
Abstract
Alzheimer’s disease (AD) is a common form of dementia that affects approximately one in nine people in the United States over the age of 65. As AD progresses, patients experience symptoms of dementia including increased deficits in memory and problem-solving abilities, as well as behavior changes that result from severe neuronal death. Although specific causes of the disease remain unknown, AD is primarily characterized by abnormalities in two proteins: amyloid beta (Aβ) and tau, which can appear up to years before the onset of dementia symptoms. Because of the early appearance of protein biomarkers, it is important to initiate early intervention of AD– to target tau and Aβ before significant neuronal death and cognitive decline associated with later stages of AD. Current interventions largely target late-stage AD, resulting in only symptomatic alleviation rather than slowed disease progression. Some drugs such as Aβ-targeting antibodies are currently on the market and are able to slow AD progression, but still cause many adverse effects in patients. Due to the shortcomings of current interventions, further research and development of therapies targeting Aβ and tau protein in the early stages of AD to slow disease progression is necessary. Future interventions are currently being developed to target these biomarkers early on, and hold promising results for the future of AD. Developing medications that target Aβ and tau protein in early-stage AD can lead to slowed disease progression and symptom onset, resulting in a better quality of life for AD patients.
Keywords: Alzheimer’s disease (AD), early intervention, amyloid beta (Aβ), tau, neurofibrillary tangles (NFT), mild cognitive impairment (MCI), positron emission tomography (PET), neurofilament light chain (NfL)
Introduction
AD is a neurodegenerative disease characterized by progressive cognitive decline. AD progresses in stages. Early stages of AD include preclinical AD, in which patients exhibit no symptoms, and AD characterized by mild cognitive impairment (MCI), which is a slight decline in an individual’s memory and thinking abilities, but not enough to interfere with daily life. MCI is a clinical diagnosis based primarily on cognitive symptoms, while early AD diagnosis is based on both clinical signs and the presence of AD-specific biomarkers. Late-stage AD is characterized by the onset of dementia. In this stage, patients encounter severe difficulties with memory and language abilities1,2. Most patients are diagnosed and treated in late-stage AD. However, current late-stage AD treatments are only able to target symptoms of AD rather than the neuropathological changes associated with AD, such as the formation and aggregation of Aβ plaques and NFTs. These treatments are unable to slow AD progression due to significant neuronal death in late-stage AD.
However, new diagnosis strategies and treatments that target AD biomarkers in early stages of the disease, such as the presymptomatic and MCI stages, are changing the field of AD treatment because they are able to slow disease progression unlike previous late-stage treatments. There are two primary protein hallmarks of AD which early-stage treatments target- amyloid-beta plaques, composed of Aβ protein and neurofibrillary tangles (NFT) composed of tau protein3,4. These biomarkers can appear in early-stage AD, several years before the onset of cognitive symptoms, and can be detected through positron emission tomography (PET) and various blood tests. Treating AD by targeting biomarkers in early-stage AD has the potential to slow disease progression before the onset of severe cognitive symptoms, unlike late-stage treatments that have been used in the past and have been largely ineffective5.
Interventions such as some Aβ targeting antibodies including lecanemab and donanemab have been approved by the FDA for use in early stage AD. Although they have demonstrated high efficacy in clinical trials, Aβ targeting antibodies continue to have widespread adverse effects6. Other drugs such as cholinesterase inhibitors have also been approved for use in early stages of AD, but do not target biomarkers of AD, making them ineffective for slowing disease progression. Because biomarker targeting interventions have proven to be more effective at slowing disease progression, several antibodies, vaccines and other treatments are currently being developed to slow disease progression in early-stage AD pateints. These interventions largely target tau and Aβ and hold promising results for the future of early-stage AD treatments6,7. However, there are still many research gaps in early-stage AD and possible intervention methods due to the uncertainty regarding the exact neuropathological causes of AD, and the difficulties surrounding the diagnosis of early-stage AD. More research in this area will allow for a better understanding of how to slow or prevent cognitive decline before significant neuronal death to improve the quality of life for AD patients and possibly reverse or prevent disease progression.
This review outlines the biomarkers associated with early-stage AD and how imaging techniques can detect these biomarkers to diagnose AD in early stages. The review then considers current late-stage AD drugs and evaluates their inefficacy in comparison to early-stage AD drugs, which target AD biomarkers. Lastly the review examines several early-stage treatments for AD that are currently approved and still under development, which hold a promising future for the slowing of AD progression. By analyzing biomarkers present in early-stage AD and current and future early interventions, this review aims to address the current gap in literature regarding early AD intervention to highlight the need for these interventions and stimulate further research.
Methods
This manuscript reviews primary research papers covering the biomarkers involved in AD, current intervention methods and future interventions. Literature searches were conducted largely through Pubmed, although some references were found through Google Scholar. Clinical trial searches were also conducted on clinicaltrials.gov to identify clinical studies that have been conducted to support the efficacy of drugs that were mentioned in the early and future intervention sections. Searches on pubmed were conducted through the use of keywords such as “Alzheimer’s disease” and “early interventions” as well as “amyloid beta” and “tau” and “MCI”. Papers published before the past five years were excluded, with exception to papers regarding AD interventions developed more than five years ago. Key findings regarding study results and drug efficacy were included in the review. Keywords utilized to search for clinical trials on clinicaltrials.gov were names of various AD drugs such as “lecanemab” and “donanemab” and all other included interventions. Clinical trials involving patients with severe AD were excluded.
