model drug delivery, we used a PAMAM dendrimer, which has been well characterized and has a branched nanoscale structure31. We also modeled the gold nanoparticle AuNP-VG16KRKP. Importantly, lipopolysaccharide (LPS) was excluded from the structural model to avoid immunogenic confounders unrelated to melanoma targeting.
The important variables and measurements we consider during this post-docking analysis include binding affinities (kcal/mol) and interaction types (hydrogen bonding, hydrophobic interactions, π-stacking). Once molecular docking of Vemurafenib with BRAF V600E using CB-Dock2 is achieved, we will extract the binding pocket with the strongest affinity to Chimera in order to analyze its three-dimensional docking pose. We will then proceed to the PAMAM dendrimer, which we docked to Vemurafenib, to determine its efficiency based on the affinity score. If it is weak, we will then proceed with the molecular docking of the AuNP-VG16KRKP and Vemurafenib. If this complex is efficient, we will finally dock AuNP-VG16KRKP to the integrin surface receptor and analyze the data. We will finally create a step-by-step, comprehensive visual delivery simulation that is conceptualized and visualized through schematic modeling. Overall, binding affinities must be compared to measure ligand-nanoparticle and ligand-receptor interactions. The intercalation of drug delivery was inferred from the strength of the docking and the specificity of the voltage-gated targets. The best candidate systems were determined on the basis of binding energy and interaction. No ethical approval was required as an in silico study involving only computational models and open-source molecular data. Data sources were cited appropriately, and no confidential or human subject data was used.
A computational docking strategy was used to investigate the drug-binding efficiencies and targeting capabilities of nanoparticle-based delivery systems for melanoma treatment. Both molecular docking simulations were conducted using two dedicated docking protocols, CB-Dock2 (AutoDock Vina v1.1.2, accessed March 2025) for smaller systems (e.g., Vemurafenib–BRAF V600E, PAMAM–Vemurafenib complexes) and DockThor v2023.1 for larger docking complexes (e.g., AuNP– integrin) since the site upload limit for CB-Dock2 prohibited the upload of AuNP PDB files.
For preparing the receptors, each receptor of interest was downloaded and used without changes; BRAF V600E (PDB ID: 4RZV), Integrin (PDB ID: 1L5G). The AuNP–VG16KRKP Model (PDB ID: 5WYE) was modified in UCSF Chimera to remove the lipopolysaccharide (LPS) to avoid introducing any unintended immunogenicity that was not related to the melanoma targeting.
The ligands, Vemurafenib (PubChem CID: 42611257) and PAMAM dendrimer (PubChem CID: 4140276), were downloaded from PubChem in SDF format.
For docking protocol, all docking simulations used the default blind docking options to eliminate biased sampling of binding sites, with no user-defined grid boxes or other adjustments to scoring functions. For CB-Dock2, default parameters were used (5 cavities per target, use auto blind docking mode), and no energy minimization, or constraints were used. The grid size is determined automatically and personalized based on the query ligand and previously discussed binding site. CB-Dock uses 10 Å resolution grid to sample full protein structure, while predicting the binding sites and modes to the appropriate closest grid box. There is no explicit set grid box size from the user, this is determined by the software which takes into consideration the previously identified binding site and the size of the ligand. For DockThor, when using blind docking mode, the software auto-centered the search space around the entire receptor, then scored the results using DockThor’s default function of grid size (40.0, 40.0, 40.0 Å). Blind docking was chosen to avoid bias in selection of binding sites; Default parameters were used to correspond to each software’s validated protocols, lending to reproducibility without user-defined arbitrated choices.
