
Abstract
Spinal muscular atrophy (SMA) is a rare, progressive neuromuscular disorder that affects motor neurons and leads to muscle weakness. Recent advances in treatment have resulted in several therapeutic options for SMA, including Nusinersen and Risdiplam, as well as a promising investigational drug, Apitegromab, each of which has slightly different administration methods and mechanisms of action in treating SMA. Nusinersen and Risdiplam have shown promise by stabilizing motor function and improving outcomes in SMA patients. Although Apitegromab has not received regulatory approval, several clinical trials have been completed with this drug, including emerging longer-duration trials thus far. However, there is limited long-term safety and efficacy data for these drugs due to various factors, including limited patient sample sizes. This review compares the mechanisms, efficacy, and safety of all these therapies while highlighting the need for more comprehensive real-world data and comparative studies to fully understand their impact. Findings suggest that although Nusinersen and Risdiplam improve motor function in treating SMA types 1-3, Risdiplam manifests continuous improvements, whereas Nusinersen plateaus in motor function gains after 26 months. Overall, all three drugs demonstrate potential and underscore the significance of continued research and long-term monitoring, and novel therapeutic approaches to further improve patient outcomes in SMA.
Keywords: Spinal Muscular Atrophy (SMA), Nusinersen, Risdiplam, Apitegromab, Long-term efficacy, Motor function improvement
1. Introduction
Spinal Muscular Atrophy (SMA) is a genetic recessive neurodegenerative disorder that leads to progressive weakness and worsening in certain muscles. SMA is caused by a mutation in SMN1 gene (survival motor neuron 1) which encodes the SMN proteins that play an essential role in maintaining motor neurons controlling muscle voluntary movement1. 5q SMA is the most common SMA, which represents SMN1 gene is located on chromosome 5q13.22. Non-5q SMA, different from the common 5q SMA, refers to mutations on other than SMN gene. Muscular atrophy is a hallmark symptom of SMA that is directly caused by the degeneration of alpha motor neurons in the spinal cord resulting in proximal muscle weakness that affects multiple body functions, limiting life expectancy3. SMA is a common genetic disorder in children, with an estimated incidence between 1 in 6000 and 11,0004. The symptoms of SMA can vary among different people, but most experience respiratory infections, scoliosis, and joint contractures5. SMA is classified in 5 types (type 0-4) based on their age of onset. The onset of SMA ranges from prior to birth to adulthood, and the severity of the disease decreases as the type number increases, or SMA onset is delayed6.
SMA treatments are designed to preserve motor neurons, extend lifespan, and improve motor functions, even though there is no cure for SMA. Medications like Nusinersen and Risdiplam are characterized by targeting the SMN2 gene, a backup gene for mutated SMN1 gene, to enhance its splicing. SMN2 and SMN1 are nearly identical, which indicates that SMN2 could temporarily compensate for the job of SMN1 when it is not working. Although SMN2 and SMN1 both encode for SMN protein, SMN2 produces less-stable and shortened SMN protein, which is insufficient for preventing the muscular atrophy but can slow down its progression6. Among the 3 available medications—Nusinersen, Risdiplam, and Onasemnogene abeparovec—only FDA-approved drugs for all SMA types are Nusinersen and Risdiplam, excluding SMA 0. Nusinersen and Risdiplam have similar mechanisms of action and are aimed at increasing the production of SMN protein, while Onasemnogene abeparovec is a gene therapy only approved for children under the age of 2, making its clinical use highly limited in scope. The administration methods of the three drugs vary drastically, as Nusinersen requires lumbar puncture, Risdiplam is taken orally, and Apitegromab is administered through intravenous infusion. However, due to Onasemnogene abeparovec’s restricted target population, the paper will not include it in the analysis. Instead, it will compare Nusinersen and Risdiplam to another medication that hasn’t been approved by the FDA– Apitgromab.
This review aims to evaluate and compare the long-term efficacy and safety of Nusinersen, Risdiplam, and Apitegromab in treating SMA, highlighting their strengths, limitations, and future directions. Long-term data is crucial in understanding the efficacy, safety, and durability of SMA treatments. SMA is a lifelong, progressive disease, so the extended studies for their drugs are essential in assessing how these therapies maintain over a longer period. Any potential adverse effects of the drug that might not be immediately observed in short-term studies will be shown in the long-term studies. Additionally, comparisons between the long-term efficacy and safety of drugs could also provide valuable clinical insights, and help evaluate the optimal therapeutic strategy.
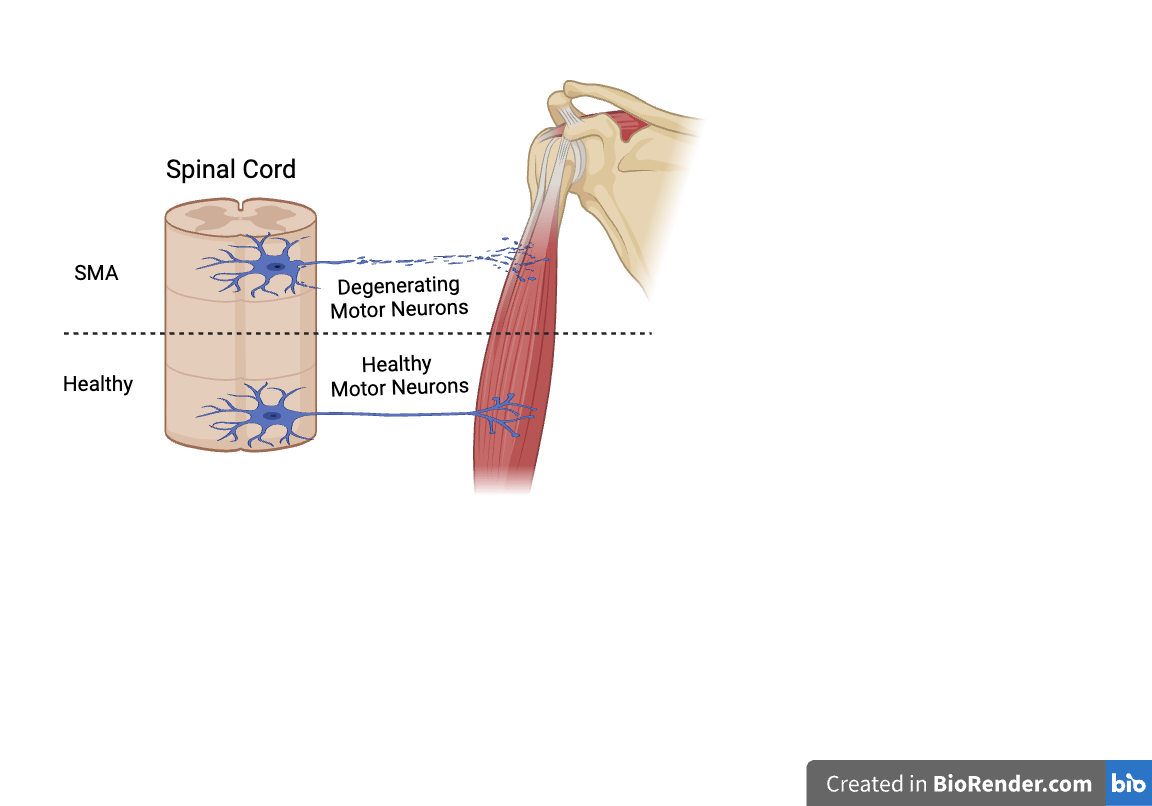
2.1: SMA Types
SMA is classified into five types based on the different ages the disease is diagnosed. Each type of SMA has distinct severity, life expectancy, and motor milestones leading to different treatment approaches. SMA 0, the most severe and rare type, starts at the prenatal period and is associated with an extremely limited lifespan. Key characteristics of SMA type 0 include reduced fetal movement, joint contractures, generalized weakness, hypotonia, and severe respiratory, and feeding difficulties. People haven’t found any effective approach to cure SMA type 0, but more emphasis is on providing supportive care. Prenatal diagnosis and selective termination during pregnancy are the conceivable prevention options for this disease8. Given the effectiveness of Nusinersen in treating SMA type 1 and the lack of curative treatments for SMA type 0, some researchers reported a case study that testifies its efficacy in SMA type 0 patients. In the case report studying an infant patient of SMA type 0, researchers tried to treat the patient after birth with Nusinersen and found that children treated with Nusinersen have shown mild improvements in motor functions but minimal improvement in respiratory drive9. This attempt illustrates that Nusinersen is considered to be a potential treatment for SMA 0, but the clinical data for SMA 0 specifically is limited, and its efficacy cannot be determined at present. Additionally, medicines like Onasemnogene Abeparvovec, and Nusinersen are highly effective in treating SMA types 1 and 2, but their efficacy for treating SMA 0 remains unclear, due to the lack of specific clinical studies targeting SMA 0 at present10.
SMA 1 appears before the age of six months, and diagnosed individuals typically have a lifespan of 8-10 months. It is also known as Werdnig-Hoffman disease and is the most common SMA type, as approximately 60% of patients are diagnosed with this type of SMA11. Patients strive to achieve motor milestones such as head control and sitting with support. They are never able to sit independently, and their swallowing and breathing function decreases over time due to the early onset of respiratory system failure which requires invasive or non-invasive (NIV) ventilatory support12. Nusinersen, Risdiplam, and Onasemnogene Abeparvovec are three medical treatments that have been approved for patients with SMA 1. One indirect comparison (MAIC) of children with SMA type I who received treatment for at least 36 months shows a better effect on using Risdiplam than treating with Nusinersen, suggesting Risdiplam may reduce 78% in mortaility and 81% reduction in the use of permanent ventilation compared to Nusinersen. However, there is a lack of head-to-head trial comparisons, and needs further confirmation and validation13. Onasemnogene Abeparvovec was only investigated in SMA type 1 patients, as it can only treat SMA in children under 2 years old14. SMA type 0 and SMA type 1 can be seen as non-sitters, people who cannot sit independently without support, typically have only 1-2 copies of SMN2.
SMA 2 onset occurs between 6-18 months of age with 70% of SMA 2 patients surviving into their mid-20s and achieving the ability to sit independently. The proximal muscles in their legs usually atrophy more than in their arms, and they develop severe scoliosis that worsens the lung’s capability and the respiratory insufficiency commonly experienced by SMA patients. In SMA type 2 patients, Nusinersen treatment experienced measurable improvements in motor function as measured by the Hammersmith Functional Motor Scale-Expanded (HFMSE) and Revised Upper Limb Module (RULM) which are widely used, validated clinical assessment tools evaluating SMA patients’ functional motor abilities15. Nusinersen may help slow the decline of the respiratory system for patients with SMA 2, though further research is needed to confirm the impact16. The SMA 2 and non-ambulatory SMA 3 patients can be seen as the sitters, as patients in these stages possess the ability to sit with or without outside support, having 3-4 copies of SMN2w17. In SMA types 2 and 3, statistically significant improvements were found in HFMSE and RULM in both Nusinersen treatments and Risdiplam treatments, compared to the baseline (p ≤ 0.001). 80.4% of patients in the Risdiplam group and 80% of patients in Nusinersen reached 3-point cutoffs after 6 months, with no significant difference between the two groups (p=0.33)18.
SMA 3, commonly known as Kugelberg-Welander disease, which begins after 18 months, is characterized by an ability to ambulate independently and a normal lifespan, but the motor function will also decrease along with the progression of the disease19. The disease course in this type of SMA is more stable, with SMA 3 patients showing almost no disease progression in a 12-month HFMSE SMA 3 patients have even shown slight increases in motor functions in 48-month HFMSE evaluations20. Given the relatively stable progression of SMA 3, treatment focuses on maintaining motor function and preventing further decline. Nusinersen is most commonly used to treat SMA type 2 and 3 patients, as it always shows a clinically meaningful improvement in their motor function, shown by the increase of medians compared to baseline in HFMSE and RULM. However, it still needs multicentric studies21. Risdiplam is another common treatment and it also has a significant improvement in patients’ motor function, and younger individuals show more effect than elders22.
SMA 4 is the least severe category of SMA. As the onset of muscle weakness occurs during adulthood, patients experience milder symptoms; and typically have a near-normal lifespan and motor function. Risdiplam and Nusinersen have been approved for treating all SMA types, including SMA 423.
In general, both Nusinersen and Risdiplam have shown clinically meaningful improvements in motor functions across all SMA types, with similarly low rates of adverse events, though slight variation might occur by type24. In contrast, Apitegromab can only treat patients with type 2 and 3 SMA and still lacks head-to-head trials with Nusinersen and Risdiplam for comparisons of efficacy and safety.