Early biomarkers and disease progression
In spite of substantial research that has been done in the field, the primary driving factors of AD remain unknown due to the complex interplay of various genetic, environmental and molecular factors that contribute to its pathogenesis. However, current research holds that the most common neuropathological hallmarks of AD are amyloid plaques and NFTs, composed primarily of abnormal accumulation of Aβ protein and tau protein, respectively8. Misfolding and self-assembly of extracellular Aβ deposits results in the formation of aggregates in neural tissue in the brain and cerebral vasculature leading to neuronal loss and synaptic dysfunction9. CSF Aβ is the most sensitive biomarker for AD and has a sensitivity of around 96.4% for AD detection10. NFTs are the result of hyperphosphorylation, resulting in abnormal buildup of tau proteins as they detach from microtubules, leading to neuronal damage11. Because of this, tau levels are elevated in individuals with AD, and total tau in the CSF has a sensitivity and specificity of approximately 90%12. Additionally, blood-based biomarkers such as Aβ42 and p-tau are present in the plasma and can serve as highly specific minimally invasive biomarkers, with sensitivity levels of approximately 88% and 79% respectively and specificity levels of 81% and 94% respectively, in patients with MCI13. Another blood-based biomarker, neurofilament light chains (NfL) located in the plasma can serve as a biomarker for axonal damage and is associated with AD14. Due to their role in maintenance of axon structure, NfLs are commonly present in higher levels in individuals with neurodegenerative disease, such as AD, which is characterized by axon degeneration15. However, although they are correlated with AD and have proven to, these biomarkers are associated with several uncertainties due to a variety of external factors being able to influence tau, Aβ and NfL levels within the CSF and plasma. Additionally, there still remains high variability in biomarker measurements among and within laboratories, meaning that there is still uncertainty regarding universal cut-off values for AD biomarkers16.
Detection through PET screening allows for clear visualization of location and severity of Aβ and tau aggregates in the CSF allowing for targeted early intervention17. Radiotracers such as F-FDDNP are used to detect both NFTs and amyloid plaques and can be used to quantify AD. The most common radiotracer for detecting Aβ plaques, 11C-PiB has a high affinity, selectivity and specificity to fibrillary Aβ, allowing for the visualization of Aβ in the neural tissue18. Additionally, the radiotracer [18F]-flortaucipir was recently approved by the FDA to be used to detect tau pathology in the neural tissue. In a diagnostic study involving terminal illness patients with varying degrees of dementia, [18F]-flortaucipir was able to successfully detect NFTs in patients with and without dementia, with high sensitivity levels of 92.3% to 100.0%, indicating that it can be used to detect neuropathological changes associated with AD, in both early and late stages19. Due to high sensitivity levels and affinity towards AD biomarkers in the CSF, PET screening should be utilized to screen patients for AD pre-symptomatically or during MCI to allow for interventions to be administered before significant cognitive decline occurs.
Because PET scans are expensive and may not be accessible to everyone, alternative diagnostic tests for early-stage detection of AD include blood tests, which target blood-based biomarkers, and are generally more cost effective and accessible than other imaging techniques. The Lumipulse G pTau217/ß-Amyloid 1-42 Plasma Ratio is the first diagnostic blood test for AD and was recently FDA approved for detection of AD in patients demonstrating symptoms, including those with MCI. The Lumipulse G pTau217/ß-Amyloid 1-42 Plasma Ratio is used to detect pTau217 and Aβ 1-4 in the human plasma20. Because this blood test can successfully detect tau and Aβ in the plasma of early-stage AD patients with MCI, and is minimally invasive, it serves as a useful tool for detection and intervention of early AD.
Both tau and Aβ deposition, measured through PET can appear up to two decades prior to severe cognitive symptoms in individuals with AD21,22 Individuals tested for cerebrospinal fluid (CSF) and blood biomarkers such as Aβ tested positive without demonstrating any cognitive symptoms, indicating that AD occurs in stages separately characterized by Aβ formation and aggregation and symptom onset, which occurs later23. Similarly, NFTs, although less commonly than Aβ, can appear pre-symptomatically as CSF tau protein concentration has been shown to increase up to fifteen years prior to symptom onset in autosomal dominant AD patients24. Tau oligomers, which are the small soluble forms of tau proteins, also appear early on in AD, several years before onset of dementia symptoms25. When using PET scans to detect tau positivity, approximately 46.5% of MCI AD patients with Aβ positivity were also tau PET positive in the temporal meta-region of interest, regions in the neural tissue with elevated tau-PET levels in AD26. Additionally, NfLs increase in the CSF and plasma ten to twenty years before symptom onset in AD patients27. The appearance of protein biomarkers such as Aβ, tau aggregates and NfL in early-stage AD illustrates the need for intervention strategies earlier in disease progression.
As AD progresses past its early-stages, biomarker concentration in the blood and CSF can begin to plateau. Aβ has higher accumulation rates in earlier stages as it follows a sigmoidal time function in individuals with AD28. After rapidly increasing, Aβ levels in the CSF level off as AD pathology increases in a preclinical AD model29. This plateau in Aβ deposition at a later age demonstrates that aggregation and growth of protein biomarkers occurs in earlier stages and plateaus in later stages of neurodegeneration, roughly around the same time cognitive symptoms arise in AD patients. Tau deposition spreads across the brain as AD progresses30. Earlier stages of AD in which individuals remain cognitively unimpaired are marked by tau protein aggregation in the transentorhinal cortex region of the brain, indicating that tau protein can act as a biomarker for pre-symptomatic AD17. The appearance and accumulation of AD biomarkers well before cognitive decline arises implies that AD pathogenesis largely occurs pre-symptomatically, signifying a need for intervention in early-stage AD, when Aβ and tau biomarker formation is occurring. This will allow for the prevention of the neuronal dysfunction and death associated with Aβ and tau aggregates.
Downfalls of treating AD in later stages
There are various downfalls to initiating intervention of moderate to severe AD, emphasizing the need for early treatment. Current treatments such as memantine that are given to late-stage AD patients are only able to target symptoms of AD rather than the neuropathological changes that are associated with AD. Treatments that are given to late-stage AD patients typically target chemicals and neurotransmitters involved in memory and cognitive function rather than protein biomarkers that appear in earlier stages such as Aβ and tau31. Memantine acts as an antagonist to inhibit overexcitation of NMDA receptors, which only provides symptomatic alleviation of AD32. When tested on moderate to severe AD patients in a randomized, double-blind, placebo-controlled study, although it relieved some cognitive symptoms of AD such as comprehension, conversation and ability to follow commands, memantine wasn’t successful in slowing disease progression, indicating synaptic dysfunction and neuronal death continued to occur. Disease progression occurred at the same rates as placebo patients in the control group33.
Cholinesterase inhibitors prevent the breakdown of acetylcholine31 (Figure 1). These drugs, although primarily prescribed to early-stage AD patients, have been shown to have decreased efficacy in late-stage AD patients, proving that initiating intervention in early-stage AD is more effective.