References
- Rebecca, V. W., Sondak, V. K., & Smalley, K. S. (2012). A brief history of melanoma: from mummies to mutations. Melanoma research, 22(2), 114–122. https://doi.org/10.1097/CMR.0b013e328351fa4d [↩]
- Islami, F., Sauer, A. G., Miller, K. D., Fedewa, S. A., Minihan, A. K., Geller, A. C., Lichtenfeld, J. L., & Jemal, A. (2020). Cutaneous melanomas attributable to ultraviolet radiation exposure by state. International Journal of Cancer, 147(5), 1385–1390. https://doi.org/10.1002/ijc.32921 [↩]
- Heistein, J., & Acharya, U. (2023). Cancer, malignant melanoma. PubMed; StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK470409/ [↩]
- Shelledy, L., & Roman, D. (2015). Vemurafenib: First-in-class BRAF-mutated inhibitor for the treatment of unresectable or metastatic melanoma. Journal of the advanced practitioner in oncology, 6(4), 361–365. https://doi.org/10.6004/jadpro.2015.6.4.6 [↩]
- Zeiderman, M. R., Egger, M. E., Kimbrough, C. W., England, C. G., Dupre, T. V., McMasters, K. M., & McNally, L. R. (2014). Targeting of BRAF resistant melanoma via extracellular matrix metalloproteinase inducer receptor. The Journal of Surgical Research, 190(1), 111–118. https://doi.org/10.1016/j.jss.2014.02.021 [↩]
- Chehelgerdi, M., Chehelgerdi, M., Allela, O. Q. B., Pecho, R. D. C., Jayasankar, N., Rao, D. P., Thamaraikani, T., Vasanthan, M., Viktor, P., Lakshmaiya, N., Saadh, M. J., Amajd, A., Abo-Zaid, M. A., Castillo-Acobo, R. Y., Ismail, A. H., Amin, A. H., & Akhavan-Sigari, R. (2023). Progressing nanotechnology to improve targeted cancer treatment: overcoming hurdles in its clinical implementation. Molecular Cancer, 22(1), 169. https://doi.org/10.1186/s12943-023-01865-0 [↩]
- Marusyk, A., & Polyak, K. (2010). Tumor heterogeneity: causes and consequences. Biochimica et biophysica acta, 1805(1), 105–117. https://doi.org/10.1016/j.bbcan.2009.11.002 [↩]
- Abedi-Gaballu, F., Dehghan, G., Ghaffari, M., Yekta, R., Abbaspour-Ravasjani, S., Baradaran, B., Dolatabadi, J. E. N., & Hamblin, M. R. (2018). PAMAM dendrimers as efficient drug and gene delivery nanosystems for cancer therapy. Applied Materials Today, 12, 177–190. https://doi.org/10.1016/j.apmt.2018.05.002 [↩]
- Hossain, A., Md. Thohid Rayhan, Md Hosne Mobarak, Hossain, I., Hossain, N., Islam, S., & Kafi, A. (2024). Advances and significances of gold nanoparticles in cancer treatment: A comprehensive review. Results in Chemistry, 8(101559), 101559–101559. https://doi.org/10.1016/j.rechem.2024.101559 [↩]
- Liu, Z., Wang, F., & Chen, X. (2008). Integrin alpha(v)beta(3)-Targeted Cancer Therapy. Drug development research, 69(6), 329–339. https://doi.org/10.1002/ddr.20265 [↩]
- Chowdhury, R., Ilyas, H., Ghosh, A., Ali, H., Arup Ghorai, Anupam Midya, Jana, N. R., Das, S., & Anirban Bhunia. (2017). Multivalent gold nanoparticle–peptide conjugates for targeting intracellular bacterial infections. Nanoscale, 9(37), 14074–14093. https://doi.org/10.1039/c7nr04062h [↩] [↩] [↩]
- Li, L., Vorobyov, I., & Allen, T. W. (2013). The different interactions of lysine and arginine side chains with lipid membranes. The Journal of Physical Chemistry. B, 117(40), 11906–11920. https://doi.org/10.1021/jp405418y [↩]
- Makowski, M., Silva, Í. C., Pais do Amaral, C., Gonçalves, S., & Santos, N. C. (2019). Advances in lipid and metal nanoparticles for antimicrobial peptide delivery. Pharmaceutics, 11(11), 588. https://doi.org/10.3390/pharmaceutics11110588 [↩]