2.2: History and mechanism of action of current SMA treatments
2.2.1 Nusinersen
Nusinersen is a therapeutic agent designed to increase the expression of SMN protein, which is reduced in SMA patients. Nusinersen does this by modulating pre-mRNA splicing of SMN2 through the 859G>C which is a genetic variant, a milder allele of SMN2, that acts to produce more full-length SMN transcript by creating a new exonic splicing enhancer (ESE) or disrupting an exonic splicing silencer (ESS)25. A patient’s amount of full-length SMN protein directly correlates with disease severity, meaning that greater protein levels result in a milder phenotype. As this medicine has been used widely by all SMA patients around the world, more than 14000 individuals have been treated with Nusinersen worldwide26.
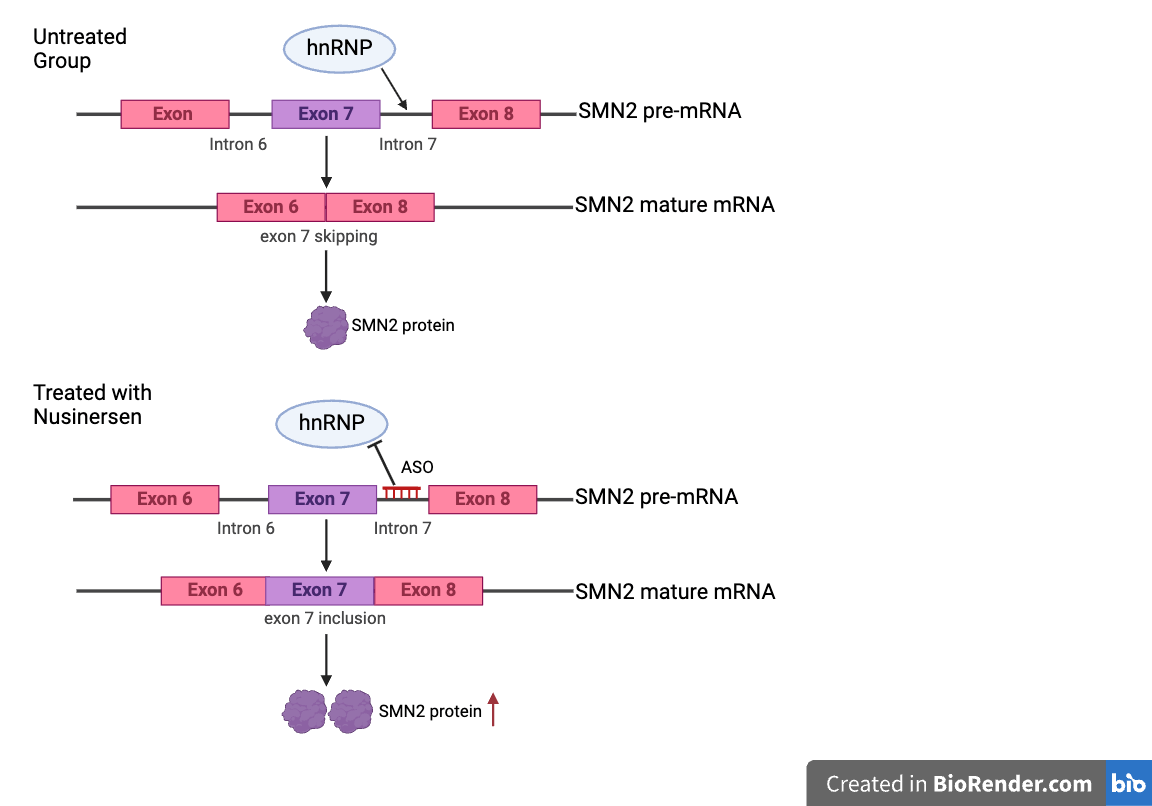
Nusinersen, sold under the brand name Spinraza, is an antisense oligonucleotide (ASO) drug that targets the SMN2 gene to increase the full-length SMN protein levels28. Its mechanism of action is SMN2 pre-mRNA splicing and facilitating the inclusion of exon 7, correcting the common skipping of exon 7 in SMN2. Nusinersen blocks the splicing repressor (heterogeneous nuclear ribonucleoproteins) hnRNP A1/A2 from binding to its target site in intron 7, restoring the recognition of exon 7, this makes SMN2 function more like the SMN1 gene, thereby increasing the number of SMN2-produced functional SMN protein production29. Nusinersen has been shown to improve motor function across all SMA types30. The drug is administered directly into the central nervous system (CNS) through intrathecal injections in a process called lumbar puncture because ASOs cannot penetrate the blood brain barrier so it has to be injected intrathecally31. The first three injections are given every 14 days, and the following are given every four months32. The efficacy of Nusinersen is commonly assessed using functional outcome measures such as the HFMSE and the Hammersmith Infant Neurological Examination Section 2 (HINE-2)33. Additionally, biomarkers found in cerebrospinal fluid (CSF) and plasma work as indicators for monitoring the treatment response. While Nusinersen has been associated with significant clinical improvements in SMA, its benefits have been particularly pronounced in ambulatory patients, as measured by RULM and HFMSE scores at 24 months following the first dose34. The efficacy of Nusinersen is various in different types of disease, including increased survival without the need for permanent respiratory support in SMA type 1 patients, and enhanced motor development slightly in types 1-335.
Before clinical trials, the mechanisms of Nusinersen were tested in in vivo assays. Researchers intrathecally injected Nusinersen into Taiwanese SMA mice and saw that Nusinersen effectively corrected the exclusion of exon 7 during splicing and significantly alleviated SMA symptoms by increasing the expression of SMA proteins. This finding testified the therapeutic potential of Nusinersen. In preclinical research using cynomolgus monkeys, ASO was distributed in the spinal cord without any adverse events, establishing the safeguard for entering future clinical trials36.
Nusinersen was the first FDA-approved treatment for SMA, receiving FDA-approval on December 23, 2016 and EMA-approval on May 30, 2017. Due to the rarity of SMA, Nusinersen was granted the “orphan drug” designation in both the U.S. and the E.U. The ENDEAR and CHERISH trials are phase 3 randomized, placebo-controlled trials including infantile-onset SMA type 1 patients and later-onset SMA type 2 and 3 patients. They were dosed every 4 months over 13 months, and dosed on days 1, 29, 85, and 274 over the span of 15 months. The trial results demonstrated that Nusinersen significantly improves motor function and survival in infants compared to placebo32. These clinical trials assessed Nusinersen’s efficacy through almost all types of SMA. In the ENDEAR trial that lasted for 13 months, 51% of patients treated with Nusinersen achieved a motor milestone, while none in the control group did. The risk of death or the need for permanent assisted ventilation was 47% lower in the Nusinersen-treated group compared to the baseline group. However, because the trial terminated earlier due to its positive outcomes, the results lack credibility in assessing Nusinersen’s long-term efficacy37. In the CHERISH trial, 57% of the children with Nusinersen treatment had a considerable improvement in motor functions from baseline to month 15 shown by HFMSE scores compared with 26% in the control group. Because of the different dosage schedule and the exclusion of patients with more advanced disease features, it limits the generalizability to the broader SMA types. Also, this trial only has 15 months, which restricts Nusinersen’s long-term efficacy38.
Despite its benefits, Nusinersen treatment poses challenges beyond its high cost. The administration of intrathecal injections can be technically difficult, especially in patients with severe scoliosis or spinal deformities, especially 6-15 year old individuals who are most vulnerable to the complication39. Furthermore, Nusinersen has been associated with several adverse events (AEs) that include post-lumbar puncture syndrome, idiopathic intracranial hypertension, procedural pain, hypokalemia, and transient hearing loss40.
2.2.2 Risdiplam
Risdiplam, a small-molecule drug that modifies SMN2 pre-messenger RNA splicing, is sold under the brand name Evrysdi. It works similarly to Nusinersen, as both treatments target the SMN2 gene trying to enhance the production of viable SMN protein by altering mRNA splicing to integrate exon 7 into the gene39. The FDA approved Risdiplam for general usage on August 7, 2020, and EMA approved it on March 30, 2021. Initially, Risdiplam was only addressed to treat SMA patients over two months, however, the FDA expanded the range to include patients younger than two months old in 202241.
Several preclinical studies helped establish mechanisms and proof-of-concept data for Risdiplam in SMA treatment. In vitro studies confirmed Risdiplam’s mechanisms as an SMN2 pre-mRNA splicing modifier, increasing SMN protein expression by including exon 7. In in vivo studies, SMA mouse models treated with Risdiplam exhibited increased SMN protein expression in CNS and peripheral tissues. SMN protein increased by 23%, 31% from baseline in muscle and heart by oral dosing, with similar increases in brain and spinal cord. Similarly, the SUNFISH trial’s clinical data showed dose-dependent, functional SMN protein increases found in the CNS42.
The FIREFISH and SUNFISH are clinical trials that assess the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of Risdiplam among all types of SMA patients. The SUNFISH is specifically focused on safety and efficacy of Risdiplam in patients with SMA Types 2 and 3. The results showed no significant differences in safety between varying doses. The trial followed patients for at least 12 weeks with escalating doses, demonstrating an increase in SMN protein levels and stabilization of motor function over 24 months. Although the Risdiplam group reported a higher number of AEs than the placebo group, these were mostly moderate, with no serious AEs across all dose levels22. In contrast to the significant decline observed in a natural history cohort, the treated group showed improvements in scores from baseline on the HFMSE, MFM32, RULM, and Spinal Muscular Atrophy Independence Scale-Upper Limb Module (SMAIS-ULM) assessments43. The long-term efficacy of Risdiplam will be discussed further in later sections of this paper.
On the other hand, the FIREFISH study is a 2 parts, open-labeled, and focused on infants between1-7 months with SMA Type 1. The treatment result demonstrates that oral Risdiplam increased functional SMN protein expression in the blood39. The results indicate that 41% of treated patients were able to sit without support after 24 months of treatment. Although none of the participants could stand or walk independently, significant motor improvements compared to the natural history patients44.
While Risdiplam’s mechanism of action is similar to that of Nusinersen, it offers some distinct advantages. It is the first oral medicine approved for SMA administered in a liquid form providing a more accessible and convenient treatment option compared to Nusinersen, as patients can take it at home instead of doing lumbar punctures45. Oral administration is a significant advantage of Risdiplam, as it allows the drug to reach systemic tissues involved in the multisystem pathogenesis of SMA and express more well-tolerated than Nusinersen. Furthermore, it showed a higher opportunity to prolong survival and improve motor function for patients, making it a potentially superior alternative to Nusinersen46. Risdiplam is certainly not a perfect solution, it has multiple restrictions for patients on their personal medical conditions, and the sample sizes are too small that might result in some unknown severe AEs47. While Risdiplam has demonstrated efficacy in increasing SMN protein levels and improving motor function, its long-term outcomes remain uncertain14. The most commonly reported AEs associated with Risdiplam across all SMA types include coughing, fever, headache, diarrhea, vomiting, upper respiratory tract infections, pneumonia, and nasopharyngitis48.The severer AEs are caused by SMA, including pneumonia, nasopharyngitis, and upper respiratory tract infections, and other AEs are drug-related that are caused by normal side effects of drugs in many clinical trials.
2.2.3 Apitegromab
Apitegromab (SRK-015) is a monoclonal antibody constructed to block the activation of myostatin, a protein that regulates skeletal muscle growth. Apitegromab aims to preserve muscle mass by binding with inactive forms of myostatin, allowing the muscles to grow49. The purpose of the Apitegromab contrasts with Nusinersen and Risdiplam, as it emphasizes improving muscle function rather than increasing SMN protein levels. Instead of directly targeting the mature forms of myostatin, Apitegromab binds to promyostatin and latent myostatin, to inhibit their activation50 . Myostatin usually signals through the ActRIIB receptor to suppress the number of protein in muscles, but this receptor also interacts with other proteins that regulates muscle growth51. Hence, Apitegromab circumvents these challenges by selectively binding to inactive myostatin forms to minimize unintended effects on related proteins52. Because of Apitegromab’s effective function in targeting muscle atrophy, myostatin has been explored as a potential therapeutic target for preventing and reversing muscle atrophy diseases53. Due to these findings, Apitegromab has received Fast Track, Orphan Drug, and Rare Pediatric Disease designations from the FDA and Orphan Medicinal Product designations from the EMA for being a possible treatment of SMA. However, the drug has not yet been approved for use in patients51.
Toxicology data in cynomolgus monkeys and rats demonstrate Apitegromab’s selectivity and safety by highlighting how it effectively inhibits promyostatin with no adverse impact on neurodevelopment, motor function, and reproduction. Based on the high sequence homology of myostatin prodomains between human and rats (94.5%), and human and monkey (95.7%), these preclinical studies support its safety when used in adult and pediatric patients with SMA54. The results of a randomized, phase 1 safety, pharmacokinetics, and pharmacodynamics study demonstrated that in both single and multiple increasing doses of Apitegromab, Apitegromab is safe and well-tolerated in healthy individuals, even at doses as high as 30 mg/kg55. These results support the accessibility of continually investigating patients with type 2 and 3 SMA, and generally align with the drug’s safety profile in TOPAZ and SAPPHIRE trials.