The cholinesterase inhibitor blocks the enzyme acetylcholinesterase (AChE), which inhibits the breakdown of ACh, a neurotransmitter associated with learning and memory, into choline and acetate. This raises levels of ACh in AD patients, allowing for improved learning, memory, and overall cognitive funciton34.
In an interventional randomized study with 61 participants with late-stage AD and dementia, patients who continued the treatment of cholinesterase inhibitors demonstrated slightly higher scores of cognitive function in a six-item screener. However, AD progression continued at the same rate, indicating that cholinesterase inhibitors are able to reduce symptoms but not stop or slow disease progression altogether when given to late-stage AD patients35. Because late-stage AD drugs are only able to slightly reduce cognitive symptoms without targeting AD pathogenesis, early intervention treatment options would be more beneficial as they can target early biomarkers to slow disease progression.
Antidepressant drugs are widely used to target and reduce dementia associated behavioral symptoms of late-stage AD, such as depression, anxiety, and agitation36. Sertraline, citalopram, escitalopram, and mirtazapine are few of the commonly used antidepressants among dementia patients. Sertraline, citalopram, and mirtazapine are selective serotonin reuptake inhibitors and mirtazapine is a tetracyclic antidepressant. However in a national cohort study in Sweden with over 18740 patients, antidepressant drugs did not reduce cognitive decline in dementia patients and even showed to be associated with faster cognitive decline, especially in severe dementia patients37. Because severe dementia is associated with late-stage AD, late-stage interventions targeting behavioral symptoms such as antidepressants are largely ineffective in controlling disease progression or slowing cognitive decline.
Furthermore, difficulties surrounding treatment of late-stage AD also stem from the issues involving the recruitment and assessment of individuals with moderate to severe AD. Many individuals with late-stage AD and their families are reluctant to enroll in clinical trials because they feel it is too late in the disease trajectory to face any improvements. Patients and families may also experience hesitancy to enroll in trials due to the advanced and meticulous care that late-stage AD patients require38. Moreover, individuals with moderate to severe AD face issues with cognition including decision-making problems, lack of cooperation and poor communication skills39. This creates difficulty when assessing subjects for late-stage AD treatments, as researchers often rely on cognition tests requiring communication from the patient to evaluate treatment efficacy.
The primary reason as to why late-stage AD treatments are less effective is due to the extent of neuronal damage that has occurred by this stage. Late-stage AD is marked by severe neuroinflammation and microglial activation, which leads to neuronal damage from cytotoxicity40. Neuronal damage increases greatly as AD progresses ultimately leading to severe neuronal death and brain atrophy, resulting in the shrinkage of the gyri by over 50%5. Because of this extreme neuronal impairment, medications that reduce hallmarks of AD like Aβ plaques and tau aggregates and are administered in late-stage AD patients often cannot reverse the neurodegeneration and cognitive decline that has already resulted from AD progression, as the neural tissue is too-far damaged for these drugs to work. This makes late-stage intervention in AD less beneficial for further research than early intervention, as early intervention can target protein hallmarks of AD before the development of significant neuronal death.
Current early intervention strategies
The development of various Aβ-targeting antibodies has helped to treat AD in its early stages41. In recent years, two of these antibodies, lecanemab and donanemab have been approved by the FDA for early-stage AD patients. Lecanemab is a humanized monoclonal IgG1 antibody medication that binds to soluble aggregated Aβ protofibrils rather than plaques (Figure 2). In an 18-month, multicenter, double-blind, phase 3 trial involving individuals 50 to 90 years of age with early AD, persons that received intravenous injections of lecanemab showed a significant reduction in cognitive decline compared to placebo treated control patients, indicating slowed disease progression. However, lecanemab is associated with adverse effects like infusion-related reactions and amyloid-related imaging abnormalities (ARIA) with edema or effusions42. with higher rates of these side effects occurring in individuals with the APOE4 allele43. After 18 months of administration of lecanemab, ARIA occurrences decrease along with dosage in non carriers of APOE444. Additionally, lecanemab may have reduced efficacy in women45. Early-stage AD patients are also treated with donanemab, a humanized monoclonal IgG1 antibody that binds to a modified form of Aβ protein, N3pE-Aβ, found in Aβ plaques (Figure 2). In a randomized clinical trial that included 1736 participants with early symptomatic Alzheimer disease and amyloid and tau, individuals that were administered donanemab showed significantly slowed disease progression and improved cognition compared to placebo groups. Groups treated with donanemab had a -6.02 change in score in the integrated Alzheimer Disease Rating Scale, while placebo groups had a change of -9.27, with lower scores indicating worse cognitive function46. Long term administration was associated with ARIA, with higher rates in individuals with the APOE4 allele47. Although lecanemab and donanemab have demonstrated efficacy clinically, it is important to note that Aβ antibody treatments are considered to be expensive medications and therefore may not be an accessible or affordable treatment option for all early-stage AD patients.