- Datta, A., Yadav, V., Ghosh, A., Choi, J., Bhattacharyya, D., Kar, Rajiv K., Ilyas, H., Dutta, A., An, E., Mukhopadhyay, J., Lee, D., Sanyal, K., Ramamoorthy, A., & Bhunia, A. (2016). Mode of action of a designed antimicrobial peptide: High potency against cryptococcus neoformans. Biophysical Journal, 111(8), 1724–1737. https://doi.org/10.1016/j.bpj.2016.08.032 [↩] [↩]
- Datta, A., Ghosh, A., Airoldi, C., Sperandeo, P., Mroue, K. H., Jiménez-Barbero, J., Kundu, P., Ramamoorthy, A., & Bhunia, A. (2015). Antimicrobial peptides: Insights into membrane permeabilization, lipopolysaccharide fragmentation and application in plant disease control. Scientific Reports, 5(1), 11951. https://doi.org/10.1038/srep11951 [↩]
- Won, T., Mohid, S. A., Choi, J., Kim, M., Krishnamoorthy, J., Biswas, I., Bhunia, A., & Lee, D. (2023). The role of hydrophobic patches of de novo designed MSI-78 and VG16KRKP antimicrobial peptides on fragmenting model bilayer membranes. Biophysical chemistry, 296, 106981. https://doi.org/10.1016/j.bpc.2023.106981 [↩]
- Schmidt, N., Mishra, A., Lai, G. H., & Wong, G. C. (2010). Arginine-rich cell-penetrating peptides. FEBS letters, 584(9), 1806–1813. https://doi.org/10.1016/j.febslet.2009.11.046 [↩]
- Shirazi, A. N., Vadlapatla, R., Koomer, A., Nguyen, A., Khoury, V., & Parang, K. (2025). Peptide-Based Inorganic Nanoparticles as Efficient Intracellular Delivery Systems. Pharmaceutics, 17(9), 1123. [↩]
- Ioanna-Aglaia Vagena, Malapani, C., Maria-Anna Gatou, Nefeli Lagopati, & Pavlatou, E. A. (2025). Enhancement of EPR effect for passive tumor targeting: Current status and future perspectives. Applied Sciences, 15(6), 3189–3189. https://doi.org/10.3390/app15063189 [↩]
- Lin, J., Zhang, H., Chen, Z., & Zheng, Y. (2010). Penetration of lipid membranes by gold nanoparticles: insights into cellular uptake, cytotoxicity, and their relationship. ACS nano, 4(9), 5421–5429. https://doi.org/10.1021/nn1010792 [↩] [↩] [↩]
- Mohid, S. A., Biswas, K., Won, T., Mallela, L. S., Gucchait, A., Butzke, L., Sarkar, R., Barkham, T., Reif, B., Leipold, E., Roy, S., Misra, A. K., Lakshminarayanan, R., Lee, D., & Bhunia, A. (2022). Structural insights into the interaction of antifungal peptides and ergosterol containing fungal membrane. Biochimica et Biophysica Acta (BBA) – Biomembranes, 1864(10), 183996. https://doi.org/10.1016/j.bbamem.2022.183996 [↩] [↩]
- Savla, R., Garbuzenko, O. B., Chen, S., Rodriguez-Rodriguez, L., & Minko, T. (2014). Tumor-targeted responsive nanoparticle-based systems for magnetic resonance imaging and therapy. Pharmaceutical research, 31(12), 3487–3502. https://doi.org/10.1007/s11095-014-1436-x [↩]
- Mundra, V., Li, W., & Mahato, R. I. (2015). Nanoparticle-mediated drug delivery for treating melanoma. Nanomedicine, 10(16), 2613–2633. https://doi.org/10.2217/nnm.15.111 [↩]
- Chang, Y., Hawkins, B. A., Du, J. J., Groundwater, P. W., Hibbs, D. E., & Lai, F. (2022). A guide to in silico drug design. Pharmaceutics, 15(1), 49. https://doi.org/10.3390/pharmaceutics1501004 [↩]
- van der Velden, W. J. C., Heitman, L. H., & Rosenkilde, M. M. (2020). Perspective: implications of ligand-receptor binding kinetics for therapeutic targeting of G protein-coupled receptors. ACS pharmacology & translational science, 3(2), 179–189. https://doi.org/10.1021/acsptsci.0c00012 [↩]