The TOPAZ trial was a Phase 2 proof-of-concept, open-label study that evaluated the safety and efficacy of Apitegromab in patients with SMA Types 2 and 3. Participants were grouped into three cohorts based on their age and motor achievements. They were treated with Apitegromab infusions and combination with Nusinersen every four weeks for 12 months. For assessing their motor function, the HFMSE, RULM, and (Revised Hammersmith Scale) RHS have been used. HFMSE is employed to test non-ambulatory individuals and RHS is used for ambulatory individuals. At the 6-month interim analysis, mean increases were found in all cohorts, and a substantial proportion of patients were found to have a significant increase in HFMSE (≥3 points)56. As the 12-month TOPAZ study indicates an improved motor function and a favorable safety condition in both types, it has been extended to test the efficacy and safety for only the non-ambulatory group to 36 months. RULM, specified to assess upper-limb function, WHO motor milestones, and PEDI-CAT are used in this study to testify the efficacy57. All of them displayed sustained improvement from baseline over 36 months, with satisfactory safety profiles58. The SAPPHIRE trial was a Phase 3, randomized, double blind, placebo-controlled study designed to assess the safety and efficacy of Apitegromab in nonambulatory patients with SMA Types 2 and 3 all of whom have been treated with either Nusinersen or Risdiplam before. 188 patients from age 2 to 21 were enrolled in this trial and were assigned evenly in three groups to receive either Apitegromab 10 mg/kg, 20 mg/kg, or placebo through IV every four weeks to 12 months59. These promising results are limited by their small sample size , and the TOPAZ trial is only in phase 2, suggesting sustained efficacy and safety, but require pending confirmation in larger Phase 3 studies like SAPPHIRE.
The FDA granted Fast Track designation to Apitegromab in May 2021, noticing its potential to address in treating SMA. This decision was influenced by promising data from the Phase 2 TOPAZ trial, which was first reported in April 2021 and demonstrated significant improvements in motor function among SMA patients receiving Apitegromab. In October 2024, the Phase 3 SAPPHIRE trial further reinforced Apitegromab’s efficacy, by reaching its primary endpoint with statistically significant improvements (mean HFMSE change: 1.8, p = 0.0192) in motor function compared to the placebo group. Based on the TOPAZ and SAPPHIRE clinical trials’ exciting data demonstrating meaningful improvement in motor function, most of the patients who participated in Phase 3 SAPPHIRE were enrolled in the ongoing ONYX trial. ONYX is another open-label phase 3 study evaluating efficacy and safety after non-ambulatory patients who finished the TOPAZ and SAPPHIRE trials. Although the FDA will not make a final decision on its approval until mid-2025, the development of Apitegromab has represented a milestone in finding effective treatments for SMA60.
Although Apitegromab has shown promising motor improvement in non-ambulatory patients, limitations still remain. The primary limitation of this drug is that it is not disease-modifying, which leads to the requirement of combinations with other drugs like Nusinersen and Risdiplam. Secondly, the majority of the clinical trials are targeting non-ambulatory patients, which limits the generalizability of a broader SMA population. Lastly, this investigational drug still needs more clinical data and more regulatory approval to enhance its credibility, as the result of the ONYX trial is pending.
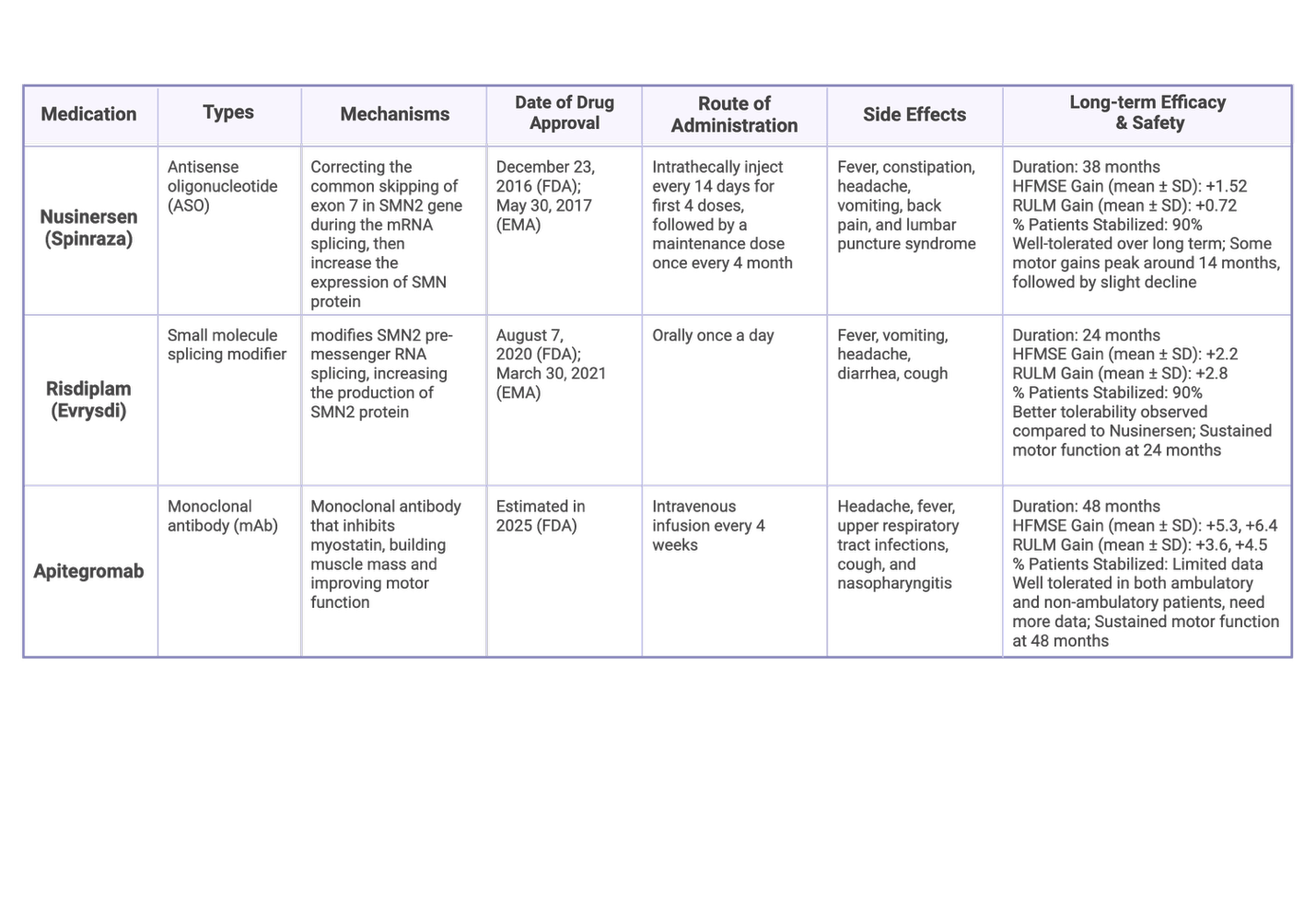
Table 1: Overview of medication option of SMA61
2.3: Long-term efficacy and safety of current SMA treatments
Long-term efficacy refers to the sustained effectiveness of a medical treatment over time. It is important to testify the drugs’ long-term manifestation because the disease is progressive, and patients’ muscle weakness will worsen over time increasing the risk to patients, and it helps identify the ways to improve their effectiveness62. There are 3 existing drugs that have been approved for use in therapies, Nusinersen (Spinraza), Onasemnogene Abeparvovec (Zolgensma), and Risdiplam (Evrysdi). The studies of these three drugs mostly focus on their advantages and setbacks, neglecting their long-term benefits and safety. Real-world data and case studies are essential for this rare disease to comprehensively explore and contrast their actual effects63. As for Apitegromab, it is a new medicine that has only been used in clinical trials but has not yet been approved for a widespread usage. Therefore, Apitegromab’s long-term experiments are extremely limited and its efficacy and safety still remains unclear55.
2.3.1 Nusinersen
Nusinersen, the most commonly used treatment for SMA, has demonstrated continued motor function benefits. Nusinersen stabilizes motor function in children with SMA types 1 to 3 maintaining up to 2-3 years, but still varies among individuals64. In a study that targets SMA type 2 and 3 patients treated with Nusinersen at least over 2 years, improvements were more pronounced in HFMSE than in RULM scores, with the most significant gains occurring at 12 months in SMA type 2 (mean HFMSE change: +1.6, p = 0.009) and at 24 months in both type 2 and 3 (mean change: +1.9, p = 0.019; +1.5, p = 0.017)65.
The efficacy of Nusinersen tends to plateau after a certain point rather than improving continuously. In a long-term observation of adult patients treated with Nusinersen for up to 30 months, the Medical Research Council strength scale (MRC median increase 1.85 points per year) exhibited improvement from baseline to 14 months but stabilized afterward (p ≤ 0.03). Similarly, the RHS increased up to 6 months (p = 0.02), but no significant changes were observed afterward, and there was no notable improvement in respiratory function66. In another European multinational observational study, Nusinersen’s efficacy in adults with 5q-SMA for up to 16 months was confirmed, but the long-term outcomes were left in uncertainty. A 38-month study, the longest Nusinersen study, showed initial motor function gains peaking at 14 months (HFMSE mean difference +1.72, 95% CI: 1.19-2.25; RULM mean difference +0.75, 95% CI: 0.43-1.07), declining at 26 months (HFMSE +1.20, 95% CI: 0.48-1.91; RULM +0.65, 95% CI: 0.27-1.03), and slightly rebounding at 38 months (HFMSE +1.52, 95% CI: 0.74-2.30; RULM +0.72, 95% CI: 0.25-1.18). The statistical significance of these changes decreased over time, with p-values of 0.032 at 14 months, 0.169 at 26 months, and 0.289 at 38 months. This suggests that motor function improvement was most pronounced at 14 months, followed by a plateau or a decline over time67. The improved motor function was observed in 72%, and stabilization in another 18% over 38 months68. While some patients experience improvement and stabilization, others experience a decline after peaking in 26 months, indicating the efficacy of Nusinersen and the need for further long-term67.
The result reported that 77% of patients were observed with at least one adverse reaction but some lacked detailed information, confirming Nusinersen’s long-term safety. The most common adverse reactions were post-lumbar puncture syndrome such as vomiting in 72 hours after lumbar puncture, headache, back pain, constipation, and infections69. There were no studies of the ENDEAR and EMBRACE trials interrupted because of Nusinersen’s AEs, the discontinuation due to AEs happens more frequently in control groups. This evidence guarantees the safety of Nusinersen70.
Overall, Nusinersen exposes notable benefits in stabilizing motor function and improving muscle strength, especially within the first half of the whole reported timeline66. However, its effects have leveled off over time rather than improved continuously. The SHINE trial and the long-term observational study confirm its initial gains in motor level but highlight the decline after reaching the climax in 26 months67. The safeness of Nusinersen is generally well-tolerated as it reveals no relevant organ toxicity, platelet declines, or coagulopathies that were universal in ASO drugs71.
2.3.2 Risdiplam
In contrast to the long-term efficacy and safety of Nusinersen, Risdiplam demonstrates a clearly lower mortality rate and the need for permanent assisted ventilation compared to other SMA treatments, even after several years. Additionally, studies indicate ongoing improvements in motor function over time with Risdiplam treatment72. The FIREFISH trial is used to assess the efficacy of Risdiplam in patients with infantile-onset SMA type 1.
In a comparative study, researchers conducted a difference between Risdiplam and Nusinersen in children with type 1 SMA who received treatment for 24 months. The study involves a SHINE trial, ENDEAR trial, and FIREFISH trial to evaluate the long-term safety and efficacy of Nusinersen across SMA types 1 to 3. Remarkably, the result found that the Risdiplam group showed a 78% reduction in mortality (MAIC HR for overall survival 0.22, 95% CI: 0.04-0.47), an 81% reduction in permanent ventilation rates (MAIC HR for event-free survival 0.19, 95% CI: 0.07-0.35), a 45 % higher possibility of achieving a HINE-2 motor milestone responses (MAIC HR 1.45, 95% CI: 1.21-1.80), and 57% reduction in the rate of severe adverse events in comparison with Nusinersen (MAIC HR 0.43, 95% CI: 0.30-0.59). Risdiplam-treated children had a lower death rate, a lower rate of permanent ventilation, a higher rate of achieving motor function responses, and a lower rate of having severe adverse events (SAEs) compared to patients treated with Nusinersen, suggesting that patients experience better treatment safety and tolerability over time. These findings, based on indirect MAIC comparison, suggest the conclusion that Risdiplam may be a superior alternative to Nusinersen in treating type 1 SMA over a long term, but head-to-head trials are needed to confirm these results due to its limitations13.