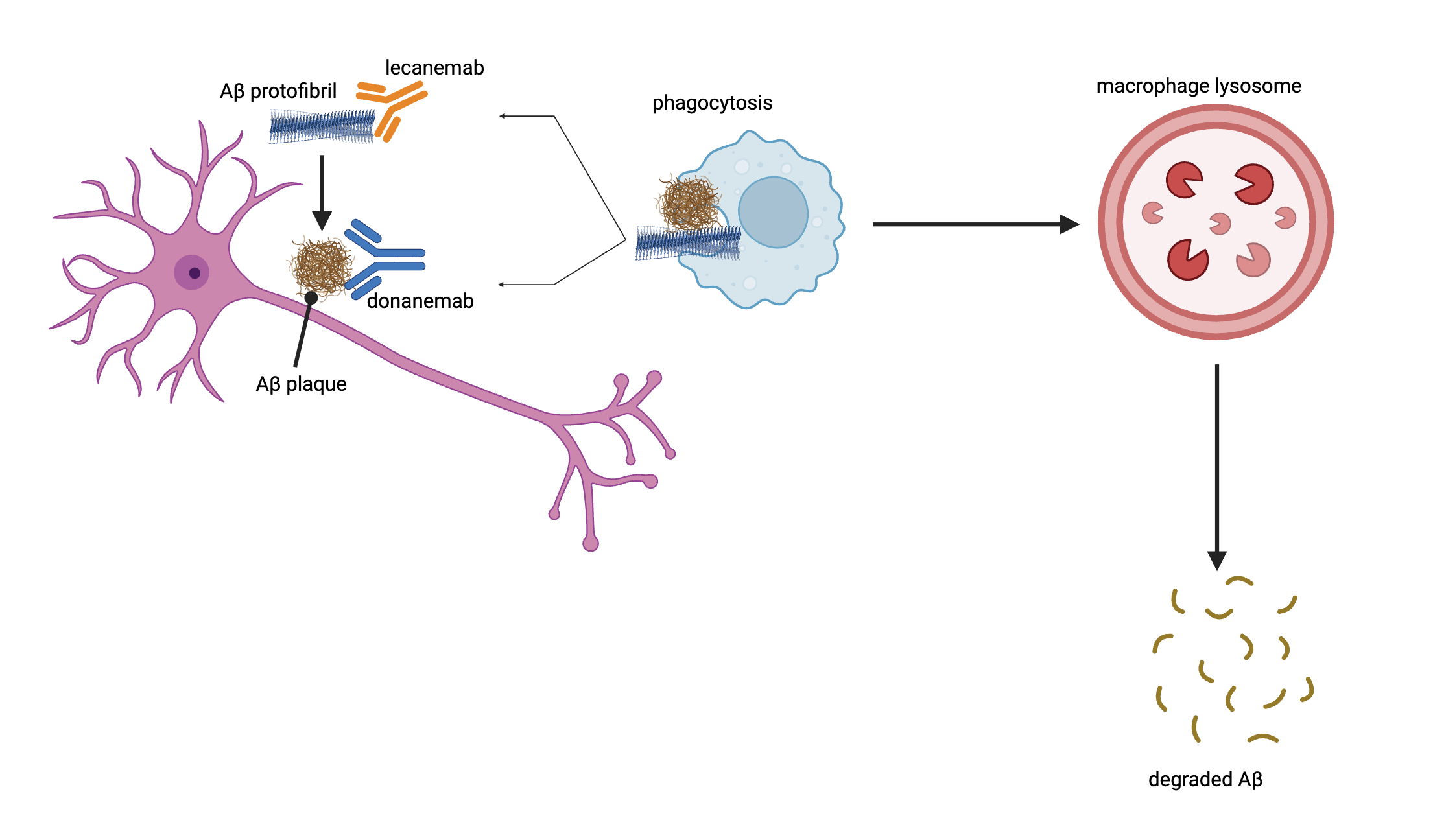
Aβ targeting antibodies, such as donanemab and lecanemab, lead to the degradation of Aβ. Lecanemab binds to the soluble Aβ protofibril before it constitutes Aβ plaque. Donanemab binds to the Aβ plaque itself. Macrophages recognize these antibodies and engulf the Aβ protofibril or the Aβ plaque. It is then degraded in the lysosome of the macrophage, helping clear Aβ build up48,49.
Cholinesterase inhibitors have also been FDA-approved and are currently being prescribed to early-stage AD patients. However, these drugs do not target the protein biomarkers associated with the neuropathology of AD. Rather, they function by preventing the breakdown of acetylcholine by inhibiting acetylcholinesterase50. Cholinesterase inhibitors have not been guaranteed to slow disease progression and are instead intended to alleviate cognitive symptoms of AD by helping to restore excitatory neuron function51. There are currently three FDA-approved cholinesterase inhibitors for AD: donepezil, galantamine, and rivastigmine. Donepezil is a cholinesterase inhibitor approved for all stages of AD that has shown to enhance cognitive abilities particularly in early-stage AD patients52. Galantamine is another cholinesterase inhibitor that is safe and tolerable in early and moderate AD patients, and is available in the form of oral tablets. AD patients given galantamine showed higher cognitive scores than placebo groups53. However, there is no distinct evidence demonstrating that galantamine is effective in patients with MCI or early-stage AD54. Rivastigmine is available in oral forms and through a transdermal patch intended for patients with early to moderate AD. When tested on patients with early-stage AD with rivastigmine showed slight improvement in cognitive performance55. Although these drugs can potentially improve MCI in early AD patients, they do not target Aβ and tau protein aggregation, allowing for continued progression of AD.
In addition to drugs, several behavioral therapies are currently being used in early-stage AD patients. Cognitive stimulation therapy (CST) is a form of behavioral therapy used for early-stage AD and dementia patients to maintain cognitive function and slow cognitive decline56. A study on men with mild to moderate dementia to test the overall effectiveness of CST shows that mild dementia groups receiving CST treatment maintained Mini-Mental State Examination (MMSE) and AD Assessment Scale scores, while groups not receiving the treatment had slight decreases in these scores over the period of the study57. Additionally, cognitive rehabilitation (CR) is also used as a form of behavioral therapy for early-stage AD patients. CR typically involves the practice of performing specific tasks to improve certain cognitive functions. Early-stage AD patients receiving CR treatment showed significant improvements in quality of life and in the orientation subscale of the MMSE when compared to the control group not receiving the treatment58.
Future early intervention strategies
In addition to FDA-approved drugs that are currently used, several drugs are undergoing development for future treatment of early-stage AD. Because tauopathy is a characteristic of early-stage AD, it is being targeted by various immunotherapy methods for early AD patients. Vaccines aim to induce antibodies that can clear tau protein aggregates. AADvac1is a vaccine intended to produce antibodies that target the microtubule-binding region of tau in patients with increased plasma phosphorylated-tau217 (p-tau) levels. Phase I trial results have shown that AADvac1 is safe and tolerable by patients. Phase II trials indicate that the drug can reduce tau levels; however, overall cognitive function remains unimpacted in AD patients, and further research is required to advance this drug as an early treatment for AD59. Another vaccine, ACI-35.030, is engineered to stimulate antibody production against p-tau. It has undergone phase II trials and has shown to be safe when tested on patients AD with MCI. The efficacy of this vaccine is still being tested, however it was able to activate the immune system of subjects between 50 and 75 years with MCI due to AD or mild AD with an antibody response against p-Tau60. Virus-like particle (VLP) based immunotherapy vaccines can be used to target p-tau in early AD patients. One of these vaccines, Qβ-PHF1, targets the phosphorylation sites Ser396 and Ser404. Qβ-PHF has been shown to diminish cognitive deficits and reduce soluble and insoluble p-tau and reactive microgliosis in rTg4510 mice61. In addition to vaccines, tau aggregation inhibitors are also on the horizon for early AD interventions by directly targeting tauopathy. For example, hydromethylthionine mesylate has been shown to prevent tau aggregation and disaggregate existing NFTs in early-stage AD patients in several phase III trials. This drug can be administered orally and does not present any major safety concerns or adverse effects when taken in low dosages62. Another tau aggregation inhibitor, curcumin has shown to reduce soluble tau protein and prevent the formation of NFTs. Using light scattering analysis, it was found that curcumin can bind to adult tau and fetal tau with dissociation constants of 3.3±0.4 and 8±1 μM, respectively, and inhibited the formation of tau oligomers63.