- Sarfraz, N., & Khan, I. (2021). Plasmonic gold nanoparticles (AuNPs): properties, synthesis and their advanced energy, environmental and biomedical applications. Chemistry, an Asian journal, 16(7), 720–742. https://doi.org/10.1002/asia.20200120 [↩]
- Kumari, A., Singla, R., Guliani, A., & Yadav, S. K. (2014). Nanoencapsulation for drug delivery. EXCLI journal, 13, 265–286. [↩]
- Tse, A., & Verkhivker, G. M. (2016). Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways. PloS one, 11(11), e0166583. https://doi.org/10.1371/journal.pone.0166583 [↩]
- Pan, X., Yi, M., Liu, C., Jin, Y., Liu, B., Hu, G., & Yuan, X. (2022). Cilengitide, an αvβ3-integrin inhibitor, enhances the efficacy of anti-programmed cell death-1 therapy in a murine melanoma model. Bioengineered, 13(2), 4557–4572. https://doi.org/10.1080/21655979.2022.2029236 [↩]
- David, L. L., Daniels, A., Habib, S., & Singh, M. (2023). Gold nanoparticles in transferrin-targeted dual-drug delivery in vitro. Journal of Drug Delivery Science and Technology, 90(105168), 105168. https://doi.org/10.1016/j.jddst.2023.105168 [↩]
- Dehkordi, A. A., Mollazadeh, S., Talaie, A., & Yazdimamaghani, M. (2025). Engineering PAMAM dendrimers for optimized drug delivery. Nano Trends, 9(100094), 100094. https://doi.org/10.1016/j.nwnano.2025.100094 [↩] [↩]
- Janaszewska, A., Lazniewska, J., Trzepiński, P., Marcinkowska, M., & Klajnert-Maculewicz, B. (2019). Cytotoxicity of dendrimers. Biomolecules, 9(8), 330. https://doi.org/10.3390/biom9080330 [↩]
- Corte-Real, M., Veiga, F., Paiva-Santos, A. C., & Pires, P. C. (2024). Improving Skin Cancer Treatment by Dual Drug Co-Encapsulation into Liposomal Systems-An Integrated Approach towards Anticancer Synergism and Targeted Delivery. Pharmaceutics, 16(9), 1200. https://doi.org/10.3390/pharmaceutics16091200 [↩]
- Farhoudi, L., Hosseinikhah, S. M., Vahdat-Lasemi, F., Sukhorukov, V. N., Kesharwani, P., & Sahebkar, A. (2024). Polymeric micelles paving the Way: Recent breakthroughs in camptothecin delivery for enhanced chemotherapy. International Journal of Pharmaceutics, 659, 124292. [↩]
- Egorova, A., Selutin, A., Maretina, M., Selkov, S., & Kiselev, A. (2022). Peptide-Based Nanoparticles for Integrin-Targeted DNA Delivery to Cancer and Uterine Leiomyoma Cells. Molecules (Basel, Switzerland), 27(23), 8363. https://doi.org/10.3390/molecules27238363 [↩]
- Barbezan, A., Rosero, W. A., Almeida, L. F., Rigo, M. E., Silva, F. F., & Rostelato, M. E. (2024). In Vivo distribution dynamics of Gold Nanoparticles: A quantitative analysis. Brazilian Journal of Radiation Sciences, 12(4A (Suppl. [↩]
- Kim, A., & Cohen, M. S. (2016). The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert opinion on drug discovery, 11(9), 907–916. https://doi.org/10.1080/17460441.2016.1201057 [↩]
- Bharti, J., Gogu, P., Pandey, S. K., Verma, A., Yadav, J. P., Singh, A. K., Kumar, P., Dwivedi, A. R., & Pathak, P. (2025). BRAF V600E in cancer: Exploring structural complexities, mutation profiles, and pathway dysregulation. Experimental Cell Research, 446(1), 114440. https://doi.org/10.1016/j.yexcr.2025.114440 [↩]
- Pachane, B. C., & Selistre-de-Araujo, H. S. (2024). The role of integrin in cancer therapy resistance. Biomedicines, 12(6), 1163. https://doi.org/10.3390/biomedicines12061163 [↩]



{kind=link}