The SUNFISH trial on the other hand evaluates the efficacy of Risdiplam in patients with later-onset SMA types 2 and 3. In a SUNFISH trial, Risdiplam led to significant improvement in both the 32-Motor Function Measure (MFM32) and RULM with later onset SMA type 2 and 3, though no significant change was observed in HFMSE. The Risdiplam group also had a higher percentage of patients achieving the Minimal Clinically Important Difference (MCID). By 12 months, Risdiplam-treated patients met the primary endpoint by showing a significant increase in the score of MFM32 compared to placebo. However, after 24 months, motor function stabilization was achieved in 58% of Risdiplam-treated patients (a change of ≥0), and 32% was achieved motor function improvement (a change of ≥3). Both improvement and stabilization are shown in the total scores of HFMSE (95% CI, mean change: +2.2 vs. +0.0), RULM (95% CI, +2.8 vs. +0.9), and MFM32 (95% CI, +1.4 vs. +0.3) relative to the comparator, but younger patients usually experience a greater improvement22. Overall, Risdiplam led to significant improvement in motor function for patients with type 2 or non-ambulant type 3 SMA, with motor function remaining stable after 12 months of treatment. Unlike Nusinersen, the safety profile of Risdiplam after 24 months remained consistent with that observed at 12 months, and over 24 months, corroborating the benefit of long-term treatment. No significant changes were observed in respiratory or nutritional outcomes over the long-term treatment, and also the quality of life (QoL), as measured by caregiver-reported SMAIS-ULM that reported independence, between the Risdiplam and placebo groups73. The limitations of the SUNFISH Part 2 study were that it excluded ambulatory patients and only represented a small age group, so its generalizability to the patient population is unclear74.
As for the safety of Risdiplam, both the Risdiplam and placebo groups experienced frequent AEs and SAEs, but only a few were considered treatment-related. The most common AEs are infections and infestations75.
While Risdiplam has demonstrated efficacy and safety in clinical trials, there is currently limited long-term data compared to Nusinersen, as a 24-month observation period may not be sufficient to fully assess its long-term effects. Further studies are needed to confirm its sustained effectiveness and safety in the longer term, and more samples are required to be studied76. Risdiplam demonstrated significant benefits in reducing mortality and improving motor function in patients with SMA, providing a strong support for its long-term efficacy.
2.3.3 Apitegromab
Apitegromab is a medicine that is used to treat SMA disease but it has not yet been licensed so it is currently only available in clinical trials. Therefore its long-term efficacy data remains limited. The primary source of long-term data for Apitegromab is the TOPAZ study, which reports outcomes in nonambulatory patients over a 36-month period. Efficacy was assessed using multiple outcome measures, including the RULM, HFMSE, and caregiver-reported assessments such as the Pediatric Evaluation of Disability Inventory-Computer Adaptive Test and the Patient-Reported Outcomes Measurement Information System. Findings from the study indicate sustained improvement from baseline prolonged to 36 months with mean changes in HFMSE of +4.0 and +4.8 (95% CI), and in RULM of 2.4 and +2.8 (95% CI), demonstrating a similar benefit compared to the 12-month results—HFMSE: +3.6 and +4.6 (95% CI); RULM: +1.3 and +1.2 (95% CI). No new safety concerns observed during the extended follow-up51. As this study is just a phase 2 study with limited sample sizes, its outcomes are referable but still need later confirmation in the phase 3 SAPPHIRE trial.
Later in a longer 48-month TOPAZ study, results emphasized a patient retention rate of over 90%, with no new safety concerns. Initial 12-month results were reported in April 2021, followed by subsequent data updates at 24, 36, and 48 months. These long-term findings consistently demonstrated sustained benefits in motor function, with no new safety concerns emerging. The mean change in HFMSE scores from baseline was 5.3 points (95% CI: 1.5-9.2) and 6.4 points (95% CI: 1.8-11.0)across different age groups, while the mean change in RULM scores was 3.6 (95% CI: 2.0-5.3)points and 4.5 points (95% CI: 2.7-6.3), respectively. Notably, 31% of participants were excluded from the four-year study due to undergoing scoliosis surgery suggesting a common confounding factor in motor function assessments77.
The safety of Apitegromab remains promising. In the 36-month TOPAZ trial, over 90% of patients experienced at least one treatment-emergent adverse event (TEAE). However, all were mild or moderate in severity, as minor amount of TEAE and death were reported. The most common TEAEs included pyrexia, nasopharyngitis, headache, vomiting, COVID-19 infection, and upper respiratory tract infections. No deaths, unexpected SAEs, or hypersensitivity reactions to Apitegromab were reported, and no patients tested positive for Apitegromab antibodies78.
In conclusion, the long-term efficacy and safety of Apitegromab remain uncertain due to limited experimental data. Its effectiveness and safety have not been fully established, emphasizing the necessity of further research and clinical trials.
3. Discussion
SMA has seen significant advancements in treatment options with the introduction of therapies such as Nusinersen, Risdiplam, and Apitegromab. However, these treatments still have several limitations and long-term safety and efficacy have yet to be determined. A critical limitation in the current SMA treatment landscape is the lack of extensive comparative studies, a bigger sample size, and a statistically robust. More studies comparing these treatments with SOCs or other FDA approved therapies could be useful. Also, many clinical trials have small sample sizes due to the disease rarity and controlled environments that may not fully represent the diverse SMA population. For instance, the FIREFISH trial evaluating Risdiplam included only 62 participants, and the TOPAZ trial only involved 58 participants79. Moreover, while clinical trials provide valuable data, they often have strict inclusion criteria that may exclude patients with comorbidities or those at advanced disease stages. This exclusion limits our understanding of how these therapies perform in the broader SMA community. Real-world evidence, beyond clinical trials, has evaluated nusinersen’s adherence in patients with SMA Types 1–3, demonstrating clinically significant motor function improvements across all types. Younger patients with Types 1 and 2 exhibited greater improvements, while older Type 3 patients primarily experienced stabilization80. Real-world data is essential to assess the effectiveness and safety of treatments in routine clinical practice, capturing a wider range of patient experiences and outcomes.
In addition, future research should focus on long-term efficacy and safety studies. Conducting extended follow-up studies to monitor any potential late-emerging adverse events and checking the sustainability of the effect. Beyond Nusinersen, Risdiplam, and Apitegromab, several other promising therapies could also improve SMA treatments. Onasemnogene Abeparvovec (Zolgensma) is one drug that has already been approved by the FDA to treat this disease. It is a one-time gene therapy that has shown significant improvements in motor function in infants with SMA type 1, but not other SMA types. Aside from pharmaceutical interventions, physical therapy could also aid in maintaining muscle function, improving mobility, and preventing complications. Ventilation assistance could also help patients with breathing difficulties, who may require non-invasive ventilation to prevent sleep apnea.
While clinical trials suggest that both Nusinersen and Risdiplam improve motor functions in patients with SMA Types 1–3, their comparative efficacy remains unclear due to the absence of direct head-to-head trials. Although Nusinersen reveals long-term benefits in SMA Types 1 and 2, its effects may plateau after 24–30 months. Also, there are no current experiments testing the efficacy of Nusinersen beyond 5 years, making it unclear whether its use is worth the risks that come with its invasive administration, which include infections, headaches, or spinal cord injury81. On the other hand, the long-term efficacy of Risdiplam is still being evaluated, exhibiting sustained efficacy for up to 2 years, with reports of fewer severe adverse events and better accessibility due to its oral administration. Clinical trials hint that Risdiplam may offer advantages over Nusinersen in SMA Type 1 due to its lower mortality rates and its respiratory conditions. This conclusion should be considered cautiously, because there are no randomized controlled trials directly compare the two. Apitegromab, an investigational drug demonstrating favorable research outcomes but still lacks extensive long-term data, real-world validation. Its current outcomes focused on non-ambulatory patients, and typically used with other SMN-enhancing therapies, limit its population and complicates its independent efficacy. Onasemnogene abeparovec has shown remarkable long-term results in infants, but as a one-time treatment, its lifelong effects on motor skills are still understudied82. Onasemnogene abeparovec is a gene therapy only for children less than 2 years old, because it is indicated for replacing patients’ SMN 1 gene with a healthy one in motor neurons, which ensures the therapy is given before the disease causes irreversible consequences6. While SMA research has greatly advanced in recent years, the sample sizes of clinical research are still not wide enough, ascribed to the small sample size for a rare disease. Head-to-head trials comparing Nusinersen, Risdiplam, and Apitegromab are highly limited, restricting the ability to directly compare their efficacy and safety across different patient subgroups; therefore, comparative effectiveness studies are needed to evaluate the long-term efficacy for each treatment.
Although current SMA treatments have improved patient outcomes, addressing the limitations of existing studies and expanding research into new therapeutic avenues are crucial. A comprehensive approach that includes comparative effectiveness research, long-term safety monitoring, and exploration of novel therapies will be essential to improve its treatment.
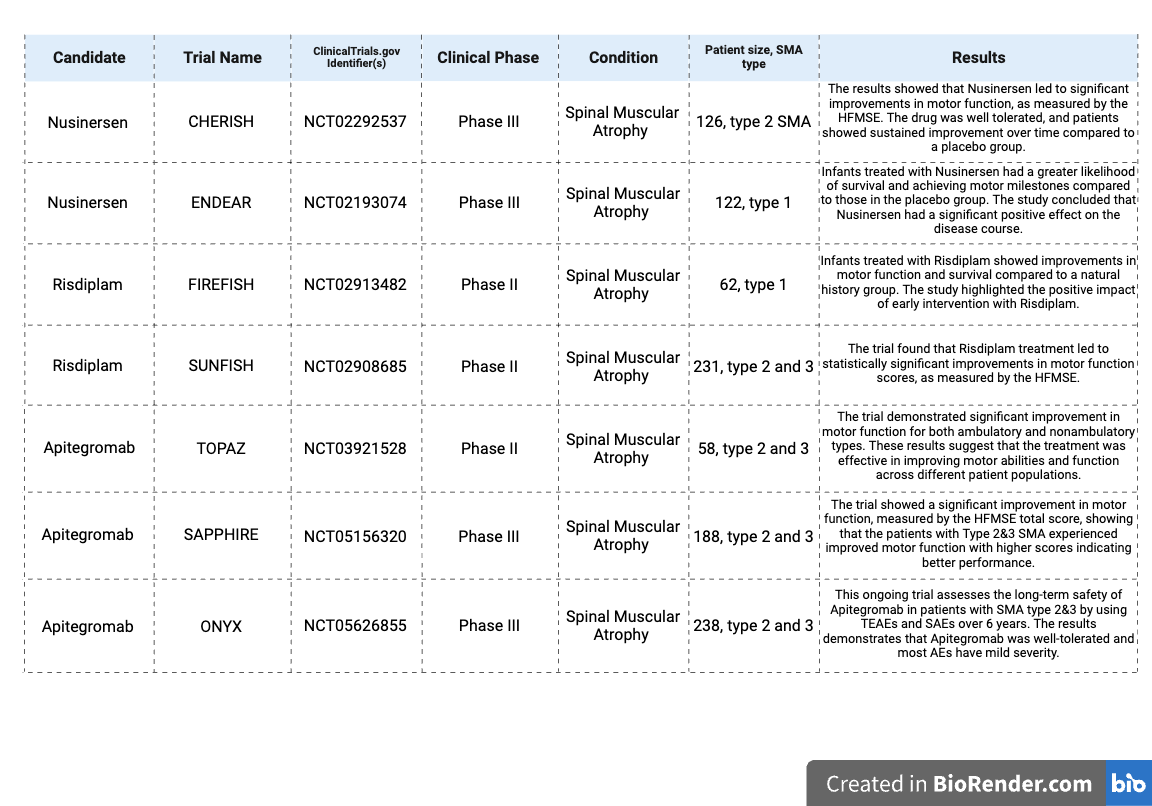
Table 2: Long-Term Clinical Trials Assessing the Efficacy of SMA Treatments83
4. Methods
A systematic literature search was performed in PubMed and ClinicalTrials.gov to identify relevant studies on mechanism, long-term efficacy, and safety of Nusinersen, Risdiplam and Apitegromab in the treatment of 5q-SMA. The search included combinations of MeSH terms and free text keywords such as “Nusinersen,” “Risdiplam,” “Apitegromab,” “Spinal muscular atrophy,” and “Long-term effect.” For subtopics 1 and 2, which cover the pathology of SMA and mechanisms of the three drugs, the search included peer-reviewed preclinical and clinical studies, as well as narratives and systematic reviews that provided detailed insight. No publication date restriction was applied here. For subtopic 3, which provides a long-term efficacy and safety analysis, peer-reviewed completed clinical trials and observational studies from the last 10 years were included. In PubMed, filters were applied to limit results to clinical trials and reviews, while on ClinicalTrial.gov, filters were applied to include only interventional and observational study types, with a minimum follow-up duration of 12 months. Studies were screened based on titles and abstracts, followed by full-text review. Inclusion criteria contained studies involving 5q-SMA patients treated with the specified drugs, showing clinical efficacy or safety outcomes. Exclusion criteria contained non-peer-reviewed articles, case reports, and trials with insufficient follow-up. Scientific and clinical data with a focus on long-term efficacy and safety of different drugs across different SMA types was primarily included in the review.