New drugs in early phases of clinical trials may hold promising results for the slowing of amyloid plaque formation. Remternetug, an experimental monoclonal antibody that targets the pyroglutamate-modified Aβ component of amyloid plaques present in AD, is currently under phase III development. In a phase 1 trial in AD patients with mild to moderate dementia, remternetug was able to reduce Aβ plaques significantly64.
New interventions on the horizon such as antisense oligonucleotides (ASOs) are being researched. ASOs target RNA transcripts and modify them, which can reduce levels of toxic tau and Aβ protein. ASOs, when tested on cortical neurons differentiated from human induced pluripotent stem cell lines with three copies of the amyloid precursor protein (APP) gene, showed significant decrease in APP65. A tau-targeting ASO, MAPTRx, inhibits expression of the microtubule associated protein tau gene. When intrathecal bolus administrations were given to patients with mild AD, dose dependent reduction of total tau levels in the CSF showed to be greater than 50% compared to baseline, 24 weeks after the final dose was administered66. This significant reduction in tau levels is promising and demonstrates a need for additional research in RNA therapy to intervene in early-stage AD. Early-stage interventions have proven to be successful in reducing levels of AD protein biomarkers such as Aβ and tau. This indicates a need for further development of drugs and therapies to treat AD in its earlier stages, as these interventions target neuropathological causes rather than symptoms of AD.

This table shows various AD interventions, which stage of AD they are used for, their status in the drug approval process and their overall efficacy.
Discussion
Early appearance and aggregation of Aβ and tau aggregates in the neural tissue of AD patients suggests early interventions targeting and blocking these AD biomarkers may be effective in preventing AD progression. Medications that treat late-stage AD provide only symptomatic relief rather than curing or slowing disease progression. Some current early interventions are less effective in early-stage AD patients and target symptomatic relief, such as cholinesterase inhibitors and behavioral therapies. Early interventions such as Aβ targeting antibodies can slow disease progression successfully. However, these antibody interventions are associated with adverse effects especially in individuals with the APOE4 allele. Furthermore, future interventions that are currently under development targeting Aβ and tau aggregates hold promising results in slowing AD progression.
Practitioners should utilize screening techniques such as PET scans and new blood tests, which have higher affordability and accessibility, as ways to detect abnormal Aβ and tau in the CSF and plasma, as potential biomarkers for AD in pre-symptomatic and MCI patients. The administration of Aβ monoclonal antibodies, although expensive, is also advisable to slow cognitive decline in early AD patients.
The ability for new pharmacological interventions to slow AD progression has vast implications for the future of AD treatment. Therapies which target the neuropathological biomarkers of AD, unlike previous symptom-targeting treatments, could improve the lives of AD patients by slowing the onset of cognitive and physical symptoms in late-stage AD. Additionally, slowing disease progression reduces burdens on caregivers and reduces societal costs associated with advanced stages of AD significantly67, furthering the need for early interventions.
Studies included in this review are largely conducted in western cohorts, particularly in Europe and North America. Although these studies have contributed largely to the understanding of AD and early interventions, it is important to consider that they may not entirely capture the diversity of variables such as age, sex, and ethnicity might influence treatment response and biomarker profiles. Because a majority of AD research is conducted in western cohorts due to higher resource availability and funding organizations, future research can aim to be more inclusive by engaging all populations affected by AD to assess the effects and benefits of early-stage AD treatments in relation to diverse demographic factors68.
Due to the potential societal and individual benefits of slowing AD progression through the developments of various biomarker-targeting early interventions, more research should be conducted in this area. Further research regarding antibodies which bind to soluble Aβ can stimulate the creation of new monoclonal antibodies that can degrade Aβ aggregates before plaque formation occurs in the neural tissue and cerebral vasculature, with fewer adverse effects than current treatments. Additionally, more research on tau aggregation and its appearance in early-stage AD can help create new vaccines, aggregation inhibitors and antibodies targeting tau tangles. ASOs which target RNA transcripts rather than the proteins themselves can also work to reduce tau and Aβ dysregulation in AD patients. Further research will allow for the creation of new intervention therapies that target protein build up associated with early-stage AD. Other modalities in early development such as stem cell therapies and gene therapies are on the horizon for the treatment of AD in early stages. Stem cell therapies have the ability to replace damaged neurons and target neuroinflammation to reverse AD progression69. Gene therapies can alter genes involved in AD pathology to possibly prevent or slow disease onset70. These advancements have the potential to ultimately slow AD progression, providing AD patients with new opportunities to restore cognitive function and improve overall quality of life.
Acknowledgments
I would like to thank Yoo Jin Jung for her guidance and mentorship throughout my writing process. I would also like to thank Anustup Garai and Gabriel Ortiz for their helpful feedback and commentary.
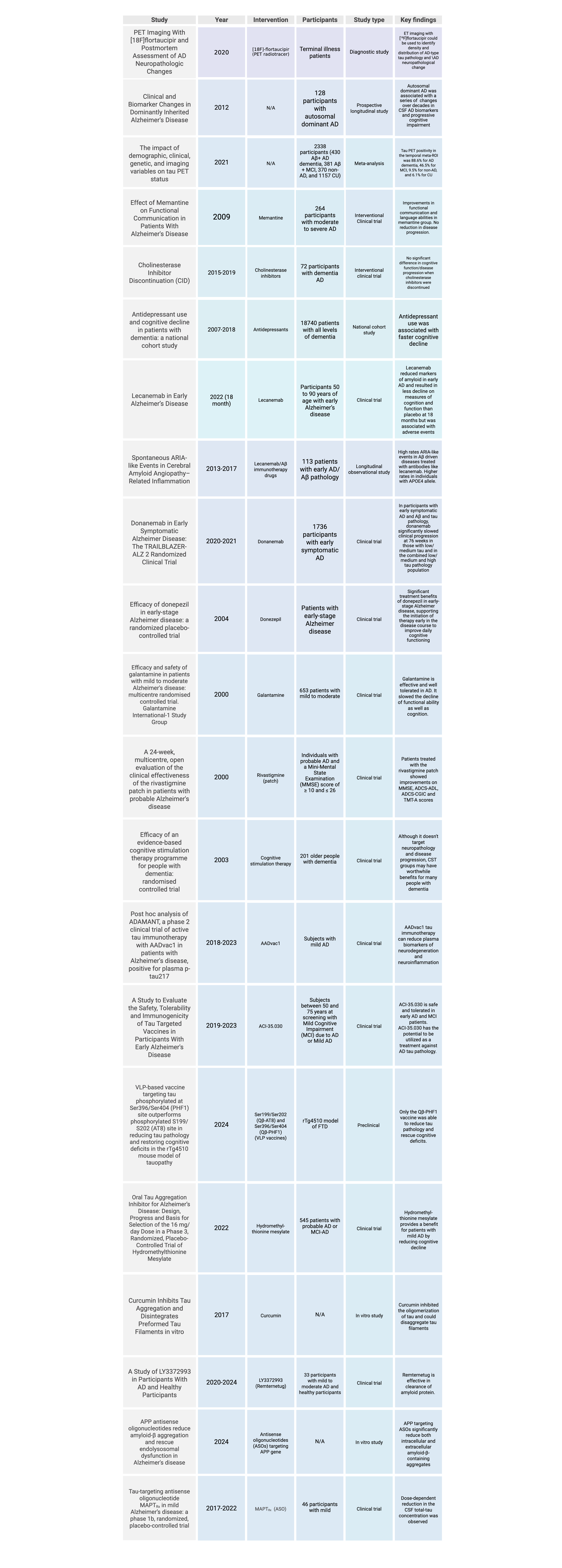
This table includes all preclinical and clinical trials, observational studies, longitudinal studies and meta-analysis included in the review. The study name along with year, intervention, participant information, type of study and key findings are mentioned.
References
- Reisberg, B., Ferris, S. H., De Leon, M. J. & Crook, T. The global deterioration scale for assessment of primary degenerative dementia. American Journal of Psychiatry 139, https://doi.org/ 10.1176/ajp.139.9.1136 (1982). [↩]
- Kumar A, S. J. L. F. et al. Alzheimer Disease. (Treasure Island (FL): StatPearls Publishing, 2024). [↩]
- Hardy, J. & Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends in Pharmacological Sciences vol. 12 Preprint at https://doi.org/10.1016/0165-6147(91)90609-V (1991). [↩]
- Wischik, C. M. et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85, https://doi.org/10.1073/pnas.85.12.4506 (1988). [↩]
- Goel, P. et al. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Frontiers in Molecular Neuroscience vol. 15 Preprint at https://doi.org/10.3389/fnmol.2022.937133 (2022). [↩] [↩]
- Zhang, H. et al. Cellular response to β-amyloid neurotoxicity in Alzheimer’s disease and implications in new therapeutics. Animal Models and Experimental Medicine vol. 6 Preprint at https://doi.org/10.1002/ame2.12313 (2023). [↩] [↩]
- Espay AJ, K. K. H. K. Lecanemab and Donanemab as Therapies for Alzheimer’s Disease: An Illustrated Perspective on the Data. eNeuro. https://doi.org/10.1523/ENEURO.0319-23.2024 (2024). [↩]
- Monteiro, A. R., Barbosa, D. J., Remião, F. & Silva, R. Alzheimer’s disease: Insights and new prospects in disease pathophysiology, biomarkers and disease-modifying drugs. Biochemical Pharmacology vol. 211 Preprint at https://doi.org/10.1016/j.bcp.2023.115522 (2023). [↩]
- Lue, L. F. et al. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. American Journal of Pathology 155, https://doi.org/10.1016/s0002-9440(10)65184-x (1999). [↩]
- Shaw, L. M. et al. Cerebrospinal fluid biomarker signature in alzheimer’s disease neuroimaging initiative subjects. Ann Neurol 65, https://doi.org/10.1002/ana.21610 (2009). [↩]
- Metaxas, A. & Kempf, S. J. Neurofibrillary tangles in Alzheimer’s disease: Elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen Res 11, https://doi.org/0.4103/1673-5374.193234 (2016). [↩]
- Sunderland, T. et al. Longitudinal stability of CSF Tau levels in Alzheimer patients. Biol Psychiatry 46, https://doi.org/10.1016/s0006-3223(99)00143-2 (1999). [↩]
- Chen, Y. R. et al. Diagnostic accuracy of blood biomarkers for Alzheimer’s disease and amnestic mild cognitive impairment: A meta-analysis. Ageing Research Reviews vol. 71 Preprint at https://doi.org/10.1016/j.arr.2021.101446 (2021). [↩]
- Mattsson, N. et al. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol 74, https://doi.org/10.1001/jamaneurol.2016.6117 (2017). [↩]
- Giacomucci, G. et al. Plasma neurofilament light chain as a biomarker of Alzheimer’s disease in Subjective Cognitive Decline and Mild Cognitive Impairment. J Neurol 269, https://doi.org/10.1007/s00415-022-11055-5 (2022). [↩]
- Mattsson, N. et al. CSF biomarker variability in the Alzheimer’s Association quality control program. Alzheimer’s and Dementia 9, https://doi.org/10.1016/j.jalz.2013.01.010 (2013). [↩]
- Therriault, J. et al. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat Aging 2, https://doi.org/10.1038/s43587-022-00204-0 (2022). [↩] [↩]
- Krishnadas, N., Villemagne, V. L., Doré, V. & Rowe, C. C. Advances in Brain Amyloid Imaging. Seminars in Nuclear Medicine vol. 51 Preprint at https://doi.org/10.1053/j.semnuclmed.2020.12.005 (2021). [↩]
- Fleisher, A. S. et al. Positron Emission Tomography Imaging with [18F]flortaucipir and Postmortem Assessment of Alzheimer Disease Neuropathologic Changes. JAMA Neurol 77, https://doi.org/10.1001/jamaneurol.2020.0528 (2020). [↩]
- FDA. FDA Clears First Blood Test Used in Diagnosing Alzheimer’s Disease. (2025). [↩]
- Jansen, W. J. et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA – Journal of the American Medical Association 313, https://doi.org/10.1001/jama.2015.4668 (2015). [↩]
- Dubois, B. et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. The Lancet Neurology vol. 13 Preprint at https://doi.org/10.1016/S1474-4422(14)70090-0 (2014). [↩]
- Salvadó, G. et al. Disease staging of Alzheimer’s disease using a CSF-based biomarker model. Nat Aging 4, https://doi.org/10.1038/s43587-024-00599-y (2024). [↩]
- Bateman, R. J. et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. New England Journal of Medicine 367, https://doi.org/10.1056/NEJMoa1202753 (2012). [↩]