References
- H. Chaytow, Y.T. Huang, T.H. Gillingwater, K.M.E. Faller. The role of survival motor neuron protein (SMN) in protein homeostasis. Cellular Molecular Life Sciences 75, 3877-3894 (2018). [↩]
- S. Kleinle, V. Scholz, A. Benet-Pagés, T. Wohlfrom, S. Gehling, F. Scharf, S. Rost, E.C. Prott, S. Grinzinger, A. Hotter, V. Haug, S. Niemeier, L. Wiethoff-Ubrig, T. Hagenacker, K. Goldhahn, A. Von Moers, M.C. Walter, P. Reilich, K. Eggermann, F. Kraft, I. Kurth, H. Erdmann, E. Holinski-Feder, T. Neuhann, A. Abicht. Closing the Gap – Detection of 5q-Spinal Muscular Atrophy by Short-Read Next-Generation Sequencing and Unexpected Results in a Diagnostic Patient Cohort. Journal of Neuromuscular Diseases 10, 835 (2023). [↩]
- K. Kant-Smits, B. Bartels, F.L. Asselman, E.S. Veldhoen, R.P.A. van Eijk, W.L. van der Pol, E.H.J. Hulzebos. The RESISTANT study (Respiratory Muscle Training in Patients with Spinal Muscular Atrophy): study protocol for a randomized controlled trial. BMC Neurology 23 (2023). [↩]
- S. Nicolau, M.A. Waldrop, A.M. Connolly, J.R. Mendell. Spinal Muscular Atrophy. Seminars in Pediatric Neurology 37 (2021). [↩]
- Johns Hopkins Medicine. Spinal Muscular Atrophy (SMA) | Johns Hopkins Medicine. (n.d.). https://www.hopkinsmedicine.org/health/conditions-and-diseases/spinal-muscular-atrophy-sma; NIH. Spinal Muscular Atrophy | National Institute of Neurological Disorders and Stroke. (n.d.). https://www.ninds.nih.gov/health-information/disorders/spinal-muscular-atrophy [↩]
- NIH. Spinal Muscular Atrophy | National Institute of Neurological Disorders and Stroke. (n.d.). https://www.ninds.nih.gov/health-information/disorders/spinal-muscular-atrophy [↩] [↩] [↩]
- H. Chaytow, Y.T. Huang, T.H. Gillingwater, K.M.E. Faller. The role of survival motor neuron protein (SMN) in rotein homeostasis. Cellular Molecular Life Sciences 75, 3877-3894 (2018).; S. Kleinle, V. Scholz, A. Benet-Pagés, T. Wohlfrom, S. Gehling, F. Scharf, S. Rost, E.C. Prott, S. Grinzinger, A. Hotter, V. Haug, S. Niemeier, L. Wiethoff-Ubrig, T. Hagenacker, K. Goldhahn, A. Von Moers, M.C. Walter, P. Reilich, K. Eggermann, F. Kraft, I. Kurth, H. Erdmann, E. Holinski-Feder, T. Neuhann, A. Abicht. Closing the Gap – Detection of 5q-Spinal Muscular Atrophy by Short-Read Next-Generation Sequencing and Unexpected Results in a Diagnostic Patient Cohort. Journal of Neuromuscular Diseases 10, 835 (2023). [↩]
- R.N. Singh, N.N. Singh. Mechanism of Splicing Regulation of Spinal Muscular Atrophy Genes. Advances in Neurobiology 20, 31-61 (2018). [↩]
- E. Tiberi, S. Costa, M. Pane, F. Priolo, R. de Sanctis, D. Romeo, F.D. Tiziano, G. Conti, G. Vento, E. Mercuri. Nusinersen in type 0 spinal muscular atrophy: should we treat?, Annals of Clinical and Translational Neurology 7, 2481-2483 (2020). [↩]
- B. Chongmelaxme, V. Yodsurang, P. Vichayachaipat, T. Srimatimanon, O. Sanmaneechai. Gene-based therapy for the treatment of spinal muscular atrophy types 1 and 2 : a systematic review and meta-analysis. Gene Therapy (2024). [↩]
- C.W. Lin, S.J. Kalb, W.S. Yeh. Delay in Diagnosis of Spinal Muscular Atrophy: A Systematic Literature Review. Pediatric Neurology 53, 293-300 (2015). [↩]
- N. Brolatti, F. Trucco, M. Ferretti, C. Avanti, P. Tacchetti, C. Panicucci, P. Striano, C. Minetti, C. Bruno, M. Pedemonte. Structured Light Plethysmography for Non-Invasive Assessment of Respiratory Pattern in Spinal Muscular Atrophy Type 1. Journal of Clinical Medicine 12 (2023). [↩]
- C. Kokaliaris, R. Evans, N. Hawkins, A. Mahajan, D.A. Scott, C.S. Sutherland, J. Nam, G. Sajeev. Long-Term Comparative Efficacy and Safety of Risdiplam and Nusinersen in Children with Type 1 Spinal Muscular Atrophy. Advances in Therapy 41, 2414-2434 (2024). [↩] [↩]
- J.W. Day, K. Howell, A. Place, K. Long, J. Rossello, N. Kertesz, G. Nomikos. Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatrics 22 (2022). [↩] [↩]
- D.R. van der Woude, R.I. Wadman, F.L. Asselman, M.A.G.C. Schoenmakers, I. Cuppen, W.L. van der Pol, B. Bartels. Exploring functional strength changes during nusinersen treatment in symptomatic children with SMA types 2 and 3. Neuromuscular Disorders 41, 1-7 (2024).; T. Chen, S. Chang, Y. Wu, Y. Yen, F. Hsu, Y. Chen, Y. Ming, H. Hsu, Y. Su, S. Wong, J. Hung, S. Chiou, Y. Jong, J. Chen. MiR34 contributes to spinal muscular atrophy and AAV9-mediated delivery of MiR34a ameliorates the motor deficits in SMA mice. Molecular Therapy. Nucleic Acids 32, 144-160 (2023). [↩]
- F. Trucco, S. Dastagir, H. Tan. Pseudo-obstructive sleep disordered breathing – definition and progression in Spinal Muscular Atrophy. Sleep Medicine 115, 61-65 (2024). [↩]
- B. Wirth, M. Karakaya, M. Kye, N. Mendoza-Ferreira. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annual review of genomics and human genetics 21, 231-261 (2020).; J. Lamadrid-González, S. Castellar-Leones, J. Contreras-Velásquez, V. Bermúdez. SMN2 Copy Number Association with Spinal Muscular Atrophy Severity: Insights from Colombian Patients. Journal of Clinical Medicine 13, 6402 (2024).; T. Prior, M. Leach, E. Finanger. Spinal Muscular Atrophy. Gene Reviews, 1-30 (2024). [↩]
- M. Ashrafi, M. Babaee, S. Hashemi Nazari, M. Barzegar, M. Ghazavi, M. Beiraghi Toosi, S. Nafissi, S. Inaloo, G. Zamani Ghaletaki, F. Fatehi, R. Heshmat, M. Ghahvechi Akbari, A. Abdi, H. Bakhtiary, H. Montazerlotfelahi, A. Abbaskhanian, S. Hosseini, H. Farshadmoghadam, S. Hosseiny, F. Shariatmadari, B. Ziaadini, M. Babaei, A. Tavasoli, S. Nikbakht, A. Momen, A. Khajeh, V. Aminzadeh, M. Mollamohammadi, M. Taghdiri, M. Nasehi, S. Memarian, R. Badv, M. Heidari, N. Jafari. Comparative efficacy of risdiplam and nusinersen in Type 2 and 3 spinal muscular atrophy patients: A cohort study using real-world data. Journal of neuromuscular diseases 11, 1190-1199 (2024). [↩]
- C.W. Lin, S.J. Kalb, W.S. Yeh. Delay in Diagnosis of Spinal Muscular Atrophy: A Systematic Literature Review. Pediatric Neurology 53, 293-300 (2015). [↩]
- B. Wirth, M. Karakaya, M. Kye, N. Mendoza-Ferreira. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annual review of genomics and human genetics 21, 231-261 (2020). [↩]
- C. Serrão, S. Domingues, C. de Campos, S. Moreira, I. Conceição, M. de Carvalho, M. Oliveira Santos. Nusinersen in adults with type 3 spinal muscular atrophy: long-term outcomes on motor and respiratory function. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 45, 2887-2891 (2024). [↩]
- E. Mercuri, C. Sumner, F. Muntoni, B. Darras, R. Finkel. Spinal muscular atrophy. Nature reviews. Disease primers 8 (2022). [↩] [↩] [↩]
- T. Prior, M. Leach, E. Finanger. Spinal Muscular Atrophy. Gene Reviews, 1-30 (2024). [↩]
- Y. Qiao, Y. Chi, J. Gu, Y. Ma. Safety and Efficacy of Nusinersen and Risdiplam for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Brain Sciences 13, 1419 (2023). [↩]
- T. Prior, A. Krainer, Y. Hua, K. Swoboda, P. Snyder, S. Bridgeman, A. Burghes, J. Kissel. A positive modifier of spinal muscular atrophy in the SMN2 gene. American journal of human genetics 85, 408-413 (2009). [↩]
- Biogen. Official Patient Site | SPINRAZA® (nusinersen). https://www.spinraza.com/?cid=PPC-GOOGLE-Branded_HCP_Exact~S~PH~UB~NER~HCP~BR-nusinersen-NA-p74842344700&gclsrc=aw.ds&&gclid=CjwKCAiAzba9BhBhEiwA7glballvvQxJuBfojb5-X5vsmORozB26QPeAIY-3p-de_Qtl-AaPF7PdyhoCJRsQAvD_BwE&gad_source=1 (2024). [↩]
- T. Prior, A. Krainer, Y. Hua, K. Swoboda, P. Snyder, S. Bridgeman, A. Burghes, J. Kissel. A positive modifier of spinal muscular atrophy in the SMN2 gene. American journal of human genetics 85, 408-413 (2009).; Y. Qiao, Y. Chi, J. Gu, Y. Ma. Safety and Efficacy of Nusinersen and Risdiplam for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Brain Sciences 13, 1419 (2023). [↩]
- C. Wurster, A. Ludolph. Antisense oligonucleotides in neurological disorders. Therapeutic advances in neurological disorders 11 (2018).; Y. Qiao, Y. Chi, J. Gu, Y. Ma. Safety and Efficacy of Nusinersen and Risdiplam for Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Brain Sciences 13, 1419 (2023). [↩]
- R. Finkel, C. Chiriboga, J. Vajsar, J. Day, J. Montes, D. De Vivo, M. Yamashita, F. Rigo, G. Hung, E. Schneider, D. Norris, S. Xia, C. Bennett, K. Bishop. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet (London, England) 388, 3017-3026 (2016).; L. Marasco, G. Dujardin, R. Sousa-Luís, Y. Liu, J. Stigliano, T. Nomakuchi, N. Proudfoot, A. Krainer, A. Kornblihtt. Counteracting chromatin effects of a splicing-correcting antisense oligonucleotide improves its therapeutic efficacy in spinal muscular atrophy. Cell 185, 2057-2070.e15 (2022).; Y. Hua, T. Vickers, H. Okunola, C. Bennett, A. Krainer. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. American journal of human genetics 82, 834-848 (2008). [↩]
- M. Claborn, D. Stevens, C.Walker, B. Gildon. Nusinersen: A Treatment for Spinal Muscular Atrophy. The Annals of pharmacotherapy 53, 61-69 (2019). [↩]
- E. Neil, E. Bisaccia. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. The Journal of Pediatric Pharmacology and Therapeutics : JPPT 24, 194 (2019). [↩]
- FDA. Drug Trials Snapshots: SPINRAZA | FDA. https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-spinraza (2017). [↩] [↩]
- F. Ruythooren, P. Moens. Spinal Muscular Atrophy Scoliosis in the Era of Background Therapies-A Review of the Literature. Journal of clinical medicine 13 (2024). [↩]
- P. Andrés-Benito, J. Vázquez-Costa, N. Ñungo Garzón, M. Colomina, C. Marco, L. González, C. Terrafeta, R. Domínguez, I. Ferrer, M. Povedano. Neurodegeneration Biomarkers in Adult Spinal Muscular Atrophy (SMA) Patients Treated with Nusinersen. International Journal of Molecular Sciences 25 (2024). [↩]
- G. Athanasiadis, D. Speed, M. Andersen, E. Appel, E. E, N. Grarup, I. Brandslund, M. Jørgensen, C. C, P. Bjerregaard, T. Hansen, A. Albrechtsen. Estimating narrow-sense heritability using family data from admixed populations. Heredity 124, 751-762 (2020). [↩]
- Y. Hua, T. Vickers, H. Okunola, C. Bennett, A. Krainer. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. American journal of human genetics 82, 834-848 (2008).; J. Qiu, L. Wu, R. Qu, T. Jiang, J. Bai, L. Sheng, P. Feng, J. Sun. History of development of the life-saving drug “Nusinersen” in spinal muscular atrophy. Frontiers in Cellular Neuroscience 16, 942976 (2022). [↩]
- R. Finkel, E. Mercuri, B. Darras, A. Connolly, N. Kuntz, J. Kirschner, C. Chiriboga, K. Saito, L. Servais, E. Tizzano, H. Topaloglu, M. Tulinius, J. Montes, A. Glanzman, K. Bishop, Z. Zhong, S. Gheuens, C. Bennett, E. Schneider, W. Farwell, D. De Vivo. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. The New England journal of medicine 377, 1723-1732 (2017). [↩]
- E. Mercuri, B. Darras, C. Chiriboga, J. Day, C. Campbell, A. Connolly, S. Iannaccone, J. Kirschner, N. Kuntz, K. Saito, P. Shieh, M. Tulinius, E. Mazzone, J. Montes, K. Bishop, Q. Yang, R. Foster, S. Gheuens, C. Bennett, W. Farwell, E. Schneider, D. De Vivo, R. Finkel. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. New England Journal of Medicine 378, 625-635 (2018). [↩]
- G. Baranello, B. Darras, J. Day, N. Deconinck, A. Klein, R. Masson, E. Mercuri, K. Rose, M. El-Khairi, M. Gerber, K. Gorni, O. Khwaja, H. Kletzl, R. Scalco, T. Seabrook, P. Fontoura, L. Servais. Risdiplam in Type 1 Spinal Muscular Atrophy. The New England journal of medicine 384, 915-923 (2021). [↩] [↩] [↩]
- E. Neil, E. Bisaccia. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. The Journal of Pediatric Pharmacology and Therapeutics : JPPT 24, 194 (2019).; W. Zhuang, M. Lu, Y. Wu, Z. Chen, M. Wang, X. Wang, S. Guan, W. Lin. Safety Concerns with Nusinersen, Risdiplam, and Onasemnogene Abeparvovec in Spinal Muscular Atrophy: A Real-World Pharmacovigilance Study. Clinical drug investigation 43, 949-962 (2023). [↩]
- M. Oskoui, J. Day, N. Deconinck, E. Mazzone, A. Nascimento, K. Saito, C. Vuillerot, G. Baranello, N. Goemans, J. Kirschner, A. Kostera-Pruszczyk, L. Servais, G. Papp, K. Gorni, H. Kletzl, C. Martin, T. McIver, R. Scalco, H. Staunton, W. Yeung, P. Fontoura, E. Mercuri. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). Journal of Neurology 270, 2531-2546 (2023). [↩]
- A. Poirier, M. Weetall, K. Heinig, F. Bucheli, K. Schoenlein, J. Alsenz, S. Bassett, M. Ullah, C. Senn, H. Ratni, N. Naryshkin, S. Paushkin, L. Mueller. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacology Research & Perspectives 6, eoo447 (2018). [↩]
- M. Oskoui, J. Day, N. Deconinck, E. Mazzone, A. Nascimento, K. Saito, C. Vuillerot, G. Baranello, N. Goemans, J. Kirschner, A. Kostera-Pruszczyk, L. Servais, G. Papp, K. Gorni, H. Kletzl, C. Martin, T. McIver, R. Scalco, H. Staunton, W. Yeung, P. Fontoura, E. Mercuri. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). Journal of Neurology 270, 2531-2546 (2023). [↩]
- R. Masson, M. Mazurkiewicz-Bełdzińska, K. Rose, L. Servais, H. Xiong, E. Zanoteli, G. Baranello, C. Bruno, J. Day, N. Deconinck, A. Klein, E. Mercuri, D. Vlodavets, Y. Wang, A. Dodman, M. El-Khairi, K. Gorni, B. Jaber, H. Kletzl, E. Gaki, P. Fontoura, B. Darras, J. Volpe, J. Posner, U. Kellner, R. Quinlivan, M. Gerber, O. Khwaja, R. Scalco, T. Seabrook, A. Koch, I. Balikova, I. Joniau, G. Accou, V. Tahon, S. Wittevrongel, E. De Vos, R. de Holanda Mendonça, C. Matsui, A. Fornazieri Darcie, C. Machado, M. Kiyoko Oyamada, J. Martini, G. Polido, J. Rodrigues Iannicelli, J. Caires de Oliveira Achili Ferreira, C. Hu, X. Zhu, C. Qian, L. Shen, H. Li, Y. Shi, S. Zhou, Y. Xiao, Z. Zhou, S. Wang, T. Sang, C. Wei, H. Dong, Y. Cao, Z. Wen, W. Li, L. Qin, N. Barisic, I. Celovec, M. Galiot Delic, P. Ivkic, N. Vukojevic, I. Kern, B. Najdanovic, M. Skugor, J. Tomas, O. Boespflug-Tanguy, S. De Lucia, A. Seferian, E. Barreau, N. Mnafek, H. Peche, A. Grange, D. Trang Nguyen, D. Milascevic, S. Tachibana, E. Pagliano, S. Bianchi Marzoli, D. Santarsiero, M. Garcia Sierra, G. Tremolada, M. Arnoldi, M. Vigano, C. Dosi, R. Zanin, V. Schembri, N. Brolatti, G. Rao, E. Tassara, S. Morando, P. Tacchetti, M. Pedemonte, E. Priolo, L. Sposetti, G. Comi, A. Govoni, S. Osnaghi, V. Minorini, F. Abbati, F. Fassini, M. Foa, A. Lopopolo, M. Pane, C. Palermo, M. Pera, G. Amorelli, C. Barresi, G. D’Amico, L. Orazi, G. Corratti, D. Leone, A. Laura, R. De Sanctis, B. Berti, N. Kimura, Y. Takeshima, H. Shimomura, T. Lee, F. Gomi, T. Morimatsu, T. Furukawa, U. Stodolska-Koberda, A. Waskowska, J. Kolendo, A. Sobierajska-Rek, S. Modrzejewska, A. Lemska, E. Melnik, S. Artemyeva, N. Leppenen, N. Yupatova, A. Monakhova, Y. Papina, O. Shidlovsckaia, E. Litvinova, C. Enzmann, E. Galiart, K. Gugleta, C. Wondrusch Haschke, H. Topaloglu, I. Oncel, N. Ertugrul, B. Konuskan, B. Eldem, S. Kadayifçilar, I. Alemdaroglu, S. Sari, N. Bilgin, A. Karaduman, F. Sarikaya, R. Graham, P. Ghosh, D. Casavant, A. Levine, R. Titus, A. Engelbrekt, L. Ambrosio, A. Fulton, A. Baglieri, C. Dias, E. Maczek, A. Pasternak, S. Bres, T. Duong, R. Gee, S. Yuong. Safety and efficacy of risdiplam in patients with type 1 spinal muscular atrophy (FIREFISH part 2): secondary analyses from an open-label trial. The Lancet. Neurology 21, 1110-1119 (2022). [↩]
- G. Baranello, B. Darras, J. Day, N. Deconinck, A. Klein, R. Masson, E. Mercuri, K. Rose, M. El-Khairi, M. Gerber, K. Gorni, O. Khwaja, H. Kletzl, R. Scalco, T. Seabrook, P. Fontoura, L. Servais. Risdiplam in Type 1 Spinal Muscular Atrophy. The New England journal of medicine 384, 915-923 (2021).; W. Zhuang, M. Lu, Y. Wu, Z. Chen, M. Wang, X. Wang, S. Guan, W. Lin. Safety Concerns with Nusinersen, Risdiplam, and Onasemnogene Abeparvovec in Spinal Muscular Atrophy: A Real-World Pharmacovigilance Study. Clinical drug investigation 43, 949-962 (2023). [↩]
- V. Aponte Ribero, Y. Martí, S. Batson, S. Mitchell, K. Gorni, N. Gusset, M. Oskoui, L. Servais, C. Sutherland. Systematic Literature Review of the Natural History of Spinal Muscular Atrophy: Motor Function, Scoliosis, and Contractures. Neurology 101, e2103 (2023).; J. Kakazu, N. Walker, K. Babin, K. Trettin, C. Lee, P. Sutker, A. Kaye, A. Kaye. Risdiplam for the Use of Spinal Muscular Atrophy. Orthopedic reviews 13 (2021). [↩]
- L. Yu, L. Liu. Exploration of adverse events associated with risdiplam use: Retrospective cases from the US Food and Drug Administration Adverse Event Reporting System (FAERS) database. PloS one 19 (2024). [↩]
- E. Mercuri, C. Sumner, F. Muntoni, B. Darras, R. Finkel. Spinal muscular atrophy. Nature reviews. Disease primers 8 (2022).; R. Masson, M. Mazurkiewicz-Bełdzińska, K. Rose, L. Servais, H. Xiong, E. Zanoteli, G. Baranello, C. Bruno, J. Day, N. Deconinck, A. Klein, E. Mercuri, D. Vlodavets, Y. Wang, A. Dodman, M. El-Khairi, K. Gorni, B. Jaber, H. Kletzl, E. Gaki, P. Fontoura, B. Darras, J. Volpe, J. Posner, U. Kellner, R. Quinlivan, M. Gerber, O. Khwaja, R. Scalco, T. Seabrook, A. Koch, I. Balikova, I. Joniau, G. Accou, V. Tahon, S. Wittevrongel, E. De Vos, R. de Holanda Mendonça, C. Matsui, A. Fornazieri Darcie, C. Machado, M. Kiyoko Oyamada, J. Martini, G. Polido, J. Rodrigues Iannicelli, J. Caires de Oliveira Achili Ferreira, C. Hu, X. Zhu, C. Qian, L. Shen, H. Li, Y. Shi, S. Zhou, Y. Xiao, Z. Zhou, S. Wang, T. Sang, C. Wei, H. Dong, Y. Cao, Z. Wen, W. Li, L. Qin, N. Barisic, I. Celovec, M. Galiot Delic, P. Ivkic, N. Vukojevic, I. Kern, B. Najdanovic, M. Skugor, J. Tomas, O. Boespflug-Tanguy, S. De Lucia, A. Seferian, E. Barreau, N. Mnafek, H. Peche, A. Grange, D. Trang Nguyen, D. Milascevic, S. Tachibana, E. Pagliano, S. Bianchi Marzoli, D. Santarsiero, M. Garcia Sierra, G. Tremolada, M. Arnoldi, M. Vigano, C. Dosi, R. Zanin, V. Schembri, N. Brolatti, G. Rao, E. Tassara, S. Morando, P. Tacchetti, M. Pedemonte, E. Priolo, L. Sposetti, G. Comi, A. Govoni, S. Osnaghi, V. Minorini, F. Abbati, F. Fassini, M. Foa, A. Lopopolo, M. Pane, C. Palermo, M. Pera, G. Amorelli, C. Barresi, G. D’Amico, L. Orazi, G. Corratti, D. Leone, A. Laura, R. De Sanctis, B. Berti, N. Kimura, Y. Takeshima, H. Shimomura, T. Lee, F. Gomi, T. Morimatsu, T. Furukawa, U. Stodolska-Koberda, A. Waskowska, J. Kolendo, A. Sobierajska-Rek, S. Modrzejewska, A. Lemska, E. Melnik, S. Artemyeva, N. Leppenen, N. Yupatova, A. Monakhova, Y. Papina, O. Shidlovsckaia, E. Litvinova, C. Enzmann, E. Galiart, K. Gugleta, C. Wondrusch Haschke, H. Topaloglu, I. Oncel, N. Ertugrul, B. Konuskan, B. Eldem, S. Kadayifçilar, I. Alemdaroglu, S. Sari, N. Bilgin, A. Karaduman, F. Sarikaya, R. Graham, P. Ghosh, D. Casavant, A. Levine, R. Titus, A. Engelbrekt, L. Ambrosio, A. Fulton, A. Baglieri, C. Dias, E. Maczek, A. Pasternak, S. Bres, T. Duong, R. Gee, S. Yuong. Safety and efficacy of risdiplam in patients with type 1 spinal muscular atrophy (FIREFISH part 2): secondary analyses from an open-label trial. The Lancet. Neurology 21, 1110-1119 (2022). [↩]
- M. Nakatani, Y. Takehara, H. Sugino, M. Matsumoto, O. Hashimoto, Y. Hasegawa, T. Murakami, A. Uezumi, S. Takeda, S. Noji, Y. Sunada, K. Tsuchida. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. The FASEB Journal 22, 477-487 (2008). [↩]
- D. Barrett, S. Bilic, Y. Chyung, S. Cote, R. Iarrobino, K. Kacena, A. Kalra, K. Long, G. Nomikos, A. Place, J. Still, L. Vrishabhendra. A Randomized Phase 1 Safety, Pharmacokinetic and harmacodynamic Study of the Novel Myostatin Inhibitor Apitegromab (SRK-015): A Potential Treatment for Spinal Muscular Atrophy. Advances in Therapy 38, 3203-3222 (2021).; R. Nofsinger. Scholar Rock Reports Apitegromab Meets Primary Endpoint in Phase 3 SAPPHIRE Study in Patients with Spinal Muscular Atrophy (SMA) – Scholar Rock, Inc. https://investors.scholarrock.com/news-releases/news-release-details/scholar-rock-reports-apitegromab-meets-primary-endpoint-phase-3/ (2024). [↩]