- Lasagna‐Reeves, C. A. et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. The FASEB Journal 26, https://doi.org/10.1096/fj.11-199851 (2012). [↩]
- Ossenkoppele, R. et al. The impact of demographic, clinical, genetic, and imaging variables on tau PET status. Eur J Nucl Med Mol Imaging 48, https://doi.org/10.1007/s00259-020-05099-w (2021). [↩]
- Hofmann, A. , H. L. M. , L. M. et al. Comparative neurofilament light chain trajectories in CSF and plasma in autosomal dominant Alzheimer’s disease. Nat Commun https://doi.org/10.1038/s41467-024-52937-8 (2024). [↩]
- Wybitul M, B. A. L. N. H. C. T. V. G. A. Trajectories of amyloid beta accumulation – Unveiling the relationship with APOE genotype and cognitive decline. Neurobiol Aging 44–53 https://doi.org/10.1016/j.neurobiolaging.2024.03.007 (2024). [↩]
- Rother, C. et al. Experimental evidence for temporal uncoupling of brain Aβ deposition and neurodegenerative sequelae. Nat Commun 13, https://doi.org/10.1038/s41467-022-34538-5 (2022). [↩]
- Frontzkowski, L. et al. Earlier Alzheimer’s disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat Commun 13, https://doi.org/10.1038/s41467-022-32592-7 (2022). [↩]
- Balázs, N., Bereczki, D. & Kovács, T. CHOLINESTERASE INHIBITORS and MEMANTINE for the TREATMENT of ALZHEIMER and NON-ALZHEIMER DEMENTIAS. Ideggyogy Sz 74, https://doi.org/10.18071/isz.74.0379 (2021). [↩] [↩]
- Xia, P., Chen, H. S. V., Zhang, D. & Lipton, S. A. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. Journal of Neuroscience 30, https://doi.org/10.1523/JNEUROSCI.2488-10.2010 (2010). [↩]
- Effect of Memantine on Functional Communication in Patients With Alzheimer’s Disease. NCT00469456 (2009). [↩]
- Colovic, M. B., Krstic, D. Z., Lazarevic-Pasti, T. D., Bondzic, A. M. & Vasic, V. M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr Neuropharmacol 11, https://doi.org/10.2174/1570159X11311030006 (2013). [↩]
- Cholinesterase Inhibitor Discontinuation (CID). NCT02248636 (2020). [↩]
- Arbus, C. et al. Antidepressant use in Alzheimer’s disease patients: Results of the REAL.FR cohort. Int Psychogeriatr 22, https://doi.org/10.1017/S1041610209990780 (2010). [↩]
- Mo, M. , A. T. , H. M. T. et al. Antidepressant use and cognitive decline in patients with dementia: a national cohort study. BMC Med https://doi.org/10.1186/s12916-025-03851-3 (2025). [↩]
- Cardenas, V., Rahman, A., Zhu, Y. & Enguidanos, S. Reluctance to Accept Palliative Care and Recommendations for Improvement: Findings From Semi-Structured Interviews With Patients and Caregivers. American Journal of Hospice and Palliative Medicine 39, https://doi.org/10.1177/10499091211012605 (2022). [↩]
- Fernández, M., Gobartt, A. L. & Balañá, M. Behavioural symptoms in patients with Alzheimer’s disease and their association with cognitive impairment. BMC Neurol 10, https://doi.org/10.1186/1471-2377-10-87 (2010). [↩]
- Wang, C. et al. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Frontiers in Immunology vol. 14 Preprint at https://doi.org/10.3389/fimmu.2023.1117172 (2023). [↩]
- Rafii, M. & A. P. Amyloid-lowering immunotherapies for Alzheimer disease: current status and future directions. https://doi.org/10.1038/s41582-025-01123-5 (2025). [↩]
- van Dyck, C. H. et al. Lecanemab in Early Alzheimer’s Disease. Supplementary data. N Engl J Med https://doi.org/10.1056/NEJMoa2212948 (2022). [↩]
- Cummings, J. et al. Lecanemab: Appropriate Use Recommendations. Journal of Prevention of Alzheimer’s Disease vol. 10 Preprint at https://doi.org/10.14283/jpad.2023.30 (2023). [↩]
- Antolini, L. et al. Spontaneous ARIA-like Events in Cerebral Amyloid Angiopathy–Related Inflammation. Neurology 97, https://doi.org/10.1212/WNL.0000000000012778 (2021). [↩]
- Andrews D, D. S. C. H. S. M. C. D. The higher benefit of lecanemab in males compared to females in CLARITY AD is probably due to a real sex effect. Alzheimer’s Association https://doi.org/10.1002/alz.14467 (2025). [↩]
- Sims, J. R. et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 330, https://doi.org/10.1001/jama.2023.13239 (2023). [↩]
- Pereira da Silva AM, F. L. V. F. et al. Efficacy and APOE ε4-stratified risk of donanemab in Alzheimer’s disease: A systematic review and meta-analysis of randomized clinical trials. Journal of Alzheimer’s Disease https://doi.org/10.1177/13872877251361044 (2025). [↩]
- Zhao, L. et al. Macrophage-mediated degradation of β-amyloid via an apolipoprotein e isoform-dependent mechanism. Journal of Neuroscience 29, https://doi.org/10.1523/JNEUROSCI.5302-08.2009 (2009). [↩]
- Volloch, V. & Rits-Volloch, S. Effect of Lecanemab and Donanemab in Early Alzheimer’s Disease: Mechanistic Interpretation in the Amyloid Cascade Hypothesis 2.0 Perspective. Journal of Alzheimer’s Disease 93, https://doi.org/10.3233/JAD-230164 (2023). [↩]
- Tabet, N. Acetylcholinesterase inhibitors for Alzheimer’s disease: Anti-inflammatories in acetylcholine clothing! Age and Ageing vol. 35 Preprint at https://doi.org/10.1093/ageing/afl027 https://doi.org/10.1093/ageing/afl027 (2006). [↩]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Molecular Medicine Reports vol. 20 Preprint at https://doi.org/10.3892/mmr.2019.10374 https://doi.org/10.3892/mmr.2019.10374 (2019). [↩]
- Seltzer, B. et al. Efficacy of donepezil in early-stage Alzheimer disease: A randomized placebo-controlled trial. Arch Neurol 61, https://doi.org/10.1001/archneur.61.12.185 (2004). [↩]
- Wilcock, G. K., Lilienfeld, S. & Gaens, E. Efficacy and safety of galantamine in patients with mild to moderate Alzheimer’s disease: Multicentre randomised controlled trial. Br Med J 321, https://doi.org/10.1136/bmj.321.7274.1445 (2000). [↩]
- Lim AWY, S. L. L. C. Galantamine for dementia due to Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst Rev https://doi.org/10.1002/14651858.CD001747 (2024). [↩]
- A 24 Week, Multicenter, Open, Evaluation of the Clinical Effectiveness of the Once-daily 10 cm^2 Rivastigmine Patch Formulation in Patients With Probable Alzheimer’s Disease (EXTRA) (EXTRA). NCT00622713 (2011). [↩]
- Spector, A. et al. Efficacy of an evidence-based cognitive stimulation therapy programme for people with dementia: Randomised controlled trial. British Journal of Psychiatry 183, https://doi.org/10.1192/bjp.183.3.248 (2003). [↩]
- Carbone, E. et al. Cognitive Stimulation Therapy for Older Adults with Mild-to-Moderate Dementia in Italy: Effects on Cognitive Functioning, and on Emotional and Neuropsychiatric Symptoms. Journals of Gerontology – Series B Psychological Sciences and Social Sciences 76, https://doi.org/10.1093/geronb/gbab007 (2021). [↩]
- Kim, S. Cognitive rehabilitation for elderly people with early-stage alzheimer’s disease. J Phys Ther Sci 27, https://doi.org/10.1589/jpts.27.543 (2015). [↩]
- Kovacech B, C. N. N. P. H. J. K. E. K. S. P. V. F. M. V. J. F. H. W. B. S. E. V. E. Z. N. Post hoc analysis of ADAMANT, a phase 2 clinical trial of active tau immunotherapy with AADvac1 in patients with Alzheimer’s disease, positive for plasma p-tau217. Alzheimers Res Ther https://doi.org/10.1186/s13195-024-01620-7 (2024). [↩]
- A Study to Evaluate the Safety, Tolerability and Immunogenicity of Tau Targeted Vaccines in Participants With Early Alzheimer’s Disease. NCT04445831 (2025). [↩]
- Hulse J, M. N. P. J. C. B. B. K. Virus-like particle (VLP)-based vaccine targeting tau phosphorylated at Ser396/Ser404 (PHF1) site outperforms phosphorylated S199/S202 (AT8) site in reducing tau pathology and restoring cognitive deficits in the rTg4510 mouse model of tauopathy. bioRxiv https://doi.org/10.1101/2024.04.05.588338 (2024). [↩]
- Wischik, C. M. et al. Oral Tau Aggregation Inhibitor for Alzheimer’s Disease: Design, Progress and Basis for Selection of the 16 mg/day Dose in a Phase 3, Randomized, Placebo-Controlled Trial of Hydromethylthionine Mesylate. Journal of Prevention of Alzheimer’s Disease 9, https://doi.org/10.14283/jpad.2022.63 (2022). [↩]
- Rane, J. S., Bhaumik, P. & Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. Journal of Alzheimer’s Disease 60, https://doi.org/10.3233/JAD-170351 (2017). [↩]
- A Study of LY3372993 in Participants With Alzheimer’s Disease (AD) and Healthy Participants. NCT04451408 (2025). [↩]
- Hung C, F. E. L. F. K. D. P. R. APP antisense oligonucleotides reduce amyloid-β aggregation and rescue endolysosomal dysfunction in Alzheimer’s disease. Brain https://doi.org/10.1093/brain/awae092 (2024). [↩]
- Mummery, C. J. et al. Tau-targeting antisense oligonucleotide MAPTRx in mild Alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nat Med 29, https://doi.org/10.1038/s41591-023-02326-3 (2023). [↩]
- Chandler JM, Y. W. M. X. D. E. J. J. Potential Impact of Slowing Disease Progression in Early Symptomatic Alzheimer’s Disease on Patient Quality of Life, Caregiver Time, and Total Societal Costs: Estimates Based on Findings from GERAS-US Study. J Alzheimers Dis https://doi.org/10.3233/JAD-231166 (2024). [↩]
- Llibre-Guerra, J. J. et al. A call for clinical trial globalization in Alzheimer’s disease and related dementia. Alzheimer’s and Dementia 19, 3210–3221 https://doi.org/10.1002/alz.12995 (2023). [↩]
- Cao, Z. , K. F. , D. J. et al. Promoting Alzheimer’s disease research and therapy with stem cell technology. Stem Cell Res Ther https://doi.org/10.1186/s13287-024-03737-w (2024). [↩]
- Ataei, B. et al. A review of the advances, insights, and prospects of gene therapy for Alzheimer’s disease: A novel target for therapeutic medicine. Gene vol. 912 Preprint at https://doi.org/10.1016/j.gene.2024.148368 (2024). [↩]



{kind=link}