- T. Crawford, J. Day, D. De Vivo, J. Krueger, E. Mercuri, A. Nascimento, A. Pasternak, E. Mazzone, T. Duong, G. Song, J. Marantz, S. Baver, D. Yu, L. Liu, B. Darras. Long-term efficacy, safety, and patient-reported outcomes of apitegromab in patients with spinal muscular atrophy: results from the 36-month TOPAZ study. Frontiers in Neurology 15 (2024). [↩] [↩] [↩]
- T. Crawford, J. Day, D. De Vivo, J. Krueger, E. Mercuri, A. Nascimento, A. Pasternak, E. Mazzone, T. Duong, G. Song, J. Marantz, S. Baver, D. Yu, L. Liu, B. Darras. Long-term efficacy, safety, and patient-reported outcomes of apitegromab in patients with spinal muscular atrophy: results from the 36-month TOPAZ study. Frontiers in Neurology 15 (2024).; T. Crawford, B. Darras, J. Day, S. Dunaway Young, T. Duong, L. Nelson, D. Barrett, G. Song, S. Bilic, S. Cote, M. Sadanowicz, R. Iarrobino, T. Xu, J. O’Neil, J. Rossello, A. Place, N. Kertesz, G. Nomikos, Y. Chyung. Safety and Efficacy of Apitegromab in Patients With Spinal Muscular Atrophy Types 2 and 3: The Phase 2 TOPAZ Study. Neurology 102, e209151 (2024).; B. Welsh, S. Cote, D. Meshulam, J. Jackson, A. Pal, J. Lansita, A. Kalra. Preclinical Safety Assessment and Toxicokinetics of Apitegromab, an Antibody Targeting Proforms of Myostatin for the Treatment of Muscle-Atrophying Disease. International Journal of Toxicology 40, 322 (2021). [↩]
- M. Pirruccello-Straub, J. Jackson, S. Wawersik, M. T. Webster, L. Salta, K. Long, W. McConaughy, A. Capili, C. Boston, N. K. Mahanthappa, K. J. Turner, A. Donovan. Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Scientific Reports 2018 8:1 8, 1-15 (2018). [↩]
- B. Welsh, S. Cote, D. Meshulam, J. Jackson, A. Pal, J. Lansita, A. Kalra. Preclinical Safety Assessment and Toxicokinetics of Apitegromab, an Antibody Targeting Proforms of Myostatin for the Treatment of Muscle-Atrophying Disease. International Journal of Toxicology 40, 322 (2021). [↩]
- D. Barrett, S. Bilic, Y. Chyung, S. Cote, R. Iarrobino, K. Kacena, A. Kalra, K. Long, G. Nomikos, A. Place, J. Still, L. Vrishabhendra. A Randomized Phase 1 Safety, Pharmacokinetic and Pharmacodynamic Study of the Novel Myostatin Inhibitor Apitegromab (SRK-015): A Potential Treatment for Spinal Muscular Atrophy. Advances in Therapy 38, 3203-3222 (2021). [↩] [↩]
- C. Hu. Scholar Rock Announces Positive Proof-of-Concept Data from TOPAZ Phase 2 Trial Interim Analysis of SRK-015 in Patients with Type 2 and Type 3 Spinal Muscular Atrophy – Scholar Rock, Inc. https://investors.scholarrock.com/news-releases/news-release-details/scholar-rock-announces-positive-proof-concept-data-topaz-phase-2 (2020). [↩]
- T. Crawford, B. Darras, J. Day, S. Dunaway Young, T. Duong, L. Nelson, D. Barrett, G. Song, S. Bilic, S. Cote, M. Sadanowicz, R. Iarrobino, T. Xu, J. O’Neil, J. Rossello, A. Place, N. Kertesz, G. Nomikos, Y. Chyung. Safety and Efficacy of Apitegromab in Patients With Spinal Muscular Atrophy Types 2 and 3: The Phase 2 TOPAZ Study. Neurology 102, e209151 (2024). [↩]
- T. Crawford, J. Day, D. De Vivo, J. Krueger, E. Mercuri, A. Nascimento, A. Pasternak, E. Mazzone, T. Duong, G. Song, J. Marantz, S. Baver, D. Yu, L. Liu, B. Darras. Long-term efficacy, safety, and patient-reported outcomes of apitegromab in patients with spinal muscular atrophy: results from the 36-month TOPAZ study. Frontiers in Neurology 15 (2024).; C. Hu. Scholar Rock Announces Positive Proof-of-Concept Data from TOPAZ Phase 2 Trial Interim Analysis of SRK-015 in Patients with Type 2 and Type 3 Spinal Muscular Atrophy – Scholar Rock, Inc. https://investors.scholarrock.com/news-releases/news-release-details/scholar-rock-announces-positive-proof-concept-data-topaz-phase-2 (2020). [↩]
- Scholar Rock. Spinal Muscular Atrophy | Scholar Rock. https://scholarrock.com/our-pipeline/neuromuscular-and-obesity/spinal-muscular-atrophy/ (2025). [↩]
- R. Nofsinger. Scholar Rock Reports Apitegromab Meets Primary Endpoint in Phase 3 SAPPHIRE Study in Patients with Spinal Muscular Atrophy (SMA) – Scholar Rock, Inc. https://investors.scholarrock.com/news-releases/news-release-details/scholar-rock-reports-apitegromab-meets-primary-endpoint-phase-3/ (2024).; Scholar Rock. Scholar Rock Submits Biologics License Application (BLA) to the U.S. FDA for Apitegromab as a Treatment for Patients with Spinal Muscular Atrophy (SMA) – Scholar Rock, Inc. https://investors.scholarrock.com/news-releases/news-release-details/scholar-rock-submits-biologics-license-application-bla-us-fda?utm_source=chatgpt.com (2025). [↩]
- M. Pane, G. Coratti, M. Pera, V. Sansone, S. Messina, A. d’Amico, C. Bruno, F. Salmin, E. Albamonte, R. De Sanctis, M. Sframeli,V. Di Bella, S. Morando, C. Palermo, A. Frongia, L. Antonaci, A. Capasso, M. Catteruccia, A. Longo, M. Ricci, C. Cutrona, A. Pirola, C. Bravetti, M. Pedemonte, N. Brolatti, E. Bertini, E. Mercuri. Nusinersen efficacy data for 24-month in type 2 and 3 spinal muscular atrophy. Annals of clinical and translational neurology 9, 404-409 (2022).; R. Günther, C. Wurster, S. Brakemeier, O. Osmanovic, O. Schreiber-Katz, S. Petri, Z. Uzelac, M. Hiebeler, S. Thiele, M. Walter, M. Weiler, T. Kessler, M. Freigang, H. Lapp, I. Cordts, P. Lingor, M. Deschauer, A. Hahn, K. Martakis, R. Steinbach, B. Ilse, A. Rödiger, J. Bellut, J. Nentwich, D. Zeller, M. Muhandes, T. Baum, J. Christoph Koch, B. Schrank, S. Fischer, A. Hermann, C. Kamm, S. Naegel, A. Mensch, M. Weber, C. Neuwirth, H. Lehmann, G. Wunderlich, C. Stadler, M. Tomforde, A. George, M. Groß, A. Pechmann, J. Kirschner, M. Türk, M. Schimmel, G. Bernert, P. Martin, C. Rauscher, G. Meyer zu Hörste, P. Baum, W. Löscher, M. Flotats-Bastardas, C. Köhler, K. Probst-Schendzielorz, S. Goldbach, U. Schara-Schmidt, W. Müller-Felber, H. Lochmüller, O. von Velsen, C. Kleinschnitz, A. Ludolph, T. Hagenacker. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study. The Lancet Regional Health – Europe 39 (2024).; C. Kokaliaris, R. Evans, N. Hawkins, A. Mahajan, D.A. Scott, C.S. Sutherland, J. Nam, G. Sajeev. Long-Term Comparative Efficacy and Safety of Risdiplam and Nusinersen in Children with Type 1 Spinal Muscular Atrophy. Advances in Therapy 41, 2414-2434 (2024).; E. Mercuri, C. Sumner, F. Muntoni, B. Darras, R. Finkel. Spinal muscular atrophy. Nature reviews. Disease primers 8 (2022).; T. Crawford, J. Day, D. De Vivo, J. Krueger, E. Mercuri, A. Nascimento, A. Pasternak, E. Mazzone, T. Duong, G. Song, J. Marantz, S. Baver, D. Yu, L. Liu, B. Darras. Long-term efficacy, safety, and patient-reported outcomes of apitegromab in patients with spinal muscular atrophy: results from the 36-month TOPAZ study. Frontiers in Neurology 15 (2024).; T. Crawford, B. Darras, J. Day, S. Dunaway Young, T. Duong, L. Nelson, D. Barrett, G. Song, S. Bilic, S. Cote, M. Sadanowicz, R. Iarrobino, T. Xu, J. O’Neil, J. Rossello, A. Place, N. Kertesz, G. Nomikos, Y. Chyung. Safety and Efficacy of Apitegromab in Patients With Spinal Muscular Atrophy Types 2 and 3: The Phase 2 TOPAZ Study. Neurology 102, e209151 (2024). [↩]
- C. Sumner, T. Crawford. Early treatment offers a lifeline for infants with SMA. Nature medicine 28, 1348 (2022). [↩]
- S. Crisafulli, B. Boccanegra, G. Vitturi, G. Trifirò, A. De Luca. Pharmacological Therapies of Spinal Muscular Atrophy: A Narrative Review of Preclinical, Clinical-Experimental, and Real-World Evidence. Brain sciences 13 (2023). [↩]
- B. Darras, C. Chiriboga, S. Iannaccone, K. Swoboda, J. Montes, L. Mignon, S. Xia, C. Bennett, K. Bishop, J. Shefner, A. Green, P. Sun, I. Bhan, S. Gheuens, E. Schneider, W. Farwell, D. De Vivo. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology 92, e2492-2506 (2019).; S. Tokunaga, H. Shimomura, T. Horibe, N. Taniguchi, T. Lee, Y. Takeshima. Experience of nusinersen treatment in advanced spinal muscular atrophy type 1: Characteristics of late responders with delayed treatment efficacy. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society 54, 171-177 (2025).; A. Łusakowska, A. Wójcik, A. Frączek, K. Aragon-Gawińska, A. Potulska-Chromik, P. Baranowski, R. Nowak, G. Rosiak, K. Milczarek, D. Konecki, Z. Gierlak-Wójcicka, M. Burlewicz, A. Kostera-Pruszczyk. Long-term nusinersen treatment across a wide spectrum of spinal muscular atrophy severity: a real-world experience. Orphanet journal of rare diseases 18 (2023). [↩]
- M. Pane, G. Coratti, M. Pera, V. Sansone, S. Messina, A. d’Amico, C. Bruno, F. Salmin, E. Albamonte, R. De Sanctis, M. Sframeli,V. Di Bella, S. Morando, C. Palermo, A. Frongia, L. Antonaci, A. Capasso, M. Catteruccia, A. Longo, M. Ricci, C. Cutrona, A. Pirola, C. Bravetti, M. Pedemonte, N. Brolatti, E. Bertini, E. Mercuri. Nusinersen efficacy data for 24-month in type 2 and 3 spinal muscular atrophy. Annals of clinical and translational neurology 9, 404-409 (2022). [↩]
- Y. Fainmesser, V. Drory, S. Ben-Shushan, A. Lavon, L. Spector, B. Abramovich, A. Abraham. Longer-term follow-up of nusinersen efficacy and safety in adult patients with spinal muscular atrophy types 2 and 3. Neuromuscular disorders : NMD 32, 451-459 (2022). [↩] [↩]
- R. Günther, C. Wurster, S. Brakemeier, O. Osmanovic, O. Schreiber-Katz, S. Petri, Z. Uzelac, M. Hiebeler, S. Thiele, M. Walter, M. Weiler, T. Kessler, M. Freigang, H. Lapp, I. Cordts, P. Lingor, M. Deschauer, A. Hahn, K. Martakis, R. Steinbach, B. Ilse, A. Rödiger, J. Bellut, J. Nentwich, D. Zeller, M. Muhandes, T. Baum, J. Christoph Koch, B. Schrank, S. Fischer, A. Hermann, C. Kamm, S. Naegel, A. Mensch, M. Weber, C. Neuwirth, H. Lehmann, G. Wunderlich, C. Stadler, M. Tomforde, A. George, M. Groß, A. Pechmann, J. Kirschner, M. Türk, M. Schimmel, G. Bernert, P. Martin, C. Rauscher, G. Meyer zu Hörste, P. Baum, W. Löscher, M. Flotats-Bastardas, C. Köhler, K. Probst-Schendzielorz, S. Goldbach, U. Schara-Schmidt, W. Müller-Felber, H. Lochmüller, O. von Velsen, C. Kleinschnitz, A. Ludolph, T. Hagenacker. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study. The Lancet Regional Health – Europe 39 (2024). [↩] [↩] [↩]
- F. Scheijmans, I. Cuppen, R. Van Eijk, C. Wijngaarde, M. Schoenmakers, D. Van Der Woude, B. Bartels, E. Veldhoen, I. Oude Lansink, E. Groen, F. Asselman, R. Wadman, W. Van Der Pol. Population-based assessment of nusinersen efficacy in children with spinal muscular atrophy: a 3-year follow-up study. Brain Communications 4, fcac269 (2022). [↩]
- R. Günther, C. Wurster, S. Brakemeier, O. Osmanovic, O. Schreiber-Katz, S. Petri, Z. Uzelac, M. Hiebeler, S. Thiele, M. Walter, M. Weiler, T. Kessler, M. Freigang, H. Lapp, I. Cordts, P. Lingor, M. Deschauer, A. Hahn, K. Martakis, R. Steinbach, B. Ilse, A. Rödiger, J. Bellut, J. Nentwich, D. Zeller, M. Muhandes, T. Baum, J. Christoph Koch, B. Schrank, S. Fischer, A. Hermann, C. Kamm, S. Naegel, A. Mensch, M. Weber, C. Neuwirth, H. Lehmann, G. Wunderlich, C. Stadler, M. Tomforde, A. George, M. Groß, A. Pechmann, J. Kirschner, M. Türk, M. Schimmel, G. Bernert, P. Martin, C. Rauscher, G. Meyer zu Hörste, P. Baum, W. Löscher, M. Flotats-Bastardas, C. Köhler, K. Probst-Schendzielorz, S. Goldbach, U. Schara-Schmidt, W. Müller-Felber, H. Lochmüller, O. von Velsen, C. Kleinschnitz, A. Ludolph, T. Hagenacker. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study. The Lancet Regional Health – Europe 39 (2024).; G. Acsadi, T. Crawford, W. Müller-Felber, P. Shieh, R. Richardson, N. Natarajan, D. Castro, D. Ramirez-Schrempp, G. Gambino, P. Sun, W. Farwell. Safety and efficacy of nusinersen in spinal muscular atrophy: The EMBRACE study. Muscle & nerve 63, 668-677 (2021). [↩]
- K. Abbas, M. Eltaras, N. El-Shahat, B. Abdelazeem, M. Shaqfeh, J. Brašić. The Safety and Efficacy of Nusinersen in the Treatment of Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Medicina (Lithuania) 58, 213 (2022). [↩]
- B. Stolte, M. Nonnemacher, K. Kizina, S. Bolz, A. Totzeck, A. Thimm, B. Wagner, C. Deuschl, C. Kleinschnitz, T. Hagenacker. Nusinersen treatment in adult patients with spinal muscular atrophy: a safety analysis of laboratory parameters. Journal of neurology 268, 4667-4679 (2021).; F. Scheijmans, I. Cuppen, R. Van Eijk, C. Wijngaarde, M. Schoenmakers, D. Van Der Woude, B. Bartels, E. Veldhoen, I. Oude Lansink, E. Groen, F. Asselman, R. Wadman, W. Van Der Pol. Population-based assessment of nusinersen efficacy in children with spinal muscular atrophy: a 3-year follow-up study. Brain Communications 4, fcac269 (2022). [↩]
- G. Sajeev, R. Evans, N. Hawkins, A. Mahajan, D. Scott, J. Nam, S. Sutherland, C. Kokaliaris. P08 Long-term comparative efficacy and safety of risdiplam versus nusinersen in children with Type 1 spinal muscular atrophy (SMA). Neuromuscular Disorders 33, S164 (2023). [↩]
- G. Acsadi, T. Crawford, W. Müller-Felber, P. Shieh, R. Richardson, N. Natarajan, D. Castro, D. Ramirez-Schrempp, G. Gambino, P. Sun, W. Farwell. Safety and efficacy of nusinersen in spinal muscular atrophy: The EMBRACE study. Muscle & nerve 63, 668-677 (2021).; M. Oskoui, J. Day, N. Deconinck, E. Mazzone, A. Nascimento, K. Saito, C. Vuillerot, G. Baranello, N. Goemans, J. Kirschner, A. Kostera-Pruszczyk, L. Servais, G. Papp, K. Gorni, H. Kletzl, C. Martin, T. McIver, R. Scalco, H. Staunton, W. Yeung, P. Fontoura, E. Mercuri. Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). Journal of Neurology 270, 2531-2546 (2023). [↩]
- Hoffmann-La Roche Ltd. View of Risdiplam (Evrysdi) | Canadian Journal of Health Technologies. https://canjhealthtechnol.ca/index.php/cjht/article/view/sr0661/269?utm_source=chatgpt.com (2021). [↩]
- D. Giess, J. Erdos, C. Wild. An updated systematic review on spinal muscular atrophy patients treated with nusinersen, onasemnogene abeparvovec (at least 24 months), risdiplam (at least 12 months) or combination therapies. European Journal of Paediatric Neurology 51, 84-92 (2024).; E. Mercuri, C. Sumner, F. Muntoni, B. Darras, R. Finkel. Spinal muscular atrophy. Nature reviews. Disease primers 8 (2022). [↩]
- J. Erdos, C. Wild. Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: A systematic review of real-world study data. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society 39, 1-10 (2022). [↩]
- T. Crawford, J. Day, D. De Vivo, J. Krueger, E. Mercuri, A. Nascimento, A. Pasternak, E. Mazzone, T. Duong, G. Song, J. Marantz, S. Baver, D. Yu, L. Liu, B. Darras. Long-term efficacy, safety, and patient-reported outcomes of apitegromab in patients with spinal muscular atrophy: results from the 36-month TOPAZ study. Frontiers in Neurology 15 (2024).; R. Nofsinger. Scholar Rock Reports Second Quarter 2024 Financial Results and Highlights Business Progress – Scholar Rock, Inc. https://investors.scholarrock.com/news-releases/news-release-details/scholar-rock-reports-second-quarter-2024-financial-results-and?utm_source=chatgpt.com (2024). [↩]
- T. Crawford, J. Day, D. De Vivo, J. Krueger, E. Mercuri, A. Nascimento, A. Pasternak, E. Mazzone, T. Duong, G. Song, J. Marantz, S. Baver, D. Yu, L. Liu, B. Darras. Long-term efficacy, safety, and patient-reported outcomes of apitegromab in patients with spinal muscular atrophy: results from the 36-month TOPAZ study. Frontiers in Neurology 15 (2024).; T. Crawford, B. Darras, J. Day, S. Dunaway Young, T. Duong, L. Nelson, D. Barrett, G. Song, S. Bilic, S. Cote, M. Sadanowicz, R. Iarrobino, T. Xu, J. O’Neil, J. Rossello, A. Place, N. Kertesz, G. Nomikos, Y. Chyung. Safety and Efficacy of Apitegromab in Patients With Spinal Muscular Atrophy Types 2 and 3: The Phase 2 TOPAZ Study. Neurology 102, e209151 (2024). [↩]
- Scholar Rock. Study Details | An Active Treatment Study of SRK-015 in Patients With Type 2 or Type 3 Spinal Muscular Atrophy | ClinicalTrials.gov. https://clinicaltrials.gov/study/NCT03921528 (2024). [↩]
- L. Elman, B. Youn, C. Proud, M. Frey, S. Ajroud-Driss, M. McCormick, D. Michelson, M. Cartwright, T. Heiman-Patterson, J. Choi, A. Chandak, A. Khachatryan, M. Martinez, A. Paradis. Real-world Adherence to Nusinersen in Adults with Spinal Muscular Atrophy in the US: A Multi-site Chart Review Study. Journal of Neuromuscular Diseases 9, 655 (2022). [↩]
- B. Darras, C. Chiriboga, S. Iannaccone, K. Swoboda, J. Montes, L. Mignon, S. Xia, C. Bennett, K. Bishop, J. Shefner, A. Green, P. Sun, I. Bhan, S. Gheuens, E. Schneider, W. Farwell, D. De Vivo. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology 92, e2492-2506 (2019).; R. Günther, C. Wurster, S. Brakemeier, O. Osmanovic, O. Schreiber-Katz, S. Petri, Z. Uzelac, M. Hiebeler, S. Thiele, M. Walter, M. Weiler, T. Kessler, M. Freigang, H. Lapp, I. Cordts, P. Lingor, M. Deschauer, A. Hahn, K. Martakis, R. Steinbach, B. Ilse, A. Rödiger, J. Bellut, J. Nentwich, D. Zeller, M. Muhandes, T. Baum, J. Christoph Koch, B. Schrank, S. Fischer, A. Hermann, C. Kamm, S. Naegel, A. Mensch, M. Weber, C. Neuwirth, H. Lehmann, G. Wunderlich, C. Stadler, M. Tomforde, A. George, M. Groß, A. Pechmann, J. Kirschner, M. Türk, M. Schimmel, G. Bernert, P. Martin, C. Rauscher, G. Meyer zu Hörste, P. Baum, W. Löscher, M. Flotats-Bastardas, C. Köhler, K. Probst-Schendzielorz, S. Goldbach, U. Schara-Schmidt, W. Müller-Felber, H. Lochmüller, O. von Velsen, C. Kleinschnitz, A. Ludolph, T. Hagenacker. Long-term efficacy and safety of nusinersen in adults with 5q spinal muscular atrophy: a prospective European multinational observational study. The Lancet Regional Health – Europe 39 (2024). [↩]
- J. Mendell, M. Wigderson, I. Alecu, L. Yang, L. Mehl, A. Connolly. P223 Long-term follow-up of onasemnogene abeparvovec gene therapy in patients with spinal muscular atrophy (SMA) type 1. Neuromuscular Disorders 33, S91 (2023). [↩]
- R. Finkel, E. Mercuri, B. Darras, A. Connolly, N. Kuntz, J. Kirschner, C. Chiriboga, K. Saito, L. Servais, E. Tizzano, H. Topaloglu, M. Tulinius, J. Montes, A. Glanzman, K. Bishop, Z. Zhong, S. Gheuens, C. Bennett, E. Schneider, W. Farwell, D. De Vivo. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. The New England journal of medicine 377, 1723-1732 (2017).; E. Mercuri, B. Darras, C. Chiriboga, J. Day, C. Campbell, A. Connolly, S. Iannaccone, J. Kirschner, N. Kuntz, K. Saito, P. Shieh, M. Tulinius, E. Mazzone, J. Montes, K. Bishop, Q. Yang, R. Foster, S. Gheuens, C. Bennett, W. Farwell, E. Schneider, D. De Vivo, R. Finkel. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. New England Journal of Medicine 378, 625-635 (2018).; G. Baranello, B. Darras, J. Day, N. Deconinck, A. Klein, R. Masson, E. Mercuri, K. Rose, M. El-Khairi, M. Gerber, K. Gorni, O. Khwaja, H. Kletzl, R. Scalco, T. Seabrook, P. Fontoura, L. Servais. Risdiplam in Type 1 Spinal Muscular Atrophy. The New England journal of medicine 384, 915-923 (2021).; Hoffmann-La Roche Ltd. View of Risdiplam (Evrysdi) | Canadian Journal of Health Technologies. https://canjhealthtechnol.ca/index.php/cjht/article/view/sr0661/269?utm_source=chatgpt.com (2021).; C. Kokaliaris, R. Evans, N. Hawkins, A. Mahajan, D.A. Scott, C.S. Sutherland, J. Nam, G. Sajeev. Long-Term Comparative Efficacy and Safety of Risdiplam and Nusinersen in Children with Type 1 Spinal Muscular Atrophy. Advances in Therapy 41, 2414-2434 (2024).; Scholar Rock. Spinal Muscular Atrophy | Scholar Rock. https://scholarrock.com/our-pipeline/neuromuscular-and-obesity/spinal-muscular-atrophy/ (2025). [↩]



{kind=